
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDeuterium equilibrium isotope effects in a supramolecular receptor for the hydrochalcogenide and halide anions†
Hazel A. Fargher ,
Russell A. Nickels,
Thaís P. de Faria,
Michael M. Haley*,
Michael D. Pluth* and
Darren W. Johnson*
,
Russell A. Nickels,
Thaís P. de Faria,
Michael M. Haley*,
Michael D. Pluth* and
Darren W. Johnson*
Department of Chemistry & Biochemistry, Materials Science Institute, and Knight Campus for Accelerating Scientific Impact, University of Oregon, Eugene, OR 97403-1253, USA. E-mail: haley@uoregon.edu; pluth@uoregon.edu; dwj@uoregon.edu
First published on 4th August 2021
Abstract
We highlight a convenient synthesis to selectively deuterate an aryl C–H hydrogen bond donor in an arylethynyl bisurea supramolecular anion receptor and use the Perrin method of competitive titrations to study the deuterium equilibrium isotope effects (DEIE) of anion binding for HS−, Cl−, and Br−. This work highlights the utility and also challenges in using this method to determine EIE with highly reactive and/or weakly binding anions.
Molecular recognition and host–guest binding in both biological and synthetic systems are often driven by a mixture of competitive and additive primarily non-covalent interactions. Understanding the role of each of these forces in a host–guest system can reveal insights into the driving forces behind binding and help inform on the molecular design of future hosts.1–3 Equilibrium isotope effects (EIE), also referred to as binding isotope effects (BIE) in structural molecular biology,4 measure the effect of isotopic substitution on supramolecular interactions through changes in the vibrational energy of the substituted bond. These studies can be used to elucidate the complex non-covalent forces involved in host conformational changes and host–guest binding.5–8
Examples from structural molecular biology have demonstrated that EIEs can reveal mechanistic information in enzyme–ligand binding events.4,9 Isotopic substitution in synthetic supramolecular systems has been used both for labelling purposes and for studying individual non-covalent interactions. For example, Bergman, Raymond, and coworkers used deuterium equilibrium isotope effects (DEIE) to study benzylphosphonium cation guest binding in a self-assembled supramolecular complex in aqueous solution.10 From these DEIE studies, the authors found that attractive cation⋯π interactions in the interior of the host were important for promoting guest binding, and that C–H⋯π and π⋯π interactions were relatively small contributors. In another example, Shimizu and coworkers studied the DEIE on the strength of C–H⋯π interactions in their molecular balances.11 Both computational and experimental results showed that the strength of C–H⋯π and C–D⋯π interactions were about equal, settling the debate on which interaction is stronger and easing concerns about using deuteration for spectroscopic and labelling applications.
Previously, we used DEIE to study Cl− binding with the arylethynyl bisurea anion receptor 1H/D (Fig. 1) in DMSO-d6.12 We found an experimental DEIE of 1.019 ± 0.010, which matched the computationally-predicted DEIE of 1.020. Further computational analysis determined that the DEIE was due to a distorted N–H⋯Cl− hydrogen bond geometry, which resulted in changes in the C–H/D bond vibrational energy in the host–guest complex. In addition, Paneth and coworkers performed a computational study with 1H and other hydrogen bonding supramolecular Cl− receptors to determine the EIE of 35/37Cl− binding in these hosts.13 Because isotope effects, both equilibrium and kinetic, originate solely from changes in the vibrational energy of the isotopically labelled bond, the EIE arising from this study came from changes in the vibrational energies of the bonds in the supramolecular hosts when participating in hydrogen bonding with Cl− isotopes. Indeed, a linear relationship was observed between the hydrogen bond donor (D) D–H bond lengths in the host–guest complex and the computed 35/37Cl EIE.
