
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
The factors that govern the allosteric chemical sensing of polythiophene chemosensors: scope and limitation toward signal-amplification sensing†
Tomoaki Tsuchiyaa,
Hiroaki Mizunoa and
Gaku Fukuhara *ab
*ab
aDepartment of Chemistry, Tokyo Institute of Technology, 2-12-1 Ookayama, Meguro-ku, Tokyo 152-8551, Japan. E-mail: gaku@chem.titech.ac.jp
bJST, PRESTO, 4-1-8 Honcho, Kawaguchi, Saitama 332-0012, Japan
First published on 14th September 2021
Abstract
The newly designed polythiophene chemosensors (PT1 and PT2) were synthesized via the Suzuki–Miyaura polymerization with appropriate yields. The photophysical properties of PTs thus obtained were examined by means of UV/vis, fluorescence, excitation spectroscopy, and time-correlated single-photon-counting method. The π–π* transitions around 400–600 nm and the emissions in the range of 400–650 nm were observed. The binding behavior of PTs was also investigated upon the interaction of tetrabutylammonium or tetrabutylphosphonium isophthalate, affording the binding constants (K) of 5790–8310 M−1, which were quite smaller than those observed in the corresponding repeating unit. The comprehensive analyses of the UV/vis data and theoretical calculation supports revealed the origins of scope and limitation toward signal-amplification sensing. The present results obtained herein will guide the development of new amplification chemosensors.
Introduction
Chemical sensing of target molecules with chemosensors has become a recent trend in the fields of chemistry due to some applications in the precise detection of hazardous explosives and toxic vapors (VOCs)1–3 and biomarkers relating to serious diseases or cancers.4–6 The previous chemosensors have been achieved based on the “lock-and-key” model, which was proposed as activations of enzymes by Emil Fischer since the late 1800's,7 and thus, many sophisticated reports have been exemplified so far.8–19 Nevertheless, unluckily, such a model is not as almighty since target analytes have become complicated structurally and conformationally, e.g., peptides, proteins, and carbohydrates.20–24 This current research implies the necessity of an alternative to the conventional lock-and-key principle-based chemosensor construction.25A smarter sensing method seems to be a signal amplification, e.g., polymerase chain reaction (PCR)26 and enzyme-linked immunosorbent assay (ELISA),27 the former of which has been well-known owing to the Covid-19 disaster in recent years. Recently, as a signal-amplification reporter, polymer chains turn out to be good candidates. In a pioneering work in 1995, Swager et al. demonstrated the prominently increased sensitivity of a target molecule using conjugated polymers, which is called as the molecular wire approach.28,29 Indeed, a crown ether cavity was incorporated in the conjugated polymer chains, i.e., a type of cyclophane receptor repeating unit, causing pseudorotaxane complexation upon the interaction of the target paraquat guest with the enhancement in sensitivity by a factor of 64 compared to the corresponding repeating unit. The amplification by the molecular wire effect is ascribable to a facile energy migration onto the conjugated polymer chains. On the other hand, we proposed a novel amplification method as “supramolecular allosteric signal-amplification sensing” (SASS) in 2015, which is shown in Fig. 1a.21–24 The SASS approach consists of the following three steps: (1) supramolecular complexation of a target analyte with a recognition site in a chemosensor skeleton, (2) allosteric propagation of binding information upon the complexation of the analyte through a polymer chain, and (3) signal amplification caused by an enhanced complexation stability of the analyte into an induced-fit site. The key mechanisms of SASS are assignable to the synchronized conformational changes of polymer backbones and the subsequent allosteric complexation of an analyte, which is quite different from the previous molecular wire approach relating to the complex excited-state behavior. Therefore, the SASS strategy appears to be simpler as an idea, but the molecular design of polymers may be rather critical.
 | ||
| Fig. 1 (a) The basic idea of supramolecular allosteric signal-amplification sensing (SASS). (b) Chemical structures of polythiophene chemosensors (PT0, PT1, and PT2) and analytes (TBA and TBP), in which the triad monomer (TM) unit is highlighted in red. | ||
Very recently, to further develop a new SASS system, we synthesized functional polythiophene (PT0), including the triad monomer unit (TM) composed of bisthiourea as the molecular recognition tweezer moiety, binaphthyl as the signal transducer skeleton, and bithiophene as a part of the signal reporter unit, and then, examined the amplification sensing behavior of phthalic acids and phthalates (see the structures in Fig. 1b).30 In fact, the PT0 sensor showed extremely large binding constants (K) enhanced by a factor of 5–15 compared to the TM sensor, for which the applicable syn–anti conformational changes of the polythiophene (PT) chain are responsible. However, we did not completely understand whether the direct connection between the bithiophene units possessing the molecular recognition moiety is really proper because the synchronized backbone changes are undoubtedly the key point from the viewpoint of the SASS mechanism.
