
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceTetrazol-Cu(I) immobilized on nickel ferrite catalyzed green synthesis of indenopyridopyrimidine derivatives in aqueous media†
Zahra Ghanbari and
Hossein Naeimi *
*
Department of Organic Chemistry, Faculty of Chemistry, University of Kashan, Kashan, 87317, I. R. Iran. E-mail: naeimi@kashanu.ac.ir; Fax: +98-31-55912397; Tel: +98-31-55912388
First published on 22nd September 2021
Abstract
After the initial study of different protocols in the synthesis of indeno[2′,1′:5,6]pyrido[2,3-d]pyrimidines, herein, a new method is presented using cheaper and more accessible starting materials to produce high-efficiency products. In this protocol, the novel nanocatalyst is very effective in the progression of the reaction and increasing the efficiency. This green approach in aqueous media has several advantages as compared with other methods, such as easier work-up, very mild reaction conditions, reusability of the catalyst, and eco-friendliness. The products of this four-component condensation were evaluated using IR, 1H NMR, 13C NMR spectra, and C. H. N. analyses, and the catalyst structure was confirmed by FT-IR, XRD, SEM, EDX, TGA and VSM techniques.
1. Introduction
The preparation of effective biological compounds with new and simpler methods can be considered a valuable goal for organic chemists. A protocol that brings higher product efficiency despite selecting more available materials is highly desirable. In previous studies, the indenopyridopyrimidine compounds have proven their special therapeutic effects; for example, tuberculostatic,1 anticonvulsants,2 calcium channel antagonists,3 anticancer,4 antimicrobial,5 antiviral,6 diuretic and potassium-sparing,7 antifolate,8 and antiinflammatory9 activities. Previous methods used to prepare this product include three-component reactions using uracil derivatives.10–13 From a variety of synthetic routes, the choice of multi-component reactions (MCRs), as simple, economical, environmentally friendly, and applicable procedures, is desirable to obtain favored drug combinations. This suitable pathway easily prepares various highly functionalized heterocyclic compounds without generating many waste products that are often produced in multi-step reactions.14–27 The combination of two reaction paths according to the properties of the material used can help in achieving the desired product. With this view, we have considered the preparation of indenopyridine,28 pyridopyrimidines,29 and pyrimidines,30 in addition to the consideration of the usual preparation method for indenopyridopyrimidines.31,32 With this approach, the product is obtained using barbituric acid and ammonium acetate, instead of uracil, in a four-component reaction.The use of copper complexes in various chemical reactions, such as alkyne–azide cycloaddition,33 hydroboration reactions,34 allylic alkylation reactions,35 direct addition of terminal alkynes to imines,36 and β-boration of α,β-unsaturated esters,37 has led us to the use of these compositions as a catalyst in this reaction.
The choice of magnetic nanoparticles as a catalyst platform in chemical reactions provides a convenient separation of the heterogeneous catalyst at the end of the reaction using an external magnetic field. On the other hand, these valuable platforms have the ability to link with different functional groups for the desired purposes.38
Herein, for the first time, we report a new method for the synthesis of indenopyridopyrimidine compounds using NiFe2O4@SiO2-tetrazol-Cu(I) as the catalyst.
2. Experimental
2.1. Materials
All commercially available reagents were used without further purification and purchased from the Merck Chemical Company in high purity. All of the used solvents were purified by standard procedure.2.2. Apparatus
IR spectra were obtained as KBr pellets on a PerkinElmer 781 spectrophotometer and on an impact 400 Nicolet FT-IR spectrophotometer. 1H NMR and 13C NMR were recorded in DMSO-d6 solvents on a Bruker DRX-400 spectrometer with tetramethylsilane as the internal reference. The elemental analyses (C. H. N.) were carried out using a Vario EL Model EA 1108 analyzer.FE-SEM analysis was carried out using a Jeol SEM instrument (model – VEGA/TESCAN) combined with an INCA instrument for energy-dispersive X-ray spectroscopy scanning electron microscopy (EDS-SEM), with scanning electron electrode at 15 kV. The XRD patterns were recorded on an X-ray diffractometer (Bruker, D8 ADVANCE, Germany). The magnetic properties of the nanoparticles were measured using a vibrating sample magnetometer (VSM, PPMS-9T) at 300 K, Kashan University, Iran. The BANDELIN ultrasonic HD 3200 with probe model KE 76, of 6 mm diameter was used to produce ultrasonic irradiation. Thermogravimetric analysis (TGA) was performed on a Mettler TA4000 system TG-50 at a heating rate of 10 K min−1 under a N2 atmosphere. Melting points were obtained with a Yanagimoto micro melting point apparatus and are uncorrected. The purity determination of the substrates and reaction monitoring were accomplished by TLC on silica-gel polygram SILG/UV 254 plates (from Merck Company).
2.3. Catalyst preparation
The spectral information of various products, including IR, NMR, and C.H.N. analyses are given below.
5-(4-Chlorophenyl)-1H-indeno[2′,1′:5,6]pyrido[2,3-d] pyrimidine-2,4,6(3H)-trione (1a). White powder; mp: 258 °C, decompose; IR (KBr) (νmax/cm−1): 1413, 1558 (C
![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C, Ar), 1634, 1693 (CO), 3145 (C–H, sp2 stretch), 3265, 3444 (NH). 1H NMR (DMSO-d6, 400 MHz) δ (ppm): 7.45–7.66 (d, 2H, J = 8.0 Hz), 7.51–7.52 (m, 2H), 7.67–7.75 (d, 2H), 7.80–7.82 (d, 2H, J = 8.0 Hz), 9.95 (s, 1H). 13C NMR (100 MHz, DMSO-d6) δ (ppm): 123.52, 125.91, 127.94, 128.68, 130.23, 132.23, 135.48, 139.16, 149.25, 162.75, 165.13, 170.00, 79.00; anal. calcd for C20H10N3O3Cl: C, 64; H, 2.67; N, 11.2%; found: C, 63.91; H, 2.70; N, 11.23%.
