
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
The role of substituted pyridine Schiff bases as ancillary ligands in the optical properties of a new series of fac-rhenium(I) tricarbonyl complexes: a theoretical view†
Rosaly Morales-Guevara ab,
Juan A. Fuentesc,
Dayán Paez-Hernández*ab and
Alexander Carreño*ab
ab,
Juan A. Fuentesc,
Dayán Paez-Hernández*ab and
Alexander Carreño*ab
aUniversidad Andres Bello, Programa de Doctorado en Físicoquímica Molecular, Facultad de Ciencias Exactas, Santiago, Chile. E-mail: alexander.carreno@unab.cl; d.paez@unab.cl
bLaboratory of Organometallic Synthesis, Center of Applied NanoSciences (CANS), Facultad de Ciencias Exactas, Universidad Andres Bello, República 330, Santiago, Chile. E-mail: alexander.carreno@unab.cl; d.paez@unab.cl
cLaboratorio de Genética y Patogénesis Bacteriana, Facultad de Ciencias de la Vida, Universidad Andres Bello, República 330, Santiago, Chile
First published on 18th November 2021
Abstract
Over the last few years, luminescent Re(I) tricarbonyl complexes have been increasingly proposed as fluorophores suitable for fluorescence microscopy to visualize biological structures and cells. In this sense, incorporating an asymmetrical pyridine Schiff base (PSB) as the ancillary ligand strongly modifies the staining and luminescent properties of Re(I) tricarbonyl complexes. In this work, we analyzed two series of Re(I) tricarbonyl complexes with their respective PSB ligands: (1) fac-[Re(CO)3(2,2′-bpy)(PSB)]1+ and (2) fac-[Re(CO)3(4,4′-bis(ethoxycarbonyl)-2,2′-bpy)(PSB)]1+, where the PSB exhibits substitutions at positions 4 or 6 in the phenolic ring with methyl or halogen substituents. Thus, we performed computational relativistic DFT and TDDFT studies to determine their optical properties. The ten complexes analyzed showed absorption in the visible light range. Furthermore, our analyses, including zero-field splitting (ZFS), allowed us to determine that the low-lying excited state locates below the 3LLCT states. Interestingly, seven of the ten analyzed complexes, whose corresponding PSB harbors an intramolecular hydrogen bond (IHB), exhibited luminescent emission that could be suitable for biological purposes: large Stokes shift, emission in the range 600–700 nm and τ in the order of 10−2 to 10−3 s. Conversely, the three complexes lacking the IHB due to two halogen substituents in the corresponding PSB showed a predicted emission with the lowest triplet excited state energy entering the NIR region. The main differences in the complexes' photophysical behavior have been explained by the energy gap law and time-resolved luminescence. These results emphasize the importance of choosing suitable substituents at the 4 and 6 positions in the phenolic ring of the PSB, which determine the presence of the IHB since they modulate the luminescence properties of the Re(I) core. Therefore, this study could predict Re(I) tricarbonyl complexes' properties, considering the desired emission features for biological and other applications.
1. Introduction
Complexes based on a Re(I) tricarbonyl core with a dinitrogenated ligand (N,N such as 1,10-phenanthroline or 2,2′-bipyridine derivatives) in the equatorial position, and halogen or pyridine derivatives as ancillary ligands (X), have been extensively used for diverse applications, including solar cells,1,2 catalysis,3–8 and more recently, biological applications,9–17 including staining of bacteria, yeasts (walled cells), and proteins separated by SDS-PAGE.18,19Photophysical attributes of fac-[Re(I)(CO)3(N,N)X]n complexes (where n is 0 or 1+) can be modulated by combining the (N,N) and X ligands.10,17,19 A particular combination of ligands can affect the metalcore due to their donor or acceptor properties.20–24 Interestingly, ligands choice affects the photophysical properties and impacts the biological effect, such as therapeutical applications or antimicrobial properties, including protein stain.18,25,26
Over the last few years, luminescent Re(I) tricarbonyl complexes have been increasingly proposed as fluorophores suitable for fluorescence microscopy to visualize biological structures and cells.15,27–29 Their chemical stability,30 enhanced photostabilities (leading to lower photobleaching),31 and a relatively good cellular uptake15,29,32,33 represent attractive features in these complexes. The use of (N,N) ligands such as 2,2′-bipyridine (2,2′-bpy) or derivatives can modulate photophysical and biological properties of Re(I) complexes. For instance, when to Re(I) complexes harboring the same ancillary ligand (Br: bromide) where compared, it has been shown that fac-Re(I)(CO)3(2,2′-bpy)(Br) presents a maximum absorption at 383 nm,34 whereas fac-Re(I)(CO)3(4,4′-bis(ethoxycarbonyl)-2,2′-bpy)(Br) exhibits maximum absorption at 419 nm.35,36 In addition, it has been stated that fac-Re(I)(CO)3(2,2′-bpy)(Br) can stain yeasts (eukaryotic walled cells), whereas fac-Re(I)(CO)3(4,4′-bis(ethoxycarbonyl)-2,2′-bpy)(Br) is unable to stain the same cells under similar experimental condition.19,37 All this evidence shows that relatively small substitution can affect both photophysical and staining properties.19
The use of dinitrogenated ligands (N,N) allows incorporating ancillary ligands (X), which also modulate the complex properties. In this sense, the addition of an asymmetrical pyridine Schiff base (PSB) as the ancillary ligand strongly modifies the photophysical properties, but also the staining properties of Re(I) tricarbonyl complexes.19,37 Schiff bases are aldehyde- or ketone-like compounds, where the carbonyl group is replaced by an azomethine (–C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N–) group.38 PSB are constituted by two rings (a pyridine and a phenolic ring) connected by the azomethine group. In particular, some PSBs present intramolecular hydrogen bond (IHB) that provides stability.39 This kind of PSBs (in particular, (E)-2-((3-amino-pyridin-4-ylimino)-methyl)-4,6-di-tert-butylphenol) has been used as ancillary ligands of Re(I) tricarbonyl complexes, showing promising results as fluorophores for fluorescent microscopy, especially with walled cells (yeasts and bacteria).35,37 Thus, as stated above, the fac-Re(I)(CO)3(4,4′-bis(ethoxycarbonyl)-2,2′-bpy)(Br) is unable to stain yeasts; nevertheless, the similar complex fac-Re(I)(CO)3(4,4′-bis(ethoxycarbonyl)-2,2′-bpy)((E)-2-((3-amino-pyridin-4-ylimino)-methyl)-4,6-di-tert-butylphenol)1+, where the Br was substituted by (E)-2-((3-amino-pyridin-4-ylimino)-methyl)-4,6-di-tert-butylphenol, showed a differential staining ability, remaining retained in the cell nucleus.37
