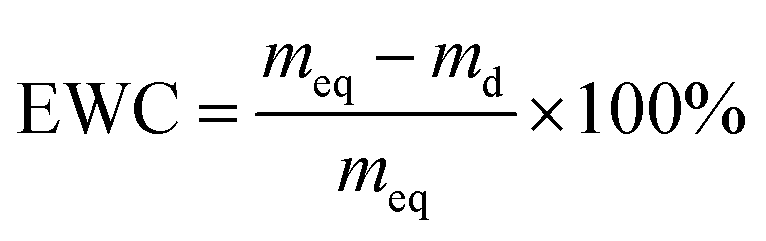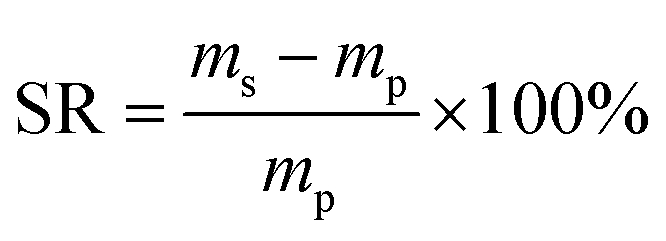DOI:
10.1039/D1RA05896G
(Paper)
RSC Adv., 2021,
11, 32988-32995
Highly tough and rapid self-healing dual-physical crosslinking poly(DMAA-co-AM) hydrogel†
Received
4th August 2021
, Accepted 22nd September 2021
First published on 7th October 2021
Abstract
Introducing double physical crosslinking reagents (i.e., a hydrophobic monomer micelle and the LAPONITE® XLG nano-clay) into the copolymerization reaction of hydrophilic monomers of N,N-dimethylacrylamide (DMAA) and acrylamide (AM) is reported here by a thermally induced free-radical polymerization method, resulting in a highly tough and rapid self-healing dual-physical crosslinking poly(DMAA-co-AM) hydrogel. The mechanical and self-healing properties can be finely tuned by varying the weight ratio of nanoclay to DMAA. The tensile strength and elongation at break of the resulting nanocomposite hydrogel can be modulated in the range of 7.5–60 kPa and 1630–3000%, respectively. Notably, such a tough hydrogel also exhibits fast self-healing properties, e.g., its self-healing rate reaches 48% and 80% within 2 and 24 h, respectively.
1. Introduction
With chemically or physically crosslinked three-dimensional (3D) network structures, polymer hydrogels possess the ability to absorb large amounts of water and resist dissolution.1–3 The excellent biocompatibility, porous structure, tunable stiffness, and biological tissue-like elasticity of polymer hydrogels impart widespread use as biomaterials in the fields of tissue engineering,4–6 gene and drug delivery systems,7–9 cell cultures,10–12 superabsorbents,13–16 biosensors,17–20 artificial e-skins,21–23 wound healing,24–26 etc. However, conventional chemically crosslinked hydrogels are always brittle and exhibit poor mechanical performance due to the absence of an energy dissipation mechanism, limiting their widespread applications.27–30 Functional biomimetic self-healing hydrogels possess remarkable performance, such as fast self-healing properties and high mechanical strength and toughness, which greatly extend their service life, especially in artificial e-skins, biosensing, and drug delivery.31–35 Different approaches for repairing the damaged network structures of hydrogels have been explored to obtain highly self-healing hydrogels. The strategies to design self-healing hydrogels are mainly based on healing agents, dynamically reversible covalent bonds, or dynamically non-covalent bonds.36–39 Up to now, the frequent use of non-covalent bonds to prepare self-healing hydrogels has primarily been based on metal–ligand interactions,40–43 host–guest interactions,44–46 hydrophobic interactions,47–50 hydrogen bonding,51–57 ionic interactions,58–62 etc.
Since non-covalent bonds are generally more susceptible to the external environment than dynamic covalent linkage, the self-healing hydrogel systems based on non-covalent interactions, or the self-healing supramolecular hydrogel systems, will be more intelligent and easier for structural control than those based on dynamically reversible covalent bonds.63,64 Among various non-covalent bonding, hydrophobic bonding is the most common for bulk self-healing materials, thus playing a dominant role in forming large biological systems.65–67 Therefore, applying hydrophobic interactions to construct robust hydrogels with superior self-healing ability and remarkable mechanical properties is highly desirable. In particular, Okay and co-workers47 presented a simple strategy to create strong hydrophobic interactions by incorporating hydrophobic chains into hydrophilic polymer network chains, resulting in self-healing hydrogels. In addition, the finite lifetime of hydrophobic associations between stearyl methacrylate (C18) blocks made the resultant hydrogel highly tough. The breakage of the hydrogel samples bearing C18 blocks occurred at elongation ratios of 3600%, and a self-healing efficiency to elongation at break of about 100% was achieved.47 However, these hydrogels suffer from low tensile strength and resilience, thus impeding their practical applications.
Hydrogels based on nanocomposite (NC)68 and dual networks (DN)69 are regarded as novel and functional self-healing materials, and NC hydrogels have drawn the most widespread attention because of their high strength and versatility. NC hydrogels can be prepared by physically crosslinking polymer chains with nanoparticles such as inorganic clay LAPONITE® XLG. The unique polymer/clay composite network structure provides NC hydrogels with self-healing capability.70–76 The interactions between the polymer chains and the adjacent clay particles are considered non-covalent or hydrogen bonding. Adjoining two such surfaces facilitates grafted chains at the two surfaces to diffuse into each other and interact with neighboring clay platelets by hydrogen bonding. Consequently, many new crosslinks are generated across the interface, which leads the cut surfaces to rejoin. Thus, the macroscopic self-healing of NC hydrogels arises from the reconstruction of the network at the interface due to the mutual diffusion of long grafted chains and their subsequent interactions with clay.70,75
This study judiciously combined the appropriate materials selection and synthetic processing to construct a highly tough and rapidly self-healing dual-physical crosslinking hydrogel material. We introduced a nanoclay (LAPONITE®) and a hydrophobic monomer micelle into the copolymerization system of acrylamide (AM) and N,N-dimethylacrylamide (DMAA) to generate a dual-physical crosslinking network through a thermally induced free-radical polymerization process. The synthetic process of the dual-physical crosslinking hydrogel is presented in Fig. 1. The generation of a dual-physical crosslinking network is realized by introducing inorganic nanoclay (leading to one physical crosslinking network) and organic hydrophobic monomer micelles (resulting in the other physical crosslinking network) into the copolymerization reaction system of hydrophilic DMAA and AM monomers. Hydrophobic and ionic interactions within the NC hydrogel matrix render it highly tough and rapidly self-healing.