C, Ar), 1634, 1693 (CO), 3145 (C–H, sp2 stretch), 3265, 3444 (NH). 1H NMR (DMSO-d6, 400 MHz) δ (ppm): 7.45–7.66 (d, 2H, J = 8.0 Hz), 7.51–7.52 (m, 2H), 7.67–7.75 (d, 2H), 7.80–7.82 (d, 2H, J = 8.0 Hz), 9.95 (s, 1H). 13C NMR (100 MHz, DMSO-d6) δ (ppm): 123.52, 125.91, 127.94, 128.68, 130.23, 132.23, 135.48, 139.16, 149.25, 162.75, 165.13, 170.00, 79.00; anal. calcd for C20H10N3O3Cl: C, 64; H, 2.67; N, 11.2%; found: C, 63.91; H, 2.70; N, 11.23%.
5-(4-Methoxyphenyl)-1H-indeno[2′,1′:5,6]pyrido[2,3-d] pyrimidine-2,4,6(3H)-trione (2a). Yellow powder; mp: 245 °C, decompose, mp (lit)41 248 °C; IR (KBr) (νmax/cm−1): 1443, 1529 (C
C, Ar), 1665, 1750 (CO), 2916 (C–H, sp3), 3243 (C–H, sp2 stretch), 3439, 3585 (NH). 1H NMR (DMSO-d6, 400 MHz) δ (ppm): 7.02–7.04 (d, 2H, J = 8.0 Hz), 7.48–7.49 (m, 2H), 7.58–7.60 (d, 2H, J = 8.4 Hz), 7.80–7.81 (m, 2H). Anal. calcd for C21H13N3O4: C, 67.92; H, 3.50; N, 11.32%; found: C, 67.91; H, 3.47; N, 11.35%.
5-(3-Methoxyphenyl)-1H-indeno[2′,1′:5,6]pyrido[2,3-d] pyrimidine-2,4,6(3H)-trione (3a). Cream powder; mp: 231 °C, decompose; IR (KBr) (νmax/cm−1): 1421, 1539 (C
C, Ar), 1635, 1711 (CO), 2922 (C–H, sp3), 3179 (C–H, sp2 stretch), 3342, 3467 (NH). 1H NMR (DMSO-d6, 400 MHz) δ (ppm): 7.04–7.06 (d, 2H), 7.23 (s, 1H), 7.33–7.35 (m, 2H), 7.51–7.52 (m, 2H), 7.68–7.69 (d, 2H). 13C NMR (100 MHz, DMSO-d6) δ (ppm): 61.13, 111.67, 112.22, 118.79, 123.57, 125.91, 127.79, 129.18, 130.20, 135.46, 149.57, 161.71, 169.76, 176.16, 179.48.
5-(3-Chlorophenyl)-1H-indeno[2′,1′:5,6]pyrido[2,3-d] pyrimidine-2,4,6(3H)-trione (4a). Yellow powder; mp: 258 °C, decompose; IR (KBr) (νmax/cm−1): 1415, 1501 (C
C, Ar), 1645, 1715, 1736 (CO), 3241 (C–H, sp2 stretch), 3342, 3515 (NH). 1H NMR (DMSO-d6, 400 MHz) δ (ppm): 7.07–7.09 (d, 2H), 7.28 (s, 1H), 7.44–7.45 (m, 2H), 7.51–7.58 (m, 2H), 7.66–7.68 (d, 2H). Anal. calcd for C20H10N3O3Cl: C, 63.93; H, 2.68; N, 11.18%; found: C, 65.12; H, 2.53; N, 10.73%.
5-(2-Fluorophenyl)-1H-indeno[2′,1′:5,6]pyrido[2,3-d] pyrimidine-2,4,6(3H)-trione (5a). Brown powder; mp: 293 °C, decompose; IR (KBr) (νmax/cm−1): 1463, 1511 (C
C, Ar), 1608, 1722 (CO), 3192 (C–H, sp2 stretch), 3418 (NH). 1H NMR (DMSO-d6, 400 MHz) δ (ppm): 7.42–7.43 (d, 2H), 7.51–7.52 (m, 2H), 7.78–7.88 (m, 2H), 8.01–8.02 (d, 2H). 13C NMR (100 MHz, DMSO-d6) δ (ppm): 123.52, 125.93, 127.56, 128.62, 130.26, 131.83, 135.53, 139.14, 149.23, 165.37, 174.18, 176.16. Anal. calcd for C20H10N3O3F: C, 66.85; H, 2.79; N, 11.70%; found: C, 66.90; H, 2.77; N, 11.66%.
5-(o-Tolyl)-1H-indeno[2′,1′:5,6]pyrido[2,3-d]pyrimidine-2,4,6(3H)-trione (6a). Cream powder; mp: 282 °C, decompose; IR (KBr) (νmax/cm−1): 1478, 1593 (C
C, Ar), 1609, 1667, 1710 (CO), 2922 (C–H, sp3), 3189 (C–H, sp2 stretch), 3313, 3413 (NH). 1H NMR (DMSO-d6, 400 MHz) δ (ppm): 2.877 (s, 3H), 7.07–7.08 (d, 2H), 7.41–743 (m, 2H), 7.51–7.52 (m, 2H), 7.73–7.74 (d, 2H). Anal. calcd for C21H13N3O3: C, 70.99; H, 3.66; N, 11.83%; found: C, 70.92; H, 3.69; N, 11.87%.
5-(2,4-Dichlorophenyl)-1H-indeno[2′,1′:5,6]pyrido[2,3-d] pyrimidine-2,4,6(3H)-trione (7a). Brown powder; mp: 285 °C, decompose, mp (lit)42 289 °C; IR (KBr) (νmax/cm−1): 1423, 1512 (C
C, Ar), 1654, 1712, 1728 (CO), 3241 (C–H, sp2 stretch), 3341, 3415 (NH). 1H NMR (DMSO-d6, 400 MHz) δ (ppm): 6.95 (s, 1H), 7.33–7.35 (d, 2H), 7.44–7.46 (m, 2H), 7.62–7.64 (d, 2H). Anal. calcd for C20H9N3O3Cl2: C, 58.68; H, 2.20; N, 10.27%; found: C, 58.65; H, 2.17; N, 10.30%.
