
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Mechanistic insights into encapsulation and release of drugs in colloidal niosomal systems: biophysical aspects†
Eva Judy a,
Manu Lopusb and
Nand Kishore*a
a,
Manu Lopusb and
Nand Kishore*a
aDepartment of Chemistry, Indian Institute of Technology Bombay, Powai, Mumbai, 400 076, India. E-mail: nandk@iitb.ac.in
bSchool of Biological Sciences, UM-DAE Centre for Excellence in Basic Sciences, University of Mumbai, Vidyanagari, Mumbai, 400 098, India
First published on 29th October 2021
Abstract
Vesicular systems such as niosomes provide an alternative to improve drug delivery systems. The efficiency of a drug delivery vehicle is strongly dependent on its components which decide its interaction with partitioned drug(s) and locus of site of partitioning. A quantitative understanding of the physical chemistry underlying partitioning of drugs in complex systems of self-assemblies such as niosomes is scarcely available. In order to obtain quantitative mechanistic insights into partitioning and release of drugs [mitoxantrone (MTX) and ketoprofen (KTP)] in systems of niosomes, we have employed ultrasensitive calorimetry, spectroscopy and microscopy to establish correlations between functionality and energetics which could provide guidance towards rational drug design and choice of suitable non-ionic surfactant-based drug delivery vehicles. Electron microscopy and dynamic light scattering (DLS) methods were used for characterization and assessing the morphology of niosomes. We present here a calorimetry-based approach in assessing the partitioning of the anticancer drugs mitoxantrone and ketoprofen in niosomes and their release to human serum albumin (HSA) employing isothermal titration calorimetry (ITC), differential scanning calorimetry (DSC) and comparison with equilibrium dialysis. The thermodynamic signatures and kinetics of release were analyzed to obtain insights into the role of the functional groups on the drugs in the partitioning process. The assessment of thermal and conformational stability of proteins during drug binding and the effect of drug delivery vehicles on proteins is also crucial. To assess these effects, DSC studies on HSA in the presence and absence of drugs and niosomes were also performed. Finally, the efficacy of the system to impact the cell viability of the MDA-MB-231 triple-negative breast carcinoma cell line was analysed using MTT assay.
1. Introduction
Drugs can be administered through oral, mucosal, transdermal or transepithelial pathways in the form of pills, capsules, ointments or injectables, or encapsulated in drug delivery systems (DDS).1–6 To overcome problems of solubility and adequate distribution, novel drug delivery carriers have been developed, such as niosomes, liposomes, microspheres, nanoparticles and microemulsions.7–11 An efficient drug delivery vehicle should be biocompatible, possess high encapsulation efficiency, and show characteristics of site-specific and controlled drug delivery with negligible leaching of drugs through the carrier.12–14Though liposomes having biodegradable characteristics have been explored extensively as drug delivery systems, their low stability in aqueous media limits their applicability.12,13,15 This has led to searches for alternate amphiphilic molecules which can form vesicular carriers. Although structurally similar to liposomes, niosomes have been reported to provide several advantages such as target-oriented drug delivery, increased transdermal drug delivery, and novel materials for drug delivery applications.16–18 Niosomes are macroscopic lamellar structures, generally unilamellar or multilamellar, based on their preparation method.19 Being amphiphilic, niosomes can encapsulate hydrophilic drugs in the core, and hydrophobic drugs within the bilayers. They can thus provide efficient encapsulation of both hydrophobic and hydrophilic drugs.20,21 Research efforts on niosomes have increased in the past decade due to their resemblance to liposomes and added advantage of increased physical and chemical stability in aqueous environments.22–24
The efficiency and drug delivery characteristics of niosomes is dependent strongly on the components that lead to their formation. The components of niosomes include non-ionic surfactants, a hydration medium, stabilizers and lipids (cholesterol). These additives are important in providing versatility,25 varied physical properties,26 mechanical strength and entrapment efficiency of niosomes.26,27 Further, niosomes with smaller size show enhanced permeation through inflamed and tumour cells with leaky vasculature and hence are used for target-oriented drug delivery.11
In spite of several such studies, the information on niosomes based drug delivery systems has remained qualitative in nature. Studies on effect of drug molecular weight on niosome size and encapsulation efficiency,28 and optimization of the process parameters for better encapsulation of drugs in niosomes29 have also enabled qualitative understanding. Loh et al.30 reviewed the use isothermal titration calorimetry (ITC) in understanding association of surfactants with polymers quantitatively, which is important in obtaining mechanistic insights into interactions. However, quantitative correlations of properties of self-assemblies with those of drugs being encapsulated in terms of their functionalities so far has mainly been limited to simple micelles.31–34 Such correlations are expected to guide selection of suitable systems in developing target-oriented drug delivery vehicles. Specifically, energetics of interactions provide role of functionality in meeting these targets. As a further step in understanding encapsulation and release properties of drugs in more complex but effective drug delivery systems, in this work we have used niosomes. Span 60 (sorbitan monostearate) has been used to prepare niosomes due to their low hydrophilic–lipophilic balance and high encapsulation efficiency, leading to the formation of small unilamellar vesicles ranging from 50 to 200 nm. Niosomes were also prepared encapsulating drugs (anti-cancer drug, mitoxantrone and anti-inflammatory drug, ketoprofen) to assess their target-oriented delivery.
The choice of suitable drug delivery systems requires a thorough understanding of role of functionality on both the drug as well as the delivery vehicle. To obtain quantitative insights into partitioning and release of drugs in niosomes, we have employed ultrasensitive calorimetry, spectroscopy and microscopy to establish structure–function–energetics relationships. A new experimental protocol is explored using ITC and monitoring interaction of the released drug from niosomes with transport protein human serum albumin (HSA). The thermodynamic signatures and kinetics of release were analyzed to obtain insights into the role of functional groups present on drugs in the partitioning process. The assessment of thermal and conformational stability of protein during drug binding and the effect of drug delivery vehicles on its structural integrity is also crucial. Differential scanning calorimetric studies of HSA in the presence and absence of drugs and niosomes were also performed to assess changes in thermal and conformational stability the protein. The effect of the vesicular assembly and drug release from niosomes was also assessed on MDA-MB-231 breast cancer cells to check viability of the systems in vitro. A comprehensive quantitative and qualitative study has been done on the formation and applicability of non-ionic vesicular systems for effective drug delivery of the chosen anti-cancer and anti-inflammatory drugs.
2. Materials and methods
2.1. Materials
Sorbitan monostearate (Span 60 abbreviated here as S60 (>98%)) was purchased from TCI Chemicals, Japan. Human serum albumin (HSA, (>97%)), dicetyl phosphate (>97%), disodium hydrogen phosphate (>99%), sodium chloride (>98%), potassium chloride (>99.5%), potassium dihydrogen phosphate (>99%), mitoxantrone (MTX, (>97%)) and ketoprofen (KTP, (>98%)) were procured from Sigma Aldrich Co. USA, whereas cholesterol (>99%), polyethylene glycol (PEG 4000, (>95%)) and solvents (chloroform and methanol (>99.8%)) were purchased from SRL Pvt. Ltd. India (Fig. 1). The hydration of niosomes and preparation of stock solutions of the drugs and protein were done in 20 mM phosphate buffer saline (PBS) at pH 7.4. The protein solutions were subjected to extensive dialysis at 4 °C with a minimum of three changes followed by determining its concentration on a Jasco V-550 double beam spectrophotometer using an extinction coefficient corresponding to A2801% = 5.8.35 The weight measurements were done on a Sartorius BP 211D digital weighing balance with a readability of 0.01 mg.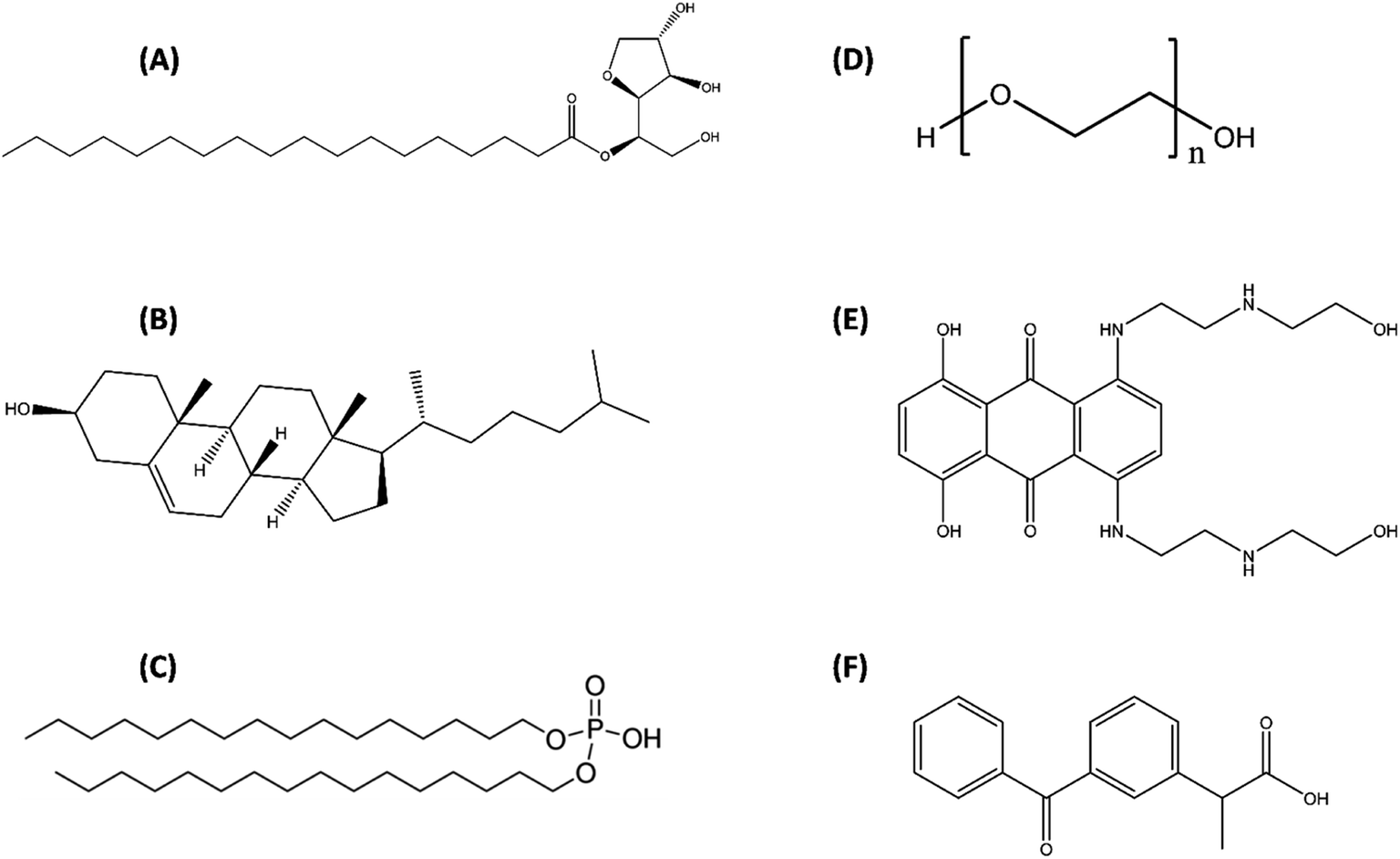 | ||
| Fig. 1 Structures of components used for preparation of niosomes (A) sorbitan monostearate (Span 60), (B) cholesterol, (C) dihexadecyl phosphate, (D) polyethylene glycol and drugs incorporated in niosomes (E) mitoxantrone (MTX) and (F) ketoprofen (KTP). | ||
2.2. Niosome preparation
The niosomes were prepared by the thin-film hydration method. In the first step, S60 and cholesterol were dissolved in a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 mol% ratio, followed by a negative stabilizer, hexadecyl phosphate, added in the ratio of 0.05 mol%. The concentration of S60 in the niosomes was maintained at 10 mM, which is higher than its CMC.36 In the formulations involving PEG, 1 mol% of the polymer was added to the vesicles in the organic phase. The components were dissolved in chloroform: methanol mixture in a ratio of 9:1 v/v and then evaporated using a rotary evaporator. The second step involved the hydration of the thin layer of pre-niosomes using PBS. Initially, the components were hydrated using PBS at 60 °C for 30 minutes at a continuous stirring rate of 250 rpm, followed by hydration at room temperature for a minimum period of 24 hours. This hydration time and a constant stirring rate were maintained for all the niosomes under preparation.