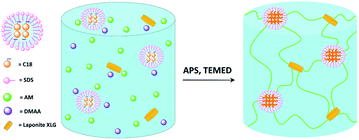 |
| | Fig. 1 Schematic illustration of the poly(DMAA-co-AM) hydrogel synthesis with dual-physical crosslinking network as generated by introducing the nanoclay and hydrophobic monomer micelles. | |
2. Experimental
2.1. Materials
DMAA and AM with the purities exceeding 99% were supplied by Tokyo Chemical Industry Co., Ltd. and Shanghai Aladdin Bio-Chem Technology Co., Ltd., respectively. Potassium persulfate (KPS, 98.0%), stearyl methacrylate (C18, 95%), NaCl (99%), N,N,N′,N′-tetramethylethylenediamine (TEMED, 99%) were obtained from Shanghai Aladdin Bio-Chem Technology Co., Ltd. Sodium dodecyl sulfate with the molecular weight of 288.38 g mol−1 was purchased from Shanghai Aladdin Bio-Chem Technology Co., Ltd. Inorganic clay “LAPONITE® XLG” nanoparticles, with the molecular formula of [Mg5.34Li0.66Si8O20(OH)4]Na0.66, possessed the diameter and thickness of 30 and 1 nm, respectively, and were purchased from Rockwood Ltd, UK. All the other chemicals were of analytic grades and supplied by Shanghai Macklin Biochemical Co., Ltd. All the chemicals were used as received unless otherwise stated.
2.2. Synthesis of the dual-network self-healing NC hydrogels
The poly(AM-co-DMAA)-based self-healing NC hydrogels were synthesized according to the procedures as follows: NaCl (0.2 g) was added to Flask 1 containing DI water (5.0 g) and then mixed with sodium dodecyl sulfate (0.35 g) under magnetic stirring at 35 °C for 1 h untilled dissolved, which was followed by cooling down to room temperature. Next, octadecyl methacrylate (0.0215 g) was added and stirred for 30 min before mixing the AM monomer (0.25 g). The inorganic LAPONITE® XLG nanoclay was charged separately into flask 2 with DI water (10 g). After rapidly dispersing the nanoclay into the aqueous solution by magnetic stirring, DMAA (2.0 g) was mixed. Finally, the reaction media in flask 1 and flask 2 were mixed by magnetic stirring, and then several drops of a promoter (in this case, TEMED) and a given amount of an initiator (namely, KPS) were stepwise added. It is worth mentioning that the copolymerization reaction media could be introduced to any models of the desired shape to form shaped hydrogels. The basic properties of the prepared samples were monitored to screen out three representative ones (designated as 0.15DPNC, 0.2DPNC, and 0.3DPNC, where the number refers to the nanoclay content with 2.0 g DMAA as the baseline at the DMAA-to-AM mass ratio of 8![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1), which would be subject to the further performance measurements.
:1), which would be subject to the further performance measurements.
2.3. Characterizations
Fourier transform infrared (FTIR) spectra were measured using an IRAffinity-1S FTIR spectrometer (Shimadzu, Japan) in the attenuated total reflectance (ATR) testing mode. The lyophilization of the hydrogel samples was carried out using a SCIENTZ-18N freezer dryer (Ningbo Scientz Biotechnology Co., Ltd., China). An Axio Scope A1 metallographic microscope was employed to monitor the self-healing effect of the hydrogel that had been deliberately cut off. The morphologies of the cross-section of lyophilized hydrogels were observed with an FEI Quanta 200 field-emission scanning electron microscope (SEM, Netherlands) at an accelerated electron energy of 10 kV. The specimen prepared for the SEM observation was described as follows: the fully swelled hydrogel was put into a refrigerator before 6 h freeze-drying treatment, and part of the freeze-dried sample was taken to the SEM holder for morphology observation after the Au-spraying had been applied to the sample surface. Mechanical properties of the prepared hydrogels were tested with an M350-CT electronic tensile machine (Testometric Co., Rochdale, UK). The specimen prepared for the mechanical test was according to the procedures as follows: the samples were fabricated into the rectangle shape with the size of 20 mm × 5 mm × 1 mm. The mechanical test was conducted at an extension rate of 20 mm min−1, and plots of strain vs. stress were recorded. We also performed a macroscopic tensile test to inspect the toughness of the sample directly as follows: long cylinder samples were knotted and stretched to examine whether the sample would be ruptured at the knotted site.
2.4. Equilibrium water content (EWC) and swelling ratio (SR) measurements
The EWC of poly(AM-co-DMAA) hydrogels was measured at the ambient temperature based on a gravimetric method. The sample was immersed in deionized water and weighed at prescribed time intervals until the swelling reached equilibrium. Note that the water was removed from the sample surface with filter paper before weighing. Then, the sample was dried to constant weight in a vacuum oven at 50 °C. The EWC and SR of hydrogels were calculated based on the equations below:
where meq and md denote the equilibrium wet weight and dry weight of hydrogels, respectively, and ms and mp are the weights of the swollen hydrogel and the as-prepared hydrogel, respectively. Each sample was measured in triplicate, and the average value was recorded.
2.5. Self-healing property evaluation
Two portions of the prepared hydrogel were placed into a clean glass vessel, and one was dyed in red with rhodamine B. Afterwards, both the portions were cut off along the middle position, and the resulting four parts intimately contacted for 1 h. The self-healed sample was tested by stretching with hands to check the interactions between the two parts integrated by self-healing. Separately, a hydrogel sample in a rectangular shape with a size of 20 mm × 5 mm × 1 mm was cut off along the middle position, and the resulting two parts contacted for 2 and 24 h. The self-healing effect was examined by the tensile test using a universal tensile machine. Besides, the microscopic morphology of the self-healed hydrogel cross-sections was also observed using a metallographic microscope to monitor the variation of the incision of the hydrogel during the self-healing process.
2.6. Rheological property analysis
The rheology test of the hydrogel was analyzed with an Anton Paar Instrument (MCR302) at 25 °C by using a parallel plate geometry with a diameter of 25 mm. The gap was set to 1 mm. The storage and loss moduli were also recorded under the alternating shearing strain between 0.01% and 100% at the constant frequency (1.0 Hz). Furthermore, a continuous step strain of this hydrogel was measured. Briefly, the hydrogel was first subjected to 500% strain and then to 1% strain at a scanning frequency of 1 rad s−1, and this process was repeated four times.
