
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceThe first amorphous and crystalline yttrium lactate: synthesis and structural features†
A. D. Yapryntseva,
A. E. Baranchikov a,
A. V. Churakova,
G. P. Kopitsabc,
A. A. Silvestrovaad,
M. V. Golikovaae,
O. S. Ivanovaa,
Yu. E. Gorshkovafg and
V. K. Ivanov*ad
a,
A. V. Churakova,
G. P. Kopitsabc,
A. A. Silvestrovaad,
M. V. Golikovaae,
O. S. Ivanovaa,
Yu. E. Gorshkovafg and
V. K. Ivanov*ad
aKurnakov Institute of General and Inorganic Chemistry of the Russian Academy of Sciences, Moscow, Russia. E-mail: van@igic.ras.ru
bPetersburg Nuclear Physics Institute of National Research Centre “Kurchatov Institute”, St. Petersburg, Russia
cGrebenshchikov Institute of Silicate Chemistry of the Russian Academy of Sciences, St. Petersburg, Russia
dNational Research University Higher School of Economics, Moscow, Russia
eMendeleev University of Chemical Technology, Moscow, Russia
fFrank Laboratory of Neutron Physics, Joint Institute for Nuclear Research, Dubna, Russia
gInstitute of Physics, Kazan Federal University, Kazan, Russia
First published on 9th September 2021
Abstract
The synthesis and crystal structure of the first molecular yttrium lactate complex, Y(Lac)3(H2O)2, is reported, where the coordination sphere of yttrium is saturated with lactate ligands and water molecules, resulting in a neutral moiety. In Y(Lac)3(H2O)2, hydrogen bonding between α-hydroxy groups and water molecules allows for the formation of 2D layers. A subtle variation in synthetic conditions, i.e. a slight increase in pH (5.5 instead of 4.5) promoted the formation of a semi-amorphous fibrous material with a presumed chemical composition of Y4(OH)5(C3H5O3)7·6H2O. The flattened fibres in this material are responsible for its good flexibility and foldability.
Introduction
Metal carboxylates attract a great deal of attention due to the wide range of their practically important properties, including high catalytic and photocatalytic activity, gas storage and sensing.1 Their well-pronounced luminescence, enhanced by aromatic antenna ligands and high chemical stability under UV-irradiation, is of primary importance for the construction of light-emitting devices.2,3 The design of novel metal carboxylates enables molecular magnets, low-temperature cooling agents, sorbents for gas separation, bioactive materials, etc.4–10The functional properties of metal carboxylates are governed by their coordination chemistry, e.g. by the coordination ability of a central atom and the coordination mode of ligands. In d-metal complexes, carboxylate ligands usually manifest a monodentate nature, often playing the role of bridging moieties.11,12 In contrast to d-metal carboxylates, in REE carboxylates, R–COO ligands show much more versatile coordination modes, including mono-, bi- or tridentate non-bridging and di- or trinuclear bridging modes. The relatively large size of REEs compared to 3d metals makes the 4-member chelating ring more stable than in 3d metal carboxylates, which favours the bidentate coordination of the ligands.11 Being hard Lewis base donors, carboxylic acids are the most suitable ligands to fill the coordination sphere of REEs.3 In turn, the large coordination numbers of REEs (6–9) (ref. 13) provide the rich coordination chemistry of REE carboxylates, demonstrating a wide variety of 1D, 2D and 3D networked coordination polymers with abundant coordination behaviours, an adjustable structure and high chemical stability.11,14
Functionalised carboxylic acids, including hydroxy carboxylic or amino acids, due to their potentially higher denticity and their ability to act as bridging ligands and to possibly form interligand hydrogen bonds, provide additional opportunities for the design of REE coordination compounds, including molecular complexes and MOFs.15,16 For example, many amino-substituted organic acids have been proposed as the ligands for gadolinium ions in the construction of new contrast-enhancing agents in clinical MRI.17,18 The coordination of lanthanides with amino acids has also been investigated in the context of creating artificial nucleases which catalyse the hydrolytic cleavage of DNA and RNA, as the coordination of peptide linkages with the metal ion affects the enzyme structure and governs its conformation.19
Among the substituted carboxylic acid residues, lactate (Lac) is a representative essential oxyanion that is involved in many metabolic interactions in living organisms. Lactate ions have been suggested to play an important role in the communication between cells.20 The detection of lactate is of prime importance in the diagnosis of cancer, diabetes, stroke, heart diseases etc.21 On the other hand, lactic acid-containing systems have been studied extensively as the tools for extraction and separation of rare earths and actinides within a TALSPEAK (Trivalent Actinide–Lanthanide Separation by Phosphorus reagent Extraction from Aqueous Komplexes) process.22–25
Thus, comprehensive information on lanthanide lactates is highly important in the search for new biosensors and theranostic agents, as well as for the improvement of industrial extraction processes. Unsurprisingly, the thermodynamic features of rare earth lactate complexes (formation constants, etc.) were the subject of extensive study during the 1960s and 1970s.25–34 These studies resulted in the identification of several structures of lanthanide lactate moieties which can form in solutions and can presumably exist in crystalline lanthanide lactates. One unexpected result of the literature survey was that information on crystalline lanthanide lactates is extremely scarce, with the following compounds reported to date: [LnNa(Lac)4]·2H2O (Ln = Sm, Eu),35, [Ln(Lac)2](H2O)2·ClO4 (Ln = Eu, Tb),36,37 La4(HL)5(Lac)2(i-PrOH)2(DMF)2·(7DMF + 5H2O) (H3L = 5-hydroxyisophthalic acid),38 Ln(TACD)(Lac)(CF3SO3)2·3H2O (Ln = Yb, Ho; TACD = tetraazacyclododecane-based ligand),39 Ln3Ag4(Lac)2(IN)8(H2O)5·2(ClO4)·2.5(H2O) [Ln = La, Lu; HIN = isonicotinic acid], Sm3Ag4(Lac)2(IN)8(H2O)4·2(ClO4)·2.5(H2O), and Eu2Ag3(S-Lac)(IN)6(H2O)4·2(ClO4)·4(H2O),40 Ln(Lac)[15MCCuIIGlyha-5]Cl2 (Ln = La, Y, Ce, Pr, Nd, Sm, Eu, Gd, Tb, or Dy; 15MCCuIIGlyha-5 = copper(II)-glycinehydroximate 15-metallacrown-5 ligand).41 As expected, in known REE lactates, a lactate ligand shows a predominantly bidentate coordination mode;39–41 in some cases, the hydroxy group is also included in the coordination environment of a lanthanide.38,40 A lactate ligand tends to act as a bridge connecting two,40 or even three,38 rare earth ions. Interestingly, examples of other rare earth α-hydroxy carboxylates are even more limited, e.g. the structure of lanthanide 2-hydroxy isobutyrates was described by Chen et al.42
Note that none of the known crystalline rare earth lactates can be regarded as a neutral molecular lactate complex, where the coordination sphere of a REE is saturated only with lactate ligands and water molecules resulting in a neutral molecular moiety. This fact seems to be somewhat odd, since the existence of such molecular complexes was recently predicted.33 In their most recent paper, Roy et al. commented on this fact as follows: ‘While lanthanide lactate complexes have been known for some time, there are no crystal structures of the isolated species to compare our optimized structures’.33
In order to fill the gap in rare earth lactate chemistry, the aim of the current research was to synthesise an yttrium lactate containing no additional organic ligands. When planning the synthetic strategy, account was taken of some recently reported principles in rare earth carboxylate chemistry, namely the importance of high ligand-to-metal molar ratio,11 as well as the use of mild hydrothermal conditions facilitating the formation of REE complexes with polydentate substituted carboxylates.36,37,43 Special attention was paid to avoid resinification reactions. This strategy enabled the preparation of the first molecular yttrium lactate, while a subtle increase in pH resulted in a semi-amorphous basic yttrium lactate, which revealed an unusual 1D shape of the particles.