N–) group.38 PSB are constituted by two rings (a pyridine and a phenolic ring) connected by the azomethine group. In particular, some PSBs present intramolecular hydrogen bond (IHB) that provides stability.39 This kind of PSBs (in particular, (E)-2-((3-amino-pyridin-4-ylimino)-methyl)-4,6-di-tert-butylphenol) has been used as ancillary ligands of Re(I) tricarbonyl complexes, showing promising results as fluorophores for fluorescent microscopy, especially with walled cells (yeasts and bacteria).35,37 Thus, as stated above, the fac-Re(I)(CO)3(4,4′-bis(ethoxycarbonyl)-2,2′-bpy)(Br) is unable to stain yeasts; nevertheless, the similar complex fac-Re(I)(CO)3(4,4′-bis(ethoxycarbonyl)-2,2′-bpy)((E)-2-((3-amino-pyridin-4-ylimino)-methyl)-4,6-di-tert-butylphenol)1+, where the Br was substituted by (E)-2-((3-amino-pyridin-4-ylimino)-methyl)-4,6-di-tert-butylphenol, showed a differential staining ability, remaining retained in the cell nucleus.37
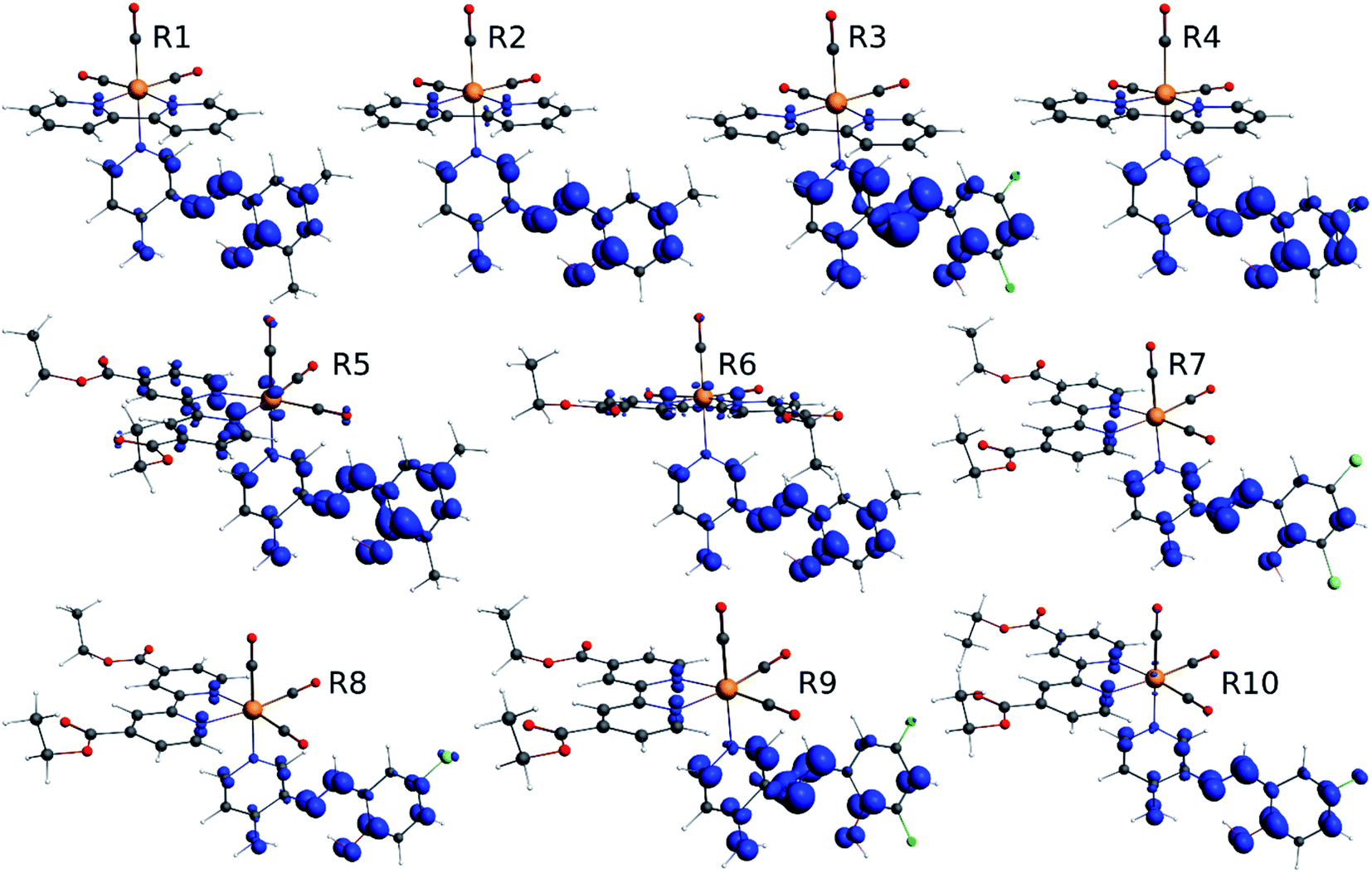
Our group has already synthesized and characterized some PSBs, derivated from (E)-2-((3-amino-pyridin-4-ylimino)-methyl)-4,6-di-tert-butylphenol,35 such as (E)-2-(((4-aminopyridin-3-yl)imino)methyl)-4,6-difluorophenol (PSB3), (E)-2-(((4-aminopyridin-3-yl)imino)methyl)-4-fluorophenol (PSB4), (E)-2-(((4-aminopyridin-3-yl)imino)methyl)-4,6-dichlorophenol (PSB5), and (E)-2-(((4-aminopyridin-3-yl)imino)methyl)-4-chlorophenol (PSB6) (Scheme 1). We previously characterized these PSBs regarding their structural, optical, electronical and antimicrobial properties.39–42 On the other hand, the PSBs (E)-2-(((4-aminopyridin-3-yl)imino)methyl)-4,6-di-methylphenol (PSB1), and (E)-2-(((4-aminopyridin-3-yl)imino)methyl)-4-methylphenol (PSB2), to our knowledge, are new (Scheme 1). In the present study, we worked with two series of Re(I) tricarbonyl complexes with their respective PSB ligands (PSB1 to PSB6, Scheme 1), which are summarized in Table S1:† (1) fac-[Re(CO)3(2,2′-bpy)(PSB)]1+ (Fig. 1, four complexes) and (2) fac-[Re(CO)3(4,4′-bis(ethoxycarbonyl)-2,2′-bpy)(PSB)]1+ (Fig. 2, six complexes). We determined the optical properties of all complexes by computational relativistic DFT and TDDFT analyses,43–47 showing that the ten complexes presented absorption in the visible light range. To perform these calculations, we used the B3LYP exchange–correlation functional48–50 with the triple-ζ quality double plus polarization function (TZ2P)51 to include the polarized effects for all atoms.35 We found that the complexes harboring PSB that could form IHB (i.e. PSB1, PSB2, PSB4, and PSB6) exhibited luminescent emission suitable for biological purposes: large Stokes shift, emission in the range 600–700 nm and τ in the order of 10−2 to 10−3 s. By contrast, complexes constituted by PSB lacking the IHB (i.e., PSB3 and PSB5) exhibited a predicted emission with the lowest triplet excited state energy entering the NIR region. Since positions 4 and 6 in the phenolic ring in the PSB participate in forming the IHB, we postulate that choosing suitable substituents at these positions could be relevant to modulate the photophysical behavior of this kind of complexes.
 | ||
| Scheme 1 Chemical structure of the pyridine Schiff bases: (E)-2-(((4-aminopyridin-3-yl)imino)methyl)-4,6-dimethylphenol (PSB1), (E)-2-(((4-aminopyridin-3-yl)imino)methyl)-4-methylphenol (PSB2), (E)-2-(((4-aminopyridin-3-yl)imino)methyl)-4,6-difluorophenol (PSB3), (E)-2-(((4-aminopyridin-3-yl)imino)methyl)-4-fluorophenol (PSB4), (E)-2-(((4-aminopyridin-3-yl)imino)methyl)-4,6-dichlorophenol (PSB5), and (E)-2-(((4-aminopyridin-3-yl)imino)methyl)-4-chlorophenol (PSB6). | ||
 | ||
| Fig. 1 Optimized geometry of the Re(I) tricarbonyl complexes R1 to R4 (series 1). | ||
 | ||
| Fig. 2 Optimized geometry of the Re(I) tricarbonyl complexes R5 to R10 (series 2). | ||
2. Computational methods
All the proposed structures were optimized in the framework of the density functional theory (DFT) using the Amsterdam Density Functional (ADF) code,52 where the scalar and the spin–orbit relativistic effects were incorporated using the two-component Hamiltonian with the zeroth-order regular approximation (ZORA).53–55 For relativistic calculations performed on organometallic chemical systems including heavy atoms (such as rhenium), ZORA expands the Dirac equation in E/(2mc2 − V), and contains corrections in 1/c2 to all orders, and can be written as the sum of a scalar relativistic (SR) part and a SOC part.56The quality of the obtained minimum was corroborated with frequency calculations, being positive in all proposed complexes. The hybrid B3LYP exchange–correlation functional, using the local density approximation to the correlation functional (which stands for Becke, 3-parameter, Lee–Yang–Parr),48–50 was used with the standard Slater type orbital (STO) basis set with the triple-ζ quality double plus polarization function (TZ2P)51 to include the polarized effects for all atoms. Implicit solvation effects on the geometry and optical properties were considered using a dielectric continuum model (COSMO)57–60 with dichloromethane as solvent to compare our results with previously reported experimental data with similar Re(I) complexes.34–36,61
Excitation energy and absorption spectra were calculated using the scalar relativistic time-dependent density functional theory (SR-TDDFT), considering 60 excitations with ground state optimized and the vertical transition to excited singlet state (Fig. 3).62,63 A similar calculation considering 40 excitations (with triplet state optimized) was used as the basis for the self-consistent two-component spin–orbit coupling TDDFT (SOC-TDDFT)64 within the ZORA Hamiltonian, where 6 spin-mixed excitations were calculated using the SR-TDDFT results as input.27,65,66 Since the T1 state is the lowest in the triplet manifold, the T1 geometry can be optimized like a regular ground state. However, with triplet spin multiplicity (unrestricted DFT) in a regular geometry optimization via the most popular generalized gradient approximated potentials GGA with nonlocal exchange and correlation corrections with the PBE functional and the SR-ZORA Hamiltonian without symmetry to reproduce the possible symmetry breaking in the excited triplet state. The emission analysis of R1 to R10 was studied by the model of spin-forbidden phosphorescence (vertical transition from triplet state optimized to singlet ground state T1 → S0) (Fig. 3).62,63 Usually, the triplet state is lower in symmetry than the ground state; thus, it is not recommended to consider symmetry restrictions. In this part of the work, we are interested in electronic transitions between the visible and ultraviolet regions.