3. Results and discussion
3.1. Structural characterization
We first carried out a preliminary screening test on the prepared series of hydrogel samples and selected representative ones for the subsequent systematic characterizations, and the selected three samples were designated as 0.15DPNC, 0.2DPNC, and 0.3DPNC, where the number represents the content of the nanoclay (vs. 2.0 g DMAA, at the DMAA-to-AM mass ratio of 8:1). Fig. 2 shows the FITR spectra of these representative samples. While the peak at 1603 cm−1 corresponds to N–H (amide II) bending vibrations, the peaks at 3393, 2922, 1252, and 1660 cm−1 can be assigned to the stretching vibrations of N–H, CH2–, C–O, and C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O (amide I), respectively. These absorption peaks present the characteristics of the poly(AM-co-DMAA) skeletons, thus verifying the successful synthesis of the poly(AM-co-DMAA) hydrogel. With a change in the nanoclay content, no significant variation can be noted in the FTIR spectra, suggesting that physical rather than chemical interactions exist between the nanoclay and poly(AM-co-DMAA) chains. The chemical structure of the synthesized the poly(AM-co-DMAA) hydrogel was also characterized by 1H NMR analyses. Fig. S1–S3† display the 1H NMR spectra of 0.15DPNC, 0.2DPNC, and 0.3DPNC, respectively. For all these hydrogels, the spectra can be divided into three main regions: 2.8–3.6 ppm (6H) attributed to the methyl protons of the amide function, 2.2–2.6 ppm (1H) indexed into the poly(AM-co-DMAA) backbone methine protons, and 0.8–1.9 ppm (2H) corresponding to the poly(AM-co-DMAA) backbone methylene protons.77–79
O (amide I), respectively. These absorption peaks present the characteristics of the poly(AM-co-DMAA) skeletons, thus verifying the successful synthesis of the poly(AM-co-DMAA) hydrogel. With a change in the nanoclay content, no significant variation can be noted in the FTIR spectra, suggesting that physical rather than chemical interactions exist between the nanoclay and poly(AM-co-DMAA) chains. The chemical structure of the synthesized the poly(AM-co-DMAA) hydrogel was also characterized by 1H NMR analyses. Fig. S1–S3† display the 1H NMR spectra of 0.15DPNC, 0.2DPNC, and 0.3DPNC, respectively. For all these hydrogels, the spectra can be divided into three main regions: 2.8–3.6 ppm (6H) attributed to the methyl protons of the amide function, 2.2–2.6 ppm (1H) indexed into the poly(AM-co-DMAA) backbone methine protons, and 0.8–1.9 ppm (2H) corresponding to the poly(AM-co-DMAA) backbone methylene protons.77–79
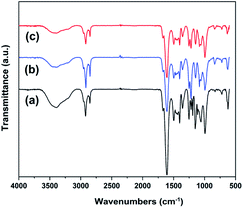 |
| | Fig. 2 FTIR spectra of the poly(AM-co-DMAA) hydrogels: (a) 0.15DPNC, (b) 0.2DPNC, and (c) 0.3DPNC. | |
3.2. Swelling properties
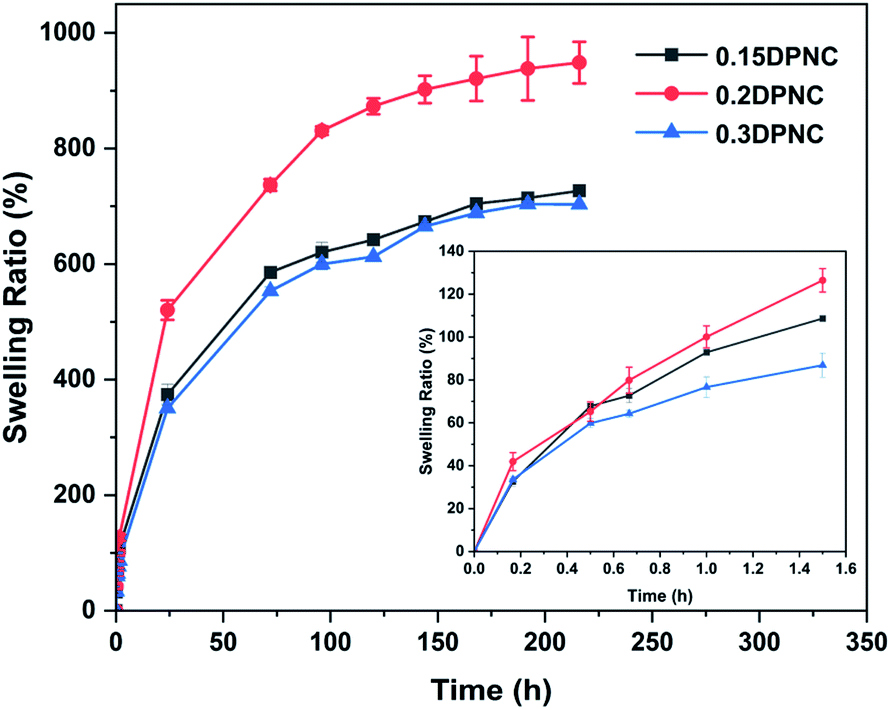
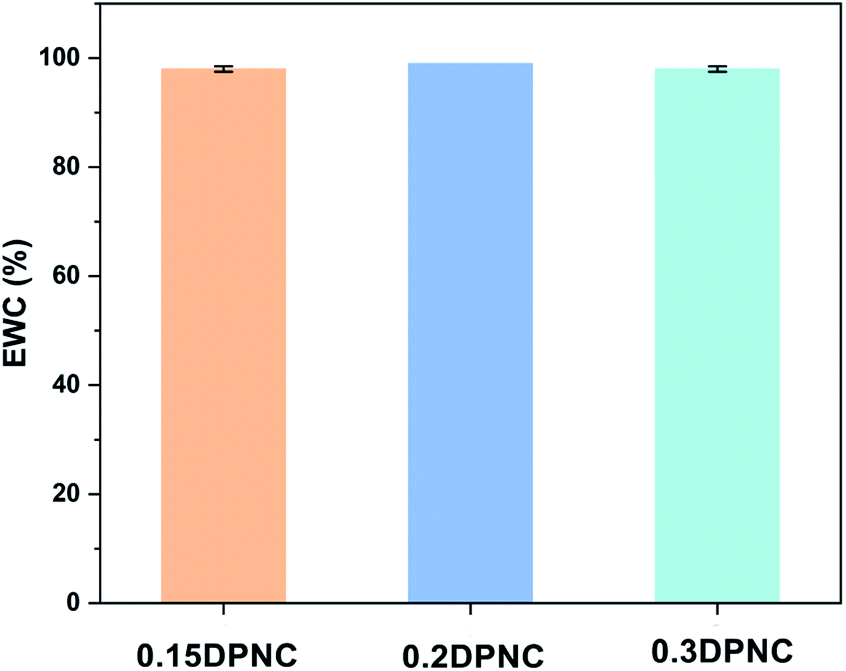
The swelling behavior of the hydrogels is one of the critical performance indexes for biomaterials. The swelling ratio is usually adopted to evaluate the absorbability and structural stability of hydrogel materials. The swelling behavior of the poly(AM-co-DMAA) hydrogels is evaluated through a long-term immersion process in water, and the results are provided in Fig. 3–5. A 4-fold increase in the volume of the swelled hydrogel relative to the as-obtained hydrogel can be noted. The drying treatment of the as-obtained hydrogel leads to the formation of a plastic sheet (Fig. 3). These findings indicate that the prepared hydrogel possesses an appreciable water absorption capacity. With an increase in the clay content, the hydrogel swelling ratio increases initially and then decreases. The hydrogels swelling ratio in this process is shown in Fig. 4. After swelling in water for 1 h, the swelling ratio of the 0.2DPNC hydrogel reaches 100% of the original mass, much higher than that of other poly(AM-co-DMAA) hydrogels, thus implying that the 0.2DPNC hydrogel absorbs water at a higher speed. The EWCs calculated for the typical hydrogels are shown in Fig. 5. The 0.2DPNC sample exhibits the highest EWC (99%) among the investigated samples, confirming the excellent water absorption capacity that likely results from the compact double crosslinking network (formed by the inorganic nanoclay and hydrophobic organic monomer micelles) and the presence of a microporous structure with the ability to hold a large amount of water. A further increase in the nanoclay content yields the 0.3DPNC sample, but the water absorption capacity becomes lowered because the nanoclay overload reduces the number of macropores.