:1 mol% ratio, followed by a negative stabilizer, hexadecyl phosphate, added in the ratio of 0.05 mol%. The concentration of S60 in the niosomes was maintained at 10 mM, which is higher than its CMC.36 In the formulations involving PEG, 1 mol% of the polymer was added to the vesicles in the organic phase. The components were dissolved in chloroform: methanol mixture in a ratio of 9:1 v/v and then evaporated using a rotary evaporator. The second step involved the hydration of the thin layer of pre-niosomes using PBS. Initially, the components were hydrated using PBS at 60 °C for 30 minutes at a continuous stirring rate of 250 rpm, followed by hydration at room temperature for a minimum period of 24 hours. This hydration time and a constant stirring rate were maintained for all the niosomes under preparation.
The drugs encapsulation was done during the second step of niosome preparation. The drugs MTX (1 mM) and KTP (5 mM) were added to the PBS buffer during rehydration and incorporated in the pre-niosomes. During hydration, depending on hydrophobic character, the drugs partition in the layers of the vesicles. The niosomes (in the absence and presence of drugs) were then subjected to vortexing and sonication for 30 min each to obtain vesicles of uniform size.37,38 The excess drugs were removed by dialysis before being subjected to analysis by biophysical methods. Span 60 niosomes and Span 60 with PEG niosomes were prepared, abbreviated as S60 and S60-PEG. The drug encapsulated systems were abbreviated as S60 + MTX, S60 + KTP, S60-PEG + MTX and S60-PEG + KTP. After preparation, the vesicles (in the presence and absence of drugs) were stored at 4 °C and used within ten days to avoid the complexities related to vesicle stability and drug release.
2.3. Characterization of niosomes
The characterization of niosomes prepared by thin-film hydration was done using electron microscopy and dynamic light scattering measurements. The techniques provide information on the shape, size, surface charge and distribution of the prepared niosomes.
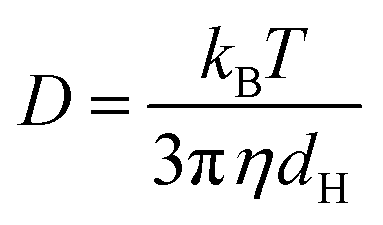 | (1) |
Here kB is the Boltzmann constant, T is the temperature in Kelvin, η is the viscosity of the solvent, and D is the translational diffusion coefficient.39 The zeta potential (ζ) determines the surface charge on the niosomes by calculating its electrophoretic mobility (μe), using the equation given below.
 | (2) |
Here η is the viscosity of solvent is, f(Ka)is Henry's function, ξr and ξ0 are the relative permittivity and permittivity in vacuum respectively.40
2.4. Drug encapsulation, release and interaction with HSA
The encapsulation and kinetics of drug release were studied based on thermodynamic parameters, giving an account of the drug that partitions into the empty niosome vesicles upon addition externally. These thermodynamic parameters can also account for the interactions taking place between the drug and the niosome bilayers. The drug partitioning experiments were designed for 25 injections, each releasing a volume of 10 μl into the cell, having a volume of 940 μl. Each injection had a duration of 20 s with an equilibration time of 240 s between two successive injections. The stirring rate in all the experiments was kept at 250 rpm to maintain the overall homogeneity of the solution. For each main experiment, respective dilution experiments were performed and subtracted to assess the final heats of interaction during the partitioning of the drugs in the niosomes.2.4.1.1. Drug release by spectroscopic methods. The drug release was studied using the dialysis method (membrane cut off: 12.5 kDa) and assessed by spectroscopic methods (absorbance and fluorescence), and further compared with the calorimetric observations. The absorbance and fluorescence measurements were performed on a Jasco V-550 double beam spectrophotometer and Cary Eclipse fluorimeter, respectively. The in vitro release studies were performed by sealing the drug encapsulated niosomes in a dialysis bag. The dialysis sac was washed and thoroughly cleaned using distilled water, followed by placing the drug encapsulated niosomal suspensions in it. The dialysis bags were then sealed and suspended into 50 ml of PBS buffer maintained at pH 7.4. The solutions were then placed in an incubator at 37.0 ± 0.5 °C and shaking at 100 rpm. The buffer samples were withdrawn at requisite intervals and replaced with fresh buffer solutions to maintain adequate sink conditions. The samples were then diluted and analysed at 234 nm and 610 nm using UV-visible spectroscopy and at 260 nm and 660 nm by fluorescence spectroscopy, respectively, for ketoprofen and mitoxantrone release. The cumulative percentage of drug release was calculated accordingly.
2.4.1.2. Drug release by ITC. To assess the kinetics and determine the thermodynamic parameters related to drug release, isothermal titration experiments were performed on Nano ITC obtained from TA Instruments, USA (injection volume: 250 μl, cell volume: 940 μl). The ITC experiments were designed for a total of 4 injections, each devolving a volume of 50 μl per injection. The cell was filled with PBS buffer, and the syringe was filled with drug encapsulated niosomes. The duration of the injection was kept at 20 s, but the interval between two successive injections was kept at 3600 s. The stirring rate was maintained at 250 rpm to maintain the homogeneity of the solution. The change in heat signatures provides information regarding the release of the encapsulated drugs in the niosomes into the buffer.
 and standard molar change in entropy
and standard molar change in entropy  . Through the modified ITC protocol to assess the kinetics of drug release, the associated values of rate constant (k) were also determined.
. Through the modified ITC protocol to assess the kinetics of drug release, the associated values of rate constant (k) were also determined.2.5. Drug encapsulation efficiency
The non-encapsulated drug was removed from the niosomal formulation by dialysis, followed by the analysis of the entrapment efficiency of niosomes (S60 and S60-PEG). The drug entrapment efficiency was calculated using the following equation.
 | (3) |
The entrapment efficiency of the niosomes for MTX and KTP was calculated by disrupting the drug containing vesicles with 90% ethanol and analysing the solution by absorbance spectroscopy at 610 nm for MTX and 260 nm for KTP, respectively.41
2.6. Differential scanning calorimetry (DSC)
The differential scanning calorimetric experiments were performed on a Nano DSC (cell volume of 300 μl) procured from TA Instruments, USA. A pressure of 3 bar and a scanning rate of 1 K min−1 were maintained in all the experiments. The thermal unfolding of HSA in the presence and absence of niosomes (with or without incorporated drugs) was done by DSC. The main experiments were rectified by subtracting baselines obtained at the same scan rate by filling both the cells with buffer or buffer with additive(s) (except protein). The baseline corrected DSC profiles were analysed using NanoAnalyse software, and the corresponding values for transition temperature (Tm) and calorimetric enthalpies (ΔHcal) were determined. The thermal transitions in protein were observed to be irreversible.2.7. Circular dichroism spectroscopy
The circular dichroism experiments were performed on a JASCO CD-180 spectropolarimeter to assess conformational changes occurring in the protein samples upon addition of niosomes, with or without encapsulated drugs. The secondary and tertiary structures of proteins were determined using far UV-CD (260 nm to 180 nm) and near UV-CD (360 nm to 260 nm), respectively. The concentrations of the protein and the path length maintained for far and near UV-CD spectra were 5 μM and 0.2 cm, respectively and 20 μM and 1 cm, respectively. The instrument was purged thoroughly using nitrogen gas during experiments to maintain an inert environment. The molar ellipticity of the protein [θ] was calculated using the following equation.
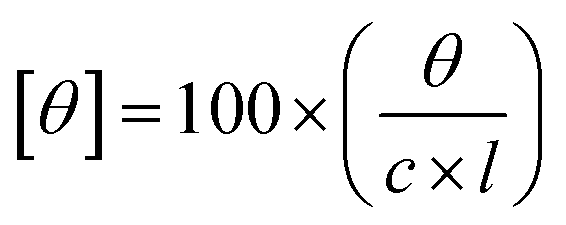 | (4) |
Here ‘c’ is concentration of the protein, ‘θ’ is specific rotation and ‘l’ is path length in cm.42
2.8. Cell viability
Human triple-negative breast cancer cells, MDA-MB-231, from American Type Culture Collection (ATCC, USA), were maintained in a humidified atmosphere with 5% CO2 at 37 °C in Dulbecco's Modified Eagle Medium (DMEM) supplemented with 10% fetal bovine serum (FBS) and 1% (penicillin (100 IU)/streptomycin (0.1 mg ml−1) solution). This aggressive cancer cell line was used to explore the biological activity of the vesicles alone and the drug-encapsulated vesicles. The cells were seeded at a density of 5000 cells per well in 96-well, surface-treated plates and allowed to adhere to the surface for 24 h. Subsequently, the medium was replaced with fresh medium containing the desired drug or vesicle of interest (MTX, KTP, S60 + MTX, S60 + KTP, S60-PEG + MTX, S60-PEG + KTP, S60 vesicle, or S60-PEG vesicle) for 72 h. The empty vesicles were prepared from non-ionic surfactants, polyethylene glycol (PEG) and cholesterol. After incubation of the cells for a period of three days, the cell viability was determined using an MTT assay.43 Briefly, MTT (3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl tetrazolium bromide) (0.5 mg ml−1 final concentration) was added to each well and incubated for 4 h at 37 °C. The MTT-containing media was then discarded, and 100 μL of dimethylsulfoxide (DMSO) was added to each well and incubated for 10 min. The absorbance of the samples was measured at 570 nm in a Tecan multimode reader (TECAN infinite 200 PRO, Tecan, Switzerland) and the percentage cell viability was calculated as described elsewhere.44,453. Results and discussion
3.1. Characterization of niosomes
The characterization of the prepared niosomes in the presence and absence of drugs was done by a combination of electron microscopy (TEM and SEM) and dynamic light scattering experiments (particle size measurements and zeta potential measurements) to assess the structural and morphological changes. The preparation of niosomes was done by the thin-film hydration method, leading to two variants of non-ionic surfactant vesicles with two encapsulated drugs, leading to a total of six systems under analysis.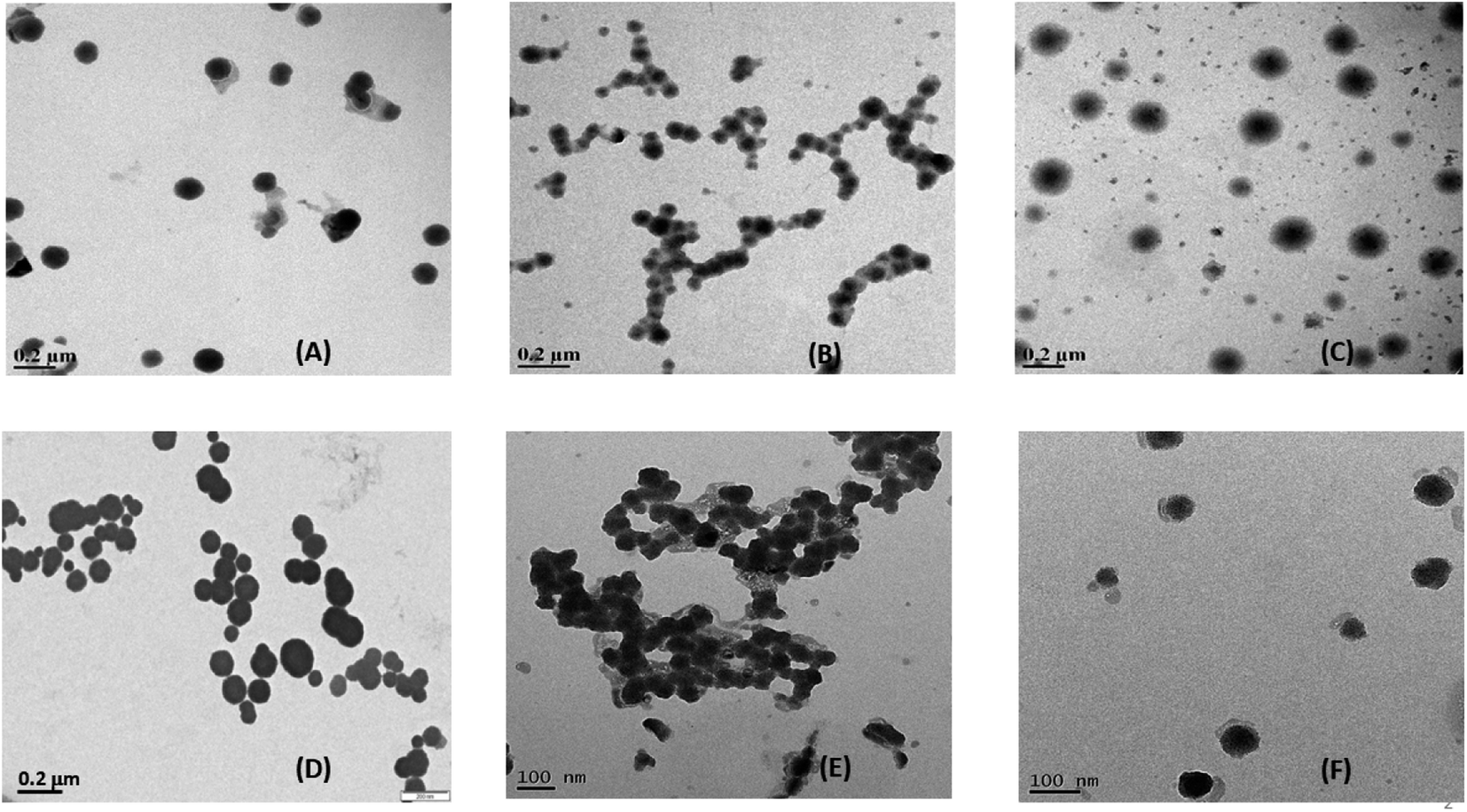 | ||
| Fig. 2 TEM images of (A) S60 niosomes, (B) S60 + MTX niosomes, (C) S60 + KTP niosomes, (D) S60-PEG niosomes, (E) S60-PEG + MTX niosomes and (F) S60-PEG + KTP niosomes. | ||
The particle size was analysed using imageJ software for the niosomes (in the presence and absence of drugs). The average particle size was observed to be ranging from 100–250 nm (Table S1†). The morphology of the niosomes was also assessed using scanning electron microscopy which indicated spherical niosomes with varied distribution (Fig. S1†).