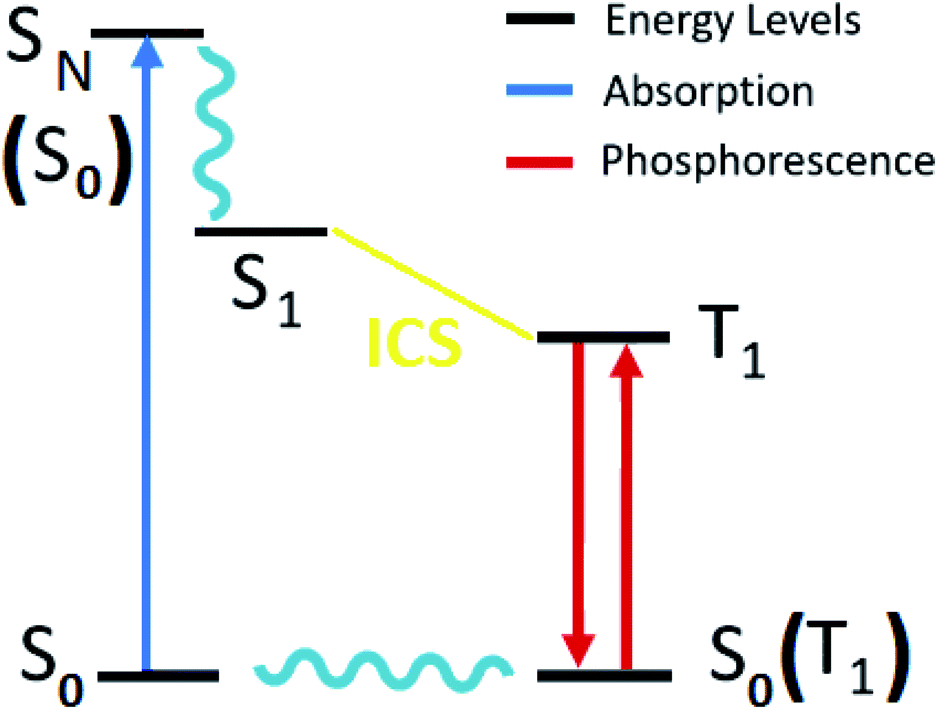 | ||
| Fig. 3 Diagram of the electronic states involved in the theoretical protocol used to predict optical properties of Re(I) complexes via TDDFT. | ||
The radiative rate ki and radiative lifetime τi from sub-state i (i = 1, 2, 3) of the T1 state to the ground state were calculated from the excitation energy ΔEi and the transition dipole moment Mi with SOC included (eqn (1)).67,68
 | (1) |
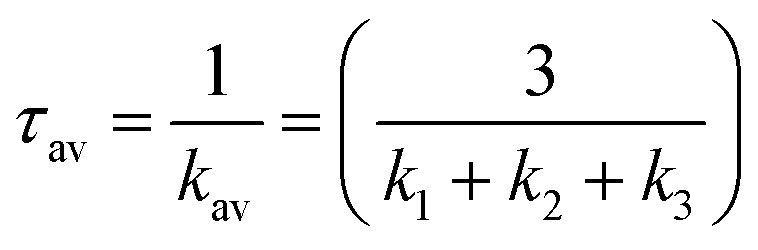 | (2) |
From the Boltzmann distribution, the τav can be obtained as follows in eqn (3):73–75
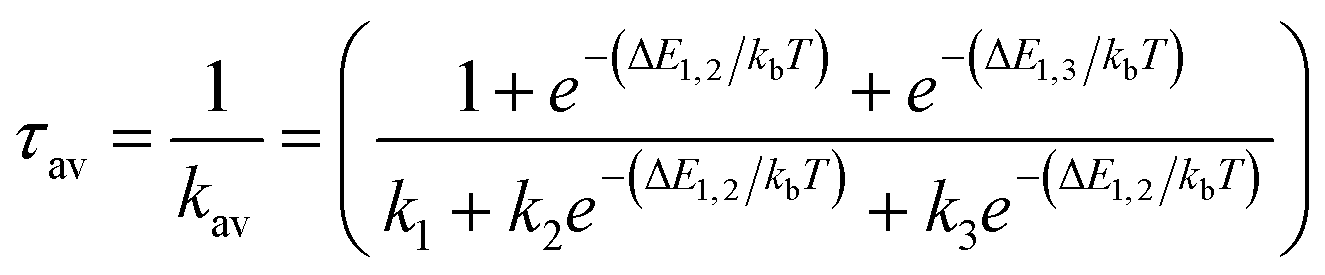 | (3) |
All calculated radiative rate ki (or radiative lifetime τi) was multiplied (or divided) by the square of the refractive index of dichloromethane.
2.1 Zero-field splitting
The luminescence properties of organometallic Re(I) tricarbonyl complexes are crucially affected by relativistic effects.47,76 In a non- or scalar-relativistic picture, triplet states are three-fold degenerate.43,77 The spin–orbit effect on this degeneracy can be quantified by the zero-field splitting (ZFS) resulting from the dipolar interaction between magnetic moments of unpaired electrons (Fig. 4), which is relevant to clarify the nature of the emissive state.78,79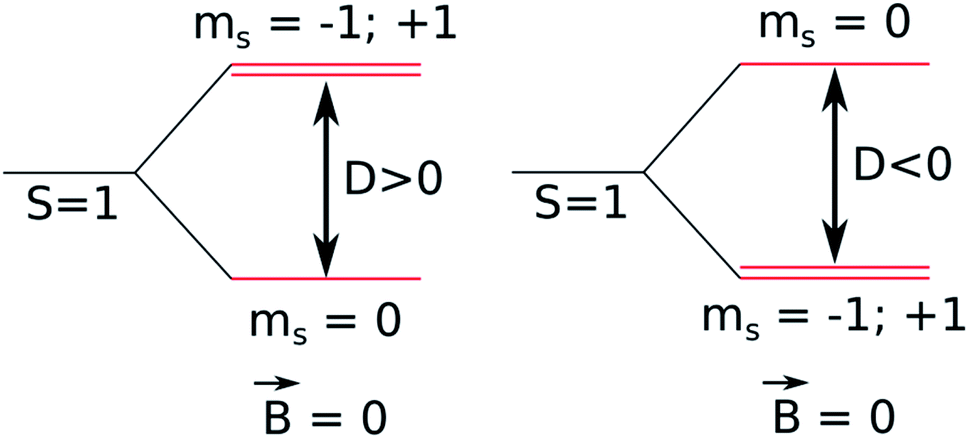 | ||
| Fig. 4 Schematic diagram of a triplet state zero-field splitting (ZFS). | ||
ZFS, in combination with emission lifetime (τr), is an important parameter to characterize the first triplet excited state of these transition metal complexes.80,81
3. Results and discussion
3.1 Geometry structures in the ground state
The optimized structures of all the Re(I) complexes are shown in Fig. 1 (series 1) and Fig. 2 (series 2). These structures and the geometry parameters were obtained by comparing the experimental data of similar Re(I) complexes, including fac-[Re(CO)3(4,4′-dimethyl-2,2′-bpy)(E)-2-(((4-aminopyridin-3-yl)imino)methyl)-4,6-di-tert-butylphenol)]1+,82 and fac-Re(CO)3(4,4′-bis(ethoxycarbonyl)-2,2′-bpy)Br.35,82 As depicted in Fig. 1 and 2, the Re(I) adopts a distorted octahedral coordination geometry with the three carbonyls of all complexes distributed with facial coordination. The equatorial Re–C (around 1.932 Å) and axial Re–C (around 1.939 Å) bond distances showed no significant differences. Experimental results obtained for similar Re(I) complexes show equivalent distances.11,17,83 In the case of CO carbonyl ligands, the axial CO distance (around 1.159 Å) is shorter than that of the equatorial (around 1.162 Å) bond (Tables S2 and S3 in the ESI†). This result is attributed to the ligand-to-metal back bonding effect in both axial and carbonyl ligands at equatorial positions.84–86 The Re(I)–(N,N) ligand (where N,N is 2,2′-bpy or 4,4′-bis(ethoxycarbonyl)-2,2′-bpy) distances are approximately 2.192 Å. These distances are shorter than the Re(I)–PSB (ancillary ligand) distances (approximately 2.249 Å, Tables S2 and S3 in the ESI†), confirming the deviation of the Re(I) coordination sphere from the ideal octahedron.
In the case of R1, R2, R5 and R6, calculations were in agreement with experimental data reported for a similar fac-[Re(CO)3(4,4′-dimethyl-2,2′-bpy)((E)-2-((3-amino-pyridin-4-ylimino)-methyl)-4,6-di-tert-butylphenol)]1+ complex, where Re(I)–(N,N) bond distance is 2.18 (6) Å and 2.13 (5) Å, shorter than the Re(I)–PSB bond distance 2.22 (5) Å.82 Regarding the azomethine group (CN), bond distances are approximately 1.307 Å (comparable to that described for a similar PSB,87). By contrast, R3, R7 and R9, where the IHB is not observed in their corresponding PSB (i.e., PSB3 for R3 and R9, and PSB5 for R7), the distance CN was around 1.289 Å. This result can be explained by the presence of halogen substitutions, particularly at position 6 of the phenolic ring, which enhances the electron-withdrawing effect (inductive effect88–91). Furthermore, the presence of the halogen could participate in forming a halogen–carbon permanent dipole, favoring the interaction with the vicinal OH group in the phenolic ring (as previously reported92,93) (Fig. 5), potentially affecting the photoluminescent behavior (see below).
 | ||
| Fig. 5 Numbering atoms of the PSB ancillary ligands. (A) PSB1, PSB2, PSB4 and PSB6; (B) PSB3 and PSB5. | ||
Analysis of frequencies calculations for all complexes showed three characteristic bands associated with carbonyl groups coordinated to the rhenium core (Table S4 and Fig. S1† in the ESI). In all cases, C–O bands were found around 2033 cm−1, and convolution bands at 1967 cm−1 and 1951 cm−1. These results can be explained by the symmetry loss of the complexes under study. The first band (around 2033 cm−1) was assigned to the symmetric vibration mode of the CO. On the other hand, the convolution bands (around 1967 cm−1 and 1951 cm−1) were assigned to the antisymmetrical vibration mode of the CO (wide band). This is a consequence of the trans effect of the ancillary ligand nature, which increases the CO ligand's force constant, lowering the difference between both antisymmetric bands.65
The study of geometry optimization obtained by the proposed calculation protocol showed a good agreement with previously reported experimental data obtained by FTIR and UV-Vis,34,35 and X-ray,82 for similar complexes. Thus, this geometry optimization provides robust support for absorption and emission calculations, as described below.