5-(3-Nitrophenyl)-1H-indeno[2′,1′:5,6]pyrido[2,3-d] pyrimidine-2,4,6(3H)-trione (8a). White powder; mp: 279 °C, decompose; IR (KBr) (νmax/cm−1): 1418, 1561 (C
C, Ar), 1369, 1512 (NO), 1619, 1711 (CO), 2909 (C–H, sp3), 3158 (C–H, sp2 stretch), 3509 (NH). 1H NMR (DMSO-d6, 400 MHz) δ (ppm): 7.13–7.16 (m, 2H), 7.33–7.35 (d, 2H), 7.46–7.47 (d, 2H), 7.50–7.55 (m, 2H), 7.66 (s, 1H). 13C NMR (100 MHz, DMSO-d6) δ (ppm): 121.27, 123.46, 125.86, 127.76, 129.46, 130.24, 134.23, 135.36, 147.63, 149.46, 168.93, 170.57, 177.77. Anal. calcd for C20H10N4O5: C, 62.18; H, 2.59; N, 18.13%; found: C, 62.11; H, 2.57; N, 18.17%.
5-(2-Chlorophenyl)-1H-indeno[2′,1′:5,6]pyrido[2,3-d] pyrimidine-2,4,6(3H)-trione (9a). Yellow powder; mp: 228 °C, decompose; IR (KBr) (νmax/cm−1): 1429, 1541 (C
C, Ar), 1619, 1668 (CO), 3219 (C–H, sp2 stretch), 3336, 3412 (NH). 1H NMR (DMSO-d6, 400 MHz) δ (ppm): 7.07–7.09 (d, 2H), 7.31–7.33 (m, 2H), 7.50–7.52 (m, 2H), 7.62–7.64 (d, 2H). 13C NMR (100 MHz, DMSO-d6) δ (ppm): 123.52, 126.23, 127.63, 128.65, 130.23, 131.83, 135.19, 139.11, 149.20, 168.77, 174.16, 176.44.
5-(4-Bromophenyl)-1H-indeno[2′,1′:5,6]pyrido[2,3-d] pyrimidine-2,4,6(3H)-trione (10a). Cream powder; mp: 242 °C, decompose; IR (KBr) (νmax/cm−1): 1447, 1559 (C
C, Ar), 1681, 1719 (CO), 3072 (C–H, sp2 stretch), 3235, 3527 (NH). 1H NMR (DMSO-d6, 400 MHz) δ (ppm): 7.36–7.38 (d, 2H, J = 8.0 Hz), 7.53–7.55 (m, 2H), 7.67–7.69 (d, 2H, J = 8.0 Hz), 7.724 (d, 2H). 13C NMR (100 MHz, DMSO-d6) δ (ppm): 120.62, 123.50, 125.88, 126.75, 127.81, 130.18, 135.46, 139.13, 141.65, 147.57, 149.23, 168.63, 174.27, 176.64. Anal. calcd for C20H10N3O3Br: C, 57.14; H, 2.38; N, 10%; found: C, 57.11; H, 2.41; N, 10.03%.
5-(5-Methylfuran-2-yl)-1H-indeno[2′,1′:5,6]pyrido[2,3-d] pyrimidine-2,4,6(3H)-trione (11a). Red brown powder; mp: 339 °C, decompose; IR (KBr) (νmax/cm−1): 1423, 1531 (C
C, Ar), 1631, 1733 (CO), 2931 (C–H, sp3), 3130 (C–H, sp2 stretch), 3242, 3425 (NH). 1H NMR (DMSO-d6, 400 MHz) δ (ppm): 2.73 (s, 3H), 7.45–7.47 (d, 2H), 7.67–7.70 (m, 2H), 7.84–7.87 (d, 2H). Anal. calcd for C19H11N3O4: C, 66.08; H, 3.19; N, 12.17%; found: C, 66.03; H, 3.11; N, 12.21%.
5-(4-Nitrophenyl)-1H-indeno[2′,1′:5,6]pyrido[2,3-d] pyrimidine-2,4,6(3H)-trione (12a). Dark brown powder; mp: 343 °C, decompose, mp (lit)41 345 °C; IR(KBr) (νmax/cm−1): 1433, 1531 (C
C, Ar), 1317, 1553 (NO), 1621, 1739 (CO), 3205 (C–H, sp2 stretch), 3242, 3428 (NH). 1H NMR (DMSO-d6, 400 MHz) δ (ppm): 7.754–7.775 (d, 2H, J = 8.0 Hz), 7.85–7.86 (m, 2H), 8.18–8.20 (m, 2H), 8.37–8.40 (d, 2H, J = 8.4 Hz).
3. Results and discussion
3.1. Preparation and characterization of the NiFe2O4@SiO2-tetrazol-Cu(I) catalyst
The preparation of the nickel ferrite nanoparticles using the previously reported method were performed by co-precipitation of NiCl2 and FeCl3 in basic solution at 80 °C.43 Scheme 1 shows the preparation method of the final catalyst. Fourier transform infrared (FT-IR) spectra, X-ray diffraction (XRD), vibrating sample magnetometer (VSM), thermo-gravimetric analysis (TGA), energy-dispersive X-ray spectroscopy (EDX) and scanning electron microscopy (SEM) techniques were utilized to confirm the catalyst structure. | ||
| Scheme 1 Synthesis of the NiFe2O4@SiO2-tetrazol-Cu(I). | ||
The FT-IR spectra of NiFe2O4 NPs, NiFe2O4@SiO2, NiFe2O4@SiO2-epoxide and NiFe2O4@SiO2-tetrazol are shown in Fig. 1. The stretching modes of absorbed water appeared in the bands within the range of 3400–1600 cm−1. The vibrations of Fe–O and Ni–O bonds were observed at 590 cm−1 and 433 cm−1, respectively. In all spectra, the absorptions of the Fe–O and Ni–O bonds give characteristic bands at 429–630 cm−1. The peaks at 1023, 1149, 1151 cm−1 are related to the Si–O–Si stretching vibrations in the NiFe2O4@SiO2, NiFe2O4@SiO2-epoxide and NiFe2O4@SiO2-tetrazol spectra; therefore, SiO2 coated the surface of NiFe2O4.