 |
| | Fig. 3 (a–f) Photo images of the as-prepared 0.2DPNC hydrogel sample (a and d), the swelled 0.2DPNC hydrogel sample after water absorption equilibrium (b and e), and the corresponding oven-dried sample (c and f). | |
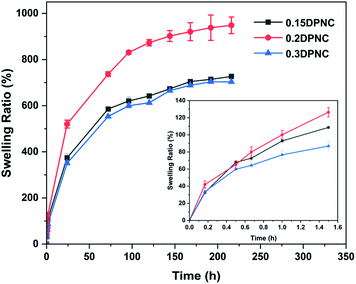 |
| | Fig. 4 Plots of the swelling rate of the hydrogels as a function of the swelling duration. | |
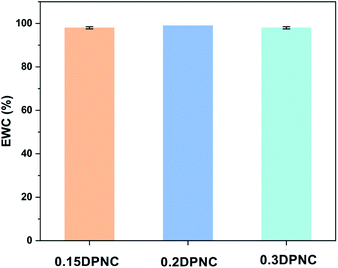 |
| | Fig. 5 EWCs estimated for the three representative 0.15DPNC, 0.2DPNC, and 0.3DPNC hydrogel samples. | |
3.3. Morphology observation
The SEM morphologies of the cross-section of the typical poly(AM-co-DMAA) hydrogels are presented in Fig. 6. It can be noted that these hydrogel samples exhibit a honeycomb-like porous structure. Having an average pore size of approximately 50 μm, the porous structure exhibits uniformity, homogenous distribution, and high density. The higher concentration of the crosslinker would give rise to a more compact crosslinking network. The incorporation of the nanoclay can improve the composite hydrogel toughness and, consequently, contribute to maintaining structural integrity during the swelling process. The compact crosslinking network contributes to the impressive toughness of the NC hydrogel. Notably, incorporating the nanoclay into the poly(AM-co-DMAA) makes the pore wall surface rougher, revealing the dispersion of the inorganic nanoclay within the polymer matrix. As a result, the strengthening effect of the inorganic nanoclay can be effectively imparted to the composite hydrogel.
 |
| | Fig. 6 (a–c) SEM images of the representative 0.15DPNC (a), 0.2DPNC (b), and 0.3DPNC (c) hydrogels. | |
3.4. Mechanical property evaluation
The mechanical properties of the hydrogel materials usually dictate their practical application potential. In this regard, we first tested the knotted long cylinder hydrogel sample by stretching with hands to see whether it would rupture at the knotted site and evaluate the toughness (Fig. 7). The good toughness can be revealed by the finding that the knotted long cylinder hydrogel can be stretched to a significant extent that it becomes many times longer than that before stretching.
 |
| | Fig. 7 Photo images illustrating the robustness of the as-prepared 0.2DPNC hydrogel: the 0.2DPNC hydrogel strip-tied knot before (left) and after (right) stretching with hand. | |
A universal tensile machine was employed further to assess the mechanical properties of the typical samples. The specimens with a rectangular shape and a size of 20 mm × 5 mm × 1 mm were prepared for the uniaxial extension test (Fig. 8). The photo image (on the right side) presents that the typical hydrogel can be highly stretched. The strain–stress plots in Fig. 8 show that the 0.15DPNC sample with the lowest nanoclay content performs the worst in the tensile test by considering its lowest tensile strength as a result of the smallest number of the crosslinking sites afforded by the nanoclay. With an increase in the nanoclay content, the tensile strength of the resulting 0.2DPNC sample is markedly increased, along with a noticeable improvement in elongation at break (exceeding 3000%). A further increase in the nanoclay content degrades the tensile properties of the resultant 0.3DPNC sample, as caused by the overhigh content of the nanoclay that would substantially increase the contact area between the polymer chains and nanoclay, consequently restraining the molecular chains to a great extent.