3.2 Optical properties
Time-dependent density functional theory (TDDFT) calculations94–96 were performed to predict and characterize excited states of R1 to R10 complexes. In addition, we have carried out a study of the vertical transitions associated with the absorption of the proposed complexes. These singlet–singlet transitions occur without a change in molecular geometry (vertical) and can be mapped within the framework of the TDDFT. The assignments in terms of orbitals are intended to understand the nature of the transition, ligand-centered or charge-transferring. The theoretical calculations predicted three kinds of absorption bands in all complexes, using dichloromethane as solvent. R1, R2, and R4 (Table 1) presented a first band between 270–280 nm, a second band was located at 310–312 nm, and a third band was found at 393–398 nm. Nevertheless, we observed only two bands in R3 located around 278–279 nm and 396 nm. This difference presented by R3 could be attributed to the electron-donating effect of the phenol ring due to two fluorine substituents at positions 4 and 6 (compare R3 with R1, R2 or R4 in Fig. 1 and Table S1 in the ESI†). In addition, and as stated above, the presence of a halogen in position 6 precludes the formation of the IHB.| Complex | λ (nm) | f | Transition | Assignment* |
|---|---|---|---|---|
| a bpy: 2,2′-bpy. | ||||
| R1 | 394 | 0.4434 | H → L+1 (96%) | LLCT/MLCT(π → π*/d → π*) |
| 312 | 0.4878 | H−6 → L (51%) | LbpyLbpyCT(π → π*) | |
| 270 | 0.3378 | H−1 → L+4 (36%) | LLCT/MLCT(π → π*/d → π*) | |
| H−2 → L+5 (13%) | MLCT(d → π*) | |||
| R2 | 393 | 0.4774 | H → L+1 (97%) | LLCT/MLCT(π → π*/d → π*) |
| 311 | 0.3654 | H−6 → L (65%) | LbpyLbpyCT(π → π*) | |
| 280 | 0.4279 | H−5 → L+1 (77%) | LLCT/MLCT(π → π*/d → π*) | |
| R3 | 396 | 0.4003 | H → L+1 (95%) | LLCT/MLCT(π → π*/d → π*) |
| 279 | 0.2794 | H−7 → L+1 (52%) | LLCT(π → π*) | |
| 278 | 0.5497 | H−5 → L+1 (40%) | LLCT/MLCT(π → π*/d → π*) | |
| R4 | 398 | 0.3026 | H → L+1 (72%) | LLCT/MLCT(π → π*/d → π*) |
| 310 | 0.3191 | H → L+3 (41%) | LLbpyCT/MLbpyCT(π → π*/d → π*) | |
| H−6 → L (41%) | LbpyLbpyCT(π → π*) | |||
| 280 | 0.4969 | H−5 → L+1 (58%) | LLCT/MLCT(π → π*/d → π*) | |
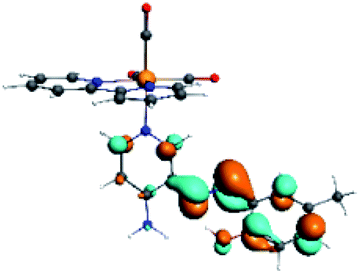
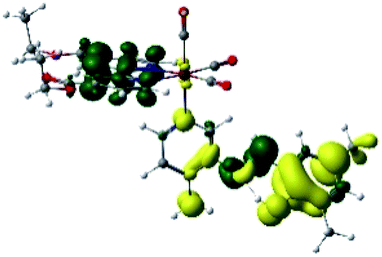
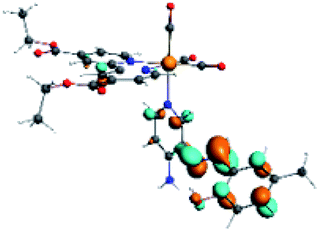
The assignments and nature of the described transitions are summarized in Table 1. In R1 to R4 complexes, the calculated UV-Vis spectrum centered around 395 nm, similar to other cationic Re(I) complexes,23,97–100 assigned to HOMO → LUMO+1 with a mixture of π → π* and MLCT. Table 2 shows the isosurfaces of the molecular orbitals and the electron density differences maps for these first series (R1 to R4), respectively, where the calculated transitions showed that the compositions of the excitation associated with the LLCT (π → π* corresponding to PSB ancillary ligand, Scheme 1) and d → π* transitions involved the metals and PSB (Table 1).101–103 The most important orbitals involved in electronic transitions are qualitatively represented in Fig. S2 to S5 in the ESI.†
| Complex | HOMO | LUMO+1 | Density diff. |
|---|---|---|---|
| R1 | 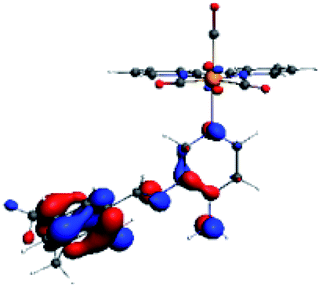 |
 |
 |
| R2 |  |
 |
 |
| R3 | 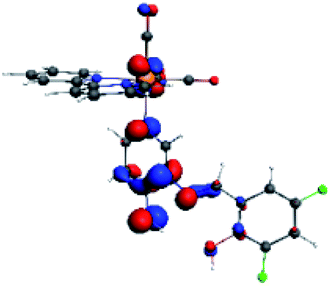 |
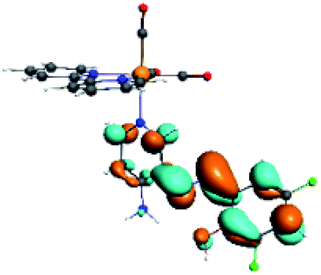 |
 |
| R4 | 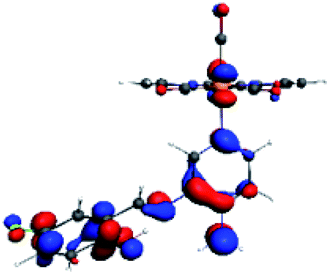 |
 |
 |
The difference electron density maps of Table 2 show that in R1, R2 and R4 the electron density is transferred to the azomethine π system (green color). In the molecular orbital analysis, it is shown that the acceptor orbital is naturally the empty π-antibonding. For R1, R2 and R4, the donor portion of the PSB (yellow color) is mainly centered at the pyridine moiety. On the other hand, the donor orbital (π) in R3 changes from the phenolic ring to the pyridinic ring, does not involve the phenolic ring given the electron-withdrawing effect of the two F atoms. This result can be attributed to a lower electron density of the phenolic ring in R3, compared with R1, R2 and R4 (Table 2). In addition, the presence of the halogen at position 6 changes the LLCT character, displacing it from the phenolic ring to the pyridine ring. Furthermore, the LUMO+1 orbital exhibits mainly a π-antibonding character centered at the azomethine group in R1 to R4 complexes (Table 2), explaining why the IHB has a significant effect on transition energies, besides the effect of the molecular rigidity on the triplet lifetime (see below).
On the other hand, R5 to R10 presented three absorption bands around 287 nm, 332 nm and 394 nm (Table 3). Regarding the transition around 394 nm, typical for cationic Re(I) complexes, as stated above, R5 to R10 showed a composition HOMO → LUMO+2, where HOMO and LUMO+2 is mainly centered among the ancillary ligand (PSB). In R5 and R6, a minority metal-centered composition was observed in comparison with R7 to R10. A second composition for 394 nm was found for R5 (HOMO → LUMO+3) and R6, R7 and R9 (HOMO → LUMO+1). Table 4 shows the isosurfaces of the molecular orbitals and the electron density differences maps for series 2 (R5 to R10) (the most important orbitals plots for R5 to R10 are qualitatively represented in Fig. S6 to S11 in the ESI†).
| Complex | λ (nm) | f | Transition | Assignment* |
|---|---|---|---|---|
| a deeb: 4,4′-bis(ethoxycarbonyl)-2,2′-bpy. | ||||
| R5 | 392 | 0.4383 | H → L+2 (42%) | LLCT/LLdeebCT/MLdeebCT(π → π*/π → π*/d → π*) |
| H → L+3 (31%) | ||||
| 330 | 0.5594 | H−7 → L (52%) | LdeebLdeebCT(π → π*) | |
| 283 | 0.2532 | H−5 → L+2 (63%) | LLCT/LLdeebCT/MLdeebCT(π → π*/π → π*/d → π*) | |
| R6 | 395 | 0.2921 | H → L+2 (46%) | LLCT/MLCT(π → π*/d → π*) |
| H → L+1 (38%) | LLdeebCT/MLdeebCT(π → π*/d → π*) | |||
| 333 | 0.4156 | H−6 → L (83%) | LdeebLdeebCT(π → π*) | |
| 279 | 0.3421 | H−5 → L+2 (74%) | LLCT/MLCT(π → π*/d → π*) | |
| R7 | 388 | 0.3695 | H → L+2 (47%) | LLCT/MLCT(π → π*/d → π*) |
| H → L+1 (46%) | LLdeebCT/MLdeebCT(π → π*/d → π*) | |||
| 332 | 0.5483 | H−7 → L (49%) | LdeebLdeebCT(π → π*) | |
| H−3 → L+1 (42%) | MLdeebCT(d → π*) | |||
| 286 | 0.2954 | H−6 → L+2 (61%) | LLCT/MLCT(π → π*/d → π*) | |
| R8 | 400 | 0.386 | H → L+2 (73%) | LLCT/MLCT(π → π*/d → π*) |
| 332 | 0.4896 | H−6 → L (45%) | LdeebLdeebCT(π → π*) | |
| H−2 → L+1 (34%) | MLdeebCT(d → π*) | |||
| 318 | 0.2338 | H−3 → L+2 (40%) | LLCT/MLCT(π → π*/d → π*) | |
| H−3 → L+1 (38%) | MLdeebCT(d → π*) | |||
| R9 | 387 | 0.3142 | H → L+1 (48%) | LLdeebCT/MLdeebCT(π → π*/d → π*) |
| H → L+2 (43%) | LLCT/MLCT(π → π*/d → π*) | |||
| 332 | 0.5538 | H−7 → L (55%) | LdeebLdeebCT(π → π*) | |
| 275 | 0.277 | H−5 → L+2 (45%) | LLCT/MLCT(π → π*/d → π*) | |
| R10 | 401 | 0.4929 | H → L+2 (83%) | LLCT/MLCT(π → π*/d → π*) |
| 333 | 0.4639 | H−6 → L (81%) | LdeebLdeebCT(π → π*) | |
| 281 | 0.3865 | H−5 → L+2 (35%) | LLCT/MLCT(π → π*/d → π*) | |
| H−5 → L+1 (27%) | LLdeebCT/MLdeebCT(π → π*/d → π*) | |||
| Complex | HOMO | LUMO+3 | LUMO+2 | Density diff. |
|---|---|---|---|---|
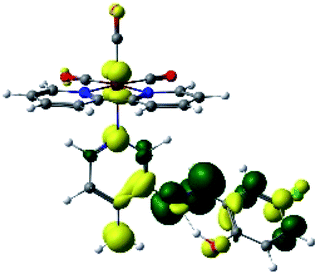
| R5 | 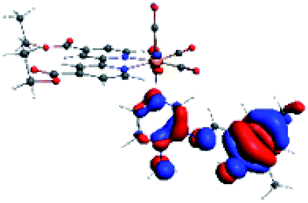 |
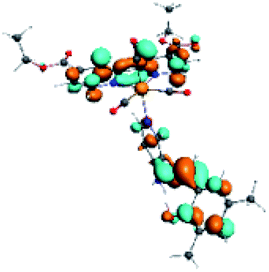 |
 |
 |
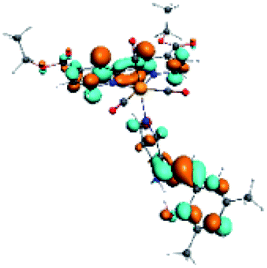
| R6 | HOMO | LUMO+1 | LUMO+2 | Density diff. |
 |
 |
 |
 |
|
| R7 |  |
 |
 |
 |
| R8 | HOMO | LUMO+2 | Density diff. | |
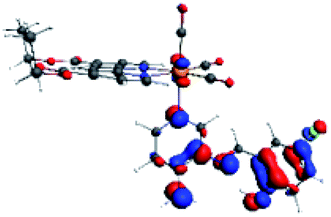 |
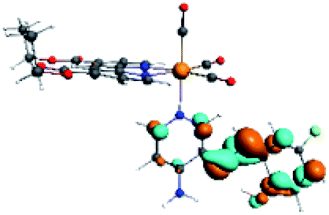 |
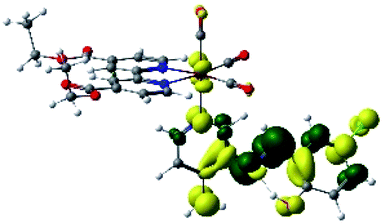 |
||
| R9 | HOMO | LUMO+1 | LUMO+2 | Density diff. |
 |
 |
 |
 |
|
| R10 | HOMO | LUMO+2 | Density diff. | |
 |
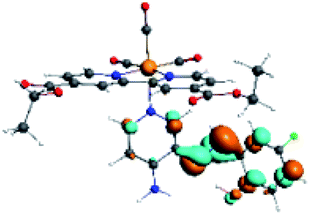 |
 |
In addition, the HOMO orbital for R7 and R9 is more localized in the phenolic ring due to a lower electron density, compared with the same ring in R5, R6, R8 and R10 (Table 4). The LUMO+2 orbital showed mainly a π-antibonding character centered at the azomethine group in R5 to R10 complexes (Table 4). Thus, as well as with R3, the IHB has an important effect on the transition energies for R7 and R9 complexes.