In the present study, to expand the scope of the SASS using the functional TM unit, we want to elaborate on the effects on how to connect TM via monothiophene (PT1) or bithiophene spacer (PT2) shown in Fig. 1b. The comprehensive sensing studies on PT1 and PT2 by comparing to those obtained in PT0 enable us to conclude the SASS mechanistic insights into the factors that govern the amplification sensing outcomes. Since the synthetic chemistry of PTs has considerably been established,31 the results obtained from this study can easily feedback to the chemistry of PT-based chemosensors.32,33
Experimental
Materials
All chemicals and solvents were employed as received. Spectroscopic-grade chloroform was used for spectroscopy without further purification. Tetrabutylammonium (TBA) and tetrabutylphosphonium isophthalates (TBP) were prepared according to the appropriate protocol.34,35 Concentrations of the synthesized PTs are expressed as monomer unit throughout this work. All spectra were measured in a 1 cm cell in CHCl3 at 25 °C unless stated otherwise.Instruments
Melting points were measured by a Büchi M-560 apparatus. HRMS spectra were recorded on a Bruker ESI microTOF II instrument. 1H, 13C, 2D-COSY, and -HMQC NMR spectra (400 or 100 MHz) were measured in CDCl3 containing tetramethylsilane (TMS) as an internal standard with an ECX400 NMR spectrometer. UV/vis spectra were measured in a quartz cell (1 cm path length) by recording on a JASCO V-650 spectrometer equipped with an ETCS-761 temperature controller. Fluorescence spectra were measured in the same cell using a JASCO FP-8500 spectrophotometer equipped with an ETC-815 temperature controller. Fluorescence lifetime decays were observed in the same cell by a Hamamatsu Quantaurus-Tau single-photon-counting instrument fitted with an LED excitation light (340 nm). IR spectra were measured on a JASCO FT/IR-4700 spectrometer. The molecular weights of the PTs were determined using polystyrene standards through an analytical GPC with a TOSOH TSKgel α-4000 column (condition: 40 °C, 0.5 mL min−1, DMF).Synthesis of monomer
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :10 EtOAc/n-hexane) of the residue afforded the dimerized product as a yellow oil (1.19 g, 2.64 mmol (containing 14% impurities)). The yellow oil (1.19 g, 2.64 mmol) and n-bromosuccinimide (0.99 g, 5.58 mmol) in a 50 mL round-bottomed flask were dissolved in THF (4 mL) and acetic acid (4 mL), and the mixture was stirred for 4 h at room temperature. Then, the solution was extracted with diethyl ether and saturated aqueous sodium bicarbonate solution. The organic layer obtained was dried over anhydrous Na2SO4, filtered, and evaporated under a high vacuum. Flash column chromatography (7:3 n-hexane/EtOAc) of the residue gave the brominated product as an orange oil (1.30 g, 2.14 mmol (containing 14% impurities)). The orange oil (1.30 g, 2.14 mmol) and pyridinium p-toluenesulfate (0.23 g, 0.92 mmol) were dissolved in dry MeOH (9 mL), and the mixture was stirred overnight at 50 °C. Then, the solution was extracted with dichloromethane and water. The organic layer obtained was dried over anhydrous MgSO4, filtered, and evaporated under high vacuum. Flash column chromatography (7:3–3:7 n-hexane/EtOAc) of