Materials and methods
Materials
Yttrium chloride hexahydrate (99.9%, Lanhit), L-lactic acid (HLac, 80%, Sigma) and hexamethylenetetramine (HMT, 99+%, Alfa Aesar) were used without further purification.Synthesis of yttrium lactates
60 ml of aqueous clear solution of yttrium chloride hexahydrate (1.7 × 10−2 M), HMT (2.4, 5.6 or 12 × 10−2 M) and L-lactic acid (0.5, 2.5, 4.7, 6.6 or 8.5 × 10−2 M) was placed into a closed 100 ml glass vessel and heated at 100 °C for 24 h. The solid product was separated from the mother liquor on a glass filter, washed several times with distilled water and then dried at 50 °C overnight.Characterisation
Powder X-ray diffraction (XRD) patterns of the samples were recorded on a Bruker D8 Advance diffractometer (CuKα radiation, Ni-filter, LYNXEYE detector, Bragg–Brentano reflection geometry). The microstructure of the samples was investigated using high-resolution scanning electron microscopy (Carl Zeiss NVision 40 equipped with an Oxford Instruments X-Max EDX detector) at accelerating voltages in the range of 1–10 kV. The yttrium content in the solid samples was determined by reverse complexometric titration.44 Briefly, the samples were dissolved in an excess of hot EDTA and free EDTA was titrated by magnesium sulfate with Arsenazo I indicator. Carbon, hydrogen and nitrogen content in the samples was determined using a Carlo Erba Instruments EA 1108 CHN analyser. The IR spectra of the samples were recorded on an ALPHA FTIR Spectrometer (Bruker) in the range of 4000–300 cm−1, with a resolution of 0.5 cm−1, in an attenuated total reflection mode. Luminescence spectra of the powders were recorded using a Perkin Elmer LS-55 luminescence spectrophotometer with 0.5 nm resolution, at room temperature. The thermal behaviour of basic yttrium lactate was studied using a combined TGA/DSC/DTA (thermogravimetric analysis/differential scanning calorimetry/differential thermal analysis) analyser SDT Q-600, the samples were heated to a temperature of 800 °C (heating rate of 10° min−1) in an air flow (250 ml min−1).SEM, CHN, powder and single crystal X-ray diffraction measurements were performed using the equipment of the Joint Research Centre for Physical Methods of Research at Kurnakov Institute of General and Inorganic Chemistry of the Russian Academy of Sciences (JRC PMR IGIC RAS).
Small angle neutron scattering (SANS) investigations were performed using the YuMO time-of-flight spectrometer at the IBR-2 pulsed reactor (Dubna, Moscow region, Russia). Standard data acquisition time per sample was 30 min. Two ring wire He3-detectors47 at distances of 4 m and 13 m from the sample position were used in the experiment. Scattered intensity (differential cross section per sample volume) was measured as a function of the momentum transfer modulus q = (4π/λ) sin(θ/2), where θ is the scattering angle and λ is the incident neutron wavelength. An incident neutron beam distribution provided an available wavelength range of 0.5–8 Å, which corresponds to a momentum transfer (q) range of 0.007–0.3 Å−1. The raw data were treated using SAS software.48 The raw SANS data were converted to an absolute scale by normalisation to the incoherent scattering cross section of a standard vanadium sample. The data were corrected taking account of scattering from the setup and the empty cell. The corrected small-angle neutron scattering curves were presented using an absolute scale with background subtraction.49
Results and discussion
Low-temperature hydrothermal treatment (100 °C) of the solutions containing yttrium chloride (1.7 × 10−2 M), HMT (2.4–6.6 × 10−2 M) and L-lactic acid (0.5–8.3 × 10−2 M) resulted in the formation of several different solid products (Fig. 1). At low lactic acid concentrations (0.005 M), white precipitates were formed; at high lactic acid concentrations (0.08 M), needle-like, whitish crystals 1 cm length and a few millimetres thick were obtained. At intermediate lactic acid concentrations (0.025–0.065 M), the synthesis resulted in the formation of white, monolithic gels.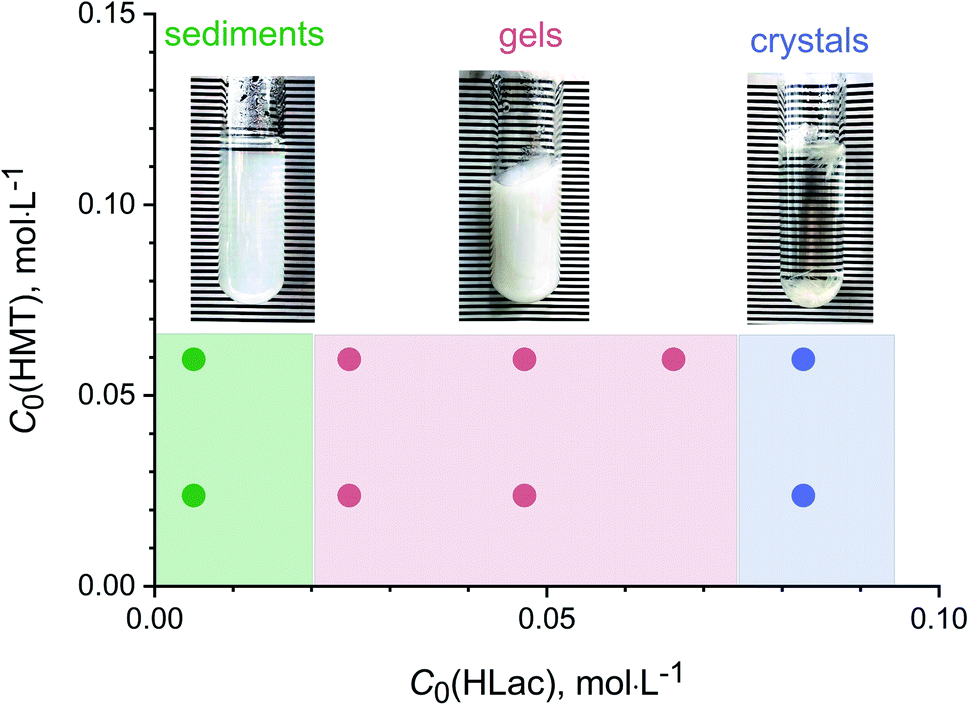 | ||
| Fig. 1 The products formed upon low-temperature hydrothermal treatment of solutions containing yttrium chloride (1.7 × 10−2 M), HMT (0.024, 0.066 M) and L-lactic acid (0.005–0.083 M). | ||
Fig. 2 shows typical powder X-ray diffraction patterns of the samples prepared from the reaction mixtures containing various amounts of L-lactic acid. Hydrolysis of yttrium chloride in the presence of a small amount (0.005 M) of lactic acid resulted in an X-ray amorphous powder. The position of the maximum of the halo (9–10° 2θ) in its diffraction pattern (Fig. 2a) indicates a probable short-range ordering (∼10 Å) in the amorphous compound obtained. The X-ray diffraction pattern of the powder prepared with a higher lactic acid concentration (0.047 M) contained three wide, low-angle diffraction maxima (5.8°, 7.4°, 8.9° 2θ), indicating low crystallinity of this product and corresponding to interlayer distances of ∼15, 12 and 10 Å (Fig. 2b). These diffraction peaks cannot be attributed to any known crystalline substance. At higher L-lactic acid concentrations in the reaction mixtures (0.08 M), a previously unknown crystalline compound 1 was formed (Fig. 2c).
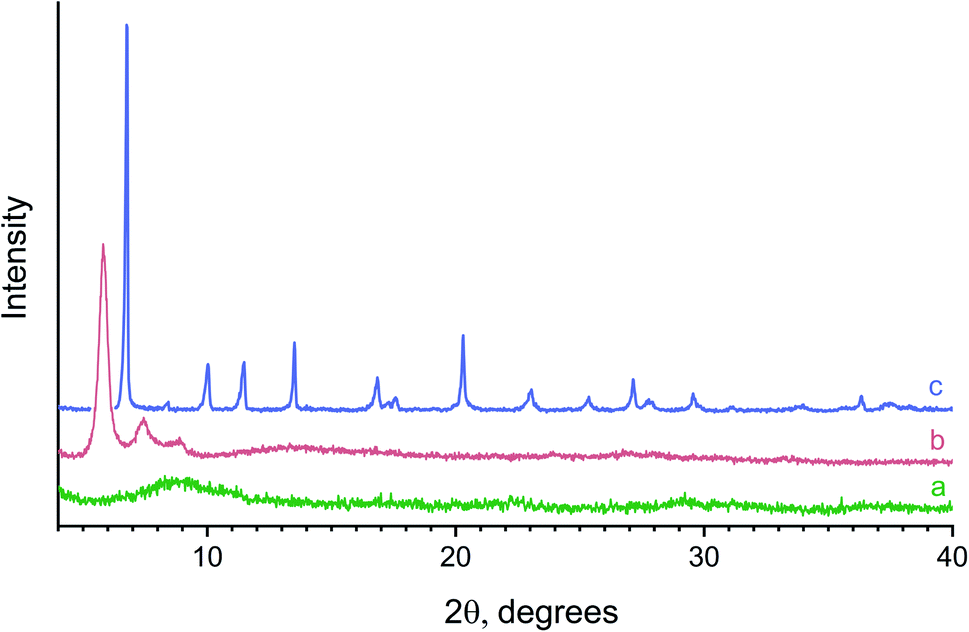 | ||
| Fig. 2 Powder X-ray diffraction patterns of the solids formed upon hydrothermal treatment of mixed solutions containing yttrium chloride (1.7 × 10−2 M), HMT (6.6 × 10−2 M) and L-lactic acid (a – 5 × 10−3 M, b – 4.7 × 10−2 M and c – 8.3 × 10−2 M). | ||
While rare earth lactate complexes in the solutions have been extensively studied,25,30–34 to date, no crystal structures of rare earth lactates have been reported. Only layered structure of [Ln(Lac)2](H2O)2[ClO4] (Ln = Eu and Tb) complex,36,37 is known. This compound comprises host cationic Ln(Lac)2 layers and interlayer perchlorate ions connected to the host through hydrogen bonds.