The experimental absorption of precursors of the complexes analyzed in the present study, which have Br instead of PSB, was already reported. The precursor for series 1 complexes (i.e., fac-Re(I)(CO)3(2,2′-bpy)Br) showed a λmax = 383 nm in acetonitrile;34,61 whereas the precursor for series 2 (i.e., fac-Re(I)(CO)3(4,4′-bis(ethoxycarbonyl)-2,2′-bpy)Br) exhibited a λmax = 419 nm in dichloromethane or acetonitrile.35,36 When Br was replaced by a PSB harboring an IHB (i.e., (E)-2-(((4-aminopyridin-3-yl)imino)methyl)-4,6-di-tert-butylphenol), similar to those analyzed in this manuscript, an experimental blue-shifted absorption was observed compared with their respective precursors, which was attributed to the presence of the ancillary ligand (i.e., fac-[Re(I)(CO)3(2,2′-bpy)((E)-2-(((4-aminopyridin-3-yl)imino)methyl)-4,6-di-tert-butylphenol)]1+ [acetonitrile, λmax = 361 nm] and fac-[Re(I)(CO)3(4,4′-bis(ethoxycarbonyl)-2,2′-bpy)((E)-2-(((4-aminopyridin-3-yl)imino)methyl)-4,6-di-tert-butylphenol)]1+ [dichloromethane or acetonitrile, λmax = 361 nm].34,35 The theoretical protocol used in the present manuscript, considered a blue-shift aborsorption of complexes harboring this kind of PSBs with respect to their corresponding precursors to perform calculations.
3.3 Theoretical study of emission
We have considered, as usually happens, that the emissions occur after a process of geometric reorganization in the triplet state (much faster in general than the radiative mechanisms); thus, the geometry of this state has been optimized, and the SOC-TDDFT calculations have been performed considering this new geometry. Then, we first analyzed the geometry optimizations of the singlet and triplet states for all complexes (Tables S5 and S6 in the ESI†).In the singlet state, the O–H bond distance (Table S1 in the ESI†) were observed around 1.008 Å for PSB1, PSB2, PSB4 and PSB6; and, where the IHB is not present, this distance was shorter (≈0.980 Å) (i.e., for PSB3 and PSB5). On the other hand, the H⋯N (IHB) bond distance was observed around 1.696 Å for PSB1, PSB2, PSB4 and PSB6; whereas the H⋯F (PSB3) and H⋯Cl (PSB5) bond distances were obtained at 2.127 Å and 2.268 Å, respectively. For R3, R7 and R9 we observed an interaction between the hydrogen of the –OH group in the phenolic ring and neighbored halogen in the same ring (PSB3 or PSB5). The C14C15OH bond angle in PSB1, PSB2, PSB4 and PSB6 (S0 state) were observed around −0.30°; for PSB3 and PSB5, in the corresponding R3, R7 and R9, appeared at −179.37° (Tables S5 and S6 in the ESI†; and numbering atoms in Fig. 5). In R1, R2, R4, R5, R6, R8 and R10, the formation of IHB was observed; therefore, the distances and bond angles showed the structural differences indicated above, compared to R3, R7 and R9. The geometric parameters of the triplet state presented a similar behavior when compared with the R1 to R10 complexes.
In a previous study, the photoluminescence of seven fac-Re(I) tricarbonyl complexes showed similar maximum emission wavelengths, regardless of the ancillary ligand tested (including pyridine, imidazole, or triphenylphosphine).17 By contrast, we found differences in the emission of R1, R2, R4, R5, R6, R8, and R10 with R3, R7, and R9, showing that, in our case, the ancillary ligand does exert an impact on the photoluminescent properties. This result can be mainly attributed to the presence of IHB in the corresponding ancillary ligand, as described above, remarking the importance of this intramolecular interaction regarding the emission behavior.
Regarding the geometry of R3, R7 and R9, we found differences between the singlet S0 and triplet state T1 in each case, involving changes in bond distances and angles. These differences between the states can be attributed to a structural relaxation from the lowest triplet excited state (T1) to the ground state (S0). By contrast, the presence of the IHB precludes this relaxation; accordingly, R1, R2, R4, R5, R6, R8 and R10, only show slight geometrical differences between the singlet S0 and triplet state T1.
In order to calculate the emission lifetime from the T1 state, a SOC-TDDFT calculation was done on the triplet from the electronic singlet ground state on that geometry.104 Typically, the three lowest sub-states of the spin–orbit coupling TDDFT calculation are important, since they resemble the three sub-states in a triplet state.105,106 There will be a slight energy difference between the three sub-states because of spin–orbit coupling, the so-called zero-field splitting (ZFS). Based on the described methodology, the emission calculated for R1, R2, R4, R5, R6, R8 and R10 was observed in 622–673 nm (Table 5). Experimental data previously obtained for fac-[Re(I)(CO)3(2,2′-bpy)((E)-2-(((4-aminopyridin-3-yl)imino)methyl)-4,6-di-tert-butylphenol)]1+, a complex that only differs from R1 in the substitution at positions 4 and 6 in the phenolic ring, presenting tert-butyls instead methyls, shows λex = 366 nm and λem = 610 nm in acetonitrile.34 On the other hand, emission experimental data of fac-[Re(I)(CO)3(4,4′-bis(ethoxycarbonyl)-2,2′-bpy)((E)-2-(((4-aminopyridin-3-yl)imino)methyl)-4,6-di-tert-butylphenol)]1+ which is similar to R5, but presents tert-butyls instead of methyls at positions 4 and 6 in the phenolic ring, exhibit λex = 365 nm, λem = 660 nm in dichloromethane.35 These experimental data support the calculations presented for R1, R2, R4, R5, R6, R8 and R10 (Fig. S12 and S13 in the ESI†).
| Complex | λ (nm) | τr (s) | ZFS (cm−1) | Transition | Assignmenta |
|---|---|---|---|---|---|
| a bpy: 2,2′-bpy; deeb:4,4′-bis(ethoxycarbonyl)-2,2′-bpy. | |||||
| R1 | 643 | 5.04 × 10−2 | 0.08 | L+1 → H | LLCT |
| R2 | 654 | 7.40 × 10−2 | 0.08 | L+1 → H | LLCT |
| R3 | 869 | 2.76 × 10−1 | 0.00 | L+1 → H | LLCT |
| 540 | 4.36 × 10−4 | 7.90 | L → H (T2) | LbpyLCT | |
| R4 | 673 | 5.59 × 10−2 | 0.00 | L+1 → H | LLCT |
| R5 | 625 | 1.36 × 10−3 | 13.87 | L+1 → H | LdeebLCT |
| R6 | 625 | 3.15 × 10−3 | 0.40 | L+1 → H | LLCT |
| L+2 → H | LdeebLCT | ||||
| R7 | 726 | 3.64 × 10−2 | 0.40 | L+1 → H | LLCT |
| 641 | 3.33 × 10−3 | 12.14 | L → H (T2) | LdeebLCT | |
| R8 | 622 | 2.23 × 10−2 | 1.05 | L+1 → H | LLCT |
| L+2 → H | LdeebLCT | ||||
| R9 | 771 | 8.90 × 10−2 | 0.16 | L+1 → H | LLCT |
| 649 | 9.77 × 10−4 | 7.78 | L → H (T2) | LdeebLCT | |
| R10 | 637 | 2.53 × 10−2 | 0.08 | L+1 → H | LLCT |
| L+2 → H | LdeebLCT | ||||
For R3, R7 and R9, the predicted emission was identified at 869 nm, 726 nm and 771 nm, respectively (Table 5). The lowest triplet excited states in R3, R7, R9 are mainly due to π → π* transitions of the PSB (di-halogenated ligand in these cases) (Fig. S12 and S13 in the ESI†). The lowest triplet excited state energy falls into the NIR region. This result could be explained by the effect of halide substituents at positions 4 and 6 in the phenolic ring, with a reduction of the excited energy as a consequence and, on the other hand, the absence of the IHB, which substantially reduces the vibronic relaxation, diminishing the emission probability. In general, complexes exhibiting high wavelength emission (i.e., close to the NIR) are less desirable for confocal microscopy due to a lower quantum efficiency (i.e., low detection capability), potentially producing dimmer images.27,100 However, these complexes could be more suitable for other purposes, such as OLEDs,23,107,108 or photosensitizers for singlet oxygen generation,109 among other applications.