| System | Hydrodynamic diameter (dH)/(nm) | Zeta potential/(mV) |
|---|---|---|
| Span 60 niosomes | 83.9–116.1 | −(45.99 ± 0.96) |
| (Span 60 + MTX) niosomes | 60.7–107.0 | −(38.71 ± 1.32) |
| (Span 60 + KTP) niosomes | 116.1–204.6 | −(47.90 ± 0.99) |
| (Span 60 + PEG) niosomes | 65.8–160.5 | −(44.23 ± 0.97) |
| (Span 60 + PEG + MTX) niosomes | 332.7–460.0 | −(39.26 ± 1.02) |
| (Span 60 + PEG + KTP) niosomes | 125.8–221.9 | −(48.40 ± 1.10) |
The zeta potential of the niosomes is observed to be negative in the presence and absence of drugs with an average value of −(45.99 ± 0.96) mV indicating the presence of anionic polar groups on the outer surface. This negative potential is also imparted due to the presence of a negative charge stabilizer, dicetyl phosphate which prevents the aggregation and clumping of the vesicles. Similar zeta potential values obtained for S60-PEG niosomes indicate that the polar ethylene groups are assimilated around the niosomes structure providing it stability rather than interacting with external components in the solvent environment.47 The stabilizer also plays an effect role in S60-PEG niosomes in imparting it stability and charge.
The incorporation of drugs in the niosomes causes minor alteration in zeta potential values. Similar zeta potential values of the drug incorporated niosomes indicate absence of drugs on the outer regions of the niosomes suggesting their effective incorporation into the bilayers. Thus, the drugs added during niosome preparation are incorporated into the bilayers of the niosome, depending on their hydrophilic or hydrophobic characteristics and also functionality. This encapsulation of drugs protects it from the harsh external environment and also helps in sustained release depending on the sink conditions.
3.2. Partitioning of drugs in niosomes
As a first step, partitioning of drugs in empty niosomes was explored. ITC experiments were done to study the partitioning of MTX and KTP into niosomes without any preloading. Fig. S3(A) and (B)† represent ITC profiles accompanying titration of 3.5 mM MTX and 7.5 mM KTP into S60 and S60-PEG niosomes at 37 °C. The profiles correspond to titration experiments performed before and after a period of seven days. The experiments were designed to assess the difference in the partitioning of drugs, if niosomes show structural modifications after 7 days. The values of partitioning constant observed for the titration of drugs into S60 and S60-PEG niosomes are of the order of 103 and 104, respectively which indicate weak to moderate encapsulation of the drugs into these self-assemblies. The partitioning of MTX in S60 niosomes show weak two site interaction as analysed by employing independent site models. Other models provided large fitting errors. This indicates that even after a period of 7 days, S60 niosomes offer equivalent partitioning environment. Further insights into the nature of partitioning were assessed through a comparison of enthalpies of interaction. This indicates that even after 7 days, the structure of S60 niosome is not altered to a large extent as reflected by insignificant variation in values of drug partitioning. However, a comparison of the enthalpies of partitioning indicate difference in the nature of interaction between the drug moiety and the niosome structure. The values of enthalpies of partitioning of MTX in the two sites initially are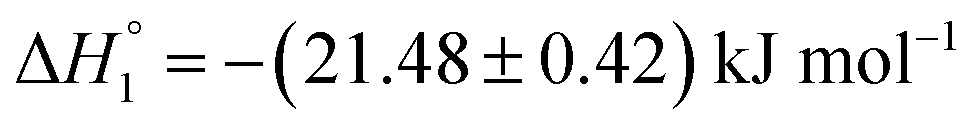 and
and 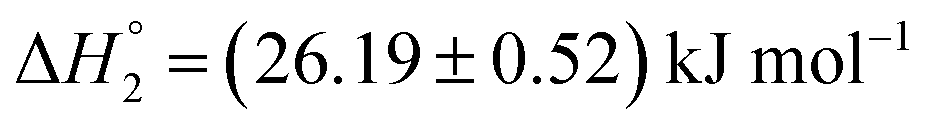 , respectively. When assessed after a period of 7 days, these values change to
, respectively. When assessed after a period of 7 days, these values change to 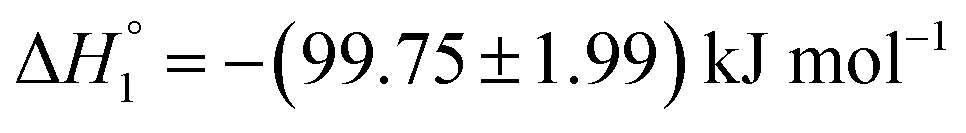 and
and 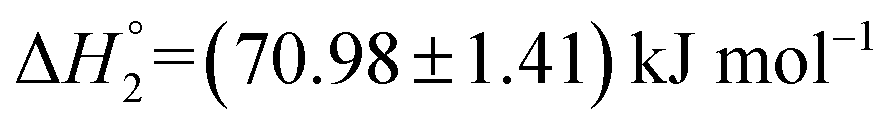 . This change in the enthalpic parameters indicates that the minor changes in the structure of the S60 niosomes leads to strengthened exothermic and endothermic interactions between the functional groups present on the drug and the exposed components of the niosome at both the binding events respectively.
. This change in the enthalpic parameters indicates that the minor changes in the structure of the S60 niosomes leads to strengthened exothermic and endothermic interactions between the functional groups present on the drug and the exposed components of the niosome at both the binding events respectively.
Spontaneous processes with an enthalpic penalty are always compensated by entropy gain. The interaction of MTX with S60-PEG niosomes shows a weak partitioning initially and also after a period of seven days. The enthalpies of partitioning do not have appreciable difference (Table 2), suggesting that the presence of PEG in the formulation, provides a structural compactness to the niosome even after period of 7 days. The partitioned drug interacts with the exposed sections of PEG and S60 in a similar manner even after 7 days as indicated by comparison of enthalpic and entropic values. The partitioning process of MTX in S60-PEG also shows enthalpic-entropic compensation. The trends of MTX partitioning in S60 and S60-PEG niosomes also varies as seen in the lower panel of the ITC profiles (Fig. S3†), which indicates a difference in mode of interaction of the drug. The partitioning of KTP was also observed to be quite weak (of the order of 103) with S60 and S60-PEG niosomes initially and also after a period of 7 days (Fig. S4†). The order of partitioning constant remains at 103, indicating a very weak surface interaction between KTP and surface groups of the niosomes.
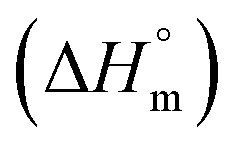 , change in standard molar entropy
, change in standard molar entropy  and stoichiometry (n) accompanying the partitioning of mitoxantrone and ketoprofen in empty S60 and S60-PEG niosomes at 37 °C
and stoichiometry (n) accompanying the partitioning of mitoxantrone and ketoprofen in empty S60 and S60-PEG niosomes at 37 °C
| System | K/(M−1) |
|
|
n |
|---|---|---|---|---|
| MTX in S60 (initial fresh preparation) | (0.66 ± 0.02) ×104 | −(21.48 ± 0.42) | −(3.9 ± 0.08) | 0.10 ± 0.00 |
| (0.40 ± 0.02) ×104 | (26.19 ± 0.52) | (153.5 ± 3.1) | 0.10 ± 0.00 | |
| MTX in S60 (after 7 days of preparation) | (0.10 ± 0.01) ×104 | −(99.75 ± 1.90) | −(264.2 ± 5.2) | 0.10 ± 0.00 |
| (0.17 ± 0.04) ×104 | (70.98 ± 1.41) | (291.1 ± 5.8) | 0.11 ± 0.00 | |
| MTX in S60-PEG (initial fresh preparation) | (1.03 ± 0.01) ×103 | −(100.0 ± 2.00) | −(277.7 ± 5.5) | 0.10 ± 0.00 |
| (1.45 ± 0.05) ×103 | (78.47 ± 1.56) | (323.8 ± 6.5) | 0.12 ± 0.00 | |
| MTX in S60-PEG (after 7 days of preparation) | (0.60 ± 0.07) ×105 | −(97.31 ± 1.94) | −(222.2 ± 4.4) | 0.10 ± 0.00 |
| (0.65 ± 0.01) ×105 | (95.77 ± 1.91) | (401.0 ± 8.0) | 0.10 ± 0.00 | |
| KTP in S60 (initial fresh preparation) | (1.00 ± 0.02) ×103 | −(100.0 ± 0.20) | −(265.0 ± 1.41) | 0.10 ± 0.00 |
| (1.02 ± 0.01) ×103 | (93.59 ± 1.87) | (359.6 ± 7.19) | 0.11 ± 0.00 | |
| KTP in S60 (after 7 days of preparation) | (1.00 ± 0.04) ×103 | −(81.93 ± 1.63) | −(206.7 ± 4.1) | 0.10 ± 0.00 |
| (1.04 ± 0.01) ×103 | (75.72 ± 1.51) | (301.9 ± 6.0) | 0.11 ± 0.00 | |
| KTP in S60-PEG (initial fresh preparation) | (1.00 ± 0.02) ×103 | −(100.0 ± 1.92) | −(265.0 ± 5.3) | 0.10 ± 0.00 |
| (1.03 ± 0.00) ×103 | (94.57 ± 1.89) | (362.6 ± 7.3) | 0.11 ± 0.00 | |
| KTP in S60-PEG (after 7 days of preparation) | (1.47 ± 0.05) ×103 | −(86.36 ± 1.72) | −(229.0 ± 4.6) | 0.10 ± 0.00 |
| (1.50 ± 0.01) ×103 | (84.00 ± 1.68) | (342.6 ± 6.9) | 0.10 ± 0.00 |
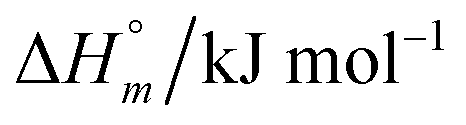
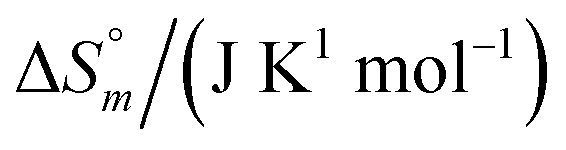
The partitioning of KTP in S60 and S60-PEG niosomes show an exothermic as well as an endothermic event as reported in Table 2. The interaction of KTP with S60 niosomes is accompanied with enthalpy change of  and
and  which are similar to those observed for the same system, when assessed after a period of 7 days
which are similar to those observed for the same system, when assessed after a period of 7 days 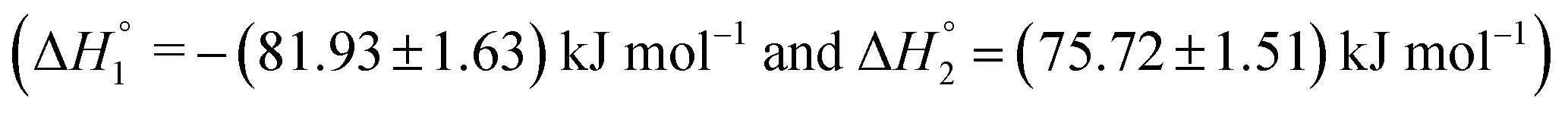 . This indicates that the incorporated drug interacts with the outer head regions as well as the inner hydrophobic tails thus indicating an exothermic and endothermic event. The extent of interaction of the drug with S60 niosomes did not increase after a period of 7 days as indicated by the partitioning constants and enthalpic values suggesting the stability and rigidity of the S60 niosomes even after 7 days. The partitioning of KTP in S60-PEG niosomes also follows similar trend as observed with S60 niosomes. The values of partitioning constant are of the order of 103, with either freshly prepared or even after a period of 7 days. The observed thermodynamic signatures also show no variation in the partitioning of drugs during the analysis (Table 2). This indicates that KTP does not partition extensively into the bilayers, leading to its partitioning on the surface of the niosomes. The exothermic enthalpy of partitioning indicates interaction between the polar –COOH groups and the head regions of Span 60 and PEG (in S60-PEG systems) whereas the endothermic enthalpy of interaction could be due to the interaction between the phenyl and hydrophobic components of Span 60 and cholesterol. Thus, the partitioning of the drug KTP on the niosomes occurs near the head regions rather than in the core of the niosomes. The ITC data for drug partitioning of MTX and KTP in the niosomes fits to two independent site models. The interaction of drugs with niosomes can yield exothermic as well as endothermic events depending on the mode and extent of partitioning. The first partitioning event indicates incorporation of the drug near the head regions of the niosome as suggested by the exothermic mode of interaction. The second interaction indicates encapsulation of the drug near the outer palisade region of the bilayer.
