
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceTowards understanding of electrolyte degradation in lithium-mediated non-aqueous electrochemical ammonia synthesis with gas chromatography-mass spectrometry†
Rokas Sažinas ,
Suzanne Zamany Andersen,
Katja Li,
Mattia Saccoccio,
Kevin Krempl,
Jakob Bruun Pedersen,
Jakob Kibsgaard,
Peter Christian Kjærgaard Vesborg,
Debasish Chakraborty and
Ib Chorkendorff*
,
Suzanne Zamany Andersen,
Katja Li,
Mattia Saccoccio,
Kevin Krempl,
Jakob Bruun Pedersen,
Jakob Kibsgaard,
Peter Christian Kjærgaard Vesborg,
Debasish Chakraborty and
Ib Chorkendorff*
Department of Physics, Technical University of Denmark, Kongens Lyngby, 2800, Denmark. E-mail: ibchork@fysik.dtu.dk
First published on 23rd September 2021
Abstract
Lithium-mediated electrochemical ammonia synthesis (LiMEAS) in non-aqueous media is a promising technique for efficient and green ammonia synthesis. Compared to the widely used Haber–Bosch process, the method reduces CO2 emissions to zero due to the application of green hydrogen. However, the non-aqueous medium encounters the alkali metal lithium and organic components at high negative potentials of electrolysis, which leads to formation of byproducts. To assess the environmental risk of this synthesis method, standardized analytical methods towards understanding of the degradation level and consequences are needed. Here we report on the implementation of an approach to analyze the liquid electrolytes after electrochemical ammonia synthesis via high-resolution gas chromatography-mass spectrometry (GCMS). To characterize the molecular species formed after electrolysis, electron ionization high-resolution mass spectrometry (EI-MS) was applied. The fragmentation patterns enabled the elucidation of the mechanisms of byproduct formation. Several organic electrolytes were analyzed and compared both qualitatively and quantitatively to ascertain molecular composition and degradation products. It was found that the organic solvent in contact with metallic electrodeposited lithium induces solvent degradation, and the extent of this decomposition to different organic molecules depends on the organic solvent used. Our results show GCMS as a suitable technique for monitoring non-aqueous electrochemical ammonia synthesis in different organic electrolytes.
Introduction
Lithium-mediated electrochemical ammonia synthesis (LiMEAS) is a promising alternative to the traditional complex thermochemical Haber–Bosch process,1–5 which predominantly requires high temperatures (400–500 °C) and pressures (150–200 bar) coupled with a steam reforming plant for hydrogen (H2) production.6 On the other hand, the LiMEAS is thermodynamically driven by an electrical potential instead of high temperatures and pressures, and the chemical reactivity of lithium towards nitrogen gas (N2).7 By utilization of green electricity from e.g. wind or solar energy sources, the process can be considered a renewable alternative. This enables softer operation conditions in a modular fashion, similar to a flow reactor,8 and leads to lower capital costs for the process without greenhouse gas emissions compared to the 1.4% of global CO2 emissions for ammonia (NH3) production by the Haber–Bosch process. The LiMEAS could be operated on a local level e.g. individual farms or greenhouses, thereby further eliminating the need for transportation and storage. This alternative decentralized NH3 production method turns against the centralized nature of Haber–Bosch making ammonia accessible at a local scale and employing renewable energy sources e.g. wind or solar. The production of H2 (for example, water splitting) may overcome some of the issues associated with the traditional Haber–Bosch process, such as the large amount of CO2 from steam reforming9 emissions and high cost.2A typical LiMEAS cell consists of a noble metal anode e.g. platinum (Pt) and transition metal cathode which does not interact or alloy with lithium (Li) e.g. molybdenum (Mo).10,11 The electrodes are usually submerged in the non-aqueous organic electrolyte with or without a membrane or separator. The electrolyte is composed of a conducting Li salt and a solvent, typically lithium perchlorate (LiClO4)12 and tetrahydrofuran (THF), respectively.10,13 Other mostly ether-based solvents, such as dimethoxyethane (DME) or diethyleneglycol dimethyl ether or diglyme (DG) can also be used for LiMEAS. Thus, the influence of the electrolyte on the electrochemical ammonia synthesis and the overall stability have to be addressed in order to overcome limited performance and inhibition of the nitrogen reduction reaction (N2RR).
Due to the complex nature of LiMEAS, no reports on the aging have been introduced so far which can be related to the used materials and components, as well as to the interactive reactions. However, due to the instability issues previously noted,5,11 there is reason to believe that undesirable organic lithium compounds are formed during the chemical reactions of LiMEAS, leading to degradation of the electrolyte. Li metal is both chemically and electrochemically unstable and reacts with traces of moisture, oxygen and organic solvent. Even at room temperature, Li reacts with ethanol (EtOH) forming lithium ethoxide (LiOEt).14 These reactions can lead to hydrolysis and oxidation products inside the electrolyte. Furthermore, under electrochemical operation conditions, a ring opening reaction of the cyclic THF can occur.14 The electrochemical reduction of the organic electrolyte at the anode of lithium ion batteries (LIB) results in the formation of the solid electrolyte interphase (SEI).15 This layer is composed of inorganic and organic decomposition products and is most likely necessary for long-term performance, as it is ideally only permeable for lithium ions, thus protecting the highly reactive anode against further reduction reactions.16–18 The reaction of Li with the solvent leads to the formation of protective SEI, but byproducts dissolved in the electrolyte might encumber the system by increasing contamination.19–22 Thus, the protective nature of this passivation layer could be distinct to prevent further electrolyte decomposition. There are plenty of reports and techniques regarding the monitoring of aging in LIB cells.15,23 Among all of them, the electrolyte aging can be monitored intensively using chromatography techniques including gas chromatography (GC)20,24 and high-resolution mass spectrometry (MS) for structural elucidation along with different ionization techniques.24
In this work, we report on the application of a GCMS method for analyzing and identifying the soluble side-products in the LiMEAS process. This approach with MS detection gives an insight to understand the chemical and electrochemical reactions in non-aqueous LiMEAS qualitatively and quantitatively. The aim of this report is to investigate possible degradation products formed during LiMEAS in the most widely used, efficient, and promising ether-based electrolytes, such as THF, DME and DG. The assessment of individual electrolytes and comparison between each is very important to select the most efficient and stable system for LiMEAS. This report brings the field closer to understanding of the stability and aging effects vital for advancing the technological application of LiMEAS.
Experimental methods
Electrochemical ammonia synthesis
The electrochemical measurements of this study were performed in non-aqueous electrolytes in an OMNI-LABORATORY (Vacuum Atmospheres) Ar-filled glovebox. Single compartment borosilicate glass cells were used with a volume of 10 mL. All the LiMEAS experiments were performed using 7 mL of electrolyte, with three immersed electrodes: a molybdenum foil (0.125 mm, 99.9%, Goodfellow) working electrode, a platinum-wire (99.9%, Goodfellow) pseudo-reference electrode and a platinum-mesh (99.9%, Goodfellow) counter electrode. Both counter and working electrodes had the same geometrical area of 1.5 cm2. Non-aqueous organic solvents used in this study were tetrahydrofuran (THF, anhydrous, 99.9%, inhibitor-free, Sigma-Aldrich), dimethoxyethane (DME, anhydrous, 99.9%, Sigma-Aldrich), and diethylene glycol dimethyl ether or diglyme (DG, anhydrous, 99.99%, Sigma-Aldrich). The electrolytes contained 0.5 M of lithium perchlorate (LiClO4, battery grade, 99.99%, Sigma-Aldrich) mixed with 1 vol% ethanol (anhydrous EtOH, 99.5%, AcroSeal, Sigma-Aldrich) and the appropriate solvent. The exact potential vs. standard hydrogen electrode (SHE) was determined with slightly fluctuating current. The iR drop in the electrolyte was measured by electrochemical impedance spectroscopy (EIS). It was generally in the range of 20–150 Ω for the 0.5 M solutions of LiClO4 depending on the solvent. For the chronopotentiometry (CP) experiments, the applied current was negative, 3 mA cm−2. We estimated the initial working electrode potential to be approximately −3.1 V vs. SHE under reaction conditions, which became slightly more negative during the experiment. The faradaic efficiencies were within 2–5% depending on the electrolyte. The lowest FE was obtained in DG and the highest in THF.A modified colorimetric indophenol method was used to quantify the synthesized ammonia.13,25 The UV/Vis spectroscope (UV-2600, Shimadzu) with absorbance was used to characterize the samples between 400–1000 nm. Each spectrum was analyzed after subtraction of the blank sample solution. The measurements were performed with 0.5 mL sample of the water trap and three 0.5 mL samples from the electrolyte including one sample from the electrolyte for a background spectrum. The difference between the peak around 630 nm and the trough at around 860 nm is used as maximum and minimum, respectively. A fitted curve of the difference between the peak and trough of each concentration showed a linear regression with an R2 value of 0.999. The amount of ammonia in the headspace was quantified by de-gassing the system through an ultra-pure water trap, however revealed to be negligible in the experiments of the current study. The samples were treated as described previously, to determine the ammonia concentration.13
Gas chromatography-mass spectrometry (GCMS)