In addition, we performed an exhaustive scan for R3, R7 and R9 to find other excited triplet states. In all these cases, we found a possible emission (in the range 540–649 nm) with a sufficiently long lifetime (4.36 × 10−4 to 3.33 × 10−3 s) from a second excited triplet state (Table 5, Fig. S12 and S13 in the ESI†). Furthermore, the low intensity of the observed emission of R3, R7 and R9 could be explained by a simultaneous non-radiative deactivation of the second triplet, which competes to reach the first excited state due to the lack of the IHB in the respective PSB, as discussed above. A similar phenomenon was experimentally observed with fac-Re(CO)3(4,5-diazafluoren-9-one)Br complex,27 showing that the theoretical methodology used in this work is accurate for this kind of analysis.
For all the complexes (R1 to R10), the emission can be attributed to a ligand to ligand charge transfer transition involving a π* orbital (LUMO+1 and LUMO+2) located at the (N,N) or the PSB ligand, where the π orbitals (HOMO) were found in the corresponding PSB ancillary ligands (Fig. S12 and S13 in the ESI†). Finally, in all cases, the emission mechanism suggests that, after absorption, a triplet excited state is reached, attributed to 3LLCT. This assignment was supported by the calculated ZFS43 (Table 5).
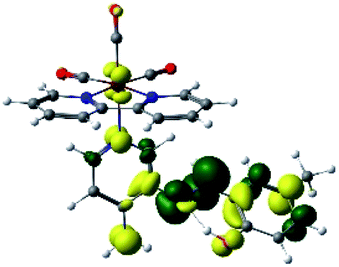
In order to confirm the nature of the emissive electronic state, the spin density distributions were obtained for all the complexes. As shown in Fig. 6, the spin density was predominantly localized on the ancillary ligand PSB, with a minimal contribution of the nitrogen atoms (in the denitrogenated N,N ligands). Thus, the spin density observed in Fig. 6 is consistent with the predominant emission character of the corresponding calculated transitions, with the ligand-centered ZFS classification (3LC) for all the complexes, except for R5. R5 classification is 3LC /3MLCT, where the spin density is distributed among the metal (N,N), and PSB.
 | ||
| Fig. 6 Spin density plots of the optimized triplet states for R1 to R10. | ||
In summary, for R1, R2, R5, R6, R8 and R10, the photophysical behavior was explained by the presence of the IHB, which stabilizes the planar E conformation. On the other hand, in the case of R3, R7 and R9, the interaction C–halogen⋯H–O might contribute to the photophysical response by the inductive effects of the halogen at position 6, as stated above. Thus, in this work, we postulate that the IHB is important regarding the emission wavelength since a potential disruption of this interaction could promote the emission shift from the visible range to the NIR range. In this context, it is possible to speculate that the IHB could be disrupted in protic solvents such as in the biological environment. Nevertheless, it has been reported that, in similar complexes, the IHB seems to resist the presence of a protic solvent. For instance, a previous study of a Re(I) tricarbonyl complexes harboring a similar pyridine Schiff base harboring IHB [(E)-2((3-amino-pyridin-4-ylimino)-methyl)-4,6-diterbutylphenol] to those shown in the present manuscript, showed no significant differences regarding their optical or luminescent properties when diluted in dichloromethane (ε = 8.93) or acetonitrile (ε = 37.5).34,35 In addition, it was reported that, in studies involving only the [(E)-2((3-amino-pyridin-4-ylimino)-methyl)-4,6-diterbutylphenol], the H-bond stability is independent of solvent polarity;39 this same result was also observed with other similar Schiff bases harboring IHB.40 Furthermore, confocal microscopy studies performed with these complexes, whose stock was diluted in DMSO, allowed obtaining successful images.37 Interestingly, in that study, the confocal microscope was set with laser excitation at 405 nm and emission of 555 to 625 nm.37 This emission is in the visible range, not in the NIR, as expected for complexes harboring the IHB. All these results argue the high stability of the IHB, even in the presence of protic solvents or in cellular environments, as previously stated.37 Finally, if indeed the IHB were to break in biological conditions, the complexes would exhibit different emissions dependent on the cellular microenvironment, e.g., cytoplasm (protic environment) or cell membranes (aprotic environments), potentially generating a differential staining. However, the studied complexes must be mandatorily tested to elucidate their behavior with cells.
4. Conclusions
Relativistic DFT and TDDFT methods were used to investigate the effect on electronic structures in two series of Re(I) tricarbonyl complexes. In each case, the electronic structure analysis showed that R3, R7 and R9 emit in the NIR region with a low emission probability from the second excited triplet state. On the other hand, R1, R2, R4, R5, R6, R8 and R10 presented emissions in the range of 622–673 nm from a 3LLCT electronic state. According to our results, an IHB in the PSB ancillary ligand is crucial to modulating photophysical properties. In this sense, the presence of a halogen at position 6 in the phenolic ring precludes the formation of the IHB, affecting the emission wavelength and lifetime. These results reinforce the concept of structure–functionality relationship in this kind of complex.The increasing development of biotechnological applications based on microorganisms (especially walled-cell models such as bacteria and fungi), including the need for their identification and diagnosis and protein staining, emphasizes the need for systematic research of new cationic Re(I) tricarbonyl complexes. This study could contribute to predicting properties of Re(I) tricarbonyl complexes to be synthesized, considering the desired emission properties for biological applications.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was funded by FONDECYT INICIO 11170637 (ANID) and Núcleo UNAB DI-02-19/N (Universidad Andres Bello). D. Páez-Hernández thanks to FONDECYT 1180017 (ANID) and R. Morales-Guevara thanks PhD fellowship ANID 21210811 (ANID is Agencia Nacional de Investigación y Desarrollo, Chile).References
- L. Veronese, E. Q. Procopio, F. De Rossi, T. M. Brown, P. Mercandelli, P. Mussini, G. D'Alfonso and M. Panigati, New J. Chem., 2016, 40, 2910–2919 RSC.
- J. R. Dilworth, Coord. Chem. Rev., 2021, 436, 213822 CrossRef CAS.
- B. L. Souza, L. A. Faustino, F. S. Prado, R. N. Sampaio, P. I. S. Maia, A. E. H. Machado and A. O. T. Patrocinio, Dalton Trans., 2020, 49, 16368–16379 RSC.
- L. A. Faustino, B. L. Souza, B. N. Nunes, A.-T. Duong, F. Sieland, D. W. Bahnemann and A. O. T. Patrocinio, ACS Sustainable Chem. Eng., 2018, 6, 6073–6083 CrossRef CAS.
- L. A. Faustino, A. E. Hora Machado and A. O. T. Patrocinio, Inorg. Chem., 2018, 57, 2933–2941 CrossRef CAS PubMed.
- J. Agarwal, E. Fujita, H. F. Schaefer 3rd and J. T. Muckerman, J. Am. Chem. Soc., 2012, 134, 5180–5186 CrossRef CAS PubMed.
- G. F. Manbeck, J. T. Muckerman, D. J. Szalda, Y. Himeda and E. Fujita, J. Phys. Chem. B, 2015, 119, 7457–7466 CrossRef CAS PubMed.
- J. K. Nganga, L. M. Wolf, K. Mullick, E. Reinheimer, C. Saucedo, M. E. Wilson, K. A. Grice, M. Z. Ertem and A. M. Angeles-Boza, Inorg. Chem., 2021, 60, 3572–3584 CrossRef CAS PubMed.
- B. L. Murphy, S. C. Marker, V. J. Lambert, J. J. Woods, S. N. MacMillan and J. J. Wilson, J. Organomet. Chem., 2020, 907 DOI:10.1016/j.jorganchem.2019.121064.
- K. Ranasinghe, S. Handunnetti, I. C. Perera and T. Perera, Chem. Cent. J., 2016, 10, 71 CrossRef PubMed.
- Y. Aimene, R. Eychenne, S. Mallet-Ladeira, N. Saffon, J. Y. Winum, A. Nocentini, C. T. Supuran, E. Benoist and A. Seridi, J. Enzyme Inhib. Med. Chem., 2019, 34, 773–782 CrossRef CAS PubMed.
- L. Henry, N. Delsuc, C. Laugel, F. Lambert, C. Sandt, S. Hostachy, A. S. Bernard, H. C. Bertrand, L. Grimaud, A. Baillet-Guffroy and C. Policar, Bioconjugate Chem., 2018, 29, 987–991 CrossRef CAS PubMed.
- S. N. Sovari, S. Vojnovic, S. S. Bogojevic, A. Crochet, A. Pavic, J. Nikodinovic-Runic and F. Zobi, Eur. J. Med. Chem., 2020, 205, 112533 CrossRef CAS PubMed.
- V. Fernández-Moreira and H. Sastre-Martín, Inorg. Chim. Acta, 2017, 460, 127–133 CrossRef.
- L. C. Lee, K. K. Leung and K. K. Lo, Dalton Trans., 2017, 46, 16357–16380 RSC.