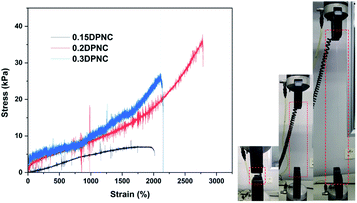 |
| | Fig. 8 Strain–stress plots for the typical composite hydrogel samples with different nanoclay contents. The photo image on the right side presents the typical 0.2DPNC hydrogel before and after the uniaxial extension test. | |
3.5. Self-healing and mechanical stretching behaviors
Smart hydrogels have become the research focus of many important areas, such as biomedicines, biochemical engineering, and environmental remediations. Self-healing hydrogels are under the spotlight due to their viability for practical biochemical engineering applications. Notably, the present hydrogel exhibits excellent self-healing performance (Fig. 9). The parts cut from the hydrogel can be healed along the contact area within 1 h, yielding an integrated hydrogel that can be highly stretched. We dyed one portion in red and connected to the other along the cut surface to present the self-healing performance. The efficient self-healing performance most likely results from the self-assembling capability of the hydrophobic octadecyl methacrylate sections in the hydrophobic micelles. The non-covalent interactions occur between the self-assembled sections/DMAA chains and the nanoclay surface, resulting in physical crosslinking points. The dual-physical crosslinking networks provide the composite hydrogel with rapid self-healing capacity.
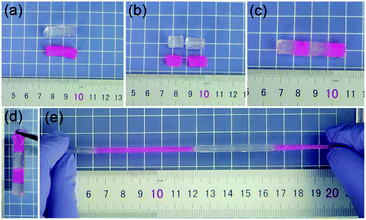 |
| | Fig. 9 Photo image demonstrating the self-healing performance of the typical 0.2DPNC hydrogel. | |
A metallographic microscope was further employed to observe the self-healing effect of the typical sample at the microscopic level (Fig. 10). A scratch was created on the thin hydrogel slice using a sharp blade, which was then monitored under the metallographic microscope. Of note, the scratch almost disappeared within 3 min. We also employed the universal tensile machine to measure the tensile properties of the self-healed samples prepared by contacting the cut parts for 2 h and 24 h. The tensile plots of the samples self-healed for 2 h and 24 h are presented in Fig. 11. After self-repaired for 2 h, the integrated hydrogel sample exhibits an elongation at break of 1503%, corresponding to the self-healing rate of 48.7%. In contrast, self-healing for 24 h yields the integrated hydrogel possessing an elongation at break of 2334%, corresponding to the self-healing rate of as high as 80%, basically the same as the original sample. Thus, these results prove the rapid self-healing properties of the present poly(AM-co-DMAA)-based nanocomposite hydrogel, different from a similar poly(AM-co-DMAA)-based nanocomposite hydrogel without self-healing properties albeit with good mechanical strength.80,81 Notably, our dual-physical crosslinking poly(DMAA-co-AM) hydrogels incorporating a small amount of clay exhibit both excellent elongation at break and self-healing properties. The properties of similar poly(AM-co-DMAA)-based nanocomposite hydrogel are listed in Table 1.
 |
| | Fig. 10 Metallographic microscope images showing the self-healing process of the typical 0.2DPNC composite hydrogel for different durations: (a) 0 min, (b) 1 min, (c) 2 min, and (d) 3 min test. | |
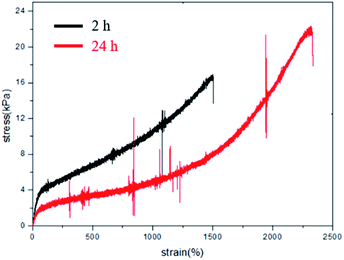 |
| | Fig. 11 Tensile test for the 0.2DPNC hydrogel integrated by self-healing for 2 h (black line) and 24 h (red line). | |
Table 1 The properties of similar poly(AM-co-DMAA)-based nanocomposite hydrogels
| Similar hydrogels |
Tensile strength |
Elongation at break |
Self-healing |
| TAD gels80 |
50–367 kPa |
500–1760% |
No |
| NCP gels81 |
18–313 kPa |
About 1600–3205% |
No |
| DPNC gels |
7.5–60 kPa |
1630–3000% |
Yes |
3.6. Rheological property investigation
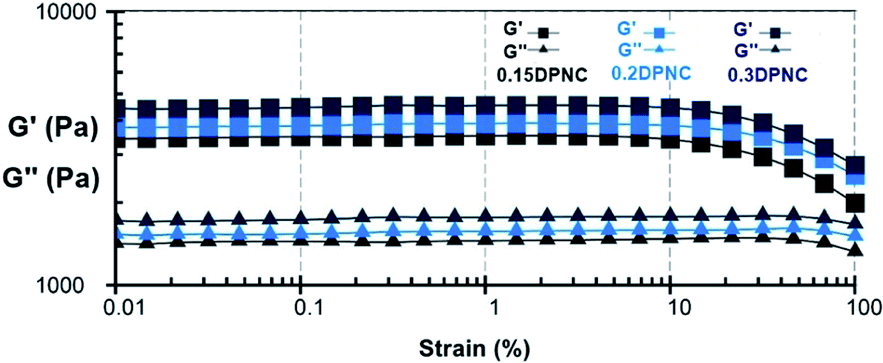
The rheological properties of the samples were examined by a dynamic rheology test. A strain amplitude sweep measurement was performed to analyze the storage modulus G′ and the loss modulus G′′ of this hydrogel vs. the oscillatory strain amplitude, with the results shown in Fig. 12. When the parameter γ is smaller than 100%, the hydrogel exhibits an elastic characteristic as reflected by the constant value of both G′ and G′′ (G′ > G′′). The G′ value decreases rapidly with the parameter γ larger than 100%, making G′′ approach the value of G′ and thus implying the collapse of the gel network.