. This indicates that the incorporated drug interacts with the outer head regions as well as the inner hydrophobic tails thus indicating an exothermic and endothermic event. The extent of interaction of the drug with S60 niosomes did not increase after a period of 7 days as indicated by the partitioning constants and enthalpic values suggesting the stability and rigidity of the S60 niosomes even after 7 days. The partitioning of KTP in S60-PEG niosomes also follows similar trend as observed with S60 niosomes. The values of partitioning constant are of the order of 103, with either freshly prepared or even after a period of 7 days. The observed thermodynamic signatures also show no variation in the partitioning of drugs during the analysis (Table 2). This indicates that KTP does not partition extensively into the bilayers, leading to its partitioning on the surface of the niosomes. The exothermic enthalpy of partitioning indicates interaction between the polar –COOH groups and the head regions of Span 60 and PEG (in S60-PEG systems) whereas the endothermic enthalpy of interaction could be due to the interaction between the phenyl and hydrophobic components of Span 60 and cholesterol. Thus, the partitioning of the drug KTP on the niosomes occurs near the head regions rather than in the core of the niosomes. The ITC data for drug partitioning of MTX and KTP in the niosomes fits to two independent site models. The interaction of drugs with niosomes can yield exothermic as well as endothermic events depending on the mode and extent of partitioning. The first partitioning event indicates incorporation of the drug near the head regions of the niosome as suggested by the exothermic mode of interaction. The second interaction indicates encapsulation of the drug near the outer palisade region of the bilayer.
3.3. Drug encapsulation efficiency
The encapsulation efficiency (see eqn (3)) indicates the amount of drug entrapped in the niosome. It can also be mentioned as the drug retention capacity of the niosomes. During the formation of niosomes, drug is added in the rehydrating medium. After formation the untrapped drug still present in the niosome suspension is removed by dialysis for a period of one hour. The amount of encapsulated drug was calculated by measuring the absorbance of the drug in this dialysate buffer and subtracting it from the absorbance of the stock drug solution. The absorbance values were analysed at 610 nm for MTX and 234 nm for KTP, respectively. The encapsulation efficiency of MTX in S60 niosomes is observed to be (60 ± 5) % whereas for S60-PEG niosomes it is estimated to be (71 ± 5) %. The values of EE of KTP in S60 and S60-PEG niosomes are estimated to be (62 ± 5) % and (65 ± 5) %, respectively. It is observed that the amount of MTX encapsulated in S60-PEG is slightly more as compared to that in S60 niosomes whereas similar amount of KTP is encapsulated in both S60 and S60-PEG niosomes. As mentioned in Section 2.2, 1 mM MTX and 5 mM KTP were incorporated during encapsulation. The EE of drugs in the niosomes is statistically not that different but when we assess using the concentrations of the drug incorporated we observe that S60 incorporated 0.6 mM MTX and 3.1 mM KTP whereas S60 + PEG incorporates 0.7 mM MTX and 3.3 mM KTP. In effect, the amount of KTP incorporated by both the niosomal systems is 5 times more than that of MTX.3.4. Drug release
The drug release from the niosomes was studied using dialysis (assessed by specroscopic methods) and isothermal titration calorimetry.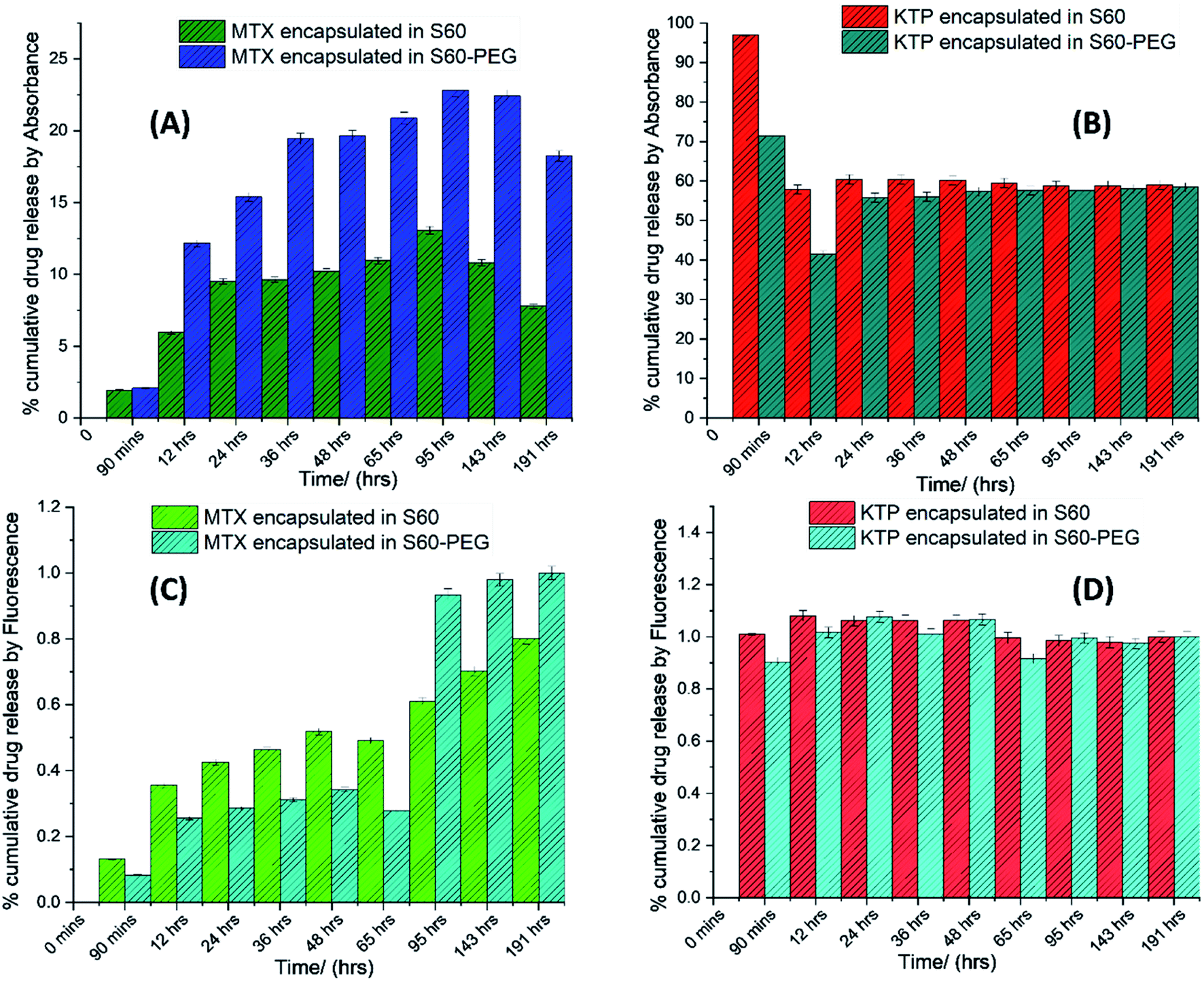 | ||
| Fig. 3 Percent cumulative drug release in (A) MTX incorporated S60 and S60-PEG niosomes, (B) KTP incorporated S60 and S60-PEG niosomes by UV-visible spectroscopy, and (C) MTX incorporated S60 and S60-PEG niosomes, (D) KTP incorporated S60 and S60-PEG niosomes by fluorescence spectroscopy. | ||
The representative cumulative plots (Fig. 3) shows the release of mitoxantrone and ketoprofen analysed using UV-visible and fluorescence spectroscopy. The release of MTX from S60 and S60-PEG niosomes is a gradual process which increases with time. It is observed that the concentration of MTX in the buffer increases in a sustained manner with time. This can be related to the concentration and position of the encapsulated drug in the bilayer of the niosomes. In case of KTP incorporated niosomes, a burst release from S60 and S60-PEG niosomes is observed as indicated by sudden increase in absorbance spectra in the range of 250 to 300 nm. An increase in absorbance at 260 nm is due to the release of KTP in the sink buffer. After a burst release (as observed by the analysis of the dialysate), there is a sustained release of the drug in the buffer (Fig. 3). The drawback of this method is that the concentrations of the drug in the buffer are so diluted that the intensities obtained by spectroscopic methods are very less.