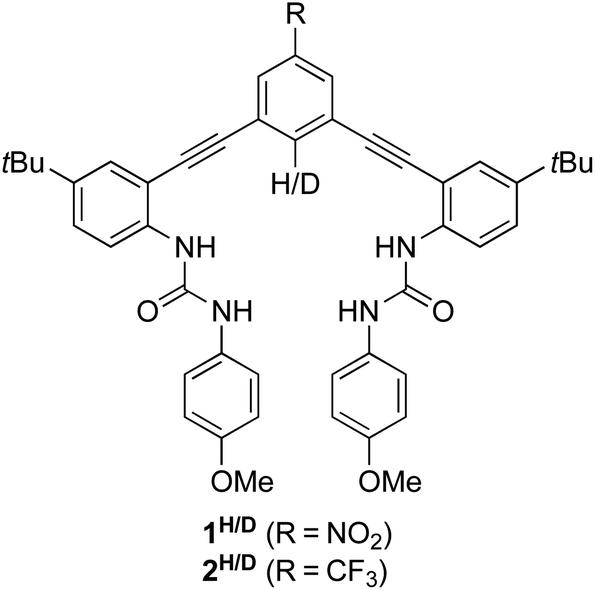 | ||
| Fig. 1 Arylethynyl bisurea receptors 1H and 1D used in our previous DEIE study of Cl− binding. Related receptors 2H and 2D are used in this study to avoid reaction of the nitro group with HS−. | ||
Previous EIE studies with receptor 1H/D have focused on Cl− binding; however, to the best of our knowledge, no work has yet investigated the EIE of hydrosulfide (HS−) binding in this or other systems. HS− is a highly reactive anion that plays crucial roles in biology. At physiological pH, HS− is favored in solution by a 3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio over its conjugate acid, hydrogen sulfide (H2S). H2S has been identified as the third physiological gasotransmitter alongside CO and NO and plays essential roles in physiological systems.15 Despite its high nucleophilicity and redox activity, HS− has been observed to be bound through non-covalent interactions in the protein crystal structure of a bacterial ion channel16 and in the turn-over state of vanadium-containing nitrogenase.17 The supramolecular chemistry of HS− is under-studied in synthetic supramolecular receptors, likely due to the inherent high reactivity of HS−. Indeed, we are aware of only three families of receptors that have been shown to reversibly bind HS−.18–21
:1 ratio over its conjugate acid, hydrogen sulfide (H2S). H2S has been identified as the third physiological gasotransmitter alongside CO and NO and plays essential roles in physiological systems.15 Despite its high nucleophilicity and redox activity, HS− has been observed to be bound through non-covalent interactions in the protein crystal structure of a bacterial ion channel16 and in the turn-over state of vanadium-containing nitrogenase.17 The supramolecular chemistry of HS− is under-studied in synthetic supramolecular receptors, likely due to the inherent high reactivity of HS−. Indeed, we are aware of only three families of receptors that have been shown to reversibly bind HS−.18–21
Recently, we used a series of arylethynyl bisurea anion receptors to investigate and demonstrate a linear free energy relationship between the polarity of a non-traditional C–H hydrogen bond donor and the solution binding energy of HS−, HSe−, Cl− and Br−.14 A major and unexpected finding of this study was that HS− demonstrated a significant increase in sensitivity towards the polarity of the C–H hydrogen donor over HSe−, Cl− and Br−. Although increasing the polarity of the C–H hydrogen bond donor did not lead to changes in selectivity between Cl−, Br−, and HSe−, we observed a 9-fold increase in selectivity for HS− over Cl−, suggesting a fresh approach to selective HS− recognition using non-covalent interactions. In this current study, we label the C–H hydrogen bond donor in an arylethynyl bisurea receptor with a deuterium atom (2H/D, Fig. 1) to further investigate this apparent preference of polar C–H H-bond donors for HS− over Cl− and Br− through DEIE.