the residue gave the desired product (250 mg, 0.89 mmol) as a white solid in 33% yield, mp: 91–92 °C; HRMS (ESI+, TOF): calculated for C14H17Br2O2S2 [M + H]+: 438.9031, found 438.9011; 1H NMR (CDCl3, 400 MHz, 20 °C) δH 6.80 (s, 2H), 3.68 (q, J = 7.3 Hz, 4H), 2.65 (t, J = 7.8 Hz, 4H), 1.87 (q, J = 7.1 Hz, 4H), 1.52 (br, 2H); 13C NMR (CDCl3, 100 MHz, 20 °C) δC 141.9, 136.2, 124.4, 108.3, 61.9, 32.3, 25.8.:2 n-hexane/EtOAc) of the residue gave the desired product (95 mg, 0.13 mmol) as a white solid in 30% yield, mp: 126–127 °C; HRMS (ESI+, TOF): calculated for C34H28Br2O2S2Na [M + Na]+: 740.9857, found 740.9898; 1H NMR (CDCl3, 400 MHz, 20 °C) δH 7.72–7.69 (m, 4H), 6.98–6.92 (m, 4H), 6.37 (s, 2H), 6.29 (s, 2H), 3.60–3.43 (m, 4H), 2.82–2.68 (m, 4H), 2.01–1.79 (m, 4H); 13C NMR (CDCl3, 100 MHz, 20 °C) δC 158.0, 149.3, 143.1, 140.8, 135.1, 129.7, 129.1, 127.2, 123.9, 115.8, 113.8, 112.0, 109.0, 103.2, 63.8, 27.3, 24.6.:3 CH2Cl2/diethyl ether) of the residue gave the desired product as a white powder (98 mg, 0.068 mmol) in 64% yield, mp: 106–107 °C; HRMS (ESI+, TOF): calculated for C62H63Br2Cl4N4O10S4 [M + H]+: 1449.0548, found 1449.6399; 1H NMR (CDCl3, 400 MHz, 20 °C) δH 7.94–7.89 (m, 4H), 7.59 (d, J = 8.9 Hz, 2H), 7.35 (s, 2H), 7.22–7.19 (m, 4H), 6.45 (s, 2H), 6.42 (s, 4H), 6.15 (s, 2H), 4.13 (t, J = 5.1 Hz, 4H), 3.86 (t, J = 5.1 Hz, 4H), 3.75–3.65 (m, 12H), 3.57–3.54 (m, 4H), 3.37 (s, 6H), 2.75–2.64 (m, 4H), 2.01–1.82 (m, 4H); 13C NMR (CDCl3, 100 MHz, 20 °C) δC 180.6, 158.0, 151.0, 140.4, 135.7, 134.9, 133.7, 131.5, 130.1, 130.0, 128.6, 128.1, 127.7, 127.3, 126.6, 124.5, 119.1, 109.2, 103.5, 72.6, 71.9, 70.7, 70.6, 70.5, 70.0, 64.5, 59.0, 27.0, 24.8.
:10 EtOAc/n-hexane) of the residue afforded the dimerized product as a yellow oil (1.19 g, 2.64 mmol (containing 14% impurities)). The yellow oil (1.19 g, 2.64 mmol) and n-bromosuccinimide (0.99 g, 5.58 mmol) in a 50 mL round-bottomed flask were dissolved in THF (4 mL) and acetic acid (4 mL), and the mixture was stirred for 4 h at room temperature. Then, the solution was extracted with diethyl ether and saturated aqueous sodium bicarbonate solution. The organic layer obtained was dried over anhydrous Na2SO4, filtered, and evaporated under a high vacuum. Flash column chromatography (7:3 n-hexane/EtOAc) of the residue gave the brominated product as an orange oil (1.30 g, 2.14 mmol (containing 14% impurities)). The orange oil (1.30 g, 2.14 mmol) and pyridinium p-toluenesulfate (0.23 g, 0.92 mmol) were dissolved in dry MeOH (9 mL), and the mixture was stirred overnight at 50 °C. Then, the solution was extracted with dichloromethane and water. The organic layer obtained was dried over anhydrous MgSO4, filtered, and evaporated under high vacuum. Flash column chromatography (7:3–3:7 n-hexane/EtOAc) of the residue gave the desired product (250 mg, 0.89 mmol) as a white solid in 33% yield, mp: 91–92 °C; HRMS (ESI+, TOF): calculated for C14H17Br2O2S2 [M + H]+: 438.9031, found 438.9011; 1H NMR (CDCl3, 400 MHz, 20 °C) δH 6.80 (s, 2H), 3.68 (q, J = 7.3 Hz, 4H), 2.65 (t, J = 7.8 Hz, 4H), 1.87 (q, J = 7.1 Hz, 4H), 1.52 (br, 2H); 13C NMR (CDCl3, 100 MHz, 20 °C) δC 141.9, 136.2, 124.4, 108.3, 61.9, 32.3, 25.8.:2 n-hexane/EtOAc) of the residue gave the desired product (95 mg, 