 | ||
| Fig. 1 FT-IR spectra of (a) NiFe2O4 NPs, (b) NiFe2O4@SiO2, (c) NiFe2O4@SiO2-epoxide, (d) NiFe2O4@SiO2–N3, and (e) NiFe2O4@SiO2-tetrazol. | ||
In the NiFe2O4@SiO2-epoxide spectrum, the bending vibration and stretching vibration of CH2 were attributed to the revealed bands at 1423 and 2922 cm−1, respectively. The absorption peaks at 1664, 2921 and 3096 cm−1 in the FT-IR spectrum of NiFe2O4@SiO2-tetrazol are related to the stretching vibrations of CN, C–H aliphatic, and C–H aromatic, respectively. The CC vibrations were observed at 1481 and 1605 cm−1. The preparation of the final catalyst was proved by these described bands.
The stretching vibrations of OH, N3, and the aliphatic C–H group are shown in the FT-IR spectrum of NiFe2O4@SiO2–N3 with absorption peaks at 3200–3600, 2080, and 2920 cm−1 respectively. The stretching vibrations of aromatic C–H, aliphatic C–H, and CN in the FT-IR spectrum of NiFe2O4@SiO2-tetrazol showed absorption peaks at 3096, 2921, and 1664 cm−1, respectively. In addition, CC vibrations appeared at 1605 and 1481 cm−1.
The final product is without the nitrile groups of phthalonitrile, indicating its reaction with N3 and conversion to tetrazole.38
Fig. 2 shows the powder XRD patterns of NiFe2O4 NPs and NiFe2O4@SiO2-tetrazol-Cu(I). A comparison of all the characteristic peaks of NiFe2O4 in the diffraction pattern with the standard XRD pattern (JCPDS file no. 03-0875) showed complete adaptability. The weaker diffraction lines in NiFe2O4@SiO2-tetrazol and NiFe2O4@SiO2-tetrazol-Cu(I) (Fig. 2b and c) are due to the NiFe2O4 coating.
 | ||
| Fig. 2 The X-ray diffraction patterns of (a) NiFe2O4, (b) NiFe2O4@SiO2-tetrazol, and (c) NiFe2O4@SiO2-tetrazol-Cu(I). | ||
The average diameter of the NiFe2O4 NPs and the final catalyst were calculated as 11 and 16 nm, respectively in the Debye–Scherrer equation. The morphologies and particle sizes of NiFe2O4 and the final catalyst were characterized in the SEM image (Fig. 3).
 | ||
| Fig. 3 SEM images of (a) NiFe2O4 NPs and (b) NiFe2O4@SiO2-tetrazol-Cu(I). | ||
The spherical morphology and uniform sizes of these nanoparticles were confirmed in this image and the average diameters of both NiFe2O4 and NiFe2O4@SiO2-tetrazol-Cu(I) were calculated to be about 8.7 nm. The EDX spectra confirmed the preparation of NiFe2O4 and NiFe2O4@SiO2-tetrazol-Cu(I) with the presence of O, Fe, and Ni, and the presence of C, N, Si and Cu, respectively, in addition to other elements in composites of these catalysts (Fig. 4).
 | ||
| Fig. 4 EDX spectrum of (a) NiFe2O4 NPs and (b) NiFe2O4@SiO2-tetrazol-Cu(I). | ||
The magnetic properties of NiFe2O4 and NiFe2O4@SiO2-tetrazol-Cu(I) nanoparticles were investigated at room temperature (Fig. 5). A vibrating sample magnetometer (VSM) reported the saturation magnetization of NiFe2O4 NPs and the final catalyst as 16.7 and 6 emu g−1, respectively. The functionalization with organic groups and coating of magnetic NPs reduced the magnetic properties. The magnetic properties of these catalysts allowed them to be separated more easily by an external magnet.
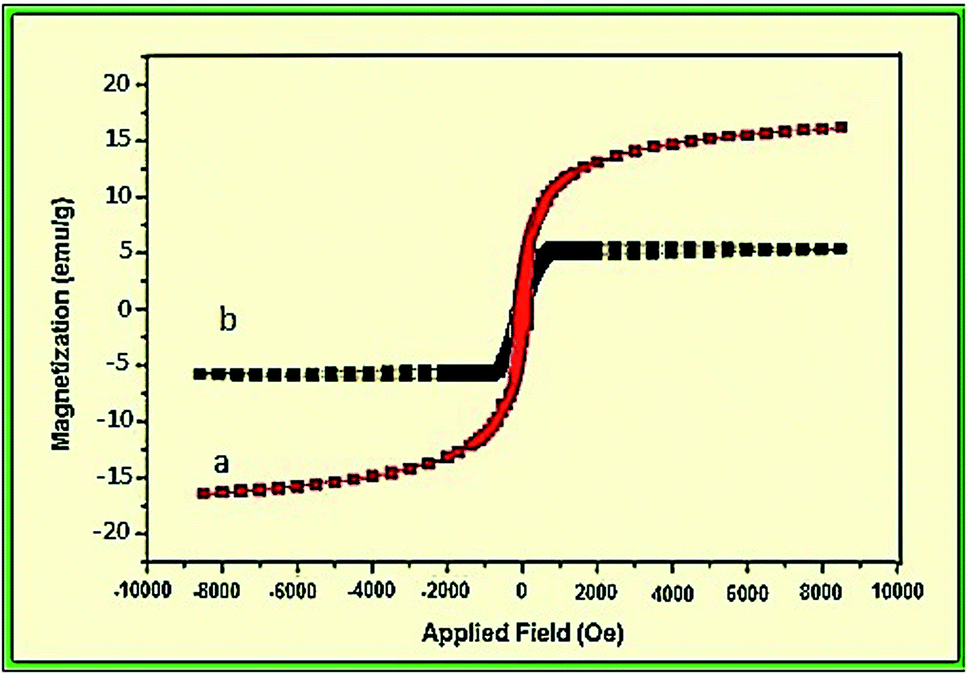 | ||
| Fig. 5 Magnetization versus applied field for (a) NiFe2O4 NPs and (b) NiFe2O4@SiO2-tetrazol-Cu(I). | ||
The thermal stability of the magnetic nanocatalyst was investigated by thermogravimetric analysis (TGA). This analysis confirmed the presence of different groups on the surface of the nanoparticles and their thermal stability (Fig. 6). No change in weight before 380 °C indicated a lack of solvent in the structure of the catalyst and, of course, its high thermal resistance.