For the single crystal experiments, more than ten crystals of compound 1 were examined and all of them exhibited a twinned nature (tripled or sextupled initial cell volumes, unstable and inaccurate cell dimensions refinement). In all the cases, attempts to resolve the twinning using APEX3 software50 failed because the close proximity of diffraction peaks resulted in the instability of integration procedures. To obtain crystals of better quality, several attempts were made to re-crystallise compound 1 from different organic solvents (methanol, DMSO, DMF) and all of them failed due to the low solubility of the compound. For all the crystals of compound 1 that were examined, the best single crystal data resulted in Y(C3H5O3)3(H2O)2 (sp. gr. P21), a = 10.5780(8), b = 5.7390(5), c = 13.2053(10) Å, β = 97.765(3)°, V = 794.31(11) Å3, Z = 2 (Fig. S1†). According to the Cambridge Structural Database, compound 1 is the first structurally characterised yttrium lactate.
The molecule of 1 is monomeric, with an yttrium coordination number equal to 8 (Fig. 3). The coordination environment of the central atom is formed by three η2-lactate ligands and two water molecules in cis-positions to each other, with an Ow–Y–Ow angle equal to 70.6(3)°. The resulting structural data (see Table 1) agree well with the theoretical calculations made by Roy et al. for the structure of lanthanum lactate complexes including La(Lac)3(H2O)2.33 The predicted interatomic distances for La–Ocarboxylate, La–Oα-hydroxy and La–Owater in the complex were 2.48 ± 0.03 Å, 2.65 ± 0.11 Å and 2.23 ± 0.44 Å;33 the differences between these values and the values presented in Table 1 are obviously due to the differences in lanthanum and yttrium atomic radii, as well as to the consideration of outer sphere water molecules in the calculations made by Roy et al.33
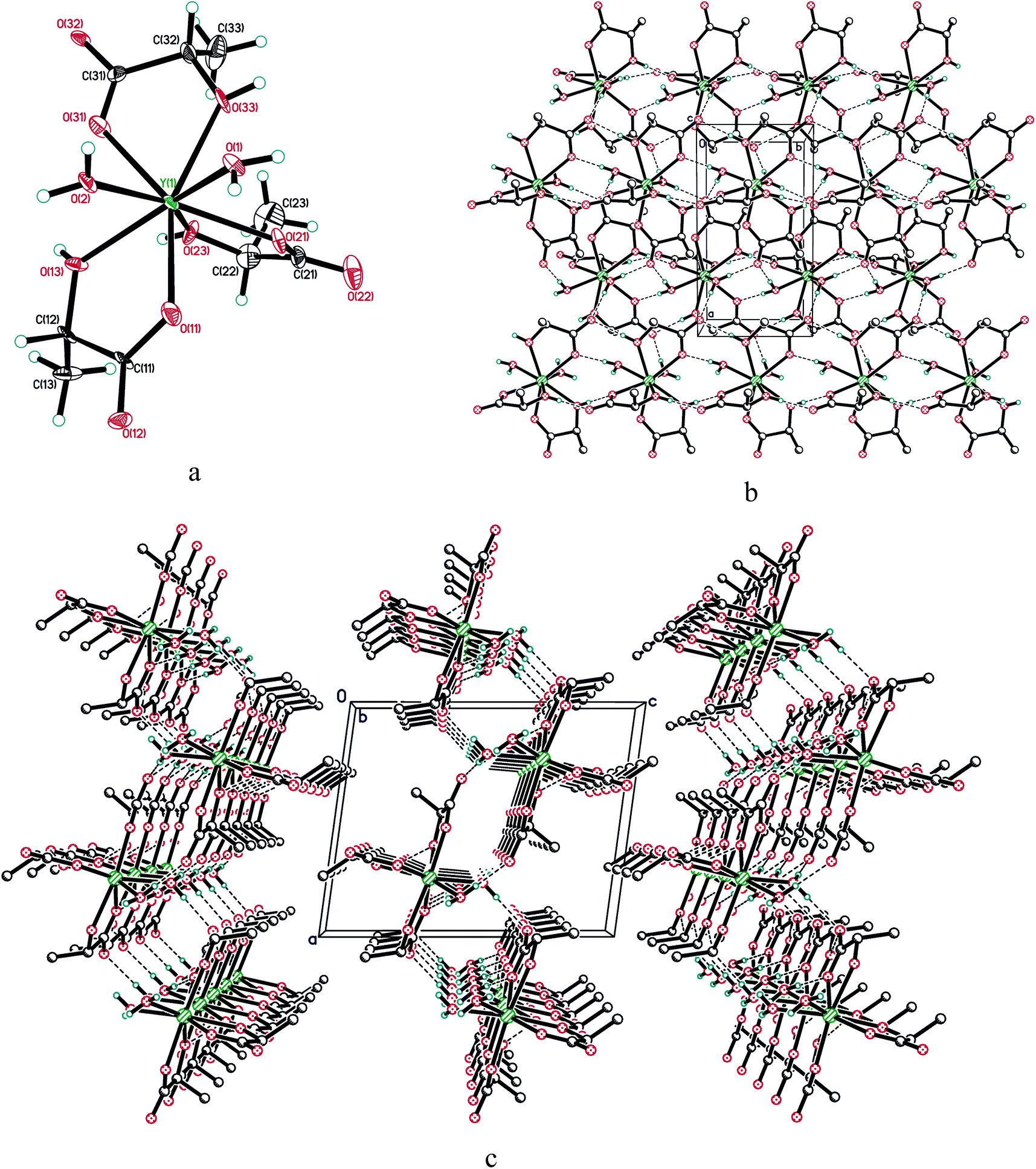 | ||
| Fig. 3 (a) Molecular structure of 1; displacement ellipsoids are shown at 50% probability level. (b) Hydrogen bonded layer in the structure of 1, viewed along the c-axis and (c) the b-axis. | ||
| Y(1)–O(11) | 2.296(9) | O(1)–Y(1)–O(2) | 70.6(3) |
|---|---|---|---|
| Y(1)–O(21) | 2.316(9) | O(11)–Y(1)–O(13) | 66.9(3) |
| Y(1)–O(31) | 2.323(9) | O(21)–Y(1)–O(23) | 65.0(3) |
| Y(1)–O(13) | 2.325(9) | O(31)–Y(1)–O(33) | 65.3(3) |
| Y(1)–O(23) | 2.369(9) | ||
| Y(1)–O(33) | 2.390(9) | ||
| Y(1)–O(1) | 2.324(9) | ||
| Y(1)–O(2) | 2.368(8) |
Both Y ← Ow distances (2.324(9) and 2.368(8) Å) lie within the range 2.276–2.652 Å that appears in the Cambridge Structural Database (ver. 5.42, November 2020 (ref. 51)) for terminal water molecules in yttrium complexes with CN = 8 (157 error-free, non-disordered entries, 450 fragments, mean 2.363 Å). The lengths of dative bonds Y ← O(H)C (2.325(9)–2.390(9) Å) are also typical for yttrium (CN = 8) compounds (2.277–2.474 Å for 28 accurate entries with 42 fragments, mean 2.381 Å). All five-membered metallocycles are planar within 0.03 Å.
As expected, Y–OC(![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O) bonds are the shortest within the polyhedra (see Table 1). Interestingly, in lactate anions, the longer the Y–OC(O) bond is, the longer the Y ← O(H)C distance is. This may indicate the active involvement of the α-hydroxy group in the coordination with yttrium. Generally, the contribution of the α-hydroxy group to the coordination of yttrium by the lactate anion is actively debated in the literature. Tian et al. reasoned that the participation of α-hydroxy oxygen atoms in the coordination of rare earths promotes the increased stability of Ln(III) lactate complexes.24 Depending on which model is used, either protonated or deprotonated α-hydroxy groups were considered to enter the coordination sphere of rare earth atoms, the hydrogen bonding of the α-hydroxy group with a water molecule playing an important role.24,25,33
O) bonds are the shortest within the polyhedra (see Table 1). Interestingly, in lactate anions, the longer the Y–OC(O) bond is, the longer the Y ← O(H)C distance is. This may indicate the active involvement of the α-hydroxy group in the coordination with yttrium. Generally, the contribution of the α-hydroxy group to the coordination of yttrium by the lactate anion is actively debated in the literature. Tian et al. reasoned that the participation of α-hydroxy oxygen atoms in the coordination of rare earths promotes the increased stability of Ln(III) lactate complexes.24 Depending on which model is used, either protonated or deprotonated α-hydroxy groups were considered to enter the coordination sphere of rare earth atoms, the hydrogen bonding of the α-hydroxy group with a water molecule playing an important role.24,25,33
In compound 1, all active hydrogen atoms of α-hydroxy groups and water molecules are involved in intermolecular hydrogen bonding, with O⋯O distances ranging within 2.642(13)–2.839(12) Å. They combine adjacent molecules in the layer perpendicular to the c-axis (Fig. 3).