Receptor 2H is a previously reported anion receptor for HS−, Cl−, and Br− and was prepared by established methods.14 Deuterium labelling of the isotopologue 2D was achieved by selective monodeuteration of intermediates through methods similar to those reported in the literature (Scheme 1).22 The diazonium salt 3 was synthesized in a 71% yield from 2,6-diiodo-4-trifluoromethylaniline.23 Dediazonation in DMF-d7 is catalyzed by FeSO4 and allows for selective synthesis of monodeuterated intermediate 4. The deuteration step proceeds through a radical pathway that uses DMF-d7 as the deuterium source. This deuteration reaction provides efficient deuterium incorporation even with up to 50% by volume H2O in the reaction solution due to the differential bond strengths in DMF and H2O.22 Sonogashira cross-coupling reaction of 4 and 4-t-butyl-2-ethynylaniline24 afforded 5 in 45% yield. Subsequent addition with 4-methoxyphenyl isocyanate gave 2D in 34% yield. Compound 2D and intermediates were characterized through 1H, 2H, 13C{1H}, and 19F NMR spectroscopy and high-resolution mass spectrometry (see ESI†).
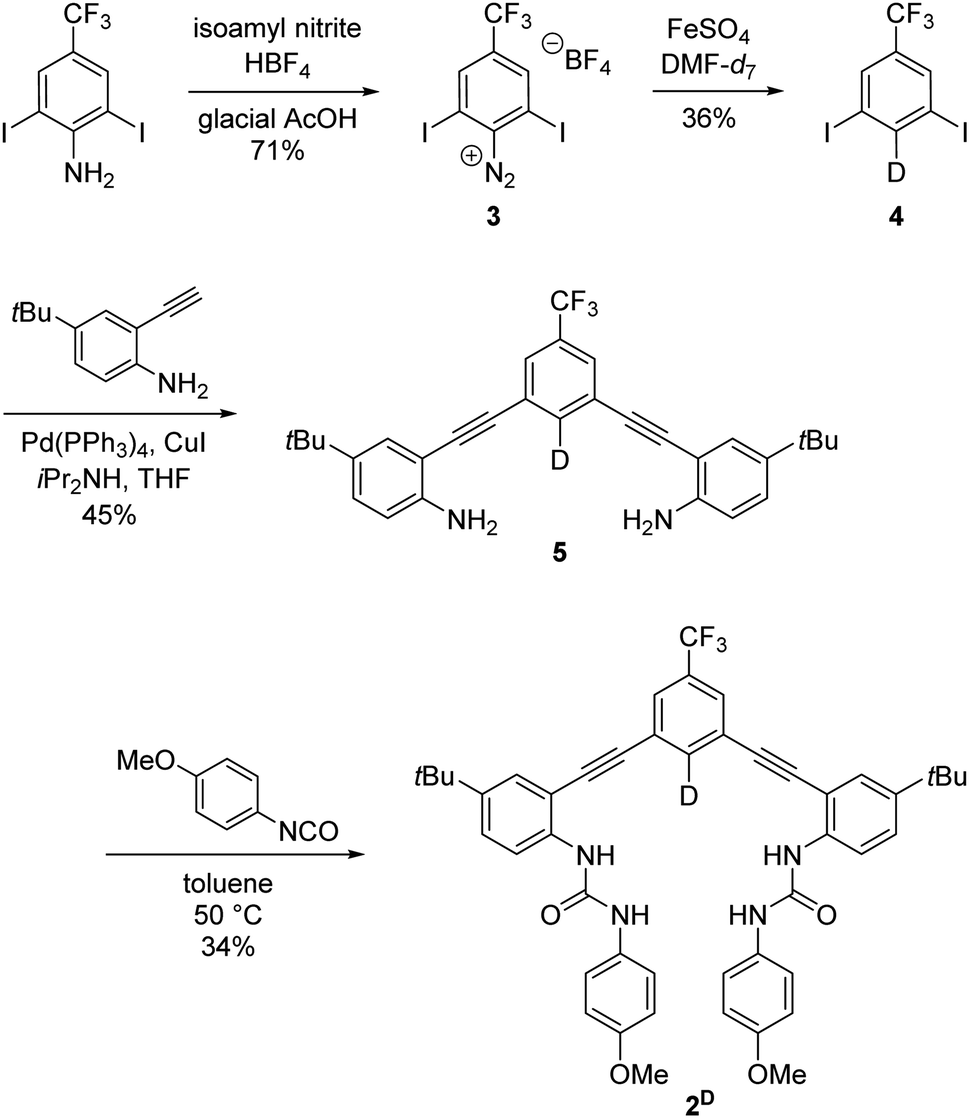 | ||
| Scheme 1 Synthetic route for the selective deuteration of anion receptor 2D. | ||
Previous work on the DEIE of Cl− binding with 1H/D in DMSO revealed an experimental isotope effect of 1.019 ± 0.010. Therefore, we expected similar small DEIEs for HS−, Cl−, and Br− binding with 2H/D. Typical methods to determine binding constants (Ka) in supramolecular systems use non-linear regression fitting of titration data. Results from this method can be affected by small errors in the known initial host and guest concentration, quality of the titration isotherm, and subsequent data fitting, which when taken together often results in 2–15% errors in Ka. To increase the precision in KHa/KDa data for this study, we used the Perrin method of competitive titrations,25 which has been shown previously to reduce errors in EIE values significantly with errors as small as 0.0004.26 In this method, a linearized plot of the chemical shifts of 2H (δH) and 2D (δD) in fast exchange with an anionic guest is fit by linear regression to eqn (1):