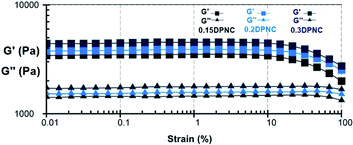 |
| | Fig. 12 G′ and G′′ values of the typical hydrogels (0.15DPNC, 0.2DPNC, and 0.3DPNC), as obtained based on the strain amplitude sweep. | |
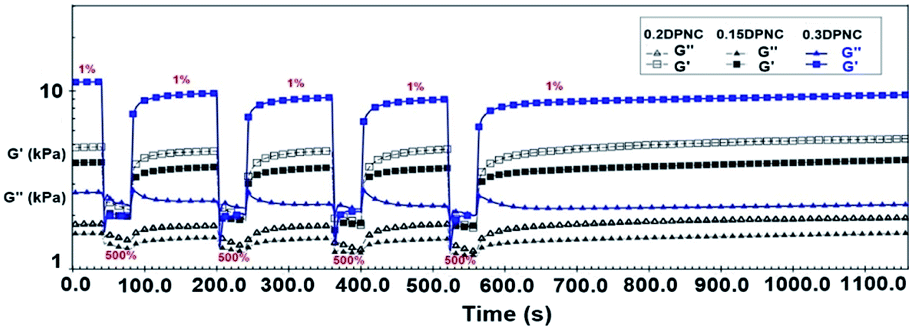
Rheological characterization was also performed further to demonstrate the robust self-healing properties of these hydrogels. Fig. 13 shows the repeated recovery of the mechanical properties of these hydrogels after being subjected to disruptive mechanical shearing forces. Initially, a small amplitude oscillatory shear (a strain of 1% at a frequency of 1 rad s−1) is applied to these hydrogels for 120 s. Under this condition, the G′ value is greater than that of G′′, and both do not change with time. This implies that the hydrogel network remains intact under small oscillatory strains. Afterward, the hydrogel is subjected to a large amplitude oscillatory shear (a strain of 500% at a frequency of 1 rad s−1) for 40 s. In this case, the G′ value decreases drastically to approach the G′′ value. This is indicative of the rupture of the gel network. Subsequently, a low amplitude strain of 1% was applied to the hydrogel within the constant regime shown in Fig. 13. The gel-like character (featuring G′ > G′′) was recovered within 10 s, along with the recovery of G′ and G′′ to their initial values within 160 s. The complete recovery of the destructed hydrogel network confirms the self-healing capacity of the hydrogel.
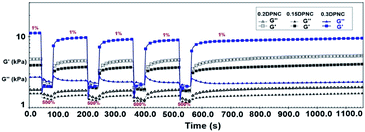 |
| | Fig. 13 G′ and G′′ values estimated based on a continuous strain sweep with the hydrogels alternatively subjected to small (1% strain) and large (500% strain) oscillation forces. | |
4. Conclusions
We have innovatively combined the appropriate raw materials selection and synthesis protocol to generate a highly tough and rapidly self-healing dual-physical crosslinking hydrogel. The dual-physical crosslinking network is formed by using hydrophobic monomer micelles and nanoclay. The central role of the nanoclay inclusion in significantly improving the mechanical properties and self-healing performance is elucidated. Incorporating an appropriate content of nanoclay gives rise to the homogeneous distribution of the physical crosslinking points, thus facilitating the efficient load stress transfer out of the hydrogel substrate and significantly reducing the destructive impact. The clay nanoadditive can be effectively incorporated into the polymer matrix, as proven by the coarsened surface of the hydrogel pore wall under SEM, thereby strengthening the wall and promoting the mechanical performance of the hydrogel material. Nevertheless, the over-load of the nanoclay degrades the mechanical properties of the NC hydrogel in that the highly concentrated nanoclay would much constrain the polymer chain motion, lowering the toughness. Notably, the optimized NC hydrogel exhibits high toughness and rapid self-healing properties, as evidenced by various characterization techniques such as metallographic microscope observation, tensile tests, dynamic rheological curve measurements. This work opens up a new avenue to construct highly tough and rapidly self-healing hydrogel materials for many practical applications of significance, such as chemical and biochemical engineering and biomedical scaffolds.
Author contributions
Yinlei Lin, Shuoqi Wang, Huawen Hu, and Guangji Li proposed the idea and supported the project. Yinlei Lin, Huawen Hu, Haichen Zhang and Shuoqi Wang carried out the experiments and wrote the paper. Shuoqi Wang, Sheng Sun, Yaoheng Liang, Yisheng Xu and Jie Luo processed the data.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
This work was supported by the National Natural Science Foundation of China (51803028), the Key Project of Guangdong Basic and Applied Basic Research Foundation (2020B1515120081), the Guangdong Provincial Education Department Special Project of Key Research Areas (2020ZDZX2066), the Guangdong Basic and Applied Basic Research Foundation (2019A1515110645), the Innovation Team of Universities of Guangdong Province (2020KCXTD011), the Engineering Research Center of Universities of Guangdong Province (2019GCZX002), the Guangdong Key Laboratory for Hydrogen Energy Technologies (2018B030322005) and the Guangdong Basic and Applied Basic Research Foundation (2019A1515110432).
Notes and references
- Y. Guo, J. Bae, Z. Fang, P. Li, F. Zhao and G. Yu, Chem. Rev., 2020, 120, 7642–7707 CrossRef CAS PubMed.
- N. R. Richbourg and N. A. Peppas, Prog. Polym. Sci., 2020, 105, 101243 CrossRef CAS.
- S. K. Das, T. Parandhaman and M. D. Dey, Green Chem., 2021, 23, 629–669 RSC.
- M. L. Pita-López, G. Fletes-Vargas, H. Espinosa-Andrews and R. Rodríguez-Rodríguez, Eur. Polym. J., 2021, 145, 110176 CrossRef.
- M. Mehrali, A. Thakur, C. P. Pennisi, S. Talebian, A. Arpanaei, M. Nikkhah and A. Dolatshahi-Pirouz, Adv. Mater., 2017, 29, 1603612 CrossRef PubMed.
- H. Zhao, M. Liu, Y. Zhang, J. Yin and R. Pei, Nanoscale, 2020, 12, 14976–14995 RSC.
- F. Mo, K. Jiang, D. Zhao, Y. Wang, J. Song and W. Tan, Adv. Drug Delivery Rev., 2021, 168, 79–98 CrossRef CAS PubMed.
- E. Mauri, G. Perale and F. Rossi, ACS Appl. Nano Mater., 2018, 1, 6525–6541 CrossRef CAS.
- M. I. Rial-Hermida, A. Rey-Rico, B. Blanco-Fernandez, N. Carballo-Pedrares, E. M. Byrne and J. F. Mano, ACS Biomater. Sci. Eng., 2021, 7, 4102–4127 CrossRef CAS PubMed.
- S. H. Lee, K. Y. Shim, B. Kim and J. H. Sung, Biotechnol. Prog., 2017, 33, 580–589 CrossRef CAS PubMed.
- M. R. Poorna, R. Jayakumar, J. Chen and U. Mony, Colloids Surf., B, 2021, 207, 111991 CrossRef CAS PubMed.
- S. Mondal, S. Das and A. K. Nandi, Soft Matter, 2020, 16, 1404–1454 RSC.