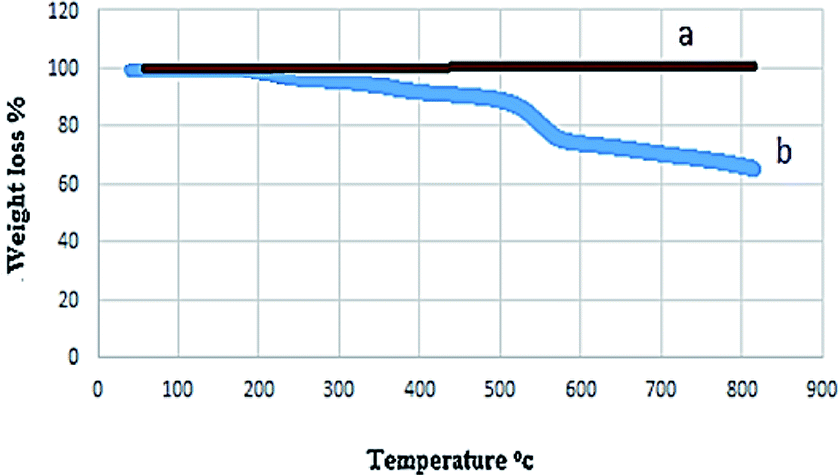 | ||
| Fig. 6 TGA curve of (a) NiFe2O4 NPs and (b) NiFe2O4@SiO2-tetrazol-Cu(I). | ||
The degradation of organic groups covering the NiFe2O4 surface was observed in weight loss (40%) at 380–800 °C in the TGA curve of NiFe2O4@SiO2-tetrazol-Cu(I). As a result, this catalyst is suitable for organic synthesis due to its thermal stability up to 380 °C.
3.2. Investigation of catalytic activity
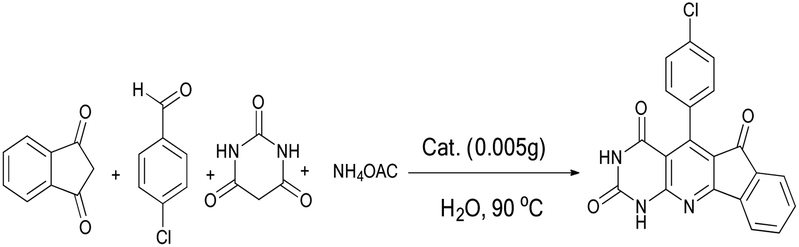
To optimize reaction parameters and characterize the catalytic efficiency of the nanocatalyst, we considered the reaction of 4-chlorobenzaldehyde (1 mmol), 1,3-indanedione (1 mmol), barbituric acid (1 mmol), and ammonium acetate (1.3 mmol) as a model reaction (Scheme 2). | ||
| Scheme 2 Synthesis of indenopyridopyrimidine. | ||
The effects of different conditions including temperature, solvent, amounts of catalyst on the reaction were determined (Tables 1–3). According to Table 1, the water solvent was shown as the most suitable solvent among the other solvents used including EtOH, MeOH, CH2Cl2 and dimethylformamide (DMF). Accordingly, the most suitable reaction temperature that increased the reaction efficiency in the shortest reaction time was determined to be 90 °C (Table 1, entry 3).
| Entry | Solvent | Temperature (°C) | Yieldb (%) |
|---|---|---|---|
| a Reaction conditions: 4-chlorobenzaldehyde (1 mmol), 1,3-indanedione (1 mmol), barbituric acid (1 mmol), and ammonium acetate (1.3 mmol).b Isolated yields. | |||
| 1 | Water | 50 | 60 |
| 2 | Water | 80 | 89 |
| 3 | Water | 90 | 92 |
| 4 | Ethanol | 50 | 55 |
| 5 | Ethanol | 60 | 79 |
| 6 | Ethanol | 70 | 85 |
| 7 | Methanol | 40 | 45 |
| 8 | Methanol | 50 | 67 |
| 9 | Methanol | 70 | 70 |
Table 2 presents the effect of catalyst amount, NiFe2O4@SiO2-tetrazol-Cu(I), on the synthesis of 1a. Here, 0.005 g of the catalyst was recorded as the best observed result and 94% of the product was obtained from the application of this amount of catalyst in the reaction under reflux conditions at two hours (Table 2, entry 8). The substrates were placed in the reaction vessel without the catalyst under the defined conditions; after eight hours no product was obtained (Table 2, entry 11).