Gas chromatography-mass spectrometry (GCMS) measurements were done with an Agilent 6890N gas chromatograph with manual liquid sample injection. The analysis of all the samples was performed immediately after LiMEAS. The samples of 0.1 μL were injected to an inlet (200 °C) and electron-ionized (30 eV) to form positive ions (e.g., M+). The injection volume of 0.1 μL was optimized to get reliable intensities of the peaks without overloading the detector. Helium (N5.0 purity, Air Liquide) was used as carrier gas with 5 mL min−1 column flow and 5 mL min−1 purge flow. Cotton fiber in the inlet was used for collecting the LiClO4 salt and any possible solid in the electrolyte upon evaporation of the injected liquid phase. Two columns connected in series were used in this study: a nonpolar Agilent CP-Volamine (30 m × 0.32 mm × 0.32 μm) and non-polar Agilent HP-5MS UI (30 m × 0.32 mm × 0.25 μm). The temperature program for the columns started at 50 °C which was held for 3 min. Afterwards, temperature was ramped with 10 °C min−1 until 100 °C with dwelling at each temperature of 1 min. The temperature was held for 2 min at 100 °C, then ramped to 120 °C with 20 °C min−1 and held for 3 min. Finally, the system was heated to 150 with 20 °C min−1, held for 3 min, and ended with 20 °C min−1 dwelling at 200 °C for 5 min. Afterwards, the system was cooled down to 50 °C. The temperature program used for GC is shown in Fig. S2 in ESI.† The GC was interfaced with an Autospec v4.0 mass spectrometer (Waters Corporation). The overall measurement time was 30 min screening the mass range from 1–200 m/z with an event time of 0.1 s in scan mode. Further detailed description of the method is given in the ESI.†EI-MS analysis
A sector mass spectrometer is directly connected to the GC system (Autospec, Waters Corporation), achieving a mass resolution of ∼30![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 and mass accuracy of 1–2 ppm. The chromatograms were analyzed with MassLynx v4.0 (Waters Corporation) software. The mass spectrometer was run in the electron impact ionization (EI) mode with the following parameters: the temperature of the ion source together with the GC inlet was set to 200 °C, and the filament was operated at a voltage of 30 eV. The detector voltage was set relative to the respective tuning results. Compound identification and corresponding structural formulae were assigned relying upon the National Institutes of Standards (NIST) Library.26 The instrument was operated in a full scan mode ranging from m/z 1 to 200. The compounds were additionally confirmed by the comparison of their retention time and fragment patterns with in-house made standards from commercially available compounds (Sigma Aldrich). The solution of 100 ppm of the compound was made in the appropriate solvent and injected into GCMS. Before each run, a mixture of ethanol and THF was used to rinse the lines and analyzed in the same manner to generate a background spectrum. In order to ensure that the observed results represent constituents of degraded electrolyte upon LiMEAS, the freshly made electrolytes containing the appropriate solvent, EtOH and LiClO4 were injected. No other compounds were found except the components. This confirmed the initial electrolytes to be clean as well as no reaction happening in the hot GC inlet part. Every recorded GCMS data set was analyzed mass-by-mass in order to evaluate the molecular masses of the molecular ions and fragmentation patterns. All the GCMS data were processed using the OpenChrom and MassLynx 4.0 software with in-house made Python script. Elemental formulae were assigned for peaks with signal-to noise (S/N) ratios larger than 3 and intensities higher than 10 times compared to the corresponding signals in the background spectrum. Firstly, total ion current (TIC) chromatograms were recorded and analyzed. Then, each sample was carefully analyzed by m/z values as extracted ion chromatograms (XIC). In an extracted-ion chromatogram (XIC or EIC), also called a reconstructed-ion chromatogram (RIC), one or more m/z values representing one or more analytes of interest are recovered (‘extracted’) from the entire data set for a chromatographic run.27 This enabled detection of many species at specific retention time in the chromatograms, which were not visible as peaks in TIC. The total sum intensity, in other words called the base peak intensity, within a mass tolerance range of interest around m/z of a particular compound is plotted at every point in the analysis. The size of the mass tolerance range typically depends on the mass accuracy and mass resolution of the data collecting instrument. This method is of a great use for revisiting data to detect previously unsuspected compounds, to highlight potential isomers, resolve suspected co-eluting substances, or to provide clean chromatograms of compounds of interest. XIC is generated by focusing upon the ions of interest in the whole data set containing the full mass spectrum over time after the fact. More discussion on the methods is given in the ESI.†
000 and mass accuracy of 1–2 ppm. The chromatograms were analyzed with MassLynx v4.0 (Waters Corporation) software. The mass spectrometer was run in the electron impact ionization (EI) mode with the following parameters: the temperature of the ion source together with the GC inlet was set to 200 °C, and the filament was operated at a voltage of 30 eV. The detector voltage was set relative to the respective tuning results. Compound identification and corresponding structural formulae were assigned relying upon the National Institutes of Standards (NIST) Library.26 The instrument was operated in a full scan mode ranging from m/z 1 to 200. The compounds were additionally confirmed by the comparison of their retention time and fragment patterns with in-house made standards from commercially available compounds (Sigma Aldrich). The solution of 100 ppm of the compound was made in the appropriate solvent and injected into GCMS. Before each run, a mixture of ethanol and THF was used to rinse the lines and analyzed in the same manner to generate a background spectrum. In order to ensure that the observed results represent constituents of degraded electrolyte upon LiMEAS, the freshly made electrolytes containing the appropriate solvent, EtOH and LiClO4 were injected. No other compounds were found except the components. This confirmed the initial electrolytes to be clean as well as no reaction happening in the hot GC inlet part. Every recorded GCMS data set was analyzed mass-by-mass in order to evaluate the molecular masses of the molecular ions and fragmentation patterns. All the GCMS data were processed using the OpenChrom and MassLynx 4.0 software with in-house made Python script. Elemental formulae were assigned for peaks with signal-to noise (S/N) ratios larger than 3 and intensities higher than 10 times compared to the corresponding signals in the background spectrum. Firstly, total ion current (TIC) chromatograms were recorded and analyzed. Then, each sample was carefully analyzed by m/z values as extracted ion chromatograms (XIC). In an extracted-ion chromatogram (XIC or EIC), also called a reconstructed-ion chromatogram (RIC), one or more m/z values representing one or more analytes of interest are recovered (‘extracted’) from the entire data set for a chromatographic run.27 This enabled detection of many species at specific retention time in the chromatograms, which were not visible as peaks in TIC. The total sum intensity, in other words called the base peak intensity, within a mass tolerance range of interest around m/z of a particular compound is plotted at every point in the analysis. The size of the mass tolerance range typically depends on the mass accuracy and mass resolution of the data collecting instrument. This method is of a great use for revisiting data to detect previously unsuspected compounds, to highlight potential isomers, resolve suspected co-eluting substances, or to provide clean chromatograms of compounds of interest. XIC is generated by focusing upon the ions of interest in the whole data set containing the full mass spectrum over time after the fact. More discussion on the methods is given in the ESI.†
The area of the most intense XIC fragment of the eluting compound was integrated and compared to the main peaks of the main components of the electrolyte, the solvent and EtOH. After integration, the relative amount of compound was estimated and compared to the amount of experimentally added 100 ppm concentration standard in THF. Some of the compounds, especially for DG system were difficult to obtain, so the amount from a standard value is missing. However, the integration of the XICs method for other compounds is in a good agreement with the experimental standards and gives confidence in the evaluated amount of the compound molecule. Further description of the method is given in the ESI.†
Results and discussion
Detection of the decomposition species formed in the liquid electrolyte
The degradation of LiMEAS organic components starts at both the positive and negative electrode–electrolyte interface. The passivating SEI layer forms, providing Li ion diffusion through it and direct electrolyte and metallic Li contact. This enables chemical reactions between the highly reactive metallic Li and the solvent in the electrolyte. The chemical and electrochemical redox processes result in decomposition products that may result in the high overpotentials reported in the literature,4 and final inhibition of N2RR with the following turn off of the LiMEAS. Here, we aim to report on the characterization of these mostly organic materials formed in LiMEAS by means of GCMS in order to promote the technique for other investigation of decomposition products in liquid organic electrolytes with application across various electrochemical synthesis processes. Three of the most promising ether-based electrolytes, tetrahydrofuran (THF), dimethoxyehtane (DME) and diglyme (DG), have been chosen in this study to characterize the soluble decomposition products.Fig. 1a shows the total ion current chromatograms (TIC) of the three selected solvents after LiMEAS. The same chromatogram with higher magnification is represented in Fig. 1b. Mainly two distinct peaks can be seen in the chromatograms, which in general present the main components of the electrolyte for LiMEAS. The first peak at ∼3 min represents ethanol (EtOH) elution, while the most intense peak is the solvent (THF; DME; DG). The small difference in retention time of EtOH is probably due to experimental or instrumental error. The set pressure of carrier He gas was not very stable for the set flow rate in the used system. This and the manual injection and program execution introduced slight differences in EtOH retention time, coupled with contamination of the GC column over time after numerous experiments. From a superficial point of view, no other products can be detected in the chromatograms, except THF. This is because TICs are very dependent on the concentration of the species. Since the solvent and EtOH concentrations are so high compared to the other components, the peak intensities of the latter ones can only be found by analyzing extracted ion chromatograms (XIC) mass-by-mass.