- J. Karges, M. Kalaj, M. Gembicky and S. M. Cohen, Angew. Chem., Int. Ed. Engl., 2021, 60, 10716–10723 CrossRef CAS PubMed.
- F. J. F. Jacobs, G. J. S. Venter, E. Fourie, R. E. Kroon and A. Brink, RSC Adv., 2021, 11, 24443–24455 RSC.
- A. Carreno, M. Gacitua, E. Solis-Cespedes, D. Paez-Hernandez, W. B. Swords, G. J. Meyer, M. D. Preite, I. Chavez, A. Vega and J. A. Fuentes, Front Chem, 2021, 9, 647816 CrossRef CAS PubMed.
- C. Otero, A. Carreno, R. Polanco, F. M. Llancalahuen, R. Arratia-Perez, M. Gacitua and J. A. Fuentes, Front Chem, 2019, 7, 454 CrossRef CAS PubMed.
- L. Sacksteder, A. P. Zipp, E. A. Brown, J. Streich, J. N. Demas and B. A. Degraff, Inorg. Chem., 1990, 29, 4335–4340 CrossRef CAS.
- R. Argazzi, C. A. Bignozzi, T. A. Heimer and G. J. Meyer, Inorg. Chem., 1997, 36, 2–3 CrossRef CAS.
- H. C. Bertrand, S. Clede, R. Guillot, F. Lambert and C. Policar, Inorg. Chem., 2014, 53, 6204–6223 CrossRef CAS PubMed.
- A. M. Maron, A. Szlapa-Kula, M. Matussek, R. Kruszynski, M. Siwy, H. Janeczek, J. Grzelak, S. Mackowski, E. Schab-Balcerzak and B. Machura, Dalton Trans., 2020, 49, 4441–4453 RSC.
- T. Klemens, A. Świtlicka, A. Szlapa-Kula, Ł. Łapok, M. Obłoza, M. Siwy, M. Szalkowski, S. Maćkowski, M. Libera, E. Schab-Balcerzak and B. Machura, Organometallics, 2019, 38, 4206–4223 CrossRef CAS.
- M. V. Werrett, G. S. Huff, S. Muzzioli, V. Fiorini, S. Zacchini, B. W. Skelton, A. Maggiore, J. M. Malicka, M. Cocchi, K. C. Gordon, S. Stagni and M. Massi, Dalton Trans., 2015, 44, 8379–8393 RSC.
- V. Fiorini, L. Bergamini, N. Monti, S. Zacchini, S. E. Plush, M. Massi, A. Hochkoeppler, A. Stefan and S. Stagni, Dalton Trans., 2018, 47, 9400–9410 RSC.
- A. Carreño, D. Páez-Hernández, C. Zúñiga, A. Ramírez-Osorio, N. Pizarro, A. Vega, E. Solis-Céspedes, M. M. Rivera-Zaldívar, A. Silva and J. A. Fuentes, Dyes Pigm., 2021, 184 DOI:10.1016/j.dyepig.2020.108876.
- T. Klemens, A. Switlicka-Olszewska, B. Machura, M. Grucela, E. Schab-Balcerzak, K. Smolarek, S. Mackowski, A. Szlapa, S. Kula, S. Krompiec, P. Lodowski and A. Chrobok, Dalton Trans., 2016, 45, 1746–1762 RSC.
- C. C. Konkankit, S. C. Marker, K. M. Knopf and J. J. Wilson, Dalton Trans., 2018, 47, 9934–9974 RSC.
- T. Klemens, A. Świtlicka, S. Kula, M. Siwy, K. Łaba, J. Grzelak, M. Szalkowski, S. Maćkowski, E. Schab-Balcerzak and B. Machura, J. Lumin., 2019, 209, 346–356 CrossRef CAS.
- T. Klemens, A. Świtlicka, B. Machura, S. Kula, S. Krompiec, K. Łaba, M. Korzec, M. Siwy, H. Janeczek, E. Schab-Balcerzak, M. Szalkowski, J. Grzelak and S. Maćkowski, Dyes Pigm., 2019, 163, 86–101 CrossRef CAS.
- H. S. Liew, C. W. Mai, M. Zulkefeli, T. Madheswaran, L. V. Kiew, N. Delsuc and M. L. Low, Molecules, 2020, 25, 4176 CrossRef CAS PubMed.
- A. Luengo, M. Redrado, I. Marzo, V. Fernandez-Moreira and M. C. Gimeno, Inorg. Chem., 2020, 59, 8960–8970 CrossRef CAS PubMed.
- A. Carreño, M. Gacitúa, J. A. Fuentes, D. Páez-Hernández, J. P. Peñaloza, C. Otero, M. Preite, E. Molins, W. B. Swords, G. J. Meyer, J. M. Manríquez, R. Polanco, I. Chávez and R. Arratia-Pérez, New J. Chem., 2016, 40, 7687–7700 RSC.
- A. Carreno, M. Gacitua, E. Schott, X. Zarate, J. M. Manriquez, M. Preite, S. Ladeira, A. Castel, N. Pizarro, A. Vega, I. Chavez and R. Arratia-Perez, New J. Chem., 2015, 39, 5725–5734 RSC.
- G. M. Hasselmann and G. J. Meyer, The Journal of Physical Chemistry B, 1999, 103, 7671–7675 CrossRef CAS.
- A. Carreño, A. E. Aros, C. Otero, R. Polanco, M. Gacitúa, R. Arratia-Pérez and J. A. Fuentes, New J. Chem., 2017, 41, 2140–2147 RSC.
- C. M. da Silva, D. L. da Silva, L. V. Modolo, R. B. Alves, M. A. de Resende, C. V. B. Martins and Â. de Fátima, J. Adv. Res., 2011, 2, 1–8 CrossRef.
- A. Carreno, M. Gacitua, D. Paez-Hernandez, R. Polanco, M. Preite, J. A. Fuentes, G. C. Mora, I. Chavez and R. Arratia-Perez, New J. Chem., 2015, 39, 7822–7831 RSC.
- A. Carreno, D. Paez-Hernandez, P. Cantero-Lopez, C. Zuniga, J. Nevermann, A. Ramirez-Osorio, M. Gacitua, P. Oyarzun, F. Saez-Cortez, R. Polanco, C. Otero and J. A. A. Fuentes, Molecules, 2020, 25, 2741 CrossRef CAS PubMed.
- A. Carreno, L. Rodriguez, D. Paez-Hernandez, R. Martin-Trasanco, C. Zuniga, D. P. Oyarzun, M. Gacitua, E. Schott, R. Arratia-Perez and J. A. Fuentes, Front Chem, 2018, 6, 312 CrossRef PubMed.
- A. Carreño, C. Zúñiga, D. Páez-Hernández, M. Gacitúa, R. Polanco, C. Otero, R. Arratia-Pérez and J. A. Fuentes, New J. Chem., 2018, 42, 8851–8863 RSC.
- K. Mori, T. P. Goumans, E. van Lenthe and F. Wang, Phys. Chem. Chem. Phys., 2014, 16, 14523–14530 RSC.
- J. M. Younker and K. D. Dobbs, J. Phys. Chem. C, 2013, 117, 25714–25723 CrossRef CAS.
- A. R. Smith, M. J. Riley, P. L. Burn, I. R. Gentle, S. C. Lo and B. J. Powell, Inorg. Chem., 2012, 51, 2821–2831 CrossRef CAS PubMed.
- J. U. Kim, I. S. Park, C. Y. Chan, M. Tanaka, Y. Tsuchiya, H. Nakanotani and C. Adachi, Nat. Commun., 2020, 11, 1765 CrossRef CAS PubMed.
- P. Cantero-López, J. Santoyo-Flores, A. Carreño, J. Fuentes, A. Vega, A. Ramirez-Osorio, A. Ortiz, L. a. Illicachi, A. Olea, J. Sánchezc and D. Páez-Hernández, Dalton Trans., 2021, 50, 13561–13571 RSC.
- K. Kim and K. D. Jordan, J. Phys. Chem., 1994, 98, 10089–10094 CrossRef CAS.
- C. S. Ashvar, F. J. Devlin, K. L. Bak, P. R. Taylor and P. J. Stephens, J. Phys. Chem., 1996, 100, 9262–9270 CrossRef CAS.
- I. J. Al-Busaidi, R. Ilmi, J. D. L. Dutra, W. F. Oliveira, A. Haque, N. K. Al Rasbi, F. Marken, P. R. Raithby and M. S. Khan, Dalton Trans., 2021, 50, 1465–1477 RSC.
- E. Van Lenthe and E. J. Baerends, J. Comput. Chem., 2003, 24, 1142–1156 CrossRef CAS PubMed.
- G. te Velde, F. M. Bickelhaupt, E. J. Baerends, C. Fonseca Guerra, S. J. A. van Gisbergen, J. G. Snijders and T. Ziegler, J. Comput. Chem., 2001, 22, 931–967 CrossRef CAS.
- J. Anton, B. Fricke and E. Engel, Phys. Rev. A, 2004, 69, 012505 CrossRef.
- E. v. Lenthe, E. J. Baerends and J. G. Snijders, J. Chem. Phys., 1993, 99, 4597–4610 CrossRef.
- Y. C. Duan, Y. Wu, X. Y. Ren, L. Zhao, Y. Geng, M. Zhang, G. Y. Sun and Z. M. Su, Dalton Trans., 2017, 46, 11491–11502 RSC.
- M. Filatov and D. Cremer, Mol. Phys., 2009, 101, 2295–2302 CrossRef.
- F. M. Bickelhaupt and E. J. Baerends, Kohn-Sham Density Functional Theory: Predicting and Understanding Chemistry, Wiley Online LIbrary, 2007 Search PubMed.
- A. W. Lange and J. M. Herbert, J. Chem. Phys., 2011, 134, 204110 CrossRef PubMed.