The absorbance and fluorescence plots of MTX incorporated niosomes show a distinctive characterization of the extent of drug incorporation in S60 and S60-PEG niosomes. As observed from the plots (Fig. 3(A) and (C)) MTX undergoes a minor burst release followed by a sustained release over the period of analysis. The release of MTX from S60-PEG niosomes is slightly more as compared to the S60 niosomes which is due to the slightly increased drug entrapment in the S60-PEG niosomes. There is a slow increase in the rate of drug release which shows a saturation plateau after 96 hours. Similarly, on assessing the drug release of KTP from S60 and S60-PEG niosomes we initially observe a substantial burst release which reaches saturation and sustained release after a period of 24 h. It is observed that the drug encapsulation of KTP in S60 and S60-PEG niosomes was on average 60% and the drug release observed from both the formulations also follows similar trend. It can be estimated that the incorporation of PEG in the studied niosomes does not affect the encapsulation tendency of KTP. Correlating the structure and partitioning variation of KTP in niosomes it is indiacted that KTP interacts with the Span 60 monomers present in the niosomes more effectively during encapsulation by the interaction of –COOH group of the drug with the hydroxyl groups present on the head regions of S60. Whereas the MTX molecules due to more hydrophobic alkyl groups partition little deeper into the bilayers interacting more with the hydrophilic tail regions of S60, PEG and also slight interactions with cholesterol. This also indicates that the KTP is present near the outer regions of the bilayers leading to burst release compared to that in case of MTX. The qualitative determination of the drug release was done by spectroscopy which was further explored by calorimetry to assess the kinetics and obtain quantitative insights into the release phenomenon.
 | ||
| Fig. 4 Representative plots of kinetic profiles of drug release of (A) MTX and (B) KTP from S60 niosomes at 37 °C. The plots were fitted to first order kinetics to determine the rate of drug release. | ||
| System | Rate constant (k) of drug release in buffer (in s−1) |
|---|---|
| Span 60 niosomes incorporated with MTX (initial drug release) | 0.042 |
| Span 60 niosomes incorporated with MTX (after a span of seven days) | 0.062 |
| Span 60 niosomes incorporated with KTP (initial drug release) | 0.080 |
| Span 60 niosomes incorporated with KTP (after a span of seven days) | 0.053 |
| Span 60 and PEG niosomes incorporated with MTX (initial drug release) | 0.088 |
| Span 60 and PEG niosomes incorporated with MTX (after a span of seven days) | 0.068 |
| Span 60 and PEG niosomes incorporated with KTP (initial drug release) | 0.068 |
| Span 60 and PEG niosomes incorporated with KTP (after a span of seven days) | 0.066 |
This is expected since after a time span of 7 days, the structural compactness of the niosomes may decrease leading to slightly enhanced release of drugs. The rate of drug release from S60 niosomes is more in case of KTP (0.080 s−1) than MTX (0.042 s−1) which is attributed to the higher concentration of the drug being loaded into the niosome. Fig. 4 shows representative plots of drug release from S60 niosomes as monitored by ITC at 37 °C showing the single injection of drug incorporated niosomes into buffer present in the cell of ITC. It shows the change in heat observed due to the release of drug in buffer. The rate of release of MTX is higher in S60-PEG niosomes (0.088 s−1) as compared to that from S60 niosomes, which is because of the higher encapsulation of drug in the S60-PEG niosomes. However, after a period of 7 days, though the rate of drug release decreases but it's in a sustained manner. The drug release of KTP from S60-PEG niosomes follow similar kinetics initially and even after a period of 7 days. The above results indicate that the driving force for drug release is mainly diffusion which is concentration dependent. A low value of the rate constant suggests sustained release of the drug for a longer duration under these conditions.
3.5. Interaction of drug loaded niosomes with human serum albumin
The interaction of drug loaded niosomes was assessed with human serum albumin (HSA), a major transport protein, to study the interaction of the components of niosomes with the protein. It acts as a carrier protein for the transport of a wide variety of substances in the body and hence its interaction with the niosome components can help in assessing their suitability as drug delivery systems. The interaction of the niosomal formulations with HSA was studied by using a combination of quantitative (ITC) and qualitative (fluorescence spectroscopy) techniques. The quantitative determination of the interaction between niosomes and HSA was assessed by two different methodologies adopted in ITC: the first methodology involved assessing the interactions by titrating the niosomes (in the presence and absence of drugs) with 0.06 mM HSA to determine the thermodynamics of the interaction and second methodology was designed to analyse the kinetics of the binding of the released drug with HSA.Fig. 5(A) represents the ITC profiles for the interaction of S60 niosomes (in the presence and absence of drugs) with HSA at 37 °C. These ITC profiles have been subtracted by their respective dilution experiments so that these represent actual heats of interaction between the niosome components and the protein.
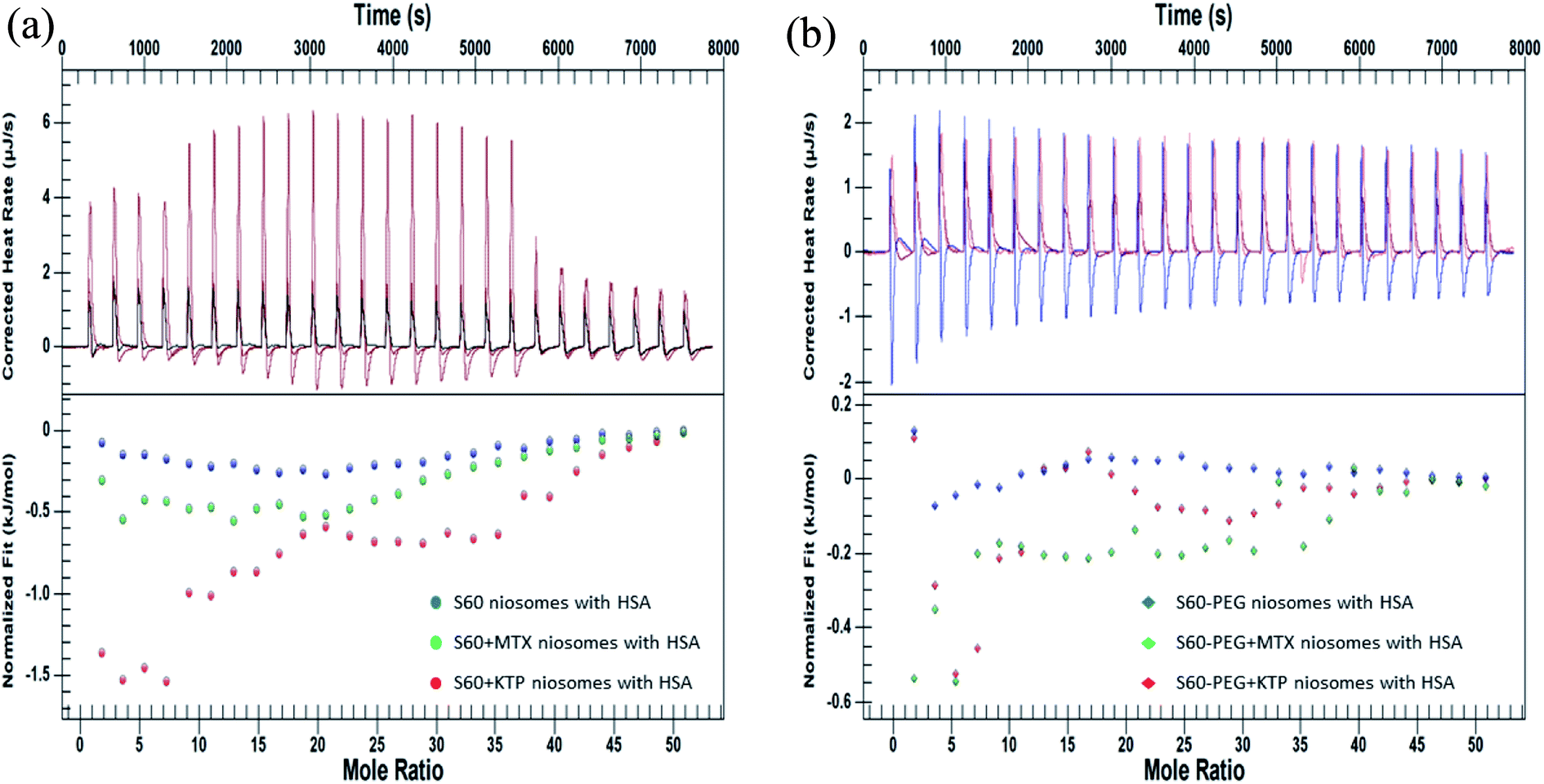 | ||
| Fig. 5 ITC profiles showing the interaction of niosomes (in the presence and absence of drugs) (a) S60, S60 + MTX and S60 + KTP and (b) S60-PEG, S60-PEG + MTX and S60-PEG + KTP with 0.06 mM HSA. | ||
 and
and 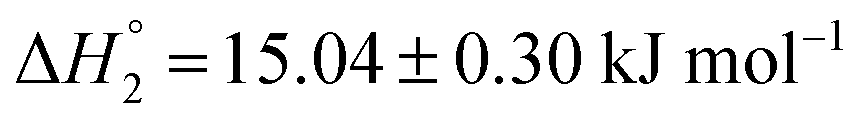 . MTX is known to bind at the site III of HSA by hydrophobic, electrostatic and hydrogen bonding interactions. The binding of MTX is known to induce conformational changes in the protein that can facilitate further binding of MTX molecules. The site III has two subdomains III A and III B which can provide binding sites for more than one MTX molecules without causing appreciable conformational alteration in the protein molecule.48 The binding of KTP incorporated S60 niosomes leads to a relatively greater change in heat signatures as compared to those with the S60 and S60 + MTX niosomes. This can be assigned to the drug released from S60 + KTP niosomes and its subsequent binding to the protein. KTP incorporated S60 niosomes on interaction with HSA show multiple site binding with two pronounced binding events of the order of 103 (suggesting weak binding of KTP to HSA). Two specific binding events with an enthalpic support of
. MTX is known to bind at the site III of HSA by hydrophobic, electrostatic and hydrogen bonding interactions. The binding of MTX is known to induce conformational changes in the protein that can facilitate further binding of MTX molecules. The site III has two subdomains III A and III B which can provide binding sites for more than one MTX molecules without causing appreciable conformational alteration in the protein molecule.48 The binding of KTP incorporated S60 niosomes leads to a relatively greater change in heat signatures as compared to those with the S60 and S60 + MTX niosomes. This can be assigned to the drug released from S60 + KTP niosomes and its subsequent binding to the protein. KTP incorporated S60 niosomes on interaction with HSA show multiple site binding with two pronounced binding events of the order of 103 (suggesting weak binding of KTP to HSA). Two specific binding events with an enthalpic support of  and
and  are observed. It is established that ketoprofen binds at the site I and site II of HSA, which is supported by the above observations.49 Hence, the drug release of KTP and MTX and interaction of the components of niosome establish that there is no alteration or disruption of protein structure and the drug released is able to bind efficiently to the carrier protein. Both the niosomal formulations show retention of the integrity of protein structure and prevent its structural and conformational modification upon interaction.
are observed. It is established that ketoprofen binds at the site I and site II of HSA, which is supported by the above observations.49 Hence, the drug release of KTP and MTX and interaction of the components of niosome establish that there is no alteration or disruption of protein structure and the drug released is able to bind efficiently to the carrier protein. Both the niosomal formulations show retention of the integrity of protein structure and prevent its structural and conformational modification upon interaction.Fig. 5(B) shows the interaction of S60-PEG niosomes (in the presence and absence of drugs) with HSA. The trends observed for the interaction of S60-PEG with HSA is similar to that observed with S60 formulations. The S60-PEG niosomes in the absence of drugs do not show pronounced interaction with serum albumin, instead it shows a very minor exothermic enthalpy of interaction approximately quantifying to −(0.15 ± 0.01) kJ mol−1 which is even lesser than the interaction of S60 niosomes with HSA. When MTX incorporated S60-PEG niosomes interact with HSA, we observe a cooperative binding process due to the binding of the released MTX at site III of the serum albumin (Fig. 5(A)). It exhibits an exothermic 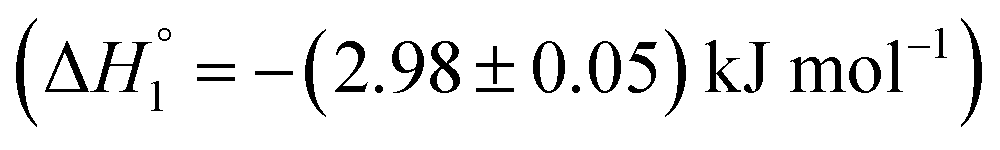 and an endothermic
and an endothermic 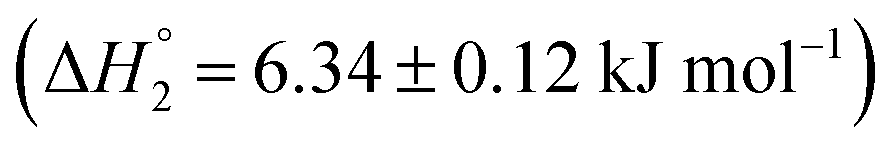 enthalpy of binding. Here also, cooperative binding suggests minor conformational changes in the protein leading to the binding of drug molecules as observed for MTX released from S60 niosomes. The released drug KTP, from the S60-PEG niosomes show two binding events exhibiting a weak and a relatively moderate affinities of the order 103 and 106. The accompanying enthalpies of interaction
enthalpy of binding. Here also, cooperative binding suggests minor conformational changes in the protein leading to the binding of drug molecules as observed for MTX released from S60 niosomes. The released drug KTP, from the S60-PEG niosomes show two binding events exhibiting a weak and a relatively moderate affinities of the order 103 and 106. The accompanying enthalpies of interaction 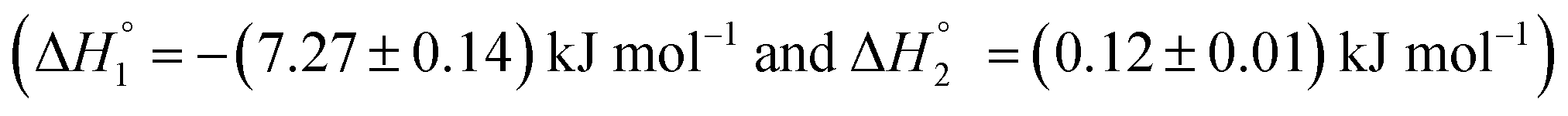 are exothermic and endothermic in nature suggesting the binding of KTP at site I and site II in a different stoichiometric ratio as compared to that of the KTP incorporated S60 niosomes.46 An overview of the stoichiometry of binding of the drug molecule n1 = 0.8 and n2 = 2.7 indicates that more than one drug molecule is able to bind to the site of HSA, suggesting a slight conformational change in the protein due to the interaction of S60-PEG. The thermodynamic parameters indicate the release of the drug from the niosomes and its subsequent binding to the model protein without creating considerable conformational changes in the protein. Hence, the thermodynamic evaluation suggests niosomes to be viable candidates for drug delivery systems.