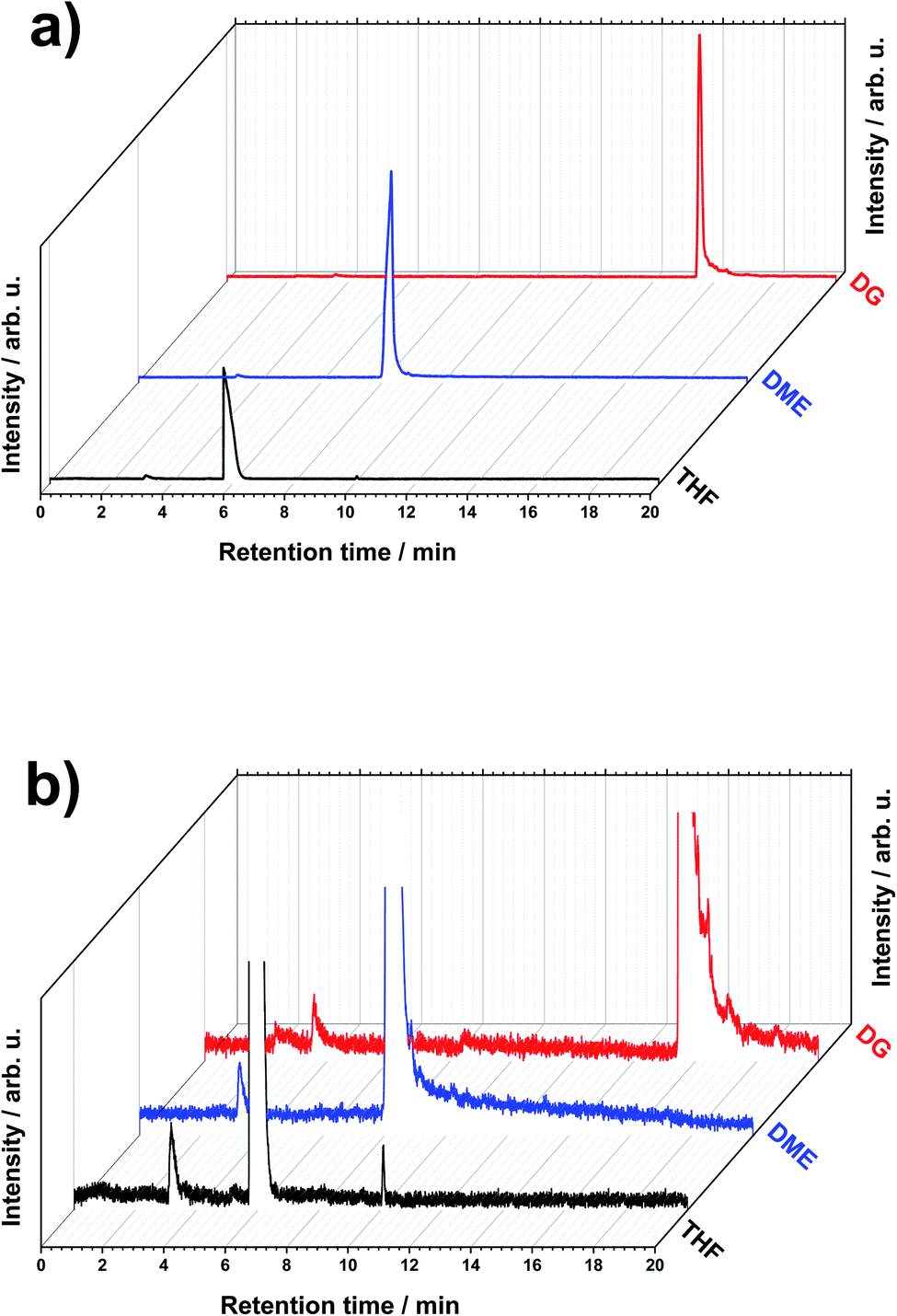 | ||
| Fig. 1 The GCMS total ion current chromatograms (TIC) (a) full and (b) with higher magnification of electrolytes after LiMEAS in different solvents: tetrahydrofuran (THF), dimethoxyethane (DME) and diglyme (DG). | ||
Upon screening XICs by m/z values from 1 to 200, other peaks start to become visible, enabling the discovery of the small amounts of various components present in the systems. No peaks at higher than 200 m/z values were found, that is why 200 m/z was chosen as an end of the scanning range. Fig. 2 shows an example of two different XICs obtained with the set m/z values of 15 and 31. These m/z values represent methyl (CH3+) and methoxyl (CH3O+) or methylene hydroxyl (CH2OH+) organic species, respectively, that are common in many fragmentation patterns of organic molecules of this study supported by NIST database center26 depending of the functional groups. The m/z 15 is found in the molecules containing methyl group, however m/z 31 is present in the molecules containing alcohol or OH functional group. Following the method, more peaks in the chromatograms appear at specific retention time. This means that there are species containing the fragmentation parts with the m/z values of 15 and 31, which elute from the GC system at different and distinct retention time. One can see that both of these m/z values are present in the decomposition of the solvents. This is probably due to the fact that the concentration of the solvent and EtOH is so high that any rational fragmentation species can be detected at the retention time of them.
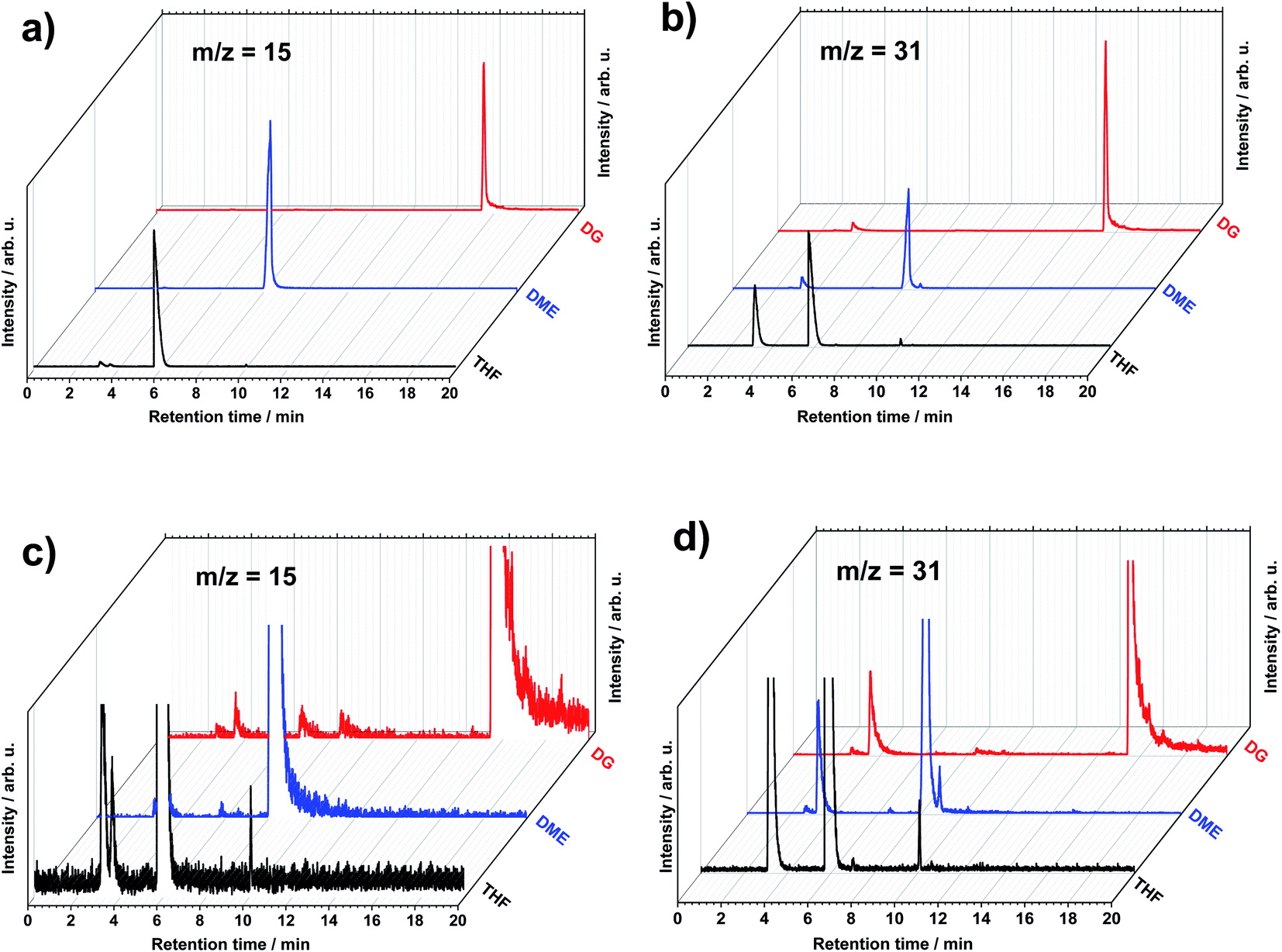 | ||
| Fig. 2 The GCMS extracted ion chromatograms (XIC) of selected (a) m/z = 15 and (b) m/z = 31 with zoom-in chromatograms as (c) and (d), respectively, in the electrolytes for LiMEAS: tetrahydrofuran (THF), dimethoxyethane (DME) and diglyme (DG). | ||
After LiMEAS in DME, the electrolyte was analyzed by GCMS providing the XIC results as shown in Fig. 3. Fig. 3a shows all the specific XICs with their individual contribution to the TIC at specific retention time, and Fig. 3b represents a mass spectrum of MS scan nr. 994 at 5.57 min retention time with thee overlaid spectrum from NIST database. One can spot that the distinct peak at ∼5.6 min, which was superficially absent in TIC (Fig. 1), is now easily distinguishable in XICs with some specific m/z values to the compound eluting in the GCMS system. In this manner, all the samples were analyzed and fragmentation particle masses recorded. The same procedure was applied for the other m/z from 1 to 200, though not all the m/z contained intensities, only the background or the spikes of contaminant molecules in the system. The rational MS fragmentation of organic molecules was always present at the specific solvent as well as EtOH retention time in all the chromatograms. This brings to the conclusion that the concentration of the solvent was so huge compared to the other components that LiMEAS decomposition products were just shadowed in TIC and impossible to detect. However, one should always remember that TIC is a sum of all the m/z at a specific time (or scan). This is why lots of m/z or peaks are shadowed in the TICs. According to Fig. 3b, the analyzed compound was in a good agreement with the database. However, it is important to remind that we used softer 30 eV ionization compared to 70 eV in NIST database due to instrument limitations – the electron energy was optimized for the instrument and tungsten (W) source filament. This explains why we see more molecular ion signal at 88 m/z compared to the main fragmentation species at 45 m/z as can be seen in the database spectrum. We also see some more species which can be explained by contaminants in the column or system in general. One should remember that the Fig. 3b represents one scan-moment, and the next one is slightly different with respect to m/z intensities. Also, m/z values at 28 and 32 which are nitrogen and oxygen in the system. And in general, NIST database is created with different MS parameters than we use, so it has to be adjusted carefully. Many, though not all, of the structures were supported by the NIST database. However, they were confirmed by the home-made standard sample analysis.