| Entry | Catalyst (mol% or g) | Time (h) | Yieldb (%) |
|---|---|---|---|
| a Reaction conditions: 4-chlorobenzaldehyde (1 mmol), 1,3-indanedione (1 mmol), barbituric acid (1 mmol), and ammonium acetate (1.3 mmol).b Yields. | |||
| 1 | NiFe2O4 NPs (3%) | 3.5 | 80 |
| 2 | NiFe2O4 NPs (2%) | 3 | 85 |
| 3 | NiFe2O4 NPs (1%) | 2.5 | 90 |
| 4 | NiFe2O4 NPs (0.5%) | 2 | 89 |
| 5 | NiFe2O4 NPs (0.3%) | 3 | 83 |
| 6 | NiFe2O4@SiO2-tetrazol-Cu(I) (0.002 g) | 2 | 80 |
| 7 | NiFe2O4@SiO2-tetrazol-Cu(I) (0.001 g) | 2.5 | 87 |
| 8 | NiFe2O4@SiO2-tetrazol-Cu(I) (0.005 g) | 2 | 94 |
| 9 | NiFe2O4@SiO2-tetrazol-Cu(I) (0.0003 g) | 3.5 | 79 |
| 10 | NiFe2O4@SiO2-tetrazol-Cu(I) (0.0002 g) | 4 | 75 |
| 11 | Without catalyst | 8 | 0 |
Each of the substituted aryl derivatives in the optimal conditions specified in the previous step presented a different yield of the product (Table 3). The high efficiency of the product and short reaction time have given a special advantage to electron-withdrawing substituents, especially the 4-nitro group with a 96% yield. However, the highest reaction time and the lowest yield were obtained for the 2-methyl group. Electron donation and the steric hindrance of the ortho position are two effective factors in generating this result. The structures of all products were confirmed by infrared (IR), 1H NMR, and C.H.N analysis.
|
|---|
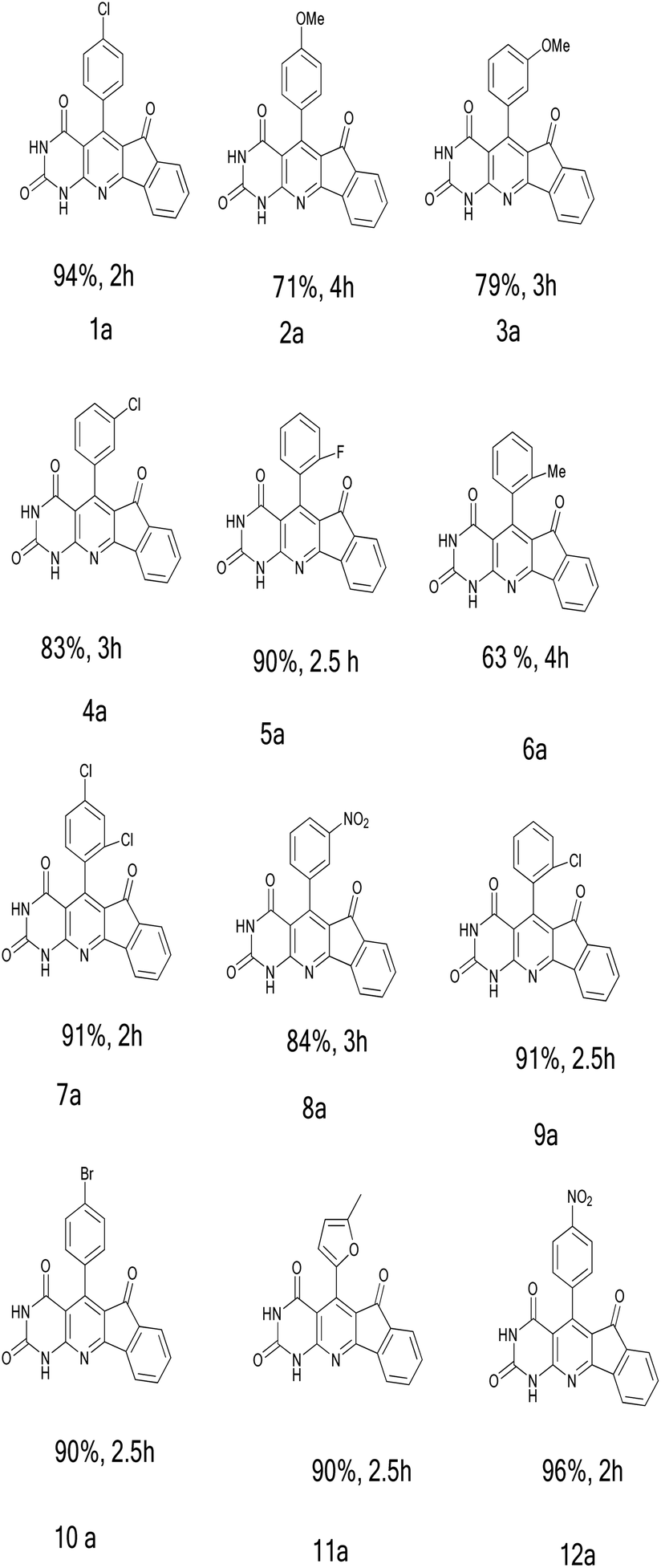 |
3.3. Proposed reaction mechanism
The mechanism shown in Scheme 3 strongly confirms the above observations and the catalyst effects. The effect of the metal complex catalyst on the carbonyl oxygen of the aldehyde provides a good electrophilic center for the nucleophilic attack of 1,3-indanedione. Thus, the intermediate I is formed from a Knoevenagel condensation. Another condensation between barbituric acid and ammonium acetate yielded the intermediate II (enamine). The interaction of two existing fragments gave the intermediate III. The desired product was achieved through intramolecular nucleophilic addition followed by the removal of water. According to observations, the role of catalytic activation in this process is very important.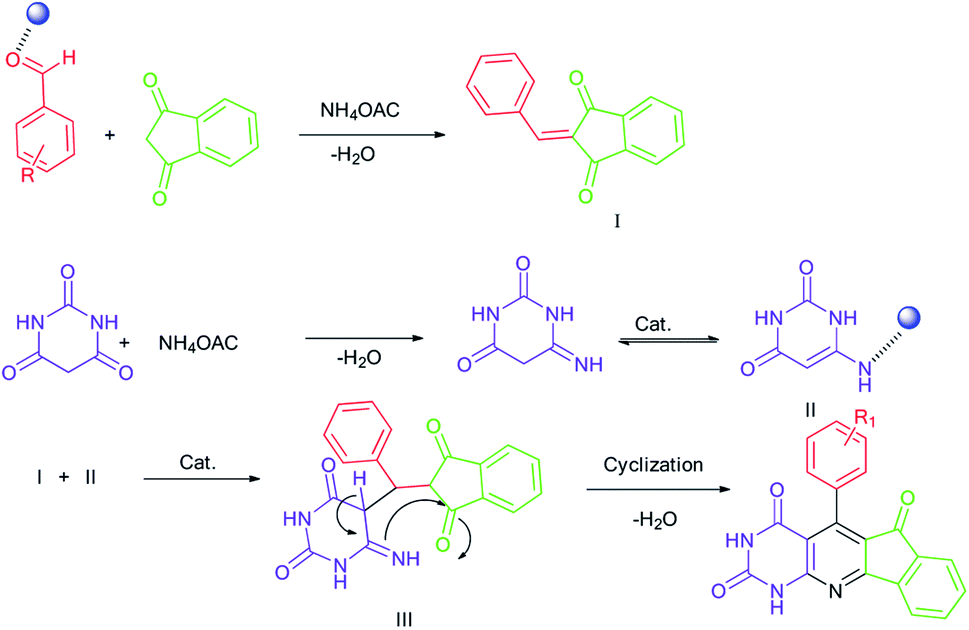 | ||
Scheme 3 The proposed reaction mechanism for the formation of 1a ( : NiFe2O4@SiO2-tetrazol-Cu(I)). : NiFe2O4@SiO2-tetrazol-Cu(I)). | ||
3.4. Reusability of catalyst
The importance of the recyclability of heterogeneous catalysts in improving their performance in other reactions requires a study on their sustainability. For this purpose, the catalyst was extracted from the reaction mixture using an external magnet and after rinsing several times with water and ethanol, it was dried at 80 °C in an oven for 9 h and reused in subsequent reactions. Satisfaction of catalyst activity after being reused six times proves its recyclability and stability in repeated reactions (Fig. 7). | ||