- C. Steinmann, K. L. Blaedel, A. S. Christensen and J. H. Jensen, PLoS One, 2013, 8, e67725 CrossRef CAS PubMed.
- A. C. Castro, H. Fliegl, M. Cascella, T. Helgaker, M. Repisky, S. Komorovsky, M. A. Medrano, A. G. Quiroga and M. Swart, Dalton Trans., 2019, 48, 8076–8083 RSC.
- C. Pac, S. Kaseda, K. Ishii and S. Yanagida, J. Chem. Soc., Chem. Commun., 1991, 787–788, 10.1039/c39910000787.
- X. Gao, S. Shi, J. L. Yao, J. Zhao and T. M. Yao, Dalton Trans., 2015, 44, 19264–19274 RSC.
- X. Qu, Y. Liu, Y. Si, X. Wu and Z. Wu, Dalton Trans., 2014, 43, 1246–1260 RSC.
- P. Cantero-Lopez, L. Le Bras, D. Paez-Hernandez and R. Arratia-Perez, Dalton Trans., 2015, 44, 20004–20010 RSC.
- A. Carreno, E. Solis-Cespedes, C. Zuniga, J. Nevermann, M. M. Rivera-Zaldivar, M. Gacitua, A. Ramirez-Osorio, D. Paez-Hernandez, R. Arratia-Perez and J. A. Fuentes, Chem. Phys. Lett., 2019, 715, 231–238 CrossRef CAS.
- X. Zarate, E. Schott, T. Gomez and R. Arratia-Perez, J. Phys. Chem. A, 2013, 117, 430–438 CrossRef CAS PubMed.
- A. Schinabeck, N. Rau, M. Klein, J. Sundermeyer and H. Yersin, Dalton Trans., 2018, 47, 17067–17076 RSC.
- T. P. Gompa, A. Ramanathan, N. T. Rice and H. S. La Pierre, Dalton Trans., 2020, 49, 15945–15987 RSC.
- C. Bizzarri, F. Hundemer, J. Busch and S. Bräse, Polyhedron, 2018, 140, 51–66 CrossRef CAS.
- A. Schinabeck, M. J. Leitl and H. Yersin, J. Phys. Chem. Lett., 2018, 9, 2848–2856 CrossRef CAS PubMed.
- G. Cioncoloni, H. M. Senn, S. Sproules, C. Wilson and M. D. Symes, Dalton Trans., 2016, 45, 15575–15585 RSC.
- S. R. De Lima, D. G. Felisbino, M. R. S. Lima, R. Chang, M. M. Martins, L. R. Goulart, A. A. Andrade, D. N. Messias, R. R. Dos Santos, F. C. Juliatti and V. Pilla, J. Photochem. Photobiol., B, 2019, 200, 111631 CrossRef CAS PubMed.
- J. Mathew and C. Chesneau, Math. Comput. Appl., 2020, 25, 65 Search PubMed.
- X. Zhang, D. Jacquemin, Q. Peng, Z. Shuai and D. Escudero, J. Phys. Chem. C, 2018, 122, 6340–6347 CrossRef CAS.
- Z. Q. Dong, J. H. Yang and B. Liu, Dalton Trans., 2021, 50, 2387–2392 RSC.
- P. Cantero-López, Y. Hidalgo-Rosa, Z. Sandoval-Olivares, J. Santoyo-Flores, P. Mella, L. Arrué, C. Zúñiga, R. Arratia-Pérez and D. Páez-Hernández, New J. Chem., 2021, 45, 11192–11201 RSC.
- J. A. Clayton, K. Keller, M. Qi, J. Wegner, V. Koch, H. Hintz, A. Godt, S. Han, G. Jeschke, M. S. Sherwin and M. Yulikov, Phys. Chem. Chem. Phys., 2018, 20, 10470–10492 RSC.
- R. G. Shirazi, D. A. Pantazis and F. Neese, Mol. Phys., 2020, 118 DOI:10.1080/00268976.2020.1764644.
- R. Boca, J. Titis, C. Rajnak and J. Krzystek, Dalton Trans., 2021, 50, 3468–3472 RSC.
- A. Świtlicka, K. Choroba, A. Szlapa-Kula, B. Machura and K. Erfurt, Polyhedron, 2019, 171, 551–558 CrossRef.
- L. Chen, Z. Wu, J. Yang and S. Zhang, Z. Kristallogr. - New Cryst. Struct., 2019, 234, 1173–1176 CAS.
- A. Carreño, M. Gacitúa, E. Molins and R. Arratia-Pérez, Chem. Pap., 2017, 71, 2011–2022 CrossRef.
- B. Machura, M. Wolff, E. Benoist and Y. Coulais, J. Organomet. Chem., 2013, 724, 82–87 CrossRef CAS.
- Y. Yue, T. Grusenmeyer, Z. Ma, P. Zhang, R. H. Schmehl, D. N. Beratan and I. V. Rubtsov, J. Phys. Chem. A, 2014, 118, 10407–10415 CrossRef CAS PubMed.
- R. K. Hocking and T. W. Hambley, Organometallics, 2007, 26, 2815–2823 CrossRef CAS.
- T. Ziegler and A. Rauk, Inorg. Chem., 2002, 18, 1755–1759 CrossRef.
- A. Carreno, S. Ladeira, A. Castel, A. Vega and I. Chavez, Acta Crystallogr., Sect. E: Struct. Rep. Online, 2012, 68, o2507–2508 CrossRef CAS PubMed.
- F. Aribi, A. Panossian, D. Jacquemin, J.-P. Vors, S. Pazenok, F. R. Leroux and M. Elhabiri, New J. Chem., 2018, 42, 10036–10047 RSC.
- C. A. Bayse, New J. Chem., 2018, 42, 10623–10632 RSC.
- F. Adasme-Carreño, C. Muñoz-Gutierrez and J. H. Alzate-Morales, RSC Adv., 2016, 6, 61837–61847 RSC.
- S. Zheng, X. Zhao, Y. Li, F. Xu and Q. Zhang, RSC Adv., 2018, 8, 4259–4272 RSC.
- Q. Jia, Q. Li, M. Luo and H.-B. Li, RSC Adv., 2018, 8, 38980–38986 RSC.
- B. V. Pandiyan, P. Deepa and P. Kolandaivel, RSC Adv., 2016, 6, 66870–66878 RSC.
- M. Rojas-Poblete, A. Carreño, M. Gacitúa, D. Páez-Hernández, W. A. Rabanal-León and R. Arratia-Pérez, New J. Chem., 2018, 42, 5471–5478 RSC.
- M. He, H. Y. V. Ching, C. Policar and H. C. Bertrand, New J. Chem., 2018, 42, 11312–11323 RSC.
- M. M. Lee, J.-L. Lin, C.-W. Chang, C.-Y. Hung, C.-W. Chen, C.-P. Hsu and S.-S. Sun, Eur. J. Inorg. Chem., 2017, 2017, 5224–5237 CrossRef CAS.
- M. Malecka, B. Machura, A. Switlicka, S. Kotowicz, G. Szafraniec-Gorol, M. Siwy, M. Szalkowski, S. Mackowski and E. Schab-Balcerzak, Spectrochim. Acta, Part A, 2020, 231, 118124 CrossRef CAS PubMed.
- F. J. F. Jacobs, G. J. S. Venter, E. Fourie, R. E. Kroon and A. Brink, RSC Adv., 2021, 11, 24443–24455 RSC.
- R. Arevalo, L. Riera and J. Perez, Inorg. Chem., 2017, 56, 4249–4252 CrossRef CAS PubMed.
- A. J. Hallett, E. Placet, R. Prieux, D. McCafferty, J. A. Platts, D. Lloyd, M. Isaacs, A. J. Hayes, S. J. Coles, M. B. Pitak, S. Marchant, S. N. Marriott, R. K. Allemann, A. Dervisi and I. A. Fallis, Dalton Trans., 2018, 47, 14241–14253 RSC.
- T. Hosseinnejad, F. Ebrahimpour-Malmir and B. Fattahi, RSC Adv., 2018, 8, 12232–12259 RSC.
- T. Klemens, A. Świtlicka-Olszewska, B. Machura, M. Grucela, H. Janeczek, E. Schab-Balcerzak, A. Szlapa, S. Kula, S. Krompiec, K. Smolarek, D. Kowalska, S. Mackowski, K. Erfurt and P. Lodowski, RSC Adv., 2016, 6, 56335–56352 RSC.
- N. N. Mohd Yusof Chan, A. Idris, Z. H. Zainal Abidin, H. A. Tajuddin and Z. Abdullah, RSC Adv., 2021, 11, 13409–13445 RSC.
- D. Escudero and D. Jacquemin, Dalton Trans., 2015, 44, 8346–8355 RSC.
- Y. Wu, S. X. Wu, H. B. Li, Y. Geng and Z. M. Su, Dalton Trans., 2011, 40, 4480–4488 RSC.
- J. Li, Q. Zhang, H. He, L. Wang and J. Zhang, Dalton Trans., 2015, 44, 8577–8589 RSC.
- N. J. Lundin, A. G. Blackman, K. C. Gordon and D. L. Officer, Angew. Chem., Int. Ed. Engl., 2006, 45, 2582–2584 CrossRef CAS PubMed.
- T. Klemens, K. Czerwinska, A. Szlapa-Kula, S. Kula, A. Switlicka, S. Kotowicz, M. Siwy, K. Bednarczyk, S. Krompiec, K. Smolarek, S. Mackowski, W. Danikiewicz, E. Schab-Balcerzak and B. Machura, Dalton Trans., 2017, 46, 9605–9620 RSC.
- N. Manav, P. E. Kesavan, M. Ishida, S. Mori, Y. Yasutake, S. Fukatsu, H. Furuta and I. Gupta, Dalton Trans., 2019, 48, 2467–2478 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1ra05737e |
| This journal is © The Royal Society of Chemistry 2021 |