- J. Zhu, W. K. Tan, X. Song, Z. Gao, Y. Wen, C. N. Ong, C. S. Loh, S. Swarup and J. Li, ACS Sustainable Chem. Eng., 2020, 8, 9425–9433 CrossRef CAS.
- B. Cheng, B. Pei, Z. Wang and Q. Hu, RSC Adv., 2017, 7, 42036–42046 RSC.
- R. M. Barajas-Ledesma, L. Hossain, V. N. L. Wong, A. F. Patti and G. Garnier, J. Colloid Interface Sci., 2021, 599, 140–148 CrossRef CAS PubMed.
- K. Kabiri, H. Omidian, M. J. Zohuriaan-Mehr and S. Doroudiani, Polym. Compos., 2011, 32, 277–289 CrossRef CAS.
- S. Jung, J. L. Kaar and M. P. Stoykovich, Mol. Syst. Des. Eng., 2016, 1, 225–241 RSC.
- P. Shen, Y. Zhang, Z. Cai, R. Liu, X. Xu, R. Li, J. Wang and D. Yang, J. Mater. Chem. C, 2021, 9, 5840–5857 RSC.
- H. H. Bay, R. Vo, X. Dai, H. Hsu, Z. Mo, S. Cao, W. Li, F. G. Omenetto and X. Jiang, Nano Lett., 2019, 19, 2620–2626 CrossRef CAS PubMed.
- I. Y. Jung, J. S. Kim, B. R. Choi, K. Lee and H. Lee, Adv. Healthcare Mater., 2017, 6, 1601475 CrossRef PubMed.
- H. Xu, Y. Xie, E. Zhu, Y. Liu, Z. Shi, C. Xiong and Q. Yang, J. Mater. Chem. A, 2020, 8, 6311–6318 RSC.
- F. Yi, F. Guo, Y. Li, D. Wang, P. Huang and S. Fu, ACS Appl. Mater. Interfaces, 2021, 13, 32084–32093 CrossRef CAS PubMed.
- H. Huang, L. Han, X. Fu, Y. Wang, Z. Yang, L. Pan and M. Xu, Adv. Electron. Mater., 2020, 6, 2000239 CrossRef CAS.
- N. Asadi, H. Pazoki-Toroudi, A. R. D. Bakhshayesh, A. Akbarzadeh, S. Davaran and N. Annabi, Int. J. Biol. Macromol., 2021, 170, 728–750 CrossRef CAS PubMed.
- Y. Yao, A. Zhang, C. Yuan, X. Chen and Y. Liu, Biomater. Sci., 2021, 9, 4523–4540 RSC.
- Y. Xiong, X. Zhang, X. Ma, W. Wang, F. Yan, X. Zhao, X. Chu, W. Xu and C. Sun, Polym. Chem., 2021, 12, 3721–3739 RSC.
- C. Creton, Macromolecules, 2017, 50, 8297–8316 CrossRef CAS.
- P. Calvert, Adv. Mater., 2009, 21, 743–756 CrossRef CAS.
- J. P. Gong and Y. Osada, Adv. Polym. Sci., 2010, 236, 203–246 CrossRef CAS.
- O. Akca, B. Yetiskin and O. Okay, J. Appl. Polym. Sci., 2020, 137, 48853 CrossRef CAS.
- S. Billiet, X. K. D. Hillewaere, R. F. A. Teixeira and F. E. Du Prez, Macromol. Rapid Commun., 2013, 34, 290–309 CrossRef CAS PubMed.
- V. Amendola and M. Meneghetti, Nanoscale, 2009, 1, 74–88 RSC.
- F. Cheng, H. Chen and H. Li, Eur. Polym. J., 2020, 124, 109448 CrossRef CAS.
- H. Rammal, A. GhavamiNejad, A. Erdem, R. Mbeleck, M. Nematollahi, S. E. Diltemiz, H. Alem, M. A. Darabi, Y. N. Ertas, E. J. Caterson and N. Ashammakhi, Mater. Chem. Front., 2021, 5, 4368–4400 RSC.
- Z. Deng, H. Wang, P. X. Ma and B. Guo, Nanoscale, 2020, 12, 1224–1246 RSC.
- L. Teng, Y. Chen, Y. Jia and L. Ren, J. Mater. Chem. B, 2019, 7, 6705–6736 RSC.
- Z. Wei, J. H. Yang, J. Zhou, F. Xu, M. Zrínyi, P. H. Dussault, Y. Osada and Y. M. Chen, Chem. Soc. Rev., 2014, 43, 8114–8131 RSC.
- L. Cai, S. Liu, J. Guo and Y. Jia, Acta Biomater., 2020, 113, 84–100 CrossRef CAS PubMed.
- D. L. Taylor and M. in het Panhuis, Adv. Mater., 2016, 28, 9060–9093 CrossRef CAS PubMed.
- L. Shi, P. Ding, Y. Wang, Y. Zhang, D. Ossipov and J. Hilborn, Macromol. Rapid Commun., 2019, 40, 1800837 CrossRef PubMed.
- Y. Hui, Z. Wen, F. Pilate, H. Xie, C. Fan, L. Du, D. Liu, K. Yang and Y. Wang, Polym. Chem., 2016, 7, 7269–7277 RSC.
- I. Hussain, S. M. Sayed, S. Liu, F. Yao, O. Oderinde and G. Fu, Eur. Polym. J., 2018, 100, 219–227 CrossRef CAS.
- L. Shi, F. Wang, W. Zhu, Z. Xu, S. Fuchs, J. Hilborn, L. Zhu, Q. Ma, Y. Wang, X. Weng and D. A. Ossipov, Adv. Funct. Mater., 2017, 27, 1700591 CrossRef.
- Y. Li, J. Li, X. Zhao, Q. Yan, Y. Gao, J. Hao, J. Hu and Y. Ju, Chem.–Eur. J., 2016, 22, 18435–18441 CrossRef CAS PubMed.
- D. Xia, P. Wang, X. Ji, N. M. Khashab, J. L. Sessler and F. Huang, Chem. Rev., 2020, 120, 6070–6123 CrossRef CAS PubMed.
- J. Jin, L. Cai, Y. Jia, S. Liu, Y. Chen and L. Ren, J. Mater. Chem. B, 2019, 7, 1637–1651 RSC.
- D. C. Tuncaboylu, M. Sari, W. Oppermann and O. Okay, Macromolecules, 2011, 44, 4997–5005 CrossRef CAS.
- D. C. Tuncaboylu, A. Argun, M. Sahin, M. Sari and O. Okay, Polymer, 2012, 53, 5513–5522 CrossRef CAS.