0.13 mmol) as a white solid in 30% yield, mp: 126–127 °C; HRMS (ESI+, TOF): calculated for C34H28Br2O2S2Na [M + Na]+: 740.9857, found 740.9898; 1H NMR (CDCl3, 400 MHz, 20 °C) δH 7.72–7.69 (m, 4H), 6.98–6.92 (m, 4H), 6.37 (s, 2H), 6.29 (s, 2H), 3.60–3.43 (m, 4H), 2.82–2.68 (m, 4H), 2.01–1.79 (m, 4H); 13C NMR (CDCl3, 100 MHz, 20 °C) δC 158.0, 149.3, 143.1, 140.8, 135.1, 129.7, 129.1, 127.2, 123.9, 115.8, 113.8, 112.0, 109.0, 103.2, 63.8, 27.3, 24.6.:3 CH2Cl2/diethyl ether) of the residue gave the desired product as a white powder (98 mg, 0.068 mmol) in 64% yield, mp: 106–107 °C; HRMS (ESI+, TOF): calculated for C62H63Br2Cl4N4O10S4 [M + H]+: 1449.0548, found 1449.6399; 1H NMR (CDCl3, 400 MHz, 20 °C) δH 7.94–7.89 (m, 4H), 7.59 (d, J = 8.9 Hz, 2H), 7.35 (s, 2H), 7.22–7.19 (m, 4H), 6.45 (s, 2H), 6.42 (s, 4H), 6.15 (s, 2H), 4.13 (t, J = 5.1 Hz, 4H), 3.86 (t, J = 5.1 Hz, 4H), 3.75–3.65 (m, 12H), 3.57–3.54 (m, 4H), 3.37 (s, 6H), 2.75–2.64 (m, 4H), 2.01–1.82 (m, 4H); 13C NMR (CDCl3, 100 MHz, 20 °C) δC 180.6, 158.0, 151.0, 140.4, 135.7, 134.9, 133.7, 131.5, 130.1, 130.0, 128.6, 128.1, 127.7, 127.3, 126.6, 124.5, 119.1, 109.2, 103.5, 72.6, 71.9, 70.7, 70.6, 70.5, 70.0, 64.5, 59.0, 27.0, 24.8.Synthesis of polymers
Results and discussion
Design and synthesis of polythiophene chemosensors (PT1 and PT2)
In order to clearly estimate the scope and limitations of the PT-based SASS, we carefully designed monothiophene- and bithiophene-spaced PT1 and PT2, respectively, enabling us to understand the effects on the SASS mechanism that may be different from those observed in the direct TM-connected PT0. As shown in Fig. S9 in ESI,† decidedly, the Hartree–Fock calculations for the optimized structures of the model compounds for both the PTs also support the validity of the molecular design shown in this study. As can be seen in Table 1, the dihedral angles of models PT1 and PT2 are 73–78° in the recognition units, which are slightly larger than those (59–64°)30 of the model PT0 but twisting syn forms. This indicates that the bisthiourea moieties in both the PTs may function as the tweezer recognition site. The synthetic scheme is shown in Fig. 2. The THP-protected 3-(thiophen-3-yl)propan-1-ol was selected as a starting material since the propyl group is known as the most suitable alkyl chain length30 for better converting molecular recognition information upon the bisthiourea site in the repeating unit to dihedral angle changes at the bithiophene scaffold. This is also supported by the calculations (see the data in Table 1). Thus, the propylated thiophene was first dimerized with n-BuLi and Fe(acac)3, brominated at the α-position of the bithiophene and deprotected. The dibromobithiophene thus obtained was tosylated with tosyl chloride. The ditosylate was reacted with the binaphthyl transducer under a high dilution condition to afford the cyclic compound. The cyclic dibromobithiophene was further reacted with the isothiocyanate to give the dibrominated triad monomer (TM) possessing the bisthiourea moiety. The dibromo-monomer was then polymerized through the Suzuki–Miyaura coupling, rather than the conventional Stille coupling for the PT synthesis,31 with diboronic pinacol ester-monothiophene and -bithiophene to afford PT1 and PT2, respectively.| Compd | Dihedral angle/° | ||||