Comparison of single crystal diffraction data collected at 150 K and the powder X-ray diffraction pattern collected at 298 K enabled an estimate to be made of thermal expansion coefficients for lattice parameters of compound 1, 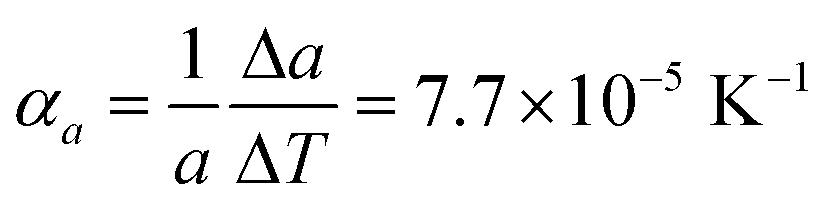 ;
; 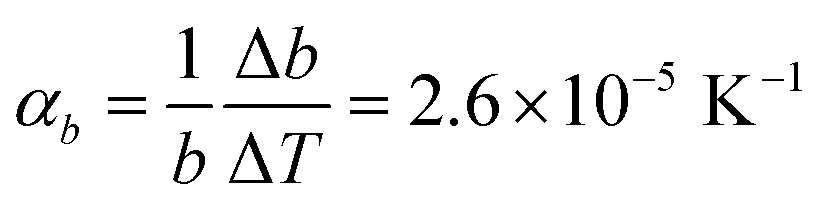 ;
; 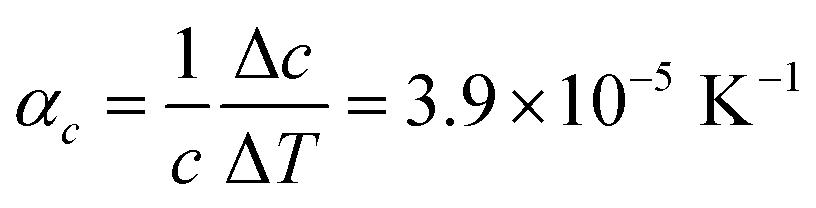 ;
; 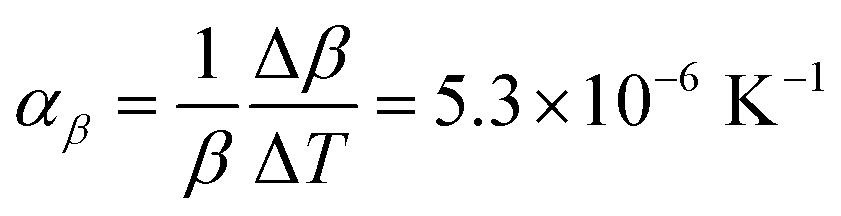 ;
;  .
.
The chemical composition of compound 1, as determined by CHN analysis and complexometric titration (Table 2), corresponds well with the chemical composition determined using single-crystal diffraction. The chemical composition of compound 1 agrees well with the lanthanum lactate complex La(Lac)3(H2O)2 predicted by Roy et al.,33 where the lanthanum first coordination sphere includes two water molecules and three lactate anions having bidentate coordination (by carboxylate and protonated α-hydroxy group).24
| L-Lactic acid concentration in the reaction mixture, M | Solid product formed | Content [wt%] | |
|---|---|---|---|
| Measured | Calculated | ||
| 8.3 × 10−2 | Yttrium lactate, 1 Y(C3H5O3)3(H2O)2 | Y 23.0 | Y 22.7 |
| C 27.9 | C 27.6 | ||
| H 5.1 | H 4.9 | ||
| O 44.0 | O 44.9 | ||
| 4.7 × 10−2 | 'Y4(OH)5(C3H5O3)7·6H2O' | Y 30.6 | Y 30.3 |
| C 21.6 | C 21.5 | ||
| H 4.5 | H 4.5 | ||
| O 43.3 | O 43.7 | ||
In their recent report, Powell and Farrell26 hypothesised the formation of yttrium lactate dihydrate, Y(Lac)3·2H2O, as a result of low-temperature (100 °C) thermal decomposition of Y(Lac)3·3H2O in air. It is assumed that, in experiments carried out for the current study, upon low-temperature hydrothermal treatment, a moderately soluble yttrium lactate trihydrate can form, which subsequently loses a water molecule and transforms to Y(Lac)3(H2O)2. Nevertheless, the exact mechanism of the formation of yttrium lactates under hydrothermal conditions needs further refinement.
With moderate L-lactic concentrations in the reaction mixtures, upon low-temperature hydrothermal treatment, the formation of monolithic gels was observed. When the gels were filtered on a glass filter, followed by washing and ambient drying, they flattened and formed paper-like, foldable, solid substances (see Fig. 4).
 | ||
| Fig. 4 Appearance of a paper-like material obtained upon the filtering of the gels formed by the low-temperature hydrothermal treatment of reaction mixtures containing yttrium chloride (1.7 × 10−2 M), HMT (6.6 × 10−2 M) and L-lactic acid (4.7 × 10−2 M). | ||
The gelation of yttrium-containing compounds is quite an unusual phenomenon. Initially, it was supposed that the gels were formed due to the polycondensation of L-lactic acid catalysed by yttrium ions, since REE compounds were reported to catalyse this reaction.52,53 However, IR-spectroscopy data did not confirm this hypothesis. The absence of the most intense absorption bands characteristic of mono- (1770 cm−1, cyclic dilactone CO valence vibration) or polylactides (1757 cm−1, valence vibration of CO of aliphatic esters)54 indicated the absence of lactide moieties in the compounds obtained. In Fig. 5, one can see that the IR spectra of crystalline yttrium lactate 1 and the paper-like material are almost identical. Within the 3500–3000 cm−1 range, OH valence vibrations of lactate ions, water molecules and hydroxide groups are observed. The most intense lines in the spectrum are located at ∼1580, 1400 and 1100 cm−1, indicating the symmetric vibrational modes of the COO− group, anti-symmetric vibrational modes of the COO− group and valence vibrational modes of the alcoholic C–OH group. These values are in a good agreement with the literature data for metal lactates55 and NpO2+ lactate complexes.56 Thus, a paper-like material is also an yttrium lactate, probably of a different chemical composition. This supposition is in line with the chemical analysis data (see Table 2).
 | ||
| Fig. 5 IR spectra of solids formed during low-temperature hydrothermal treatment of yttrium chloride (1.7 × 10−2 M), HMT (6.6 × 10−2 M) and L-lactic acid (a – 4.7 × 10−2 M; b – 8.3 × 10−2 M). AL – vibrational modes of alcoholic groups. | ||
The difference in IR-spectra of crystalline yttrium lactate 1 and a paper-like material is manifested in the broadening of ν(OH) bands (∼3500 cm−1), which indicates a more developed network of hydrogen bonds in the latter substance. The presence of vibrational bands at ∼1480 cm−1 (deformational modes of –CH2– or –CH3 groups), ∼1275 cm−1 (deformational modes of alcoholic OH groups) and 650 cm−1 (deformational modes of C–COH group) in 1, and their absence in IR spectrum of a paper-like material, indicates the higher crystallinity of the former compound and the diversity in coordination modes of lactate alcoholic OH-groups in the latter compound. The differences in spectra are also observed within a low frequency region (350–800 cm−1), where a wide absorption band at ∼700 cm−1 for the paper-like material can be attributed to Y–O valence vibrations.
A detailed analysis of the IR spectrum of the paper-like material (the splitting of antisymmetric and symmetric stretching modes of the carboxylic groups) made it possible to determine a coordination type of carboxylic groups in lactate ligands to Y3+ cations.56 Generally, a bidentate coordination of carboxylic groups of lactate ligands is accompanied by a smaller spectral splitting (≤100 cm−1), whereas a monodentate binding results in a larger spectral splitting (≥150 cm−1).56,57 The splitting of antisymmetric and symmetric stretching modes in the spectra of crystalline yttrium lactate and the paper-like material were 187 cm−1 and 169 cm−1, respectively, indicating monodentate coordination of carboxylic groups of a lactate ligand to Y3+ cations in both cases. A slightly lower splitting value for the paper-like material may indicate the presence of bridging lactate ligands. A monodentate coordination of carboxyl moiety of the lactate ligand indirectly indicates that the α-hydroxy group enters the coordination sphere of yttrium, which is in good accordance with single crystal data for compound 1.