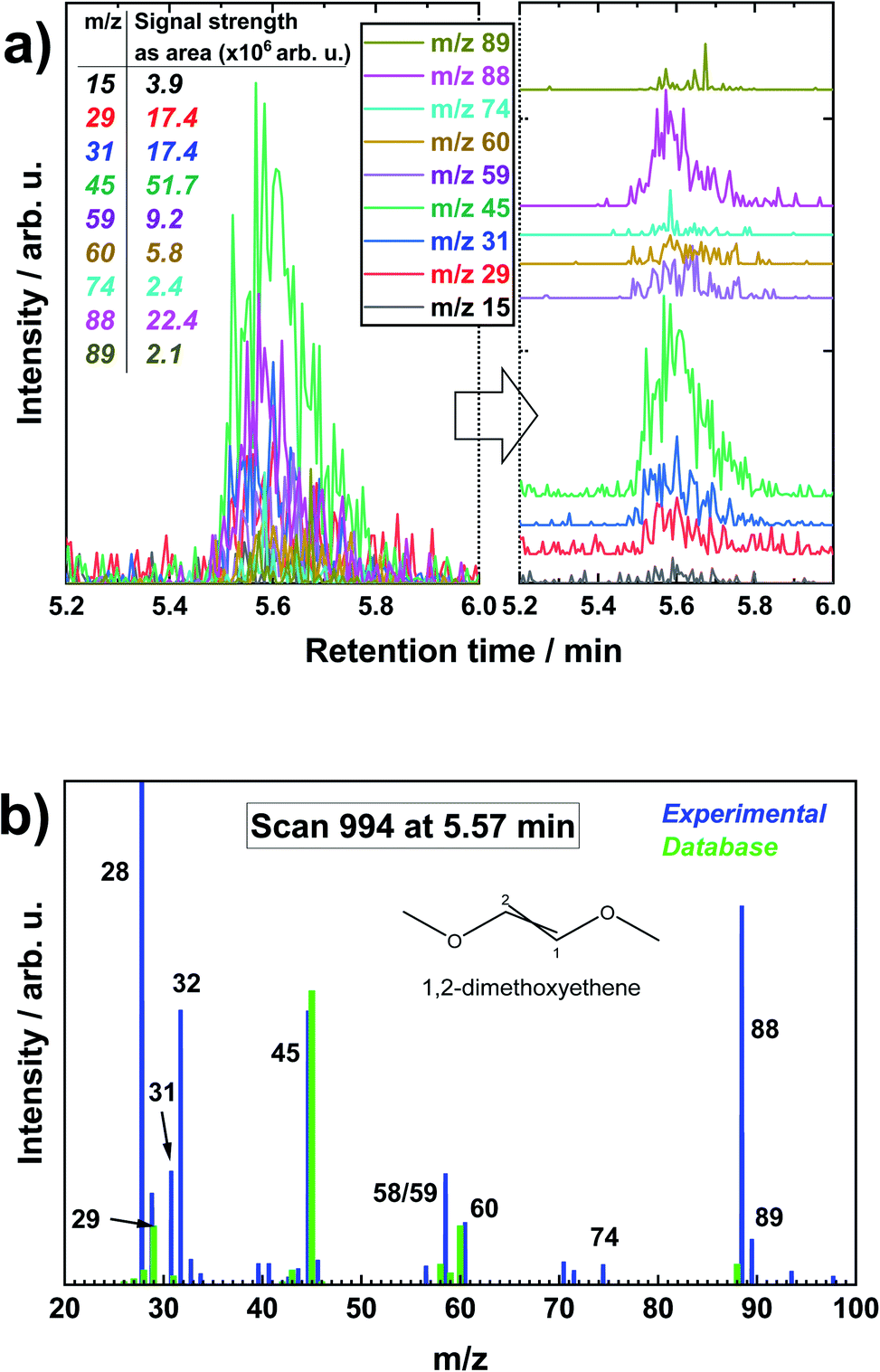 | ||
| Fig. 3 (a) The extracted ion chromatograms (XIC) vs. retention time of a specific chromatographic peak at 5.5–5.8 min retention time after LiMEAS in dimethoxyethane (DME). (b) Representative mass spectrum at 5.57 min retention time (MS scan nr. 994) with the comparison of MS spectrum from NIST database. | ||
Similar analysis results of peaks at specific retention times for THF and DG are represented in Fig. 4a and b, respectively. The peaks or specific retention times have been chosen randomly. The figures include the m/z values which had at least some intensity to evaluate. The signal strength of each m/z was evaluated by integrating the peaks and the integration results are presented as inset tables. Different m/z values have different intensity of the peaks. Some of the specific peaks are very intensive compared to the others. And this depends on the decomposing compound and ionization energy of the electrons.
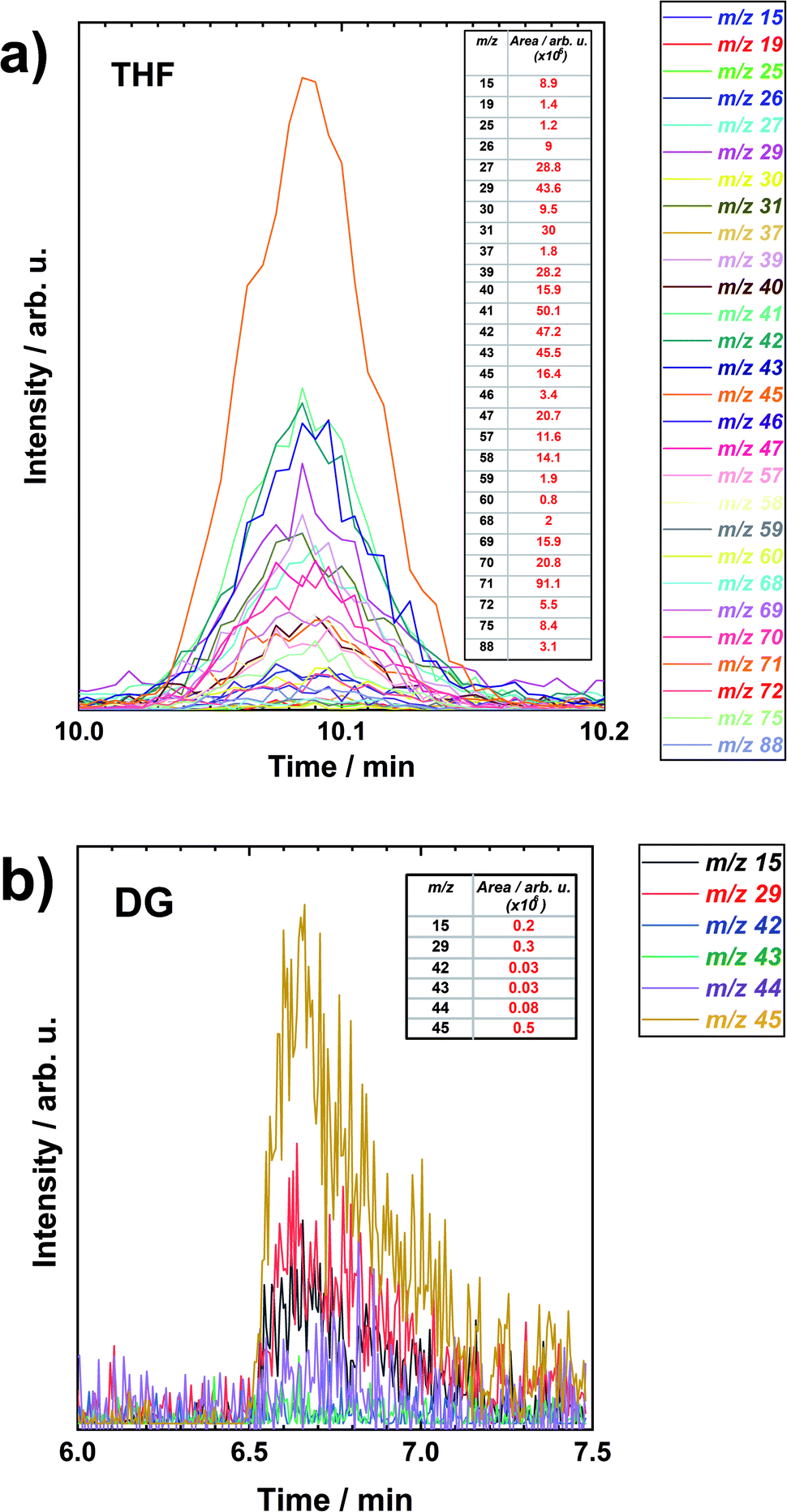 | ||
| Fig. 4 The extracted ion chromatograms (XIC) vs. retention time of a specific chromatographic peak for (a) THF and (b) DG after LiMEAS. The insets are the integrals of the peaks. | ||
The ammonia signal in GCMS is mostly overlaying with H2O signal. However, it depends on the GC program used. The current program (Fig. S2†) was optimized not for separating ammonia from H2O, but for analyzing organic volatile species in three different systems. These would be two different experimental programs for GCMS. If the GC program is modified in the way that the dwelling at 50 °C is kept for 6 min and the ramping towards 100 °C is done with 5 °C min−1 steps, ammonia can be separated from H2O. An example of such a chromatogram is given in Fig. S5 in ESI.† However, the GC program had to be optimized and used for analyzing the volatile organic decomposition products in order to save time. With this in mind, the ammonia was quantified by the well-established indophenol method described in the experimental part and literature.13,25
In the following sections, the decomposition products in each of the three different electrolytes are analyzed in more detail. The molecules of the decomposition products were constructed from the fragmentation patterns, relying heavily, but not always, on the NIST database, as the spectra are dependent on the ionization voltage, and therefore might vary in intensity and number of peaks. Also, to proceed further with the validation of the test and capability of the GCMS technique in the current application with the equipment used, we enabled the quantification methodology of the components after a typical LiMEAS in different electrolytes with in-house made standard solutions of the molecules. The consequent results were compared to the integration of the XIC results for the different components. The analysis of the standard solutions was performed individually. This brought high confidence in molecule identification since the retention time matched well. Some of the DG decomposition products were not commercially available, so the amount from a standard value is missing. However, the integration of the XICs method for other compounds agrees well with the experimental standards, providing confidence in the evaluated amount from GCMS and structure of the molecule.