|---|---|---|---|---|---|
| Recognition unitsb | Spacer unitsb | ||||
| θa | θb | θ1 | θ2 | θ3 | |
| a Basis set: HF/6-31G.b See Fig. S9 in ESI for the optimized structures with the notations.c Counter cation was omitted for simplicity during the calculation. | |||||
| PT15-mer | 73.58 | 78.28 | 173.63 | 176.42 | — |
| PT15-mer + isophthalatec | 79.51 | 67.19 | 117.97 | 157.16 | — |
| PT26-mer | 73.85 | 78.58 | 175.22 | 171.98 | 173.22 |
| PT26-mer + isophthalatec | 82.11 | 71.60 | 133.91 | 135.95 | 153.74 |
 | ||
| Fig. 2 Synthesis of novel polythiophene chemosensors (PT1 and PT2). | ||
Photophysical properties of PT1 and PT2
Next, we investigated the optical properties of PTs via UV/vis, fluorescence/excitation spectra, and fluorescence lifetime measurements in CHCl3. As shown in Fig. 3a and b, the UV/vis spectra of both the PTs broadened at around 600 nm, the edge of which was assignable to the π–π* transition of PT. Certainly, the spectra can be applicably separated to a sum of three species as a Gaussian waveform. The red dotted lines observed at the shorter wavelength almost overlap with the spectra of TM (purple solid line), in which the binaphthyl and bithiophene chromophores are contained. Thus, the blue dotted lines peaked at 324 nm, which can be ascribed to an oligomeric segment in the polymer, and the green ones that peaked at 420 nm for PT1 and 424 nm for PT2 are the π–π* transitions. According to the equation for the effective conjugation length (ne) of PT (E (eV) = 2.49 + 3.14/ne),36 ne values of the oligomer, PT1, and PT2 were determined as 2.3, 6.8, and 7.2, respectively, calculated from the extrema of the blue and green dotted lines. These ne values, except for the oligomer, are quite larger and more extended than the ne (2.8) of PT0 due to the mono- and bithiophene spacers, which can adjust as energetically stable dihedral angles (more anti) between TM and spacers. As shown in Table 1, definitely, the dihedral angles at the spacer units are 171–176°, almost anti forms.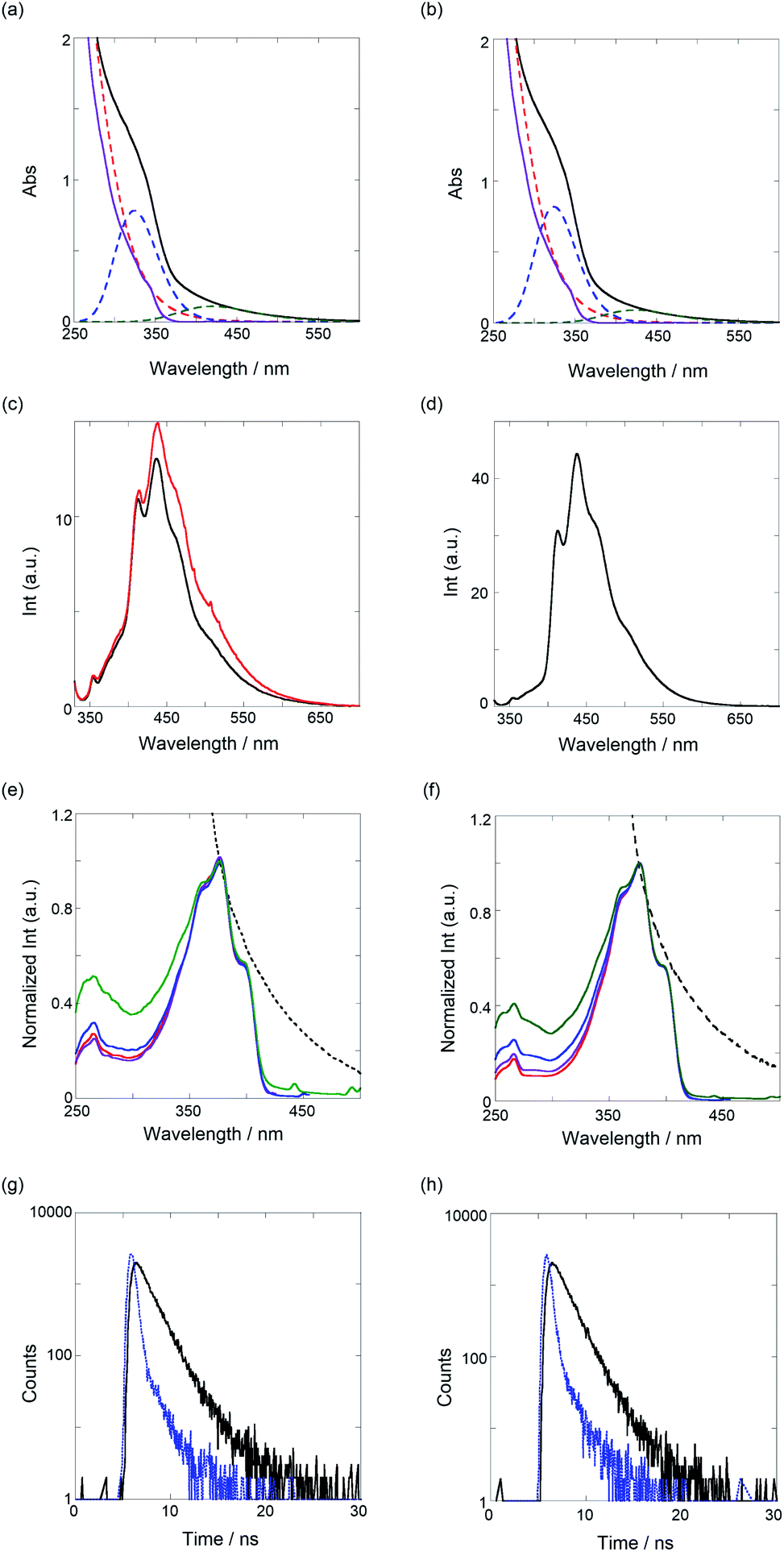 | ||
| Fig. 3 Top panels: UV/vis spectra of (a) PT1 (52.4 μM) and (b) PT2 (51.5 μM, black solid lines) and TM (normalized spectrum at 340 nm, purple solid lines) and results of waveform separation (colored dotted lines). The second panels from top: fluorescence spectra (λex 320 nm) of (c) PT1 in the absence (5.23 μM, black) and presence of TBA (21 μM, red) and (d) PT2 (5.15 μM). The third panels from top: excitation spectra of (e) PT1 (normalized UV/vis spectrum, black dotted line) and (f) PT2 (normalized UV/vis spectrum, black dotted line), monitored at 412 (red), 436–438 (purple), 465–466 (blue), and 510 nm (green). Bottom panels: time-correlated fluorescence decays (λex 340 nm, black solid lines) of (g) PT1 (5.23 μM, λem 436 nm) and (h) PT2 (5.15 μM, λem 438 nm) at room temperature, where the blue dotted lines show the instrument response function (IRF). | ||