The chemical composition of the paper-like material can be hypothesised based on general principles governing the formation of yttrium compounds in aqueous media. Compound 1 crystallised from the reaction mixtures at pH ∼ 4.5, while the gels were obtained at higher pH values, i.e. ∼5.5. This difference in pH was obviously due to the different concentration of L-lactic acid, given that in the latter case it was approximately two times lower. At pH values higher than 5, a hydrolysis of yttrium cations occurs and the formation of Y–OH moieties becomes likely and might even become uncontrollable.58 However, at relatively high pH, multidentate ligands are known to limit Y3+ hydrolysis. Within a ‘ligand controlled hydrolysis’ approach,59 different polynuclear rare earth hydroxy complexes can be obtained, bearing amino carboxylate ligands.60 In particular, rare earth amino carboxylates often comprise cubane tetranuclear [REE4(μ3-OH)4]8+ units.19 Interestingly, at nearly neutral pH (∼6) and under mild hydrothermal conditions, the interaction of rare earth ions with mono- or bidentate ligands can result in polynuclear coordination compounds, which are often regarded as layered rare earth hydroxides (LRHs) comprising infinite positively charged 2D layers of [RE(OH)3−n]n+ bonded by the ligands into laminate structures.61–63 These ion-exchangeable and exfoliatable substances have a great deal of flexibility in terms of their chemical composition61,64–66 and are now regarded as a basis for promising bioactive theranostic materials.67,68 From the findings of the current study, a paper-like material couldn't be classified as a layered rare earth hydroxide as the positions of the maxima in its diffraction pattern (see Fig. 2) are incompatible with the LRHs' structure.
Thus, it can be concluded from the current study that, at relatively high pH values and under low-temperature hydrothermal conditions, the hydrolysis of yttrium cations controlled by multidentate lactate ligands results in the formation of oligonuclear yttrium hydroxy lactate. Unfortunately, the work was unsuccessful in obtaining single crystals under corresponding synthetic conditions, since all the compounds synthesised had a semi-amorphous nature, having wide, small-angle maxima in their diffraction patterns (see Fig. 2).
Taking into account all the above considerations and the experimental data for the paper-like material, its chemical composition can be hypothesised as a basic yttrium lactate, Y4(OH)5(C3H5O3)7·6H2O. Thus, its precipitation can be described as follows:
| 4Y3+ + 3C6N4H12 + 7C3H6O3 + 29H2O → Y4(OH)5(C3H5O3)7·6H2O + 18H2CO + 12NH4+ | (1) |
Thermal analysis data corroborate the hypothesised composition of basic yttrium lactate well (Fig. 6). After heating in air, the paper-like material showed multi-stage decomposition behaviour. The first stage of thermal decomposition (up to 100 °C) was obviously due to the removal of physically bound water. The second decomposition stage (200–500 °C) was more complex and included the removal of a chemically bound water substage (100–260 °C). The weight loss (∼9.6 wt%) in this temperature range corresponds well to the removal of 8.5 water molecules per Y4(OH)5(C3H5O3)7·6H2O formula unit, yielding anticipated anhydrous yttrium oxidolactate Y4O2.5(Lac)7. At higher temperatures (up to 500 °C), the oxidation of lactate moieties occurs, probably yielding various carbonates such as Ln(C3H5O3)CO3, Ln2(CO3)3 or Ln2O(CO3)2.26 The oxidation of organic matter in the paper-like material was also indicated by a pronounced exothermic effect (maximum at 343 °C) in the DSC curve. Interestingly, the maximum rate of lactate moiety decomposition in air (∼370 °C) is observed at much higher temperatures than the maximum rate of pure lactic acid thermolysis in nitrogen flow (180–240 °C) reported by Komesu et al.69 The final decomposition stage (500–800 °C) of the paper-like material can be attributed to the decomposition of residual carbon-containing matter and the formation of yttrium oxide. The thermal analysis data seem to be similar to those obtained by Powell and Farrell26 for hypothetical yttrium lactate trihydrate, Y(Lac)3·3H2O, yet, in their report, they presented neither the results of chemical analysis of the compounds obtained, nor thermal analysis curves.
 | ||
| Fig. 6 Weight loss and DSC curves of the paper-like material formed as a result of hydrothermal treatment of yttrium chloride (1.7 × 10−2 M), HMT (6.6 × 10−2 M) and L-lactic acid (4.7 × 10−2 M) solution mixtures. | ||
One very special feature of the paper-like material was its microstructure (see Fig. 7). It consisted of fibres with an average thickness varying from 10 to 80 nm, depending on the lactic acid content of the reaction mixture (see ESI, Fig. S2†). These data are in good agreement with the particle size estimates using the Scherrer equation for the X-ray diffraction data, ∼25 nm (see Fig. 2b). According to observations derived from the current study, the concentration of HMT did not significantly affect fibre thickness. Such a microstructure obviously provides the material with superior mechanical properties, i.e. its flexibility and foldability.
 | ||
| Fig. 7 SEM images of a paper-like material formed as a result of the hydrothermal treatment of yttrium chloride (1.7 × 10−2 M), HMT (6.6 × 10−2 M) and L-lactic acid (4.7 × 10−2 M) solution mixtures. | ||
Thus, basic yttrium lactate can be classified as a 1D material composed of interlaced fibres. Interestingly, direct sol–gel methods for the synthesis of 1D gels of complex metal compounds are virtually unknown. The scarce examples of sol–gel derived 1D gels are vanadium pentoxide gels obtained from neutral VO(OH)3 species or vanadium alkoxide precursors70 and ceric hydrogen phosphate gels derived from CeO2 solutions in orthophosphoric acid.71,72 It is possible that the fibres in these gels form according to a polymeric growth mechanism, but the exact mechanism of 1D gel formation needs further clarification.
To assess the microstructure of basic yttrium lactate, low-temperature nitrogen adsorption measurements and small-angle neutron scattering experiments were performed, the latter technique being common in the study of the structure of amorphous or semi-amorphous materials.73
According to low-temperature nitrogen adsorption data, the paper-like basic yttrium lactate had a high specific surface area, of 75 m2 g−1. Fig. 8 shows the full nitrogen adsorption/desorption isotherm for the sample. The isotherm has a narrow hysteresis loop, which can be attributed to the H3 type, according to the IUPAC classification,74 indicating the presence of slit-like mesopores in the sample. The pore size distribution calculated using the BJH model indicates the presence of mesopores in the material, with the maximum at ∼6 nm.