Detection of the decomposition products in THF-based electrolyte
The LiMEAS in THF-based electrolyte after passing 90 C charge with −3 mA cm−1 constant current resulted in the compounds summarized in Fig. 5. The experiment lasted for almost 5 h and resulted in the approximately −6 V of working electrode and around 2 V of counter electrode vs. SHE. The electrochemical experimental conditions are visualized in Fig. S3a of ESI.† After the experiment was over, the amount of the measured ammonia resulted in 3–5% of Faradaic Efficiency (FE). The structures of the molecules were constructed by the fragment species at distinct retention time from XICs with the m/z values given in Table 1. The gases from the atmosphere like O2, N2, Ar, CO2 and water (H2O) are excluded from the table for convenience to read together with the main components of the electrolyte, ethanol and THF. It can be seen that the most of the products from THF decomposition were formed upon oxidation of the solvent at the counter electrode or ring opening reactions. The oxidation of THF is either dehydrogenation inducing unsaturated bonds or stem from an increase of the oxygen content in the molecule. Small molecules might be formed from ethanol transformations. However, it is important to mention that most of the products can be associated with the well-known and described initial Li reaction with THF,14 which afterwards might yield in the production of the ring-structures drawn in Fig. 5 or ring-opening reactions. We are sure that no reactivity of LiClO4 towards the electrolyte organic components occurred performing the analysis of freshly made electrolytes before LiMEAS. The most exotic result in the analysis of the decomposition products was considered to be the formation of butane (m/z = 58). This could be confused with acetone, however, acetone mass spectrum contains very small intensity m/z = 28, which is characteristic for all the molecules containing C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O functional groups. In the case of butane, there is a decent signal at 28 and 29, which correspond to CH2CH3 fragment. Regarding the compound (4) in Fig. 5, it is very difficult to evaluate which isomer is the main compound after decomposition by GCMS. Most likely, the reaction involved is just oxidation of THF, which we practically see in all of the experiments of LiMEAS with THF-based electrolyte. In order to understand how the molecules of Fig. 5 were formed, we draw the mechanisms for each product which are presented in the Fig. S4a–g in the ESI.†
O functional groups. In the case of butane, there is a decent signal at 28 and 29, which correspond to CH2CH3 fragment. Regarding the compound (4) in Fig. 5, it is very difficult to evaluate which isomer is the main compound after decomposition by GCMS. Most likely, the reaction involved is just oxidation of THF, which we practically see in all of the experiments of LiMEAS with THF-based electrolyte. In order to understand how the molecules of Fig. 5 were formed, we draw the mechanisms for each product which are presented in the Fig. S4a–g in the ESI.†
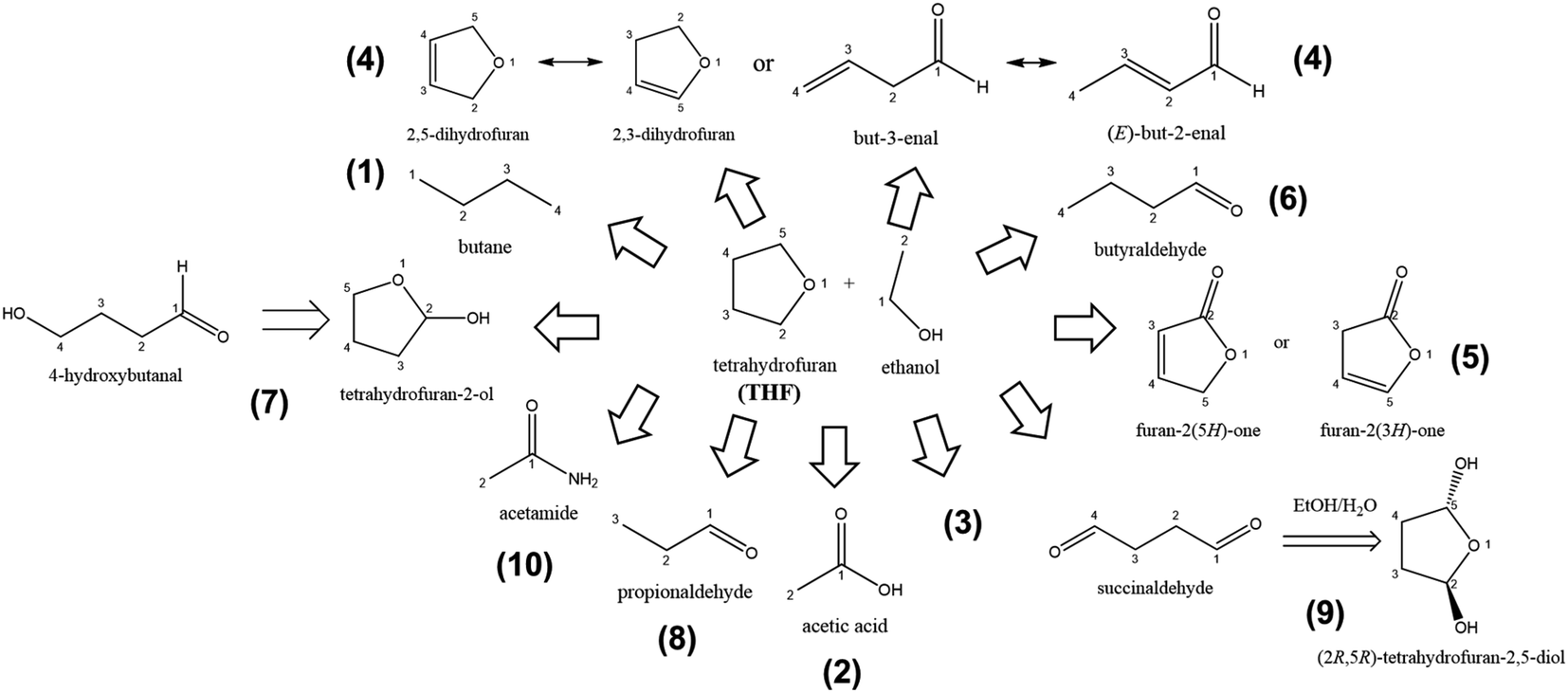 | ||
| Fig. 5 The schematic of decomposition products formed after LiMEAS in THF-based electrolyte. The compounds are summarized in Table 1. | ||
| Nr. | Retention time (min) | m/z | Compound namea | GCMS | Standard retention time (min) | From solution (ppm ± 5) | ||
|---|---|---|---|---|---|---|---|---|
| Area (arb. u.) | Amount (ppm ± 5) | |||||||
| a No chlorinated compounds have been detected, which could be associated to transformations of perchlorate anion in the electrolyte.b Water content before LiMEAS according to Karl–Fischer titration. | ||||||||
| 1 | 3.6–3.7 | 15; 29; 43; 58 | Butane | 4370 | 8 | 3.6–3.7 | 7 | |
| 2 | 4.4–4.5 | 29; 31; 43; 45; 58; 59; 60 | Acetic acid | 4297 | 8 | 4.4–4.5 | 7 | |
| 3 | 4.8–5.0 | 15; 16, 29; 45 | — | 2135 | 4 | — | — | |
| 4 | 5.1–5.3 | 27–29; 31; 37–42; 68–70 | (2,5 or 2,3)-Dihydrofuran | 25213 |
45 | 5.1–5.3 | 41 | |
| 5 | 7.0–7.1 | 31; 42; 47; 59; 72; 84 | Furan-2(5H or 3H)-one | 10514 |
19 | 7.0–7.1 | 17 | |
| 6 | 9.7–10.0 | 15; 29; 41; 42; 57; 58; 71 | Butyraldehyde | 4766 | 8 | 9.7–10.0 | 8 | |
| 7 | 10.0–10.2 | 15; 19; 25–27; 29; 30; 31; 37; 39; 40–43;45–47;57–60; 68–72; 75; 88 | 4-Hydroxybutanal or tetrahydrofuran-2-ol | 45![[thin space (1/6-em)]](https://www.rsc.org/images/entities/b_char_2009.gif) 717 717 |
82 | 10.0–10.2 | 74 | |
| 8 | 10.6–10.8 | 28; 29; 31; 41; 43; 58 | Propionaldehyde | 32693 |
59 | 10.6–10.8 | 53 | |
| 9 | 11.7–11.9 | 28; 42; 55; 87 | Succinaldehyde | 38859 |
70 | 11.7–11.9 | 63 | |
| 10 | 12.5–12.8 | 15; 28; 43; 59 | Acetamide | 9096 | 16 | 12.5–12.8 | 14 | |
| H2O | 2.2–2.5 | 16; 17; 18 | Water | 71245 |
145 | 2.2–2.5 | 36b | 150 |
The results for GCMS analysis in THF-based LiMEAS electrolyte are summarized in Table 1. The main decomposition product in THF-based LiMEAS was found to be compound (7) 4-hydroxybutanal or tetrahydrofuran-2-ol. It is difficult to distinguish which exact compound is it by GCMS because both compounds fragment into similar particles by m/z. According to the analysis outcomes described above and visualized in Fig. 4, the m/z values from MS and the names of the decomposition products were constructed. The sector MS used has a very high sensitivity, which allows distinction of many molecules from peaks with weak intensity to be constructed, as can be seen in the example shown in Fig. 5. Notably the experimental MS spectra display a good signal-to-noise ratio. The very sensitive MS analysis unambiguously allowed identifying most of the molecules after LiMEAS dissolved in the liquid electrolyte based on THF. Thus, the capability of this technique for the analysis and identification of the organic molecules is clear.