| (δ0H − δH)(δD − δfD) = DEIE(δ0D − δD)(δH − δfH) | (1) |
The slope of the linear regression is equal to the DEIE of the system. Because the linear regression only relies on chemical shift values and is independent of host and guest concentration, the precision of the method is limited to the precision of the NMR instrument and quality of data fitting.
In addition, 13C NMR spectroscopy is sensitive to isotopic labelling and can show changes in chemical shifts between isotopomers. We were able to differentiate between the 13C NMR signals for Cab, C1 and C2 for free and bound 2H and 2D (Fig. 2a) in 10% DMSO-d6/CD3CN, which were similar to those reported for 1H/D in DMSO-d6.12 Competitive 13C NMR spectroscopy titrations were performed in anaerobic and anhydrous 10% DMSO-d6/CD3CN at 25 °C with mixtures of 2H and 2D in combined concentrations between 5.71 and 13.46 mM. Aliquots of the tetrabutylammonium (TBA) salts of HS−, Cl−, and Br− were added until the system had reached saturation (Titration method A in ESI†). In an effort to decrease reactivity of HS− with 2H/D and DMSO over long periods of time and decrease oxygen and water contaminations, some titrations with HS− were performed by splitting the host solution of 2H/D between four J-young NMR tubes. For each point in the competitive titration, TBASH was added to a new solution of 2H/D inside an N2-filled glovebox shortly before obtaining a 13C NMR spectra (Titration method B in ESI†). The Cab, C1 and C2 13C NMR signals were tracked for 2H and 2D in each titration for each anion. A representative competitive titration and linearized plots for Cl− binding is shown in Fig. 2.