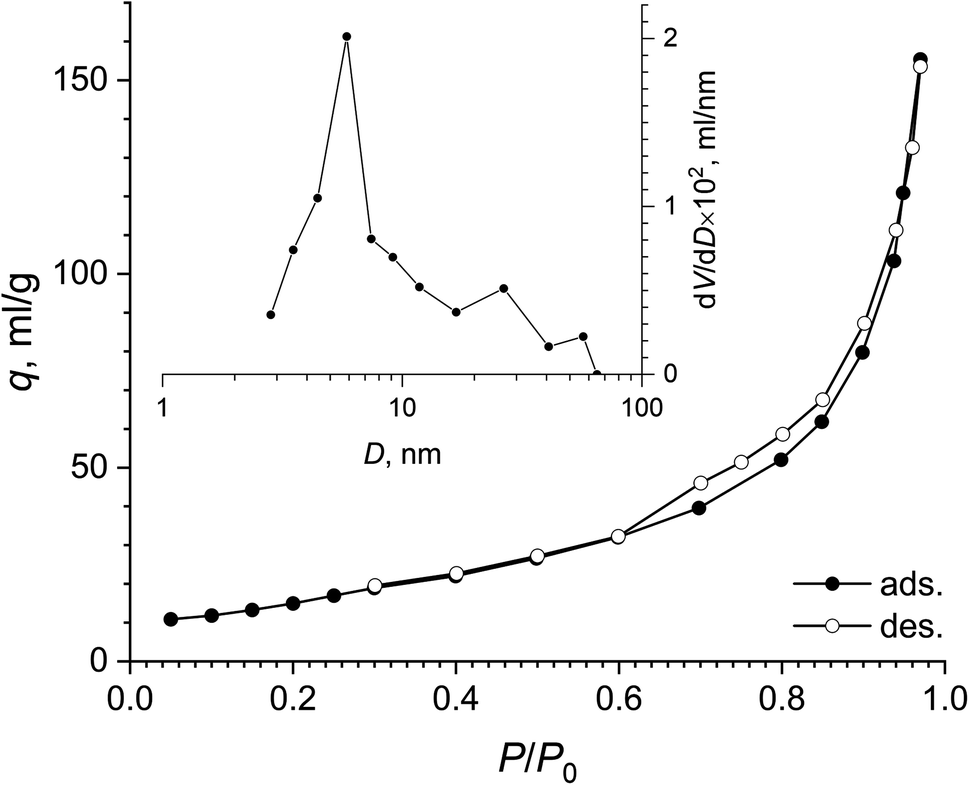 | ||
| Fig. 8 Full nitrogen adsorption–desorption isotherm and pore size distribution (inset) for the paper-like sample. | ||
Fig. S3† presents a small-angle neutron scattering curve for the basic yttrium lactate sample. The shape of the curve is typical of disordered porous systems containing two types of scatterer of different size.75–77 The exponent values calculated from the slope of the straight-line sections of the experimental 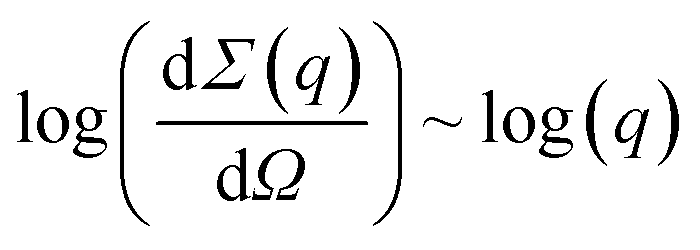 curves were n1 = 2.09 ± 0.10 and n2 = 3.98 ± 0.02. The n1 value indicates that the system consisted of randomly oriented flattened particles.73 Unfortunately, the geometrical parameters of these particles cannot be correctly estimated from the neutron scattering experimental data, since the contributions of these parameters interfere with the contribution from the structural characteristics of the aggregates of these particles.
curves were n1 = 2.09 ± 0.10 and n2 = 3.98 ± 0.02. The n1 value indicates that the system consisted of randomly oriented flattened particles.73 Unfortunately, the geometrical parameters of these particles cannot be correctly estimated from the neutron scattering experimental data, since the contributions of these parameters interfere with the contribution from the structural characteristics of the aggregates of these particles.
The value of n2 ≈ 4 (Porod scattering law78) indicates that particle aggregates possess an almost smooth surface, the surface fractal dimension being DS = 2.02 ± 0.02. The characteristic size of these aggregates Rc can be estimated from the low q region of the curve (q < 1 × 10−2 Å−1) corresponding to the transition from the Porod to the Guinier regime.79 To fit all of the experimental data, the following model was used:
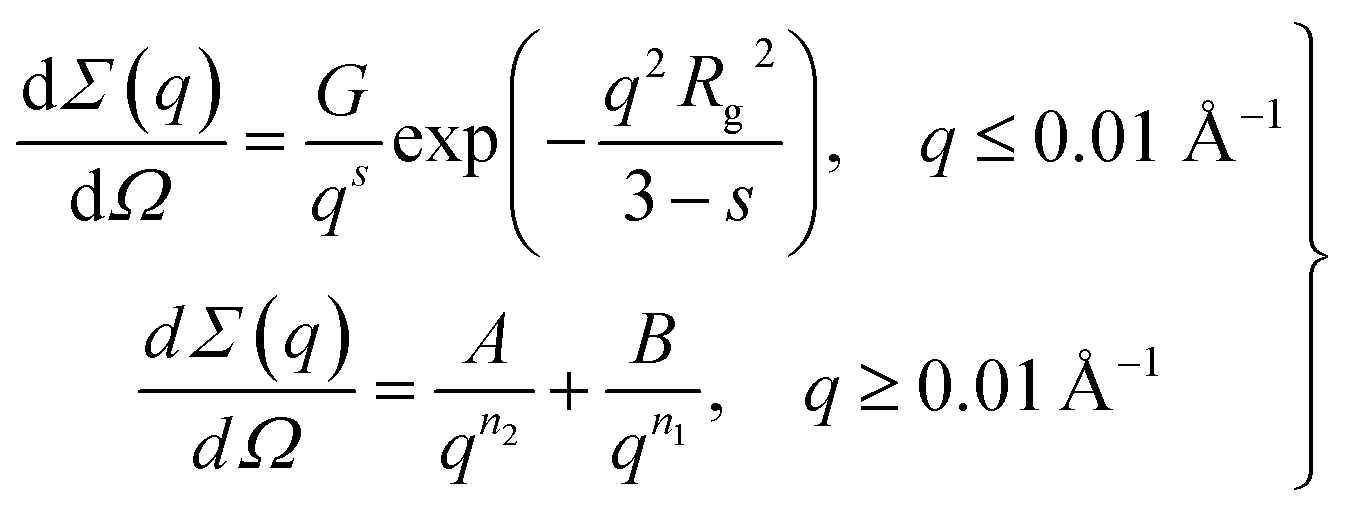 | (2) |
| Parameter | G × 102, cm−1 Å−s | s | Rg, Å | A × 107, cm−1 Å−n2 | n2 | B × 104, Å−n1 | n1 |
|---|---|---|---|---|---|---|---|
| Value | 4 ± 0.5 | 1.95 ± 0.08 | 85 ± 7 | 1.8 ± 0.4 | 3.98 ± 0.02 | 2 ± 0.2 | 2.09 ± 0.10 |
The values presented in Table 3 indicate that the paper-like sample consisted of thin ribbons (s ≈ 2) with a smooth surface (Ds = 2.02 ± 0.02) and a thickness of 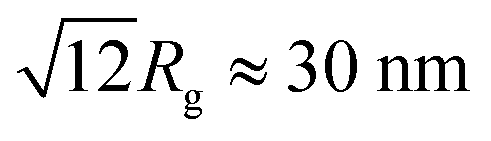 . It should be noted that this value is an upper-bound estimate. Nevertheless, this value is in good agreement with SEM data for the corresponding sample which had an average particle size of 33 nm (see Fig. 7). In turn, the X-ray diffraction pattern of basic yttrium lactate paper (see Fig. 2b) shows reflections corresponding to 1–2 nm interplanar distances. Taking into account low-temperature nitrogen adsorption data, indicating the presence of slit-like pores with a size of ∼6 nm, the structure of fibrillae which form the basic yttrium lactate paper-like material can be hypothesised (see Fig. 9).
. It should be noted that this value is an upper-bound estimate. Nevertheless, this value is in good agreement with SEM data for the corresponding sample which had an average particle size of 33 nm (see Fig. 7). In turn, the X-ray diffraction pattern of basic yttrium lactate paper (see Fig. 2b) shows reflections corresponding to 1–2 nm interplanar distances. Taking into account low-temperature nitrogen adsorption data, indicating the presence of slit-like pores with a size of ∼6 nm, the structure of fibrillae which form the basic yttrium lactate paper-like material can be hypothesised (see Fig. 9).
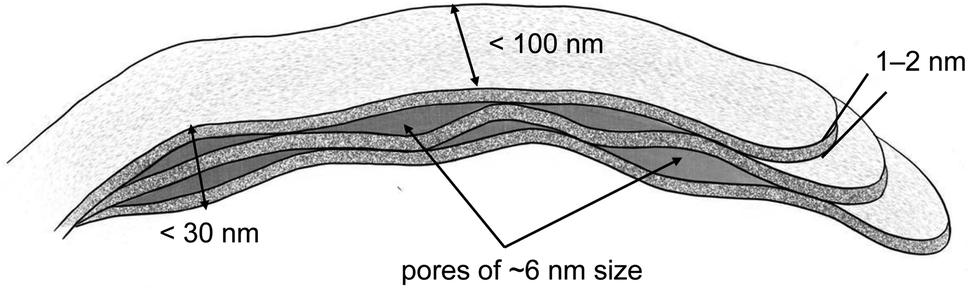 | ||
| Fig. 9 The structure of basic yttrium lactate fibres, as hypothesised from low-temperature nitrogen adsorption, small-angle neutron scattering and scanning electron microscopy data. | ||
Thin particles in the structure of basic yttrium lactate are advantageous for the preparation of ultra-small rare earth oxide nanoparticles via the thermal decomposition or combustion of this compound. The annealing of basic yttrium lactate paper in air at temperatures up to 800 °C were performed during the current study. After heating, the material changed its colour from beige (120 °C) to brown (450 °C) and white (800 °C). The sample annealed at 450 °C for 1 h consisted of nanocrystalline Y2O3, having 3 nm size, as estimated using the Scherrer equation. At 800 °C, the particle size increased to 21 nm, due to the coarsening of nanoparticles (see Fig. 10). The synthesis of ultra-small yttria nanoparticles is of special interest in view of their wide potential for biomedical applications.81,82 To date, there have been many attempts to prepare yttria nanoparticles using the sol–gel technique (e.g. ref. 82 and 83), with only a few of them succeeding in the preparation of yttria powders with a crystal size smaller than 10 nm (e.g. ref. 84 and 85).