are exothermic and endothermic in nature suggesting the binding of KTP at site I and site II in a different stoichiometric ratio as compared to that of the KTP incorporated S60 niosomes.46 An overview of the stoichiometry of binding of the drug molecule n1 = 0.8 and n2 = 2.7 indicates that more than one drug molecule is able to bind to the site of HSA, suggesting a slight conformational change in the protein due to the interaction of S60-PEG. The thermodynamic parameters indicate the release of the drug from the niosomes and its subsequent binding to the model protein without creating considerable conformational changes in the protein. Hence, the thermodynamic evaluation suggests niosomes to be viable candidates for drug delivery systems.
| System | Rate of drug release (k) (in HSA) in s−1 |
|---|---|
| Span 60 niosomes incorporated with MTX (initial drug release) | 0.068 |
| Span 60 niosomes incorporated with MTX (after a span of seven days) | 0.082 |
| Span 60 niosomes incorporated with KTP (initial drug release) | 0.099 |
| Span 60 niosomes incorporated with KTP (after a span of seven days) | 0.077 |
| Span 60 and PEG niosomes incorporated with MTX (initial drug release) | 0.068 |
| Span 60 and PEG niosomes incorporated with MTX (after a span of seven days) | 0.081 |
| Span 60 and PEG niosomes incorporated with KTP (initial drug release) | 0.078 |
| Span 60 and PEG niosomes incorporated with KTP (after a span of seven days) | 0.066 |
Since, MTX containing niosomes do not undergo a substantial burst release as compared to KTP encapsulated niosomes, we do not see a pronounced drug release from MTX containing vesicles. Since KTP binds strongly with HSA as compared to MTX, this adds to the driving force for its higher release from the vesicles into the sink conditions. The rates of drug release from niosomes in HSA are slightly higher than the drug release observed in buffer which can be assigned to the affinity of the drug for serum albumin, in addition to diffusion. These results also confirm that the niosomes can show specificity of drug release based on the sink conditions.
The innate quality of a drug delivery system is defined as its ability to sustain and protect the protein structure and functionality. For the same the assessment of thermal and conformational stability of protein during drug binding and effect of the components of drug delivery vehicles on proteins is very crucial. To assess these effects, DSC and CD studies of HSA in the presence and absence of drugs and niosomes were performed. The DSC profiles enable assessment of change in the thermal stability of the model protein HSA in the presence of the niosomal formulations whereas the conformational stability was assessed using CD spectroscopy.
3.6. Differential scanning calorimetry
In order to understand the effect of components of niosomes and the incorporated drug on the conformational integrity of the protein HSA, we performed differential scanning calorimetry. Fig. 6 represents DSC thermal unfolding profiles of HSA, in the absence and presence of empty and drug incorporated niosomes. HSA is observed to unfold in a biphasic manner with transition temperatures of (343.1 ± 5.8) K and (352.2 ± 4.8) K and calorimetric enthalpies of 425 ± 6 kJ mol−1 and 492 ± 7 kJ mol−1, respectively. These results agree with those reported in literature,50 though more than two transitions have also been reported for HSA.51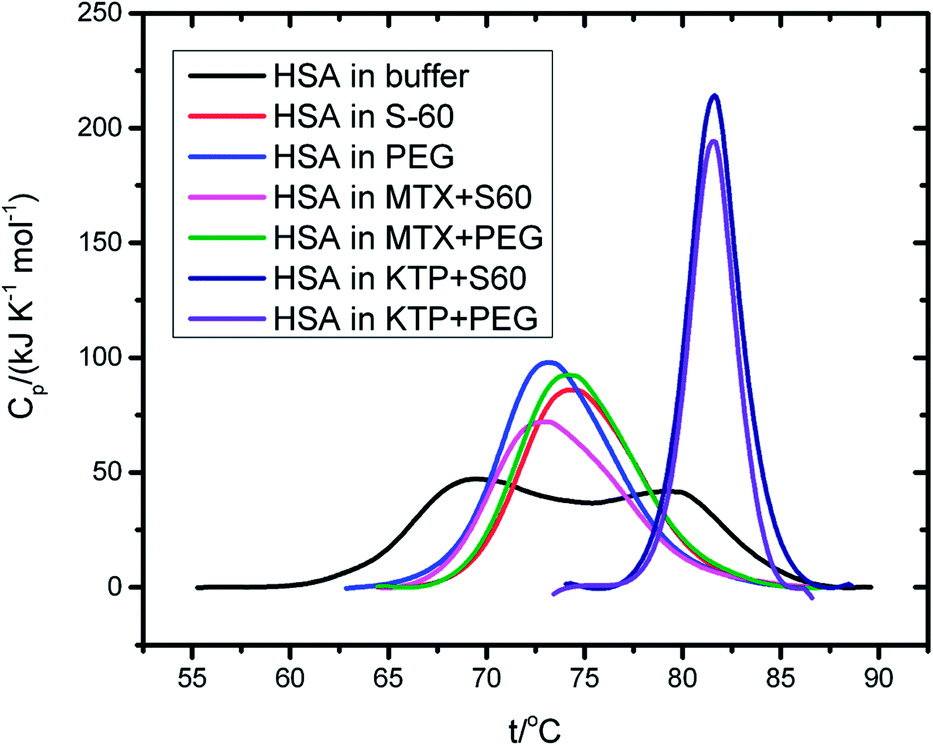 | ||
| Fig. 6 DSC thermograms of 4 mg ml−1 human serum albumin in the presence and absence of drug incorporated niosomes in the temperature range of 298.15 K to 363.15 K. | ||
In the presence of S60 niosomes (without drugs), a slight thermal stabilisation of the protein at the first transition and a slight decrease in the thermal transition at the second transition was observed with significant alteration in calorimetric enthalpies of unfolding. These results suggest that S60 based niosomes do not affect the thermal stability of the protein significantly though slight conformational change could be induced. Even the MTX incorporated S60 based niosomes provided similar results as that obtained in the absence of drugs. KTP, on the other hand induced a significant thermal stabilisation in the protein when taken together with the S60 niosomes. Here, a thermal stabilisation of 11.2 ± 0.7 K and change in calorimetric enthalpy of 707 ± 11 kJ mol−1 are observed.
Inclusion of PEG in niosomes also does not affect the thermal stability of the protein over and above that of S60 and the inclusion of mitoxantrone into the niosomes retains the biphasic thermal unfolding pattern of the protein with similar thermodynamic signatures. Here also KTP into S60 niosomes with PEG led to significant stabilisation of the protein yielding only single endotherm. All these results taken together suggest that the components of niosomes used as drug delivery vehicle do not disrupt the thermal stability and conformational integrity of the protein (Table 5).
| System | Tm (1)/K | Tm (2)/K | ΔcalH (1)/(kJ mol−1) | ΔcalH (2)/(kJ mol−1) |
|---|---|---|---|---|
| HSA | 343.1 ± 5.8 | 352.2 ± 4.8 | 425 ± 6 | 492 ± 7 |
| HSA + S60 | 346.3 ± 6.9 | 349.3 ± 6.0 | 780 ± 12 | 598 ± 9 |
| HSA + S60 + MTX | 345.0 ± 7.0 | 348.3 ± 4.9 | 786 ± 12 | 532 ± 8 |
| HSA + S60 + KTP | 354.3 ± 6.5 | — | 1132 ± 17 | — |
| HSA + S60 + PEG | 346.0 ± 5.9 | 349.9 ± 7.0 | 728 ± 11 | 688 ± 10 |
| HSA + S60 + PEG + MTX | 346.3 ± 5.7 | 349.3 ± 7.8 | 717 ± 11 | 578 ± 9 |
| HSA + S60 + PEG + KTP | 354.6 ± 7.6 | — | 1349 ± 20 | — |
3.7. Circular dichroism spectroscopy
Fig. 7 represents changes in the near UV-CD and far UV-CD regions of the spectra of HSA with and without niosomes or niosome incorporated drugs. The addition of Span 60 niosomes to HSA does not alter the secondary structure of the protein. However, when the MTX encapsulated S60 niosomes are added to the protein, HSA, though the secondary structure of the protein is not disrupted but the entire negative peak shift towards a lower wavelength with an induction of positive band at about 205 nm which is characteristic of beta turns in the protein.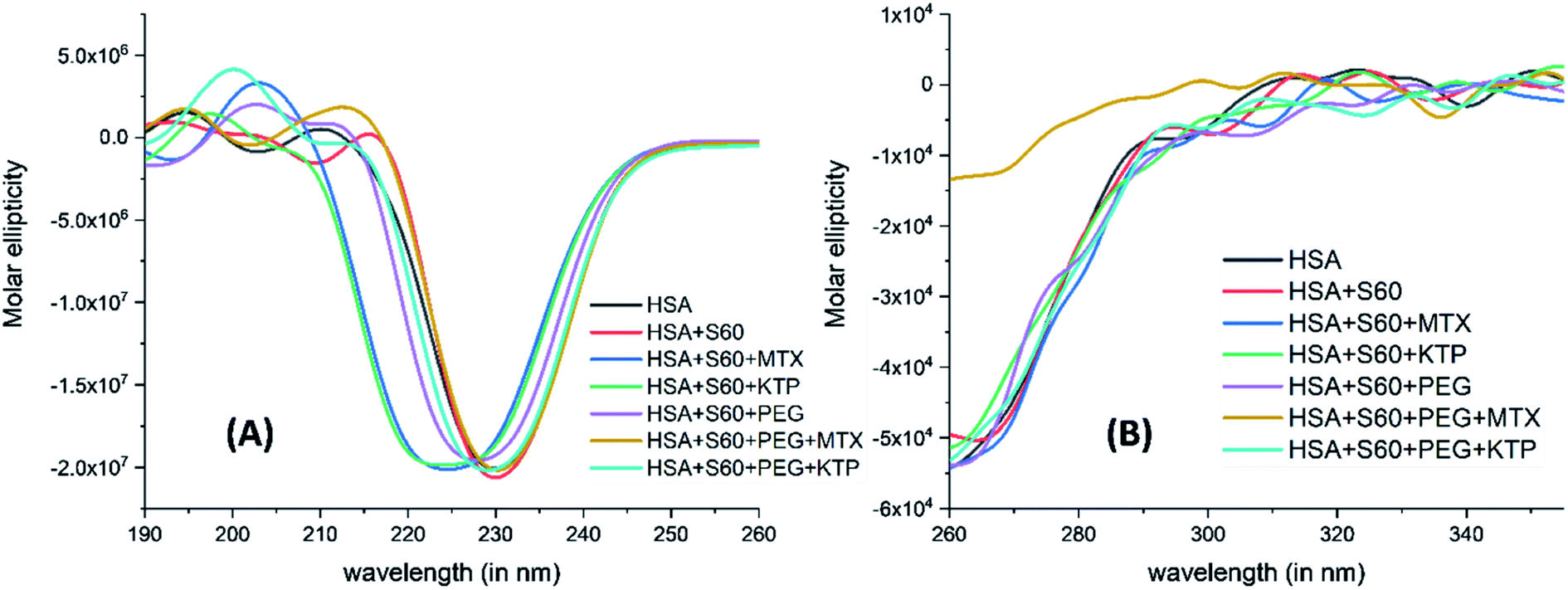 | ||
| Fig. 7 Far UV-CD (A) and near UV-CD (B) spectra of HSA in the absence and presence of drug encapsulated niosomes. | ||
A similar result is observed when the experiment is done in the presence of ketoprofen loaded S60 niosomes. With the modification of S0 niosomes with PEG, the changes in secondary structure of HSA with and without MTX and KTP are insignificant. As seen in the near UV-CD spectra, there is hardly any change in the tertiary structure of the protein with and without the drugs or niosomes. These results suggest that when the S60 based niosomes are used as drug delivery vehicles in the absence or presence of PEG they do not cause any disruption to the conformational stability of the protein. On comparison with DSC results, the niosomes in the presence and absence of MTX convert a broad thermal transition into a sharp one, though the mid-point of transition remains nearly the same. This could indicate slight conformational changes which do not affect the overall protein structure. It is only the presence of KTP which provides an extraordinary thermal stability to the protein as a result of binding but surprisingly no major alterations are seen in the tertiary and secondary structure. All these results taken together establish that the niosomal preparation based on S60 surfactant can act as a safer option to be used in the delivery of drugs.