As shown in Fig. 3c and d (black lines), the band-broadened fluorescence spectra at 400–650 nm were observed. The broad emission bands are based on fluorescent species of oligomeric chromophore segments through an energy transfer in the conjugated polymer chain.37 The excitation spectra (see Fig. 3e and f) were observed in the range of 300–420 nm, the main emissive species of which comes from a 5-mer (the extremum at 398 nm) based on the ne equation. The UV/vis and fluorescence maxima are summarized in Table 2. To further elucidate the excited-state behavior of PTs, we measured fluorescence lifetimes by the time-correlated single-photon-counting method, shown in Fig. 3g and h. The fluorescence decay profiles were reasonably fitted to a sum of two and one exponential functions to give 1.4 and 0.3 ns for PT1 and 1.3 ns for PT2, respectively, as listed in Table 2. Based on the results of the excitation spectra, the longer lived excited species (1.3–1.4 ns) is ascribable to the main emissive oligomer (ca. 5-mer) and the shorter one to another oligomeric fragment (<5-mer).
| Compd | UV/vis λmax/nm | FL λem/nm | nb,c | τ1b/ns | A1b | τ2b/ns | A2b | χ2b |
|---|---|---|---|---|---|---|---|---|
| a Measured in CHCl3 at 25 °C, unless noted otherwise.b Fluorescence lifetime (τi/ns) and relative abundance (Ai) of each component determined by the time-correlated single-photon-counting method in nondegassed CHCl3 solution at room temperature (λex 340 nm, λem 436–438 nm); a lifetime of the TM was omitted due to over the measurement limitation (∼ps) of the fluorescence lifetime apparatus employed in this study.c Number of components. | ||||||||
| PT1 | 420 | 436 | 2 | 1.4 | 0.90 | 0.3 | 0.10 | 1.1 |
| PT2 | 424 | 437 | 1 | 1.3 | 1.00 | 1.1 | ||
An interesting fluorescence enhancement was also observed upon the addition of TBA as a guest molecule, as shown in Fig. 3c (red line). In the absence of the guest molecule as an initial stage, the fluorescence was suppressed by a photoinduced electron transfer (PET) from the amines in the thiourea to the fluorescent PT chromophore. Then, the addition of a small amount of TBA (11% complexation determined by the binding constant) recovered the fluorescence intensity due to the inhibition of the intra-PET process, as was the case with PT0.30 This result implies that these PTs may be useful as a turn-on fluorescence chemosensor.
Mechanistic origins of SASS in PT1 and PT2
Last, we examined the binding behavior of both the PTs using TBA and TBP salts as model analytes and then attempted to explain the mechanistic origins of the SASS operated in PT. As shown in Fig. 4a, the gradual addition of TBA to a CHCl3 solution of PT1 at 25 °C caused slight band-broadening, hyperchromic effect, and bathochromic shift, indicating a supramolecular complexation of TBA via a perturbation of the chromophores in PT1. The TM unit can trap isophthalates through the 1:1 complexation stoichiometry with binding constants (K) determined by the Job plot.30 Thus, in Fig. 4b, a nonlinear least-squares fitting, assuming the 1:1 complexation, was subjected to the UV/vis titration to give K for TBA as 6030 M−1 (ΔG° as −21.6 kJ mol−1). Also, the same analysis of the UV/vis titration of the PTs gave the K values for TBA and TBP listed in Table 3 (Fig. S10–S12 in ESI†). Interestingly, the K values dropped sharply by a factor of 0.035–0.042, in sharp contrast to the amplification (5–15 times) of PT0. This is a rather contrasting behavior despite using the same repeating TM unit.