Table 1 summarizes all the compounds characterized by GCMS at different retention time and their amount in the THF-based electrolyte after LiMEAS. The amount of the compounds was evaluated from the standards of the solutions of the component and from integration of the peaks of XICs. The approach was chosen to integrate the peak in XIC of the most intense fragment species at different retention time and comparison with the integral of the solvent XIC peak. The latter one for THF molecule is at m/z 42. Most of the materials were formed in very small amounts and were detected less than 100 ppm even after 90 C passed through the electrolytes. One can assume that this is very promising because the electrolyte can be considered stable. It is noteworthy that the amounts estimated by the integration of the chromatographic peaks are higher than from experimental standard solutions. This is probably due to overestimation and errors in the models applied for the integration of the chromatographic peaks by MassLynx or OpenChrom software. However, the integration and experimental standard results are in a close proximity to each other.
Detection of the decomposition products in DME-based electrolyte
The structural molecular formulae of the compounds after GCMS analysis in DME-based LiMEAS are summarized in Fig. 6. The electrochemical experimental conditions are shown in Fig. S3 of ESI.† The fragment species at distinct retention time in XICs with the m/z values are given in Table 2. The atmosphere gases, such as O2, N2, Ar, CO2 and water (H2O) are excluded from the table for convenience to read together with the main components of the electrolyte, ethanol and DME. Most of the soluble decomposition products in DME form linear structure. The oxidation reaction dominates together with bond cleavage products. No ring closure or cyclization reactions can be detected. Similar to the measurement of THF decomposition in LiMEAS, the small molecules detected here were most likely produced from EtOH transformations. In the current case of DME, Li probably reacts with the methylene group in the structure of DME, resulting in the various modifications and cleavage reactions. The oxidation reactions, which involve double bond formation or introduction of carbonyl or hydroxyl functional groups on the molecular structure of DME, most likely occur on the counter electrode. Further studies need to be performed separating the electrode compartments and analyzing the decomposition compounds of the anolyte and catholyte individually. According to the observed and constructed molecules, the products are more or less expected. All the molecules have rational genesis from DME molecular structure following the path of possible oxidation or linear structure cleavage reactions. The main decomposition product in THF-based LiMEAS was found to be compound (5) (Z,E)-1,2-dimethoxyethene. The twisted double bond in the structure shows that there are isomers of the compound, which cannot be distinguished by GCMS. The mechanisms of the reactions are most likely similar to THF cases presented in Fig. 4a–g of the ESI.†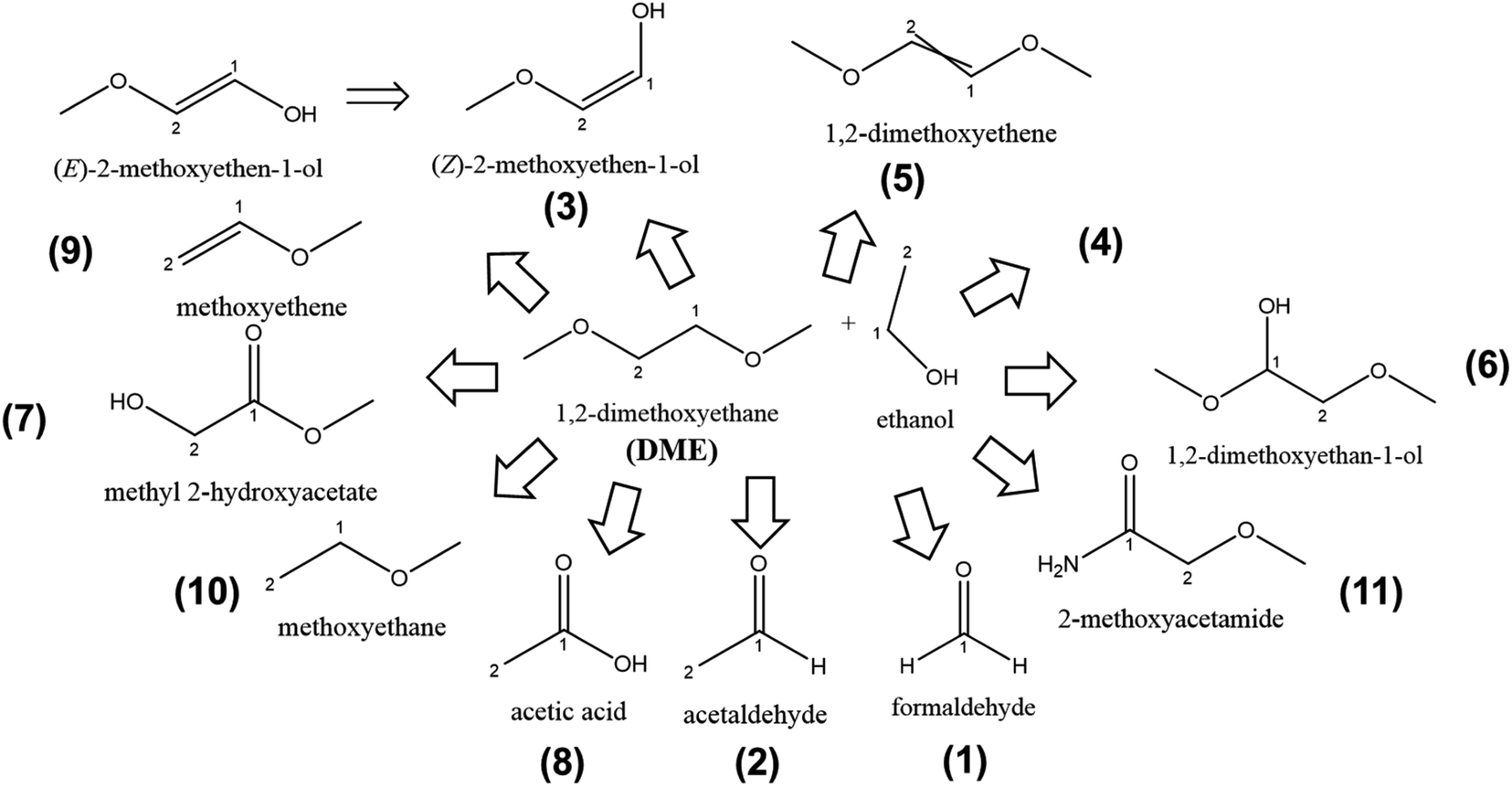 | ||
| Fig. 6 The schematic of decomposition products formed after LiMEAS with DME electrolyte. The compounds are characterized and summarized in Table 2. | ||
| Compound | Retention time (min) | m/z | Compound namea | GCMS | Standard retention time (min) | From standard solution (ppm ± 2) | ||
|---|---|---|---|---|---|---|---|---|
| Area (arb. u.) | Amount (ppm ± 2) | |||||||
| a No chlorinated compounds have been detected, which could be associated to transformations of perchlorate anion in the electrolyte.b Water content before LiMEAS according to Karl–Fischer titration. | ||||||||
| 1 | 1.4–1.7 | 29; 30 | Formaldehyde | 6114 | 35 | 1.4–1.5 | 30 | |
| 2 | 1.7–1.8 | 15; 29; 31; 32; 44 | Acetaldehyde | 1117 | 6 | 1.7–1.8 | 5 | |
| 3 | 3.3–3.4 | 15; 31; 57; 74 | 2-Methoxyethene-1-ol | 561 | 3 | 3.2–3.1 | 2 | |
| 4 | 4.8–5.0 | 15; 29; 45 | — | 4215 | 24 | — | — | |
| 5 | 5.6–5.7 | 15; 29; 31; 45; 58; 59; 60; 74; 88; 89 | (Z,E)-1,2-Dimethoxyethene | 203137 |
1177 | 5.6 | 1012 | |
| 6 | 7.8–7.9 | 19; 27; 29; 30; 31; 41; 43; 44; 47; 58; 59; 60; 74; 88; 89; 109 | 1,2-Dimethoxyethan-1-ol | 10680 |
62 | — | — | |
| 7 | 8.2–8.4 | 29; 45; 58; 90 | Methyl-2-hydroxyacetate | 7743 | 44 | 8.41 | 38 | |
| 8 | 9.2–9.3 | 29; 31; 43; 45; 58; 59; 60 | Acetic acid | 9528 | 55 | 9.1–9.3 | 47 | |
| 9 | 9.7–9.8 | 29; 43; 45; 58 | Methoxyethene | 4983 | 28 | 9.7–9.9 | 24 | |
| 10 | 10.2–10.5 | 29; 31; 43; 45; 58 | Methoxyethane | 9477 | 54 | 10.1–10.2 | 47 | |
| 11 | 14.1–14.2 | 29; 31; 45; 58; 59; 60; 88; 89 | 2-Methoxyacetamide | 6770 | 39 | 14.0–14.1 | 33 | |
| H2O | 2.2–2.5 | 16; 17; 18 | Water | 16576 |
110 | 2.2–2.5 | 48b | 107 |
The GCMS analysis performed after LiMEAS in DME-based electrolyte is given in Table 2, with the m/z values from MS and the names of the decomposition products constructed. Some of the peaks contained quite considerable amount of m/z values complicating the identification process of the product structure. The NIST database does not contain such complicated compounds which could be identified. Some of the most characteristic values of the compound (5) were presented above in Fig. 3.