 | ||
| Fig. 10 X-ray diffraction patterns of a paper-like material annealed at (a) 120, (b) 450, (c) 800 °C. | ||
Conclusions
A first successful attempt has been made to synthesise a crystalline molecular yttrium lactate complex (Y(Lac)3(H2O)2), where the coordination sphere of yttrium was saturated with lactate ligands and water molecules, resulting in a neutral molecular moiety. In a crystal structure, these molecular complexes were interconnected in 2D layers by hydrogen bonds between α-hydroxy groups of lactate ligands and water molecules. The use of similar reaction mixtures containing higher amounts of L-lactic acid, and hence having a higher pH, enabled the synthesis of a paper-like material comprising basic yttrium lactate with the supposed composition ‘Y4(OH)5(C3H5O3)7·6H2O’. This material consisted of flattened interwoven fibres with a width of ∼30 nm, which account for the flexibility and foldability of the material. The synthesised substances are the first representatives of solid state rare earth lactates.Funding
The work was supported by the Ministry of Science and Higher Education of the Russian Federation (grant agreement no. 075-15-2020-782).Conflicts of interest
There are no conflicts to declare.Acknowledgements
The graphical abstract was created using Biorender.com.References
- S. Yuan, L. Feng, K. Wang, J. Pang, M. Bosch, C. Lollar, Y. Sun, J. Qin, X. Yang, P. Zhang, Q. Wang, L. Zou, Y. Zhang, L. Zhang, Y. Fang, J. Li and H. Zhou, Adv. Mater., 2018, 30, 1704303 CrossRef PubMed
.
- S. V. Eliseeva and J.-C. G. Buenzli, Chem. Soc. Rev., 2010, 39, 189–227 RSC
- R. Janicki, A. Mondry and P. Starynowicz, Coord. Chem. Rev., 2017, 340, 98–133 CrossRef CAS
- A. J. Amoroso and S. J. A. Pope, Chem. Soc. Rev., 2015, 44, 4723–4742 RSC
- J. Wang, M. Feng, M. N. Akhtar and M.-L. Tong, Coord. Chem. Rev., 2019, 387, 129–153 CrossRef CAS
- S. Roy, A. Chakraborty and T. K. Maji, Coord. Chem. Rev., 2014, 273–274, 139–164 CrossRef CAS
- A. A. Sidorov, N. V. Gogoleva, E. S. Bazhina, S. A. Nikolaevskii, M. A. Shmelev, E. N. Zorina-Tikhonova, A. G. Starikov, M. A. Kiskin and I. L. Eremenko, Pure Appl. Chem., 2020, 92, 1093–1110 CrossRef CAS
- M. Du, C.-P. Li, C.-S. Liu and S.-M. Fang, Coord. Chem. Rev., 2013, 257, 1282–1305 CrossRef CAS
- A. S. Pronin, S. A. Semenov, D. V. Drobot, E. V. Volchkova and G. I. Dzhardimalieva, Russ. J. Inorg. Chem., 2020, 65, 1173–1185 CrossRef CAS
- M. Sertçelik and M. Durman, Russ. J. Inorg. Chem., 2020, 65, 1351–1359 CrossRef
- J. Lu and R. Wang, in Encyclopedia of Inorganic and Bioinorganic Chemistry, John Wiley & Sons, Ltd, Chichester, UK, 2012 Search PubMed
- I. P. Stolarov, N. V. Cherkashina, I. A. Yakushev, A. V. Churakov, A. B. Kornev and E. V. Fatyushina, Russ. J. Inorg. Chem., 2020, 65, 507–513 CrossRef CAS
- R. D. Shannon, Acta Crystallogr., Sect. A: Cryst. Phys., Diffr., Theor. Gen. Crystallogr., 1976, 32, 751–767 CrossRef
- Rare Earth Coordination Chemistry, ed. C. Huang, John Wiley & Sons, Ltd, Chichester, UK, 2010 Search PubMed
- P. Halder, B. Chakraborty, P. R. Banerjee, E. Zangrando and T. K. Paine, CrystEngComm, 2009, 11, 2650 RSC
- J. Yang, C. A. Trickett, S. B. Alahmadi, A. S. Alshammari and O. M. Yaghi, J. Am. Chem. Soc., 2017, 139, 8118–8121 CrossRef CAS PubMed
- J. Wahsner, E. M. Gale, A. Rodríguez-Rodríguez and P. Caravan, Chem. Rev., 2019, 119, 957–1057 CrossRef CAS PubMed
- F. Nawaz, H. Cao, Y. Xie, J. Xiao, Y. Chen and Z. A. Ghazi, Chemosphere, 2017, 168, 1457–1466 CrossRef CAS PubMed
- Z. Zheng, in Encyclopedia of Inorganic and Bioinorganic Chemistry, John Wiley & Sons, Ltd, Chichester, UK, 2012 Search PubMed
- M. Nalbandian and M. Takeda, Biology, 2016, 5, 38 CrossRef PubMed
- M. Adeva-Andany, M. López-Ojén, R. Funcasta-Calderón, E. Ameneiros-Rodríguez, C. Donapetry-García, M. Vila-Altesor and J. Rodríguez-Seijas, Mitochondrion, 2014, 17, 76–100 CrossRef CAS PubMed
- K. L. Nash, G. Johnson, D. Brigham, C. Marie, T. S. Grimes and J. C. Braley, Procedia Chem., 2012, 7, 45–50 CrossRef CAS
- K. L. Nash, Solvent Extr. Ion Exch., 2015, 33, 1–55 CrossRef CAS
- G. Tian, L. R. Martin and L. Rao, Inorg. Chem., 2010, 49, 10598–10605 CrossRef CAS PubMed
- A. Barkleit, J. Kretzschmar, S. Tsushima and M. Acker, Dalton Trans., 2014, 43, 11221–11232 RSC
- J. E. Powell and J. L. Farrell, Some Observations regarding rare-earth lactates, Ames, IA (United States), 1962 Search PubMed
- M. Sakanoue and M. Nakatani, Bull. Chem. Soc. Jpn., 1972, 45, 3429–3433 CrossRef CAS
- M. A. Gouveia and R. G. de Carvalho, J. Inorg. Nucl. Chem., 1966, 28, 1683–1688 CrossRef CAS
- G. R. Choppin, Pure Appl. Chem., 1971, 27, 23–42 CAS
- G. R. Choppin and J. A. Chopoorian, J. Inorg. Nucl. Chem., 1961, 22, 97–113 CrossRef CAS
- P. G. Manning, Can. J. Chem., 1965, 43, 3258–3263 CrossRef CAS
- A. Skerencak-Frech, F. Taube, P. L. Zanonato, M. Acker, P. J. Panak and P. Di Bernardo, Thermochim. Acta, 2019, 679, 178316 CrossRef CAS
- L. E. Roy and L. R. Martin, Dalton Trans., 2016, 45, 15517–15522 RSC
- T. S. Grimes, M. A. Nilsson and K. L. Nash, Sep. Sci. Technol., 2010, 45, 1725–1732 CrossRef CAS
- Y. Li, P. Yan, G. Hou, H. Li, P. Chen and G. Li, J. Organomet. Chem., 2013, 723, 176–180 CrossRef CAS
- Z.-R. Qu, Q. Ye, H. Zhao, D.-W. Fu, H.-Y. Ye, R.-G. Xiong, T. Akutagawa and T. Nakamura, Chem.–Eur. J., 2008, 14, 3452–3456 CrossRef CAS PubMed
- Q. Ye, D.-W. Fu, H. Tian, R.-G. Xiong, P. W. H. Chan and S. D. Huang, Inorg. Chem., 2008, 47, 772–774 CrossRef CAS PubMed
- H.-J. Chen, X.-Y. Zheng, Y.-R. Zhao, D.-Q. Yuan, X.-J. Kong, L.-S. Long and L.-S. Zheng, ACS Appl. Electron. Mater., 2019, 1, 804–809 CrossRef CAS
- R. S. Dickins, C. S. Love and H. Puschmann, Chem. Commun., 2001, 2308–2309 RSC
- Y. Qiu, Z. Liu, J. Mou, H. Deng and M. Zeller, CrystEngComm, 2010, 12, 277–290 RSC
- M. A. Katkova, G. S. Zabrodina, M. S. Muravyeva, A. S. Shavyrin, E. V. Baranov, A. A. Khrapichev and S. Y. Ketkov, Eur. J. Inorg. Chem., 2015, 2015, 5202–5208 CrossRef CAS
- X.-Y. Chen, G. S. Goff, W. C. Ewing, B. L. Scott and W. Runde, Inorg. Chem., 2012, 51, 13254–13263 CrossRef CAS PubMed
- C. N. R. Rao, S. Natarajan and R. Vaidhyanathan, Angew. Chem., Int. Ed., 2004, 43, 1466–1496 CrossRef CAS PubMed
- S. J. Lyle and M. M. Rahman, Talanta, 1963, 10, 1177–1182 CrossRef CAS
- SADABS-2016/2, Bruker AXS area detector scaling and absorption correction program, Bruker AXS Inc., Madison, Wisconsin, U.S.A., 2016 Search PubMed
- G. M. Sheldrick, Acta Crystallogr., Sect. C: Struct. Chem., 2015, 71, 3–8 Search PubMed
- A. I. Kuklin, A. K. Islamov and V. I. Gordeliy, Neutron News, 2005, 16, 16–18 CrossRef
- A. G. Soloviev, T. M. Soloveva, A. V. Stadnik, A. H. Islamov and A. I. Kuklin, Communication of JINR, Dubna, P10-2003-86, 2003, http://wwwinfo.jinr.ru/programs/jinrlib/sas/086(P10-2003-86).pdf Search PubMed.