| Fig. 7 Recyclability of NiFe2O4@SiO2-tetrazol-Cu(I) for the synthesis of 1a. | ||
4. Conclusions
We have synthesized indenopyridopyrimidine compounds by using a designed catalyst to provide a green, efficient and convenient process with easy access to starting materials. This procedure provides a new synthetic route that uses readily available compounds and is low-cost with a short reaction time and a high percentage yield of the desired products. From another standpoint, we designed a heterogeneous catalyst covered with functional groups, which affects the progression of the reaction, is reusable in other reactions and provides a credible approach to the use of this pathway. FT-IR spectroscopy, SEM, TGA, EDX analysis, and XRD confirmed the synthesis of the catalyst.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors are grateful to University of Kashan for supporting this work by Grant number 159148/85.Notes and references
- I. D. Bystryakova, I. A. Burova, G. M. Chelysheva, S. V. Zhilinkova, N. M. Smirnova and T. S. Safonova, Khim.-Farm. Zh., 1991, 25, 31–35 CAS.
- A. B. Deyanov, R. K. Niyazov, F. Y. Nazmetdinov, B. Y. Syropyatov, V. E. Kolla and M. E. Konshin, Khim.-Farm. Zh., 1991, 25, 26 CAS.
- A. Pastor, R. Alajarin, J. J. Vaquero, J. Alvarez-Builla, M. Fau de Casa-Juana, C. Sunkel, J. G. Priego, I. Fonseca and J. Sanz-Aparicio, Tetrahedron, 1994, 50, 8085–8098 CrossRef CAS.
- N. M. Evdokimov, S. V. Slambrouck, P. Heffeter, L. Tu, B. L. Calve, D. Lamoral-Theys, C. J. Hooten, P. Y. Uglinskii, S. Rogelj, R. Kiss, W. F. A. Steelant, W. Berger, J. J. Yang, C. G. Bologa, A. Kornienko and I. V. Magedov, J. Med. Chem., 2011, 54, 2012–2021 CrossRef CAS PubMed.
- A. Gangjee, A. Vasudevan, F. Queener and R. J. Kisliuk, Med. Chem., 1996, 39, 1438 CrossRef CAS PubMed.
- M. Manpadi, P. Y. Uglinskii, S. K. Rastogi, K. M. Cotter, Y. C. Wong, L. A. Anderson, A. J. Ortega, S. V. Slambrouck, W. F. A. Steelant, S. Rogelj, P. Tongwa, M. Yu. Antipin, I. V. Magedov and A. Kornienko, Org. Biomol. Chem., 2007, 5, 3865–3872 RSC.
- I. O. Donkor, C. L. Klein, L. Liang, N. Zhu, E. Bradley and A. M. Clark, J. Pharm. Sci., 1995, 84, 661–664 CrossRef CAS PubMed.
- M. N. Nasr and M. Gineinah, M. Arch. Pharm., 2002, 6, 289 CrossRef.
- A. Monge, V. Martinez-Merino, C. Sanmartin, F. J. Fernandez, M. C. Ochoa, C. Berllver, P. Artigas and E. Fernandez-Alvarez, J. Med. Chem., 1989, 24, 209–216 CAS.
- A. Rosowsky, C. E. Mota and S. F. Queener, J. Heterocycl. Chem., 1995, 32, 335–340 CrossRef CAS.
- V. E. Kolla, A. B. Deyanov, F. Y. Nazmetdinov, Z. N. Kashina and L. P. Drovosekova, Pharmaceut. Chem. J., 1993, 27, 635–636 CrossRef.
- M. Ghashang, S. Guhanathan and S. Sheik Mansoor, Res. Chem. Intermed., 2017, 43(12), 7257–7276 CrossRef CAS.