The compounds characterized by GCMS at different retention time and their amount in the DME-based electrolyte after LiMEAS are given in Table 2. The quantification of the compounds was performed from the standards and integration of the GCMS peaks as described above for THF. Most of the materials were formed in ppm amounts. The compound (5) was detected in more than 1000 ppm after 90 C passed through the electrolyte, which is unacceptable for the stability for the LiMEAS process. The decomposition of the solvent is quite considerable under the applied experimental conditions, and the electrolyte is determined to not be stable. The product (5) is formed either after the initial Li reaction with methylene group in DME or via oxidation of DME most likely on the counter electrode, as in the case of THF. The rest of the compounds formed in decent and comparable amounts to the THF experiment. As for the THF-based electrolyte after LiMEAS, the integration of GCMS and experimental results match to each other well, however the later ones are higher. On the other hand, it can be seen that the amount of the compounds in THF and DME detected after LiMEAS is the same, reaching 11 different decomposition species. This can most likely be attributed to the similar nature of the solvents as ethers, which are quite stable organic molecules. However, both of these solvents tend to form radicals and peroxides in the presence of oxygen.
Detection of decomposition compounds in DG-based electrolyte
Pursuing our aim to investigate as many candidate solvents for LiMEAS as possible to understand the system with respect to decomposition and stability, we performed the LiMEAS in DG-based liquid electrolyte according to the same experimental procedure. The electrochemical experimental conditions are given in ESI.† The soluble compounds were analyzed by means of GCMS, and the constructed structures of the constituents are summarized in Fig. 7 and Table 3. The gases from the atmosphere i.e. O2, N2, Ar, CO2 and water (H2O) are excluded from the table together with the main components of the electrolyte, ethanol and DG, because it focuses on specific species formed. It can be seen that no cyclization and only linear structure cleavage together with oxidation reactions dominate among the products formed after LiMEAS. Since DG is a molecule with quite high molecular mass, the decomposition products are constructed from many fragmentation patterns. The more molecular mass, the more complicated mass spectrum is monitored. It is also important to mention that DG decomposition process results in the largest amount of by-products after LiMEAS, in this case of 14 different compounds. The main decomposition product in DG-based LiMEAS was found to be compound (13) (2-methoxyethoxy)ethene. The most exotic molecule detected is the oxalylaldehyde (14), which can be formed after the complete collapse of the linear structure of the DG-based electrolyte, since it was not detected in the previous examples of THF and DME.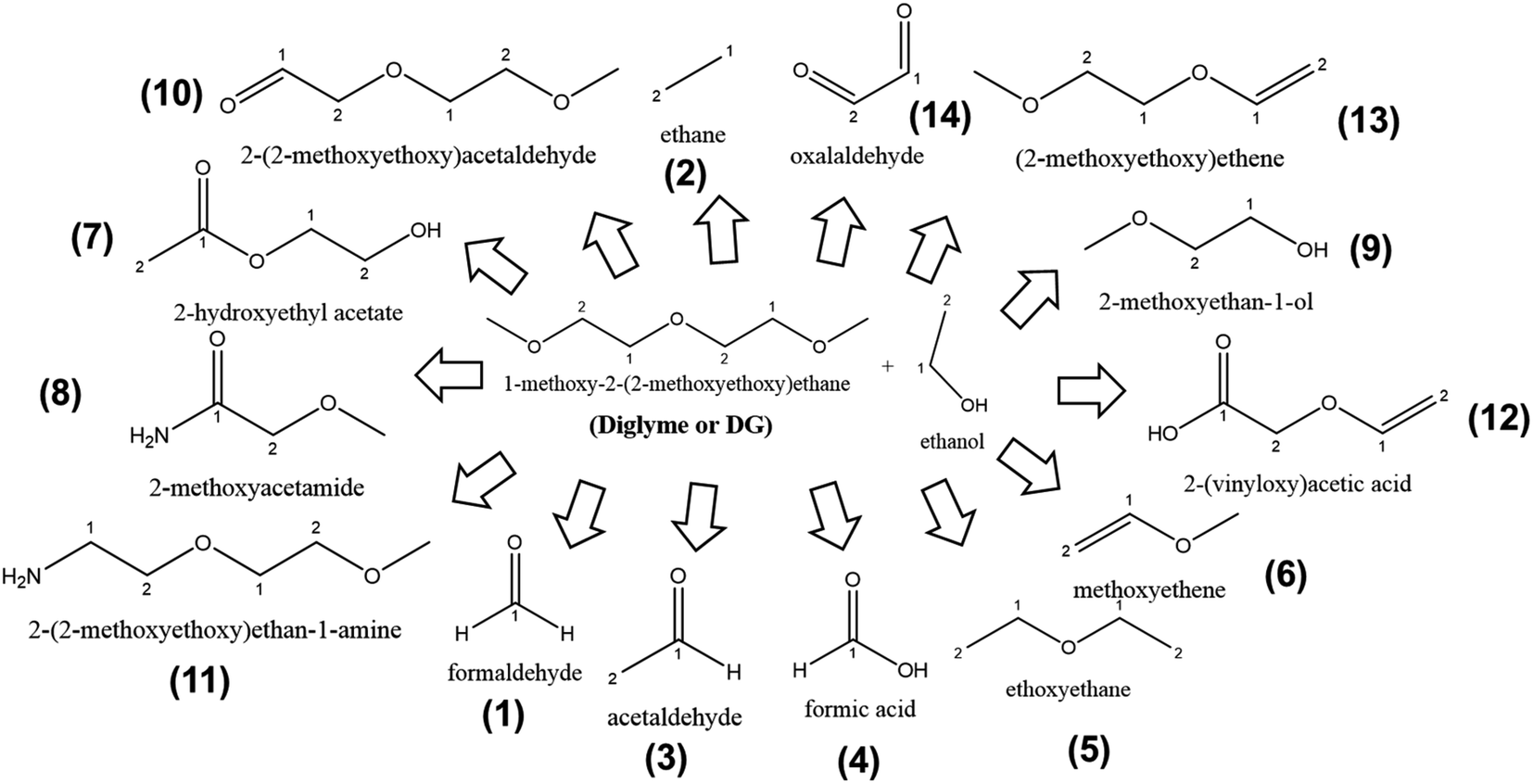 | ||
| Fig. 7 The schematic of decomposition products formed after LiMEAS with DG electrolyte. The compounds are characterized and summarized in Table 3. | ||
| Compound | Retention time (min) | m/z | Compound namea | GCMS | Standard retention time (min) | From standard solution (ppm ± 6) | ||
|---|---|---|---|---|---|---|---|---|
| Area (arb. u.) | Amount (ppm ± 6) | |||||||
| a No chlorinated compounds have been detected, which could be associated to transformations of perchlorate anion in the electrolyte.b Water content before LiMEAS according to Karl–Fischer titration. | ||||||||
| 1 | 2.2–2.4 | 28; 29; 30 | Formaldehyde | 4049 | 36 | 2.2–2.3 | 31 | |
| 2 | 2.5–2.8 | 16; 17; 18; 29 | Ethane | 10562 |
87 | 2.5–2.6 | 75 | |
| 3 | 2.6–2.7 | 15; 16; 29; 30; 31; 32; 43; 44 | Acetaldehyde | 2551 | 21 | 2.6–2.8 | 18 | |
| 4 | 6.5–6.6 | 15; 29; 42; 43; 44; 45 | Formic acid | 7732 | 64 | 6.5–6.6 | 55 | |
| 5 | 8.5–8.6 | 15; 19; 26; 27; 29; 30; 31; 42; 43; 45; 46; 47; 57; 58; 72; 75 | Diethyl ether | 1189 | 9 | 8.5–8.6 | 8 | |
| 6 | 9.1–9.2 | 29; 31; 58; 59 | Methoxyethene | 29630 |
246 | 9.1–9.2 | 211 | |
| 7 | 9.7–9.9 | 27; 31; 58; 59; 104 | 2-Hydroxyethyl acetate | 2810 | 23 | 9.7–9.8 | 20 | |
| 8 | 14.6–14.8 | 29; 58; 59; 72; 88 | 2-Methoxyacetamide | 12701 |
105 | 14.6–14.7 | 90 | |
| 9 | 16.0–16.1 | 17; 29; 31; 61; 70; 71; 72; 74; 75; 86; 88; 103; 106; 108; 109 | 2-Methoxyethan-1-ol | 23085 |
191 | 16.0–16.1 | 164 | |
| 10 | 16.5–16.6 | 15; 26; 27; 29; 31; 41; 43; 44; 45; 46; 47; 57; 58; 59; 60; 70; 71; 72; 73; 86; 88; 89; 90; 103; 106; 108; 118 | 2-(2-Methoxyethoxy)acetaldehyde | 8194 | 68 | — | — | |
| 11 | 17.2–17.4 | 15; 26; 27; 29; 31; 42; 43; 45; 57; 58; 59; 60; 86; 88; 89; 103; 106; 118 | 2-(2-Methoxyethoxy)ethan-1-amine | 3506 | 29 | — | — | |
| 12 | 17.9–18.2 | 29; 31; 43; 45; 57; 58; 70; 103; | 2-(Vinyloxy)acetic acid | 9619 | 80 | 17.9–18.2 | 68 | |
| 13 | 18.7–18.8 | 15; 29; 31; 43; 45; 57; 58; 59; 60; 88; 103 | (2-Methoxyethoxy)ethene | 259620 |
2157 | — | — | |
| 14 | 19.5–19.7 | 29; 57; 58 | Oxalaldehyde | 22112 |
183 | 19.5–19.6 | 158 | |
| H2O | 2.2–2.5 | 16; 17; 18 | Water | 10562 |
106 | 2.2–2.5 | 44b | 98 |
Many of the decomposition compounds are formed in concentrations greater than 100 ppm, especially the compound (13), which is detected with >2000 ppm after LiMEAS. This DG-based electrolyte seems to decompose the most compared to THF and DME, and is therefore the least stable solvent for the LiMEAS. It decomposes into the most numerous different products after LiMEAS, and the measured concentrations of each of them are considerably large. Most likely this can be attributed to the reactivity of Li towards methylene groups (–CH2–) adjacent to O-atoms in DG, which leads to increased decomposition of the materials. Based on this, the longer the chain in the polyether-based organic solvent in LiMEAS, the worse the stability of the electrochemical system it seems.