3.8. Inhibition of cancer cell viability
The viability of MDA-MB-231 cells was studied in the presence of free drug (MTX, KTP), empty niosomes (S60 and S60-PEG), and drug-loaded niosomes. PEG is known to increase the stability and circulation time of the niosomes from a biological point of view and hence the effect of the inclusion of PEG in niosomes can be seen by cell viability studies.52 The effect of various substituents on the cell viability as a function of concentration is shown in Fig. 8(A) and (B). It is observed that the empty vesicles (without drug encapsulation) are non-toxic to the cells. They show less inhibition on cell viability confirming that the substituents used in vesicle formation are mostly non-toxic to the cells at the studied concentrations. Among the free drugs tested, mitoxantrone showed considerable toxicity (0.21 ± 0.01 μM) whereas the anti-inflammatory drug ketoprofen enhanced the cell viability.53 The drug-loaded niosomes showed the effect of the encapsulated drugs on the cells as they are released with time from the niosomes. The mitoxantrone-containing niosomes showed lesser inhibition of the cell viability as compared to the free drug. This can be assigned to the slow release of the drug from the niosomes as well as the amount of drug being released within the time frame. With the increase in the concentration of mitoxantrone-loaded niosomes, the cell viability decreased. For example, 1 μM, 10 μM, 50 μM and 100 μM S60 + MTX reduced the viability by 9%, 41%, 63%, 73%, respectively and with 1 μM and 10 μM S60 + KTP the viability increased by 2% and for 50 μM and 100 μM S60 + KTP viability increased by 18% and 21%, respectively. S60-PEG vesicles containing MTX reduced the viability considerably. For example, 100 μM of the formulation reduced the cell viability by 72%. KTP incorporated S60-PEG showed an increase in viability (up to ∼20%). In the presence of ketoprofen loaded niosomes, initially the cell viability was not altered, but with increase in concentration of the ketoprofen loaded vesicles cell viability increased.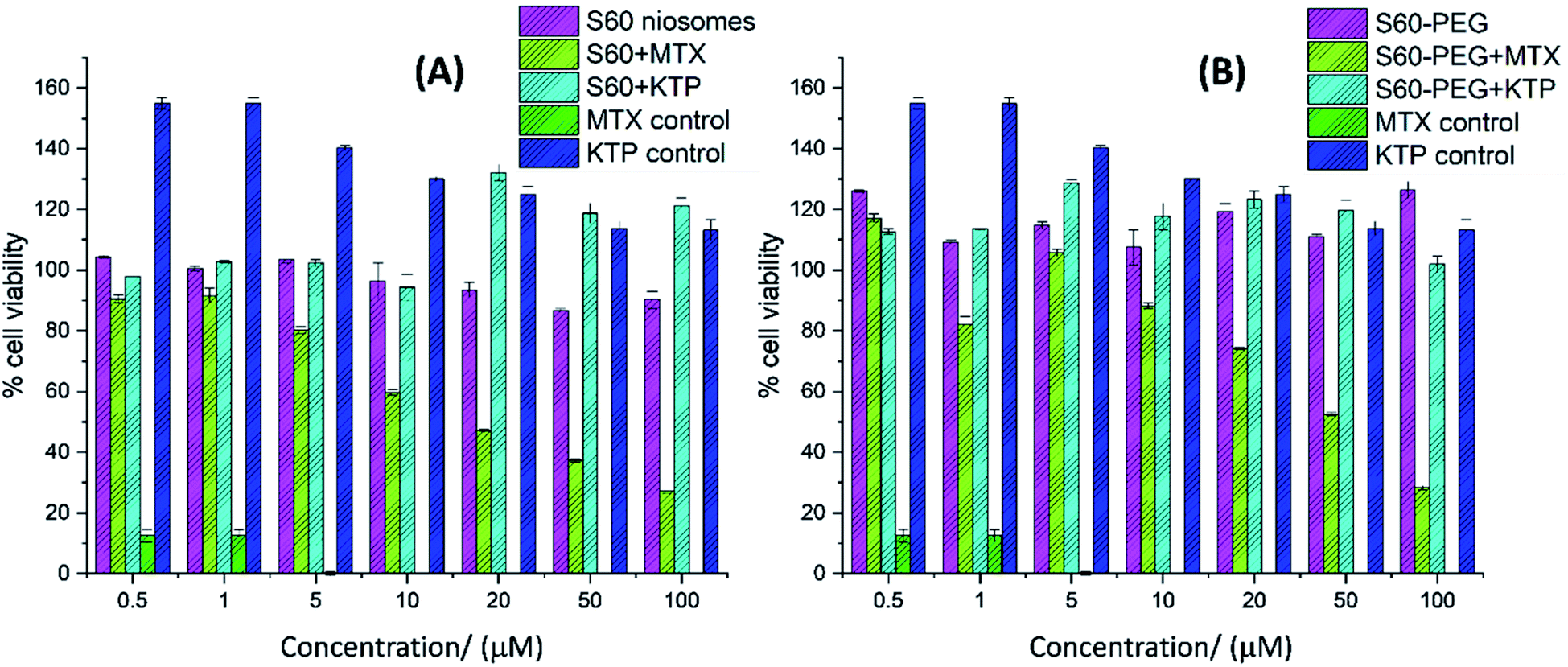 | ||
| Fig. 8 MTT assay for cell viability studies of (A) S60 and (B) S60-PEG niosomes in the absence and presence of MTX and KTP. The cell viability was calculated as a percentage of the ratio of absorbance of treated cells over control. Each point represents the mean value ± SD (n = 3). | ||
Although not up to the extent of free MTX-treated cells, the cells treated with niosomes-incorporated MTX reduced cell viability in a concentration-dependent manner. Among the encapsulated ones, the maximum decrease in cell viability was observed in case of MTX-incorporated S60 niosomes followed by MTX-incorporated S60-PEG niosomes. Since the niosomes showed sustained and slow release of drugs, they can be designed to effectively release required doses of drug molecules at designated time intervals.
The statistical analysis was performed using ANOVA calculator including Tukey HSD. The f-ratio was calculated which is the ratio of two variances that are approximately the same when the null hypothesis is true. The f-ratio value is 30.11376 (for data set in Fig. 8(A) and 31.66408 (for data set in Fig. 8(B)) and the p-value is <0.0001 and the result is significant at p < 0.01.
Niosomal systems with suitable choice of its constituent non-ionic surfactants, stabilizers, and other environmental conditions has enabled ese of preparation, characterization, and assessment of not only encapsulation, but also the release of selected drugs qualitatively and quantitatively. Further, biophysical and cell viability assays have permitted understanding efficacy of this drug delivery systems with mechanistic insights (Fig. 9).
 | ||
| Fig. 9 Schematic representation of preparation and characterization of niosomes and assessment of quantitative and qualitative aspects of drug release and its application by biophysical and cell viability assays. | ||
4. Conclusions
The partitioning and diffusion-controlled release of drugs, mitoxantrone and ketoprofen from the drug encapsulated niosomes of Span 60 and Span 60 with polyethylene glycol have been investigated. The partitioning of the anti-cancer drug, mitoxantrone and the anti-inflammatory drug, ketoprofen in empty niosomes, occurs in the bilayers of the Span 60-based surfactants in order to avoid the surface charge repulsion due to the head groups of the surfactants, cholesterol and stabilizer components present. Similar trends of partitioning of the drugs in the niosomes indicate a structural compactness and rigidity of the drug delivery systems. Weak partitioning, as indicated by ITC results, can be attributed to the surface adsorption of the drugs on the niosomes as a result of weak interactions between the drugs and surface charges on the head groups of niosomes. The release of the drugs from the drug encapsulated niosomes characterized as diffusion controlled followed slow kinetics as monitored by calorimetry and spectroscopy. The systems showed a sustained release for over a period of seven days when analysed at 37 °C at pH 7.4. A novel method of assessing drug release by using ITC was employed which not only enabled heat measurements, but also provided an assessment of kinetics of the release. The sustained release of drugs indicating that these niosome systems are efficient for drug delivery and would help in enhancement of bioavailability of the drugs in the biological system.The S60 vesicles and S60-PEG based vesicles showed stability and sustained release and can also show stability in morphology and activity for a period of minimum 10 days when stored at 4 °C. The rate of release for both the drugs was comparable with that of ketoprofen to be slightly higher than mitoxantrone which can be attributed to their structure and size. Mitoxantrone, being more hydrophobic, encapsulated deeper into the niosomes which prevents its burst release as compared to that of ketoprofen. The drug release was studied with human serum albumin as a sink to analyse the binding of the released drugs with this carrier protein. The DSC and CD results demonstrate that the components of these niosomal drug delivery vehicles do not affect the conformational and transport ability of HSA. In order to ensure selective elimination of tumour cells, multiple drug delivery systems have been explored, including conjugating potential therapeutic formulations to metal nanoparticles.54 The applicability of the niosome systems was also checked on the viability of MDA-MB-231 breast cancer cells. We show efficiency of MTX incorporated vesicles which show effective and sustained release of the drug, that suppressed the viability of the cells. Whereas release of ketoprofen showed enhanced cell proliferation.
Overall, in addition to qualitative assessment, a quantitative understanding of drug partitioning in complex self-assemblies of niosomes has been attempted by employing a novel calorimetric approach. Such studies permit correlations of structure with properties and energetics and enable assessment of effect of components of the drug delivery system on target protein. The developed ITC method has also enabled an assessment of kinetics of drug release and sustainability duration. The current research work highlights the molecular interactions with niosomal systems with insights from qualitative, quantitative and cell biology approaches. The results demonstrate effectiveness of such drug delivery systems and can also guide towards rational drug design.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors are grateful to research grant from Science and Engineering Research Board [EMR/2016/001261]. ML acknowledges financial support from UM-DAE Centre for Excellence in Basic Sciences.References
- M. Isaac and C. Holvey, Transdermal patches: the emerging mode of drug delivery system in psychiatry, Ther. Adv. Psychopharmacol., 2012, 2, 255–263 CrossRef CAS PubMed.
- S. Gholizadeh, Z. Wang, X. Chen, R. Dana and N. Annabi, Advanced nanodelivery platforms for topical ophthalmic drug delivery, Drug Discovery Today, 2021, 26, 1437–1449 CrossRef CAS PubMed.
- S. P. Bandia, S. Bhatnagar and V. V. K. Venugantia, Advanced materials for drug delivery across mucosal barriers, Acta Biomater., 2021, 119, 113–129 Search PubMed.
- M. Alagusundaram, B. Chengaiah, K. Gnanaprakash, S. Ramkanth, C. M. Chetty and D. Dhachinamoorthi, Nasal drug delivery system-an overview, Int. J. Res. Pharm. Sci., 2010, 1, 454–465 CAS.
- N. Kader, N. Ansari, R. Bharti, N. Mandavi, G. K. Sahu, A. K. Jha and H. Sharma, Novel Approaches for Colloidal Drug Delivery System: Nanoemulsion, Res. J. Pharm. Dosage Forms Technol., 2018, 10, 253–258 CrossRef.
- A. M. Vargason, A. C. Anselmo and S. Mitragotri, The evolution of commercial drug delivery technologies, Nat. Biomed. Eng., 2021, 5, 951–967 CrossRef PubMed.