- J. M. Khurana, A. Chaudhary, B. Nand and A. Lumb, Tetrahedron Lett., 2012, 53, 3018–3022 CrossRef CAS.
- S. Abdolmohammadi, S. Balalaie and M. Barari, Comb. Chem. High Throughput Screening, 2013, 16, 150 CAS.
- N. M. Evdokimov, S. V. Slambrouck, P. Heffeter, L. Tu, B. L. Calvé, D. Lamoral-Theys, C. J. Hooten, P. Y. Uglinskii, S. Rogelj, R. Kiss, W. F. A. Steelant, W. Berger, J. J. Yang, C. G. Bologa, A. Kornienko and I. V. Magedov, J. Med. Chem., 2011, 54, 2012 CrossRef CAS PubMed.
- A. Baziar and M. Ghashang, React. Kinet. Mech. Catal., 2016, 118, 463 CrossRef CAS.
- M. Ghashang, Res. Chem. Intermed., 2016, 42, 4191–4205 CrossRef CAS.
- M. Dehbashi, M. Aliahmad, M. R. Mohammad Shafiee and M. Ghashang, Synth. React. Inorg., Met.-Org., Nano-Met. Chem., 2013, 43, 1301 CrossRef CAS.
- A. Domling and I. Ugi, Angew. Chem., Int. Ed., 2000, 39, 3168 CrossRef CAS PubMed.
- M. N. Elinson, A. I. Ilovaisky, V. M. Merkulova, F. Barba and B. Batanero, Tetrahedron, 2013, 69, 7125–7130 CrossRef CAS.
- N. Chatani and T. Hanafusa, Bull. Chem. Soc. Jpn., 1990, 63, 2134–2135 CrossRef CAS.
- T. B. Poulsen, C. Alemparte and K. A. Jørgensen, J. Am. Chem. Soc., 2005, 127, 11614 CrossRef CAS PubMed.
- A. K. El-Shafei, A. A. Sultan, A. M. Soliman and E. A. Ahmed, Synth. Commun., 1995, 25, 3211–3217 CrossRef CAS.
- B. Sreedhar, A. S. Kumar and P. S. Reddy, Tetrahedron Lett., 2010, 51, 1891–1895 CrossRef CAS.
- B. V. Subba Reddy, A. Siva Krishna, A. V. Ganesh and G. G. K. S. Narayana Kumar, Tetrahedron Lett., 2011, 52, 3342–3344 CrossRef.
- I. Ugi and A. Domling, Endeavour, 1994, 18, 115 CrossRef CAS.
- S. Heck and A. Domling, Synlett, 2000, 424 CAS.
- P. K. Tapaswi and C. Mukhopadhyay, ARKIVOC, 2011, 10, 287–298 Search PubMed.
- I. O. Donkor, C. L. Klein, L. Liang, N. Zhu, E. Bradley and A. M. Clark, J. Pharm. Sci., 1995, 84, 661–664 CrossRef CAS PubMed.
- H. Naeimi and A. Didar, Ultrason. Sonochem., 2017, 34, 889–895 CrossRef CAS PubMed.
- D.-Q. Shi, Y. Li and H.-Y. Wang, J. Heterocycl. Chem., 2012, 49, 1086–1090 CrossRef CAS.
- G. I. Shakibaei, A. Feiz, H. R. Khavasi, A. A. Soorki and A. Bazgir, ACS Comb. Sci., 2011, 13(1), 96–99 CrossRef CAS PubMed.
- M. T. Ramírez-Palma, J. Segura-Arzate, G. López-Téllez and E. Cuevas-Yañez, J. Chem., 2016, 853–860 Search PubMed.
- Y. Lee and A. H. Hoveyda, J. Am. Chem. Soc., 2009, 131(9), 3160–3161 CrossRef CAS PubMed.
- J. J. Van Veldhuizen, J. E. Campbell, R. E. Giudici and A. H. Hoveyda, J. Am. Chem. Soc., 2005, 127(18), 6877–6882 CrossRef CAS PubMed.
- C. Wei and C. J. Li, J. Am. Chem. Soc., 2002, 124(20), 5638–5639 CrossRef CAS PubMed.
- V. Lillo, A. Prieto, A. Bonet, M. Mar Díaz-Requejo, J. Ramírez, P. J. Pérez and E. Fernández, Organometallics, 2009, 28(2), 659–662 CrossRef CAS.
- R. Ghosh, L. Pradhan, Y. P. Devi, S. Meena, R. Tewari, A. Kumar, S. Sharma, N. Gajbhiye, R. Vatsa and B. N. Pandey, J. Mater. Chem., 2011, 21, 13388–13398 RSC.
- K. Maaz, S. Karim, A. Mumtaz, S. K. Hasanain, J. Liu and J. L. Duan, J. Magn. Mater., 2009, 321, 1838 CrossRef CAS.
- Y. H. Deng, D. W. Qi, C. H. Deng, X. M. Zhang and D. Y. Zhao, J. Am. Chem. Soc., 2008, 130, 28–29 CrossRef CAS PubMed.
- H. R. Dehghanpour, M. H. Mosslemin and R. Mohebat, J. Chem. Res., 2018, 42(1), 35–39 CrossRef CAS.
- N. M. Evdokimov, S. V. Slambrouck, P. Heffeter, L. Tu, B. L. Calvé, D. Lamoral-Theys, C. J. Hooten, P. Y. Uglinskii, S. R. Kiss, W. F. A. Steelant, W. Berger, J. J. Yang, C. G. Bologa, A. Kornienko and I. V. Magedov, J. Med. Chem., 2011, 54(7), 2012–2021 CrossRef CAS PubMed.
- A. Elhampour, F. Nemati, H. T. Nahzomi and V. Mohagheghi, Res. Chem. Intermed., 2017, 43, 6737–6761 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1ra05889d |
| This journal is © The Royal Society of Chemistry 2021 |