Other molecules with higher m/z values than 200 or the polymeric species formed during LiMEAS could not be detected in the GCMS analyses performed in this study, due to the fact that polymers are high boiling point materials. The chromatographic columns cannot sustain temperatures higher than 300 °C. This is why it is difficult to characterize polymers and other high boiling point molecules by GCMS.
Conclusion
The organic electrolyte for Li-mediated non-aqueous ammonia synthesis is a critical component, responsible for the formation of the protective solid-electrolyte interphase layer, which enables nitrogen reduction to occur. For the ammonia synthesis reaction to be commercially viable, electrolyte and electrode stability are crucial. Therefore, much effort has to be put in the research towards understanding of electrolyte, its components, such as additives, salts and solvents in order to enhance stability and to determine their actual impact on any possible failures. This paper provides evidence of the usefulness of a high-resolution gas chromatography-mass spectrometry analytical technique to monitor and control the reliability of the electrochemical ammonia synthesis reaction. The injection of liquid electrolyte in the GCMS enabled detection of various volatile and non-volatile decomposition products, like ethane. From the liquid electrolyte analyses before and after electrolysis, the MS detector revealed several molecular organic products. All three investigated solvents decompose upon LiMEAS, however the decomposition depends on the electrolyte used. The solvent in the electrolyte the most susceptible to decomposition was determined to be DG, producing the biggest amount of by-products, while THF was revealed to be the most stable based on the lowest concentrations of decomposition products. Therefore, THF seems to be the most suitable for LiMEAS among the three electrolytes investigated. The main decomposition products for THF were (2,5 or 2,3)-dihydrofuran or but-(2 or 3)-enal, 4-hydroxybutanal or tetrahydrofuran-2-ol and succinaldehyde. DME electrolyte mostly turned to (Z,E)-1,2-dimethoxyethene and 1,2-dimethoxyethan-1-ol. While DG-based electrolyte was the most susceptible to decomposition and produced methoxyethene, 2-methoxyethan-1-ol and (2-methoxyethoxy)ethane. All of the electrolytes after LiMEAS contained increased water. In conclusion, GCMS analysis is useful to identify the vast array of gaseous and soluble molecules evolving from the electrolyte due to chemical and/or electrochemical reactivity in the lithium-mediated electrochemical ammonia synthesis process.Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
We gratefully acknowledge the funding by Villum Fonden, part of the Villum Center for the Science of Sustainable Fuels and Chemicals (V-SUSTAIN grant 9455) and E-Ammonia project funded by Innovation Fund Denmark (IFD) ref. no. 9067-00010B.References
- C. Li, T. Wang and J. Gong, Trans. Tianjin Univ., 2020, 26, 67–91 CrossRef CAS.
- I. Garagounis, A. Vourros, D. Stoukides, D. Dasopoulos and M. Stoukides, Membranes, 2019, 9(112), 1–17 Search PubMed.
- X. Cui, C. Tang and Q. Zhang, Adv. Energy Mater., 2018, 8(1800369), 1–25 Search PubMed.
- S. Z. Andersen, M. J. Statt, V. J. Bukas, S. G. Shapel, J. B. Pedersen, K. Krempl, M. Saccoccio, D. Chakraborty, J. Kibsgaard, P. C. K. Vesborg, J. Nørskov and I. Chorkendorff, Energy Environ. Sci., 2020, 13, 4291–4300 RSC.
- N. Lazouski and K. Manthiram, Trends Chem., 2019, 1, 141–142 CrossRef CAS.
- A. J. Martín, T. Shinagawa and J. Pérez-Ramírez, Chem, 2019, 5, 263–283 Search PubMed.
- E. F. McFarlane and F. C. Tompkins, Trans. Faraday Soc., 1962, 58, 997–1007 RSC.
- M. A. Shipman and M. D. Symes, Catal. Today, 2017, 286, 57–68 CrossRef CAS.
- J. G. Chen, R. M. Crooks, L. C. Seefeldt, K. L. Bren, R. M. Bullock, M. Y. Darensbourg, P. L. Holland, B. Hoffman, M. J. Janik, A. K. Jones, M. G. Kanatzidis, P. King, K. M. Lancaster, S. V. Lymar, P. Pfromm, W. F. Schneider and R. R. Schrock, Science, 2018, 360(eaar6611), 1–7 Search PubMed.
- A. Tsuneto, A. Kudo and T. Sakata, J. Electroanal. Chem., 1994, 367, 183–188 CrossRef CAS.
- A. Tsuneto, A. Kudo and T. Sakata, Chem. Lett., 1993, 22, 851–854 CrossRef.
- R. Marom, O. Haik, D. Aurbach and I. C. Halalay, J. Electrochem. Soc., 2010, 157, A972 CrossRef CAS.
- S. Z. Andersen, V. Colic, S. Yang, J. A. Schwalbe, A. C. Nielander, J. M. McEnaney, K. Enemark-Rasmussen, J. G. Baker, A. R. Singh, B. A. Rohr, M. J. Statt, S. J. Blair, S. Mezzavilla, J. Kibsgaard, P. C. K. Vesborg, M. Cargnello, S. F. Bent, T. F. Jaramillo, I. E. L. Stephens, J. K. Norskov and I. Chorkendorff, Nature, 2019, 570, 504–508 CrossRef CAS PubMed.
- G. R. Zhuang, K. Wang, Y. Chen and P. N. Ross, J. Vac. Sci. Technol., A, 1998, 16, 3041–3045 CrossRef CAS.
- S. K. Heiskanen, J. Kim and B. L. Lucht, Joule, 2019, 3, 2322–2333 CrossRef CAS.
- L. Wang, A. Menakath, F. Han, Y. Wang, P. Y. Zavalij, K. J. Gaskell, O. Borodin, D. Iuga, S. P. Brown, C. Wang, K. Xu and B. W. Eichhorn, Nat. Chem., 2019, 11, 789–796 CrossRef CAS PubMed.
- D. Aurbach, J. Electrochem. Soc., 1987, 134, 1611 CrossRef CAS.
- D. Aurbach, M. Daroux, P. Faguy and E. Yeager, J. Electroanal. Chem. Interfacial Electrochem., 1991, 297, 225–244 CrossRef CAS.
- C. Schultz, S. Vedder, B. Streipert, M. Winter and S. Nowak, RSC Adv., 2017, 7, 27853–27862 RSC.
- V. Kraft, W. Weber, M. Grützke, M. Winter and S. Nowak, RSC Adv., 2015, 5, 80150–80157 RSC.
- G. Gourdin, J. Collins, D. Zheng, M. Foster and D. Qu, J. Phys. Chem. C, 2014, 118, 17383–17394 CrossRef CAS.
- L. Yang, C. Smith, C. Patrissi, C. R. Schumacher and B. L. Lucht, J. Power Sources, 2008, 185, 1359–1366 CrossRef CAS.
- D. Liu, Z. Shadike, R. Lin, K. Qian, H. Li, K. Li, S. Wang, Q. Yu, M. Liu, S. Ganapathy, X. Qin, Q. H. Yang, M. Wagemaker, F. Kang, X. Q. Yang and B. Li, Adv. Mater., 2019, 31, e1806620 CrossRef PubMed.
- Y. P. Stenzel, F. Horsthemke, M. Winter and S. Nowak, Separations, 2019, 6, 26 CrossRef CAS.
- P. L. Searle, Analyst, 1984, 109, 549 RSC.
- V. NIST Standard Reference Database 20 and https://webbook.nist.gov.
- K. K. Murray, R. K. Boyd, M. N. Eberlin, G. J. Langley, L. Li and Y. Naito, Pure Appl. Chem., 2013, 85, 1515–1609 CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1ra05963g |
| This journal is © The Royal Society of Chemistry 2021 |