- R. Rajera, K. Nagpal, S. K. Singh and D. N. Mishra, Niosomes: a controlled and novel drug delivery system, Biol. Pharm. Bull., 2011, 34, 945–953 CrossRef CAS PubMed.
- H. E. Schaeffer and D. L. Krohn, Liposomes in topical drug delivery, Invest. Ophthalmol. Visual Sci., 1982, 22, 220–227 CAS.
- W. H. De Jong and P. J. A. Borm, Drug delivery and nanoparticles: applications and hazards, Int. J. Nanomed., 2008, 3, 133 CrossRef CAS PubMed.
- M. J. Lawrence and G. D. Rees, Microemulsion-based media as novel drug delivery systems, Adv. Drug Delivery Rev., 2000, 45, 89–121 CrossRef CAS PubMed.
- I. Akbarzadeh, M. Fatemizadeh, F. Heidari and N. M. Niri, Niosomal Formulation for Co-Administration of Hydrophobic Anticancer Drugs into MCF-7 Cancer Cells, Archives of Advances in Biosciences, 2020, 11, 1–9 CrossRef.
- D. Bhowmik, H. Gopinath, B. P. Kumar, S. Duraivel and K. P. S. Kumar, Controlled release drug delivery systems, Pharma Innovation, 2012, 1, 24–32 CAS.
- F. Yamashita and M. Hashida, Pharmacokinetic considerations for targeted drug delivery, Adv. Drug Delivery Rev., 2013, 65, 139–147 CrossRef CAS PubMed.
- J. Majumder and T. Minko, Multifunctional and stimuli-responsive nanocarriers for targeted therapeutic delivery, Expert Opin. Drug Delivery, 2021, 18, 205–227 CrossRef CAS PubMed.
- D. Guimarães, A. Cavaco-Paulo and E. Nogueira, Design of liposomes as drug delivery system for therapeutic applications, Int. J. Pharm., 2021, 601, 120571 CrossRef PubMed.
- H. Schreier and J. Bouwstra, Liposomes and niosomes as topical drug carriers: dermal and transdermal drug delivery, J. Controlled Release, 1994, 30, 1–15 CrossRef CAS.
- L. Tavano, L. Gentile, C. O. Rossi and R. Muzzalupo, Novel gel-niosomes formulations as multicomponent systems for transdermal drug delivery, Colloids Surf., B, 2013, 110, 281–288 CrossRef CAS PubMed.
- F. D. Moghaddam, I. Akbarzadeh, E. Marzbankia, M. Farid, L. Khaledi, A. H. Reihani, M. Javidfar and P. Mortazavi, Delivery of melittin-loaded niosomes for breast cancer treatment: an in vitro and in vivo evaluation of anti-cancer effect, Cancer Nanotechnol., 2021, 12, 14 CrossRef.
- N. B. Mahale, P. D. Thakkar, R. G. Mali, D. R. Walunj and S. R. Chaudhari, Niosomes: novel sustained release nonionic stable vesicular systems—an overview, Adv. Colloid Interface Sci., 2012, 183, 46–54 CrossRef PubMed.
- N. O. Sahin, Niosomes as nanocarrier systems, Nanomaterials and Nanosystems for Biomedical Applications, 2007, 67–81 Search PubMed.
- J. Rathee, R. Kanwar, D. Kaushik, D. B. Salunke and S. K. Mehta, Niosomes as efficient drug delivery modules for encapsulation of Toll-like receptor 7 agonists and IDO-inhibitor, Appl. Surf. Sci., 2020, 505, 144078 CrossRef CAS.
- X. Hu, J. Zhang, L. Deng, H. Hu, J. Hu and G. Zheng, Galactose-Modified PH-Sensitive Niosomes for Controlled Release and Hepatocellular Carcinoma Target Delivery of Tanshinone IIA, AAPS PharmSciTech, 2021, 22, 1–14 CrossRef PubMed.
- N. D. Machado, M. A. Fernández, M. Häring, C. Saldías and D. D. Díaz, Niosomes encapsulated in biohydrogels for tunable delivery of phytoalexin resveratrol, RSC Adv., 2019, 9, 7601–7609 RSC.
- M. G. Arafa, D. Ghalwash, D. M. El-Kersh and M. M. Elmazar, Propolis-based niosomes as oromuco-adhesive films: A randomized clinical trial of a therapeutic drug delivery platform for the treatment of oral recurrent aphthous ulcers, Sci. Rep., 2018, 8, 1–14 CAS.
- C. Marianecci, L. Di Marzio, F. Rinaldi, C. Celia, D. Paolino, F. Alhaique, S. Esposito and M. Carafa, Niosomes from 80s to present: the state of the art, Adv. Colloid Interface Sci., 2014, 205, 187–206 CrossRef CAS PubMed.
- A. Rogerson, J. Cummings, N. Willmott and A. T. Florence, The distribution of doxorubicin in mice following administration in niosomes, J. Pharm. Pharmacol., 1988, 40, 337–342 CrossRef CAS PubMed.
- A. Manosroi, P. Wongtrakul, J. Manosroi, H. Sakai, F. Sugawara, M. Yuasa and M. Abe, Characterization of vesicles prepared with various non-ionic surfactants mixed with cholesterol, Colloids Surf., B, 2003, 30, 129–138 CrossRef CAS.
- P. García-Manrique, N. D. Machado, M. A. Fernández, M. C. Blanco-López, M. Matos and G. Gutiérrez, Effect of drug molecular weight on niosomes size and encapsulation efficiency, Colloids Surf., B, 2020, 186, 110711 CrossRef PubMed.
- S. Joshi, R. White, R. Sahu, V. A. Dennis and S. R. Singh, Comprehensive Screening of Drug Encapsulation and Co-Encapsulation into Niosomes Produced Using a Microfluidic Device, Processes, 2020, 8, 535 CrossRef CAS.
- W. Loh, C. Brinatti and K. C. Tam, Use of isothermal titration calorimetry to study surfactant aggregation in colloidal systems, Biochim. Biophys. Acta, 2016, 1860, 999–1016 CrossRef CAS PubMed.
- E. Judy, D. Pagariya and N. Kishore, Drug partitioning in micellar media and its implications in rational drug design: insights with streptomycin, Langmuir, 2018, 34, 3467–3484 CrossRef CAS PubMed.
- E. Judy and N. Kishore, Correlating Properties of Antibiotics with Energetics of Partitioning in Colloidal Self-Assemblies and Effect on Binding of Released Drug with Target Protein, Langmuir, 2021, 37, 7203–7218 CrossRef CAS PubMed.
- R. Migliore, G. Granata, A. Rivoli, G. M. L. Consoli and C. Sgarlata, Binding affinity and driving forces for the interaction of calixarene-based micellar aggregates with model antibiotics in neutral aqueous solution, Front. Chem., 2020, 8, 1227 Search PubMed.
- S. Gediminas, D. Danielius, D. Matulis and V. Petrauskas, Thermodynamics of Interactions Between Charged Surfactants and Ionic Poly(amino acids) by Isothermal Titration Calorimetry, ACS Omega, 2019, 4, 17527–17535 CrossRef PubMed.
- A. Mukhija and N. Kishore, Prevention and disintegration of human serum albumin fibrils under physiological conditions: biophysical aspects, J. Phys. Chem. B, 2018, 122, 9896–9906 CrossRef CAS PubMed.
- L. J. Peltonen and J. Yliruusi, Surface pressure, hysteresis, interfacial tension, and CMC of four sorbitan monoesters at water–air, water–hexane, and hexane–air interfaces, J. Colloid Interface Sci., 2000, 227, 1–6 CrossRef CAS PubMed.
- A. J. Baillie, A. T. Florence, L. R. Hume, G. T. Muirhead and A. Rogerson, The preparation and properties of niosomes—non-ionic surfactant vesicles, J. Pharm. Pharmacol., 1985, 37, 863–868 CrossRef CAS PubMed.
- M. Manconi, C. Sinico, D. Valenti, G. Loy and A. M. Fadda, Niosomes as carriers for tretinoin. I. Preparation and properties, Int. J. Pharm., 2002, 234, 237–248 CrossRef CAS PubMed.
- A. L. Kholodenko and J. F. Douglas, Generalized Stokes-Einstein equation for spherical particle suspensions, Phys. Rev. E: Stat. Phys., Plasmas, Fluids, Relat. Interdiscip. Top., 1995, 51, 1081 CrossRef CAS PubMed.
- A. Sze, D. Erickson, L. Ren and D. Li, Zeta-potential measurement using the Smoluchowski equation and the slope of the current–time relationship in electroosmotic flow, J. Colloid Interface Sci., 2003, 261, 402–410 CrossRef CAS PubMed.
- Y.-Z. Huang, J.-Q. Gao, W.-Q. Liang and S. Nakagawa, Preparation and characterization of liposomes encapsulating chitosan nanoparticles, Biol. Pharm. Bull., 2005, 28, 387–390 CrossRef CAS PubMed.
- N. J. Greenfield, Using circular dichroism spectra to estimate protein secondary structure, Nat. Protoc., 2006, 1, 2876 CrossRef CAS PubMed.
- P. Verma, P. K. R. Nagireddy, S. S. Prassanawar, J. G. Nirmala, A. Gupta, S. Kantevari and M. Lopus, 9-PAN promotes tubulin-and ROS-mediated cell death in human triple-negative breast cancer cells, J. Pharm. Pharmacol., 2020, 72, 1585–1594 CrossRef CAS PubMed.
- P. Kumar, A. Nagarajan and P. D. Uchil, Analysis of cell viability by the MTT assay, Cold Spring Harbor Protocols, 2018, pdb-prot095505 CrossRef PubMed.
- J. Van Meerloo, G. J. L. Kaspers and J. Cloos, in Cancer cell culture, Springer, 2011, pp. 237–245 Search PubMed.
- D. Cosco, D. Paolino, R. Muzzalupo, C. Celia, R. Citraro, D. Caponio, N. Picci and M. Fresta, Novel PEG-coated niosomes based on bola-surfactant as drug carriers for 5-fluorouracil, Biomed. Microdevices, 2009, 11, 1115–1125 CrossRef CAS PubMed.
- L. Basiri, G. Rajabzadeh and A. Bostan, Physicochemical properties and release behavior of Span 60/Tween 60 niosomes as vehicle for α-Tocopherol delivery, LWT--Food Sci. Technol., 2017, 84, 471–478 CrossRef CAS.
- N. K. Shahper and U. K. Asad, Computational simulation of mitoxantrone binding with human serum albumin, J. Proteomics Bioinf., 2008, 1, S017–S020 Search PubMed.
- T. Barri, T. Trtić-Petrović, M. Karlsson and J. Å. Jönsson, Characterization of drug–protein binding process by employing equilibrium sampling through hollow-fiber supported liquid membrane and Bjerrum and Scatchard plots, J. Pharm. Biomed. Anal., 2008, 48, 49–56 CrossRef CAS PubMed.
- A. Michnik, K. Michalik, A. Kluczewska and Z. Drzazga, Comparative DSC study of human and bovine serum albumin, J. Therm. Anal. Calorim., 2006, 84, 113–117 CrossRef CAS.
- F. Bou-Abdallah, S. E. Sprague, B. M. Smith and T. R. Giffune, Binding thermodynamics of Diclofenac and Naproxen with human and bovine serum albumins: A calorimetric and spectroscopic study, J. Chem. Thermodyn., 2016, 103, 299–309 CrossRef CAS.
- A. Kolate, D. Baradia, S. Patil, I. Vhora, G. Kore and A. Misra, PEG—a versatile conjugating ligand for drugs and drug delivery systems, J. Controlled Release, 2014, 192, 67–81 CrossRef CAS PubMed.
- D. A. Hamdani, A. Javeed, M. Ashraf, J. Nazir, I. Altaf and M. Shahbaz, In vitro cytotoxic and genotoxic evaluation to ascertain toxicological potential of ketoprofen, Afr. J. Pharm. Pharmacol., 2014, 8, 386–391 Search PubMed.
- J. G. Nirmala, K. Rachineni, S. Choudhary, R. V Hosur and M. Lopus, Triphala polyphenols-functionalized gold nanoparticles impair cancer cell survival through induction of tubulin dysfunction, J. Drug Delivery Sci. Technol., 2021, 61, 102167 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1ra06057k |
| This journal is © The Royal Society of Chemistry 2021 |