 | ||
| Fig. 4 (a) UV/vis spectral changes of a chloroform solution of PT1 (52.4 μM, black) upon gradual addition of TBA (0.03–1.25 mM, colored lines); the dotted line represents the UV spectrum of TBA only. (b) Nonlinear least-squares fitting, assuming a 1:1 model, to determine the binding constant. UV/vis spectra of (c) PT1 (52.4 μM) and (d) PT2 (51.5 μM) in the absence (black solid lines) and presence (red solid lines) of TBA (1.25 mM for PT1 (88% complexation) and 2.18 mM for PT2 (93% complexation)) and results of waveform separation (dotted lines); the spectra without TBA (black solid and dotted lines) in (c and d) are identical to those shown in Fig. 3a and b. Full results of waveform separation for the spectra with TBA are given in Fig. S13 in ESI.† | ||
:1 complexation of isophthalates with PTsa
| Chemosensor | Analyte | K/M−1 | ΔG°b/kJ mol−1 |
|---|---|---|---|
| a The binding constants of PTs were determined using monomer unit concentration.b Gibbs energy changes at 298 K are listed. | |||
| TM | TBA | 159000 ± 11300 |
−29.5 |
| TBP | 198000 ± 39600 |
−30.2 | |
| PT1 | TBA | 6030 ± 690 | −21.6 |
| TBP | 8310 ± 1480 | −22.4 | |
| PT2 | TBA | 5790 ± 570 | −21.5 |
| TBP | 6840 ± 1130 | −21.9 | |
To elucidate the origins of the contrasting binding amplification vs. the inhibition, the UV/vis spectral changes in the absence and presence of TBA were examined. As shown in Fig. 4c and d, the spectra, upon the addition of an adequate amount of TBA (≥88% complexation), showed appreciable bathochromic shifts, indicating an extension of ne. The waveform separations by the addition of TBA strictly afforded the extrema changes from 420 nm to 429 nm for PT1 and from 424 nm to 435 nm for PT2, respectively. According to the equation, the ne value of 6.8 (7.2) was changed to 7.8 (8.7) for PT1 (for PT2). These results can be deduced from facts based on the changes in the dihedral angle of PT backbones. For instance, from the data of PT1 listed in Table 1, the angle in the tweezer increased from 73° to 79° upon the addition of TBA, and then, the other binding site slightly closed (from 78° to 67°). Thus, the averaged angle changes in the recognition unit are almost negligible (76° → 73°) before and after the interaction of TBA, for which a relaxation of the thiophene spacer unit is highly likely to be responsible. Namely, freely rotating/changing the dihedral angles between the spacers (θ1 and θ2) may inhibit a tighter tweezer mode at the neighbor recognition position by the first binding. Also, the averaged angle of 73° after the interaction of TBA is demonstrably larger than that of 31° in the PT0 for the tighter tweezer (the amplification mode), eventually causing the drastic decrease of the binding constant. This trend can also be seen in the case of PT2. The averaged angle changes at the recognition unit are almost the same as 76° before and after the addition of TBA, which is consistent with the case of PT1. These results suggest that the thiophene spacers between the recognition sites play a critical role in amplification sensing. Therefore, it can be emphasized that the binding behavior of target analytes can be drastically controlled, i.e., amplification or inhibition, just by changing the conformationally flexible spacers.
Conclusion
We have developed the newly synthesized polythiophene chemosensors (PT1 and PT2) that afford information on chemical sensing based on the conformational changes of polymer backbones upon inclusion of salt guests. The binding constants obtained in this study were rather inhibited by a factor of 0.035–0.042, and thus, very different from those observed in the previous amplification chemosensor (5–15 times). The relaxation of the recognition information through the thiophene spacer units is a critical factor that governs allosteric chemical sensing. The present study provided new guidelines for an effective molecular design of SASS-operative chemosensors, and hence, can be expanded to other polymer chemosensors that show an allosteric amplification.Conflicts of interest
The authors declare no competing financial interest.Acknowledgements
This work was supported by the generous funds of Grant-in-Aid for Scientific Research (B) (No. 19H02746) from the Japan Society for the Promotion of Science (JSPS) and Japan Science and Technology Agency (JST), PRESTO (No. JPMJPR17PA). We are grateful to Dr Makoto Harada and Prof. Tetsuo Okada at the Tokyo Institute of Technology for their helpful discussion.References
- S. J. Toal and W. C. Trogler, J. Mater. Chem., 2006, 16, 2871–2883 RSC.
- Y. Salinas, R. Martínez-Máñez, M. D. Marcos, F. Sancenón, A. M. Costero, M. Parra and S. Gil, Chem. Soc. Rev., 2012, 41, 1261–1296 RSC.
- L. Chen, D. Wu and J. Yoon, ACS Sens., 2018, 3, 27–43 CrossRef CAS PubMed.
- F. T. Andón and B. Fadeel, Acc. Chem. Res., 2013, 46, 733–742 CrossRef PubMed.
- K. Singh, A. M. Rotaru and A. A. Beharry, ACS Chem. Biol., 2018, 13, 1785–1798 CrossRef CAS.
- F. Djago, J. Lange and P. Poinot, Nat. Rev. Chem., 2021, 5, 183–196 CrossRef CAS.
- E. Fischer, Ber. Dtsch. Chem. Ges., 1894, 27, 2985–2993 CrossRef CAS.