- Y. Ishikawa, M. Furusaka, N. Niimura, M. Arai and K. Hasegawa, J. Appl. Crystallogr., 1986, 19, 229–242 CrossRef CAS
- APEX3 Crystallography Software Suite, Bruker AXS Inc., Madison, Wisconsin, U.S.A., 2016 Search PubMed.
- C. R. Groom, I. J. Bruno, M. P. Lightfoot and S. C. Ward, Acta Crystallogr., Sect. B: Struct. Sci., Cryst. Eng. Mater., 2016, 72, 171–179 CrossRef CAS PubMed
- E. A. Poryvaeva, T. A. Egiazaryan, V. M. Makarov, M. V. Moskalev, D. A. Razborov and I. L. Fedyushkin, Russ. J. Org. Chem., 2017, 53, 344–350 CrossRef CAS
- D. A. Garlotta, J. Polym. Environ., 2001, 9, 63–84 CrossRef CAS
- L. Nikolic, I. Ristic, B. Adnadjevic, V. Nikolic, J. Jovanovic and M. Stankovic, Sensors, 2010, 10, 5063–5073 CrossRef CAS PubMed
- G. Cassanas, M. Morssli, E. Fabrègue and L. Bardet, J. Raman Spectrosc., 1991, 22, 409–413 CrossRef CAS
- M. M. Maiwald, K. Müller, K. Heim, M. Trumm, N. L. Banik, J. Rothe, K. Dardenne, A. Skerencak-Frech and P. J. Panak, New J. Chem., 2020, 44, 17033–17046 RSC
- K. Nakamoto, in Infrared and Raman Spectra of Inorganic and Coordination Compounds, John Wiley & Sons, Inc., Hoboken, NJ, USA, 2009, pp. 1–273 Search PubMed
- E. Bentouhami, G. M. Bouet, J. Meullemeestre, F. Vierling and M. A. Khan, C. R. Chim., 2004, 7, 537–545 CrossRef CAS
- Z. Zheng, Chem. Commun., 2001, 2521–2529 RSC
- Z. Zheng, in Cluster Compounds of Rare-Earth Elements, 2010, pp. 109–239 Search PubMed
- A. D. Yapryntsev, A. E. Baranchikov and V. K. Ivanov, Russ. Chem. Rev., 2020, 89, 629–666 CrossRef CAS
- A. Yapryntsev, B. Abdusatorov, I. Yakushev, R. Svetogorov, A. Gavrikov, A. Rodina, Y. Fatyushina, A. Baranchikov, Y. Zubavichus and V. Ivanov, Dalton Trans., 2019, 48, 6111–6122 RSC
- A. D. Yapryntsev, A. Y. Bykov, A. E. Baranchikov, K. Y. Zhizhin, V. K. Ivanov and N. T. Kuznetsov, Inorg. Chem., 2017, 56, 3421–3428 CrossRef CAS PubMed
- A. D. Yapryntsev, K. B. Ustinovich, A. A. Rodina, V. A. Lebedev, O. I. Pokrovskiy, K. E. Yorov, A. V. Gavrikov, A. E. Baranchikov and V. K. Ivanov, J. Supercrit. Fluids, 2019, 150, 40–48 CrossRef CAS
- N. Snejko, F. Gándara, J. Perles, M. Á. Monge, E. Gutiérrez-Puebla, B. Gómez-Lor and M. Iglesias, Angew. Chem., Int. Ed., 2006, 45, 7998–8001 CrossRef PubMed
- J. Liang, R. Ma and T. Sasaki, in Structure and Bonding, ed. M. Yan and D. Wei, 2015, vol. 166, pp. 69–103 Search PubMed
- S. Eom, G. Choi, H. Nakamura and J.-H. Choy, Bull. Chem. Soc. Jpn., 2020, 93, 1–12 CrossRef CAS
- R. Ju and Q. Gu, Appl. Organomet. Chem., 2018, 32, e3926 CrossRef
- A. Komesu, P. F. Martins Martinez, B. H. Lunelli, J. Oliveira, M. R. Wolf Maciel and R. Maciel Filho, J. Chem., 2017, 2017, 1–7 CrossRef
- J. Livage, Chem. Mater., 1991, 3, 578–593 CrossRef CAS
- T. O. Kozlova, A. E. Baranchikov, D. A. Kozlov, A. V. Gavrikov, G. P. Kopitsa, A. D. Yapryntsev, K. B. Ustinovich, A. Chennevière and V. K. Ivanov, ACS Omega, 2020, 5, 17592–17600 CrossRef CAS PubMed
- K. E. Yorov, T. O. Shekunova, A. E. Baranchikov, G. P. Kopitsa, L. Almásy, L. S. Skogareva, V. V. Kozik, A. N. Malkova, S. A. Lermontov and V. K. Ivanov, J. Sol-Gel Sci. Technol., 2018, 85, 574–584 CrossRef CAS
- L. A. Feigin and D. I. Svergun, Structure Analysis by Small-Angle X-Ray and Neutron Scattering, Springer US, Boston, MA, 1987 Search PubMed
- M. Thommes, K. Kaneko, A. V. Neimark, J. P. Olivier, F. Rodriguez-Reinoso, J. Rouquerol and K. S. W. Sing, Pure Appl. Chem., 2015, 87, 1051–1069 CAS
- M. Lin, R. Klein, H. Lindsay, D. Weitz, R. Ball and P. Meakin, J. Colloid Interface Sci., 1990, 137, 263–280 CrossRef CAS
- D. Fairén-Jiménez, F. Carrasco-Marín, D. Djurado, F. Bley, F. Ehrburger-Dolle and C. Moreno-Castilla, J. Phys. Chem. B, 2006, 110, 8681–8688 CrossRef PubMed
- J. Hyeon-Lee, G. Beaucage and S. E. Pratsinis, Chem. Mater., 1997, 9, 2400–2403 CrossRef CAS
- G. Porod, Kolloid-Z., 1951, 124, 83–114 CrossRef CAS
- A. Guinier and G. Frournet Small-Angle Scattering of X-rays, John Wiley and Sons, Inc., New York, 1955 Search PubMed
- J. Teixeira, in On Growth and Form, Springer Netherlands, Dordrecht, 1986, pp. 145–162 Search PubMed
- K. S. Tang, Life Sci., 2020, 259, 118287 CrossRef CAS PubMed
- G. Rajakumar, L. Mao, T. Bao, W. Wen, S. Wang, T. Gomathi, N. Gnanasundaram, M. Rebezov, M. A. Shariati, I.-M. Chung, M. Thiruvengadam and X. Zhang, Appl. Sci., 2021, 11, 2172 CrossRef CAS
- K. Lebbou, P. Perriat and O. Tillement, J. Nanosci. Nanotechnol., 2005, 5, 1448–1454 CrossRef CAS PubMed
- R. M. Krsmanović Whiffen, D. Bregiroux and B. Viana, Ceram. Int., 2017, 43, 15834–15841 CrossRef
- A. D. Yapryntsev, L. S. Skogareva, A. E. Gol'dt, A. E. Baranchikov and V. K. Ivanov, Russ. J. Inorg. Chem., 2015, 60, 1027–1033 CrossRef CAS
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2085515. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d1ra05923h |
| This journal is © The Royal Society of Chemistry 2021 |