- R. Fredrick, A. Podder, A. Viswanathan and S. Bhuniya, J. Appl. Polym. Sci., 2019, 136, 47665 CrossRef.
- H. Chen, B. Hao, P. Ge and S. Chen, Polym. Chem., 2020, 11, 4741–4748 RSC.
- J. Chen, Q. Peng, T. Thundat and H. Zeng, Chem. Mater., 2019, 31, 4553–4563 CrossRef CAS.
- E. Su, M. Yurtsever and O. Okay, Macromolecules, 2019, 52, 3257–3267 CrossRef CAS.
- M. C. Bastings, S. Koudstaal, R. E. Kieltyka, Y. Nakano, A. C. H. Pape, D. A. M. Feyen, F. J. van Slochteren, P. A. Doevendans, J. P. G. Sluijter, E. W. Meijer, S. A. J. Chamuleau and P. Y. W. Dankers, Adv. Healthcare Mater., 2014, 3, 70–78 CrossRef CAS PubMed.
- A. Phadke, C. Zhang, B. Arman, C. C. Hsu, R. A. Mashelkar, A. K. Lele, M. J. Tauber, G. Arya and S. Varghese, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 4383–4388 CrossRef CAS PubMed.
- J. Huang, C. Wu, X. Yu, H. Li, S. Ding and W. Zhang, Macromol. Chem. Phys., 2021, 222, 2100061 CrossRef CAS.
- Y. Lin and G. Li, J. Mater. Chem. B, 2014, 2, 6878–6885 RSC.
- D. Zhao, M. Feng, L. Zhang, B. He, X. Chen and J. Sun, Carbohydr. Polym., 2021, 256, 117580 CrossRef CAS PubMed.
- Q. Wang, J. L. Mynar, M. Yoshida, E. Lee, M. Lee, K. Okuro, K. Kinbara and T. Aida, Nature, 2010, 463, 339–343 CrossRef CAS PubMed.
- T. L. Sun, T. Kurokawa, S. Kuroda, A. B. Ihsan, T. Akasaki, K. Sato, M. A. Haque, T. Nakajima and J. P. Gong, Nat. Mater., 2013, 12, 932–937 CrossRef CAS PubMed.
- N. Yuan, L. Xu, B. Xu, J. Zhao and J. Rong, Carbohydr. Polym., 2018, 193, 259–267 CrossRef CAS PubMed.
- M. Rodin, J. Li and D. Kuckling, Chem. Soc. Rev., 2021, 50, 8147–8177 RSC.
- T. Li, X. Hu, Q. Zhang, Y. Zhao, P. Wang, X. Wang, B. Qin and W. Lu, Polym. Adv. Technol., 2020, 31, 1648–1660 CrossRef CAS.
- S. Islam and G. Bhat, Mater. Adv., 2021, 2, 1896–1926 RSC.
- W. Wang, Y. Zhang and W. Liu, Prog. Polym. Sci., 2017, 71, 1–25 CrossRef.
- Y. Lin, D. He, Z. Chen, L. Wang and G. Li, RSC Adv., 2016, 6, 12479–12485 RSC.
- H. Jiang, L. Duan, X. Ren and G. Gao, Eur. Polym. J., 2019, 112, 660–669 CrossRef CAS.
- A. Sánchez-Iglesias, M. Grzelczak, T. Altantzis, B. Goris, J. Pérez-Juste, S. Bals, G. V. Tendeloo, S. H. Donaldson, Jr, B. F. Chmelka, J. N. Israelachvili and L. M. Liz-Marzán, ACS Nano, 2012, 6, 11059–11065 CrossRef PubMed.
- K. Haraguchi and T. Takehisa, Adv. Mater., 2002, 14, 1120–1124 CrossRef CAS.
- J. P. Gong, Y. Katsuyama, T. Kurokawa and Y. Osada, Adv. Mater., 2003, 15, 1155–1158 CrossRef CAS.
- K. Haraguchi, K. Uyama and H. Tanimoto, Macromol. Rapid Commun., 2011, 32, 1253–1258 CrossRef CAS PubMed.
- S. Tamesue, M. Ohtani, K. Yamada, Y. Ishida, J. M. Spruell, N. A. Lynd, C. J. Hawker and T. Aida, J. Am. Chem. Soc., 2013, 135, 15650–15655 CrossRef CAS PubMed.
- N. Y. Kostina, S. Sharifi, A. de los Santos Pereira, J. Michálek, D. W. Grijpma and C. Rodriguez-Emmenegger, J. Mater. Chem. B, 2013, 1, 5644–5650 RSC.
- Y. Lin, Z. Zeng, Y. Li, S. Sun, X. Liu, D. He and G. Li, RSC Adv., 2019, 9, 31806–31811 RSC.
- X. Yu, Y. Zheng, H. Zhang, Y. Wang, X. Fan and T. Liu, Chem. Mater., 2021, 33, 6146–6157 CrossRef CAS.
- L. Wang, G. Gao, Y. Zhou, T. Xu, J. Chen, R. Wang, R. Zhang and J. Fu, ACS Appl. Mater. Interfaces, 2019, 3, 3506–3515 CrossRef PubMed.
- K. Haraguchi, J. Ning and G. Li, Macromol. Symp., 2015, 358, 182–193 CrossRef CAS.
- J. Lutz, D. Neugebauer and K. Matyjaszewski, J. Am. Chem. Soc., 2003, 125, 6986–6993 CrossRef CAS PubMed.
- S. Shanmugam and C. Boyer, J. Am. Chem. Soc., 2015, 137, 9988–9999 CrossRef CAS PubMed.
- E. Kot and A. Bismarck, Macromolecules, 2010, 43, 6469–6475 CrossRef CAS.
- B. Xu, H. Li, Y. Wang, G. Zhang and Q. Zhang, Polym. Compos., 2016, 37, 810–817 CrossRef CAS.
- F. Shi, M. Zhong, L. Zhang, X. Liu and X. Xie, Chin. J. Polym. Sci., 2017, 35, 25–35 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1ra05896g |
|
| This journal is © The Royal Society of Chemistry 2021 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *ab,
Shuoqi Wanga,
Sheng Suna,
Yaoheng Lianga,
Yisheng Xua,
Huawen Hu
*ab,
Shuoqi Wanga,
Sheng Suna,
Yaoheng Lianga,
Yisheng Xua,
Huawen Hu