- J. H. Hartley, T. D. James and C. J. Ward, J. Chem. Soc., Perkin Trans. 1, 2000, 3155–3184 RSC.
- M. Takeuchi, M. Ikeda, A. Sugasaki and S. Shinkai, Acc. Chem. Res., 2001, 34, 865–873 CrossRef CAS PubMed.
- T. W. Bell and N. M. Hext, Chem. Soc. Rev., 2004, 33, 589–598 CAS.
- Chemosensors: principles, strategies, and applications, ed. B. Wang and E. V. Anslyn, Wiley, Hoboken, 2011 Search PubMed.
- K. P. McDonald, Y. Hua, S. Lee and A. H. Flood, Chem. Commun., 2012, 48, 5065–5075 RSC.
- G. Ghale and W. M. Nau, Acc. Chem. Res., 2014, 47, 2150–2159 CrossRef CAS PubMed.
- N. Busschaert, C. Caltagirone, W. V. Rossom and P. A. Gale, Chem. Rev., 2015, 115, 8038–8155 CrossRef CAS PubMed.
- R. Kubota and I. Hamachi, Chem. Soc. Rev., 2015, 44, 4454–4471 RSC.
- E. Yashima, N. Ousaka, D. Taura, K. Shimomura, T. Ikai and K. Maeda, Chem. Rev., 2016, 116, 13752–13990 CrossRef CAS.
- T. J. Mooibroek, J. M. Casas-Solvas, R. L. Harniman, C. M. Renney, T. S. Carter, M. P. Crump and A. P. Davis, Nat. Chem., 2016, 8, 69–74 CrossRef CAS.
- A. C. Sedgwick, L. Wu, H.-H. Han, S. D. Bull, X.-P. He, T. D. James, J. L. Sessler, B. Z. Tang, H. Tian and J. Yoon, Chem. Soc. Rev., 2018, 47, 8842–8880 RSC.
- M. Rickhaus, M. Jirasek, L. Tejerina, H. Gotfredsen, M. D. Peeks, R. Haver, H.-W. Jiang, T. D. W. Claridge and H. L. Anderson, Nat. Chem., 2020, 12, 236–241 CrossRef CAS PubMed.
- H.-J. Schneider, Int. J. Mol. Sci., 2015, 16, 6694–6717 CrossRef CAS PubMed.
- G. Fukuhara, Polym. J., 2015, 47, 649–655 CrossRef CAS.
- G. Fukuhara, J. Inclusion Phenom. Macrocyclic Chem., 2019, 93, 127–143 CrossRef CAS.
- G. Fukuhara, J. Photochem. Photobiol., C, 2020, 42, 100340 CrossRef CAS.
- G. Fukuhara, Polym. J. DOI:10.1038/s41428-021-00547-2 , in press..
- J. J. Lavigne and E. V. Anslyn, Angew. Chem., Int. Ed., 2001, 40, 3118–3130 CrossRef CAS.
- R. K. Saiki, S. Scharf, F. Faloona, K. B. Mullis, G. T. Horn, H. A. Erlich and N. Arnheim, Science, 1985, 230, 1350–1354 CrossRef CAS PubMed.
- Y. Liu, L. Zhan, Z. Qin, J. Sackrison and J. C. Bischof, ACS Nano, 2021, 15, 3593–3611 CrossRef CAS.
- Q. Zhou and T. M. Swager, J. Am. Chem. Soc., 1995, 117, 7017–7018 CrossRef CAS.
- Q. Zhou and T. M. Swager, J. Am. Chem. Soc., 1995, 117, 12593–12602 CrossRef CAS.
- T. Tsuchiya and G. Fukuhara, J. Org. Chem., 2020, 85, 13239–13245 CrossRef CAS PubMed.
- R. D. McCullough, Adv. Mater., 1998, 10, 93–116 CrossRef CAS.
- S. Das, P. Routh, R. Ghosh, D. P. Chatterjee and A. K. Nandi, Polym. Int., 2017, 66, 623–639 CrossRef CAS.
- T. Wang, N. Zhang, W. Bai and Y. Bao, Polym. Chem., 2020, 11, 3095–3114 RSC.
- A. J. Lowe and F. M. Pfeffer, Chem. Commun., 2008, 1871–1873 RSC.
- Q. A. Lo, D. Sale, D. C. Braddock and R. P. Davies, Eur. J. Org. Chem., 2019, 1944–1951 CrossRef CAS.
- T. Izumi, S. Kobashi, K. Takimiya, Y. Aso and T. Otsubo, J. Am. Chem. Soc., 2003, 125, 5286–5287 CrossRef PubMed.
- D. A. V. Bout, W.-T. Yip, D. Hu, D.-K. Fu, T. M. Swager and P. F. Barbara, Science, 1997, 277, 1074–1077 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1ra05795b |
| This journal is © The Royal Society of Chemistry 2021 |