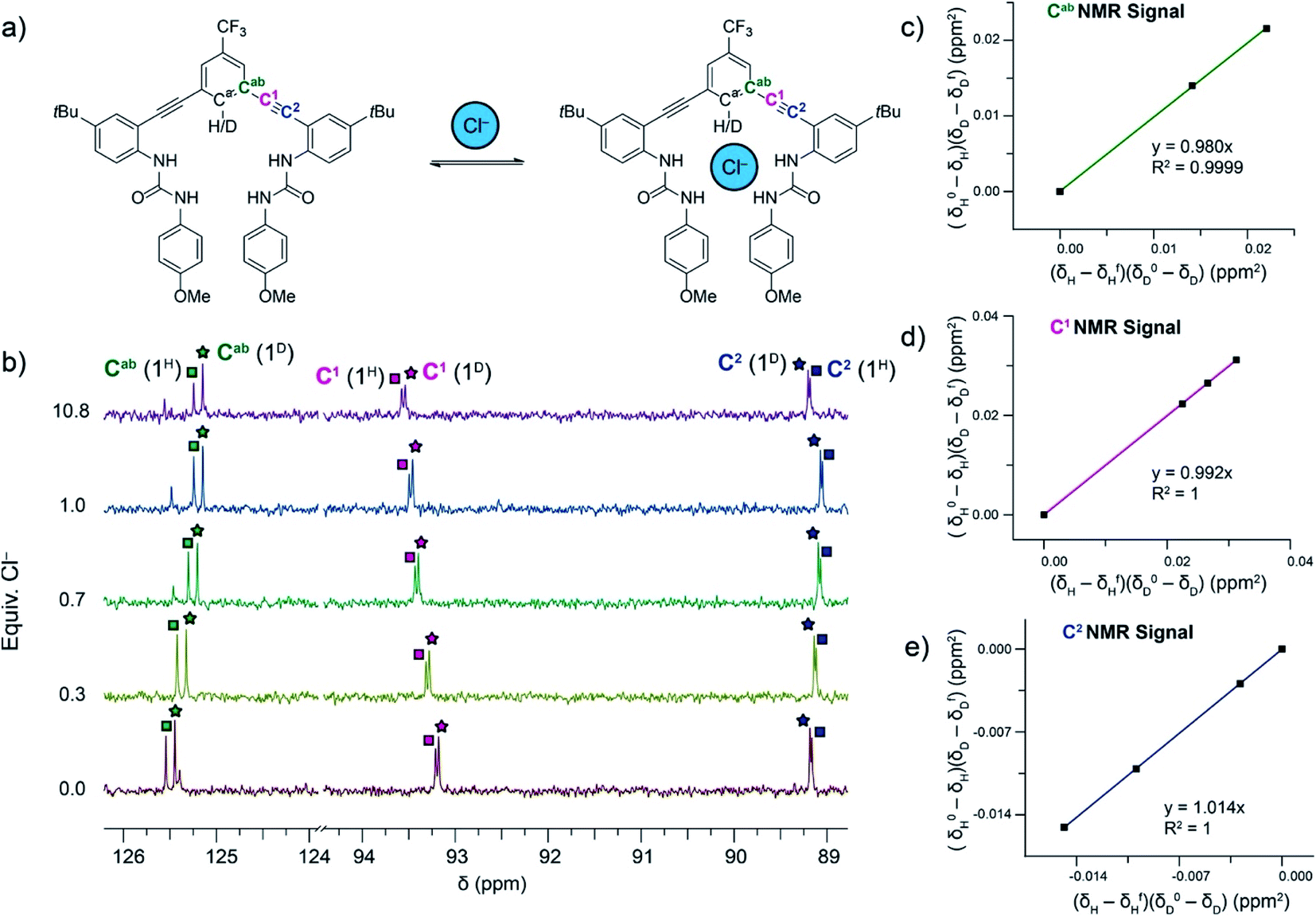 | ||
| Fig. 2 (a) Representation of the host–guest equilibrium between 2H/D and Cl−. (b) Differences in the chemical shifts between the 2H and 2D isotopologues are observed in the 13C NMR signals for the Cab, C1, and C2 carbons. 13C NMR signals for the Cab, C1, and C2 carbons in 2H and 2D are tracked throughout a titration. (c–e) Linearized plots from fitting the chemical shifts of the Cab, C1, and C2 throughout a titration to eqn (1). | ||
The DEIE data calculated from tracking the chemical shifts of the Cab, C1 and C2 13C NMR signals from Cl− and Br− binding are summarized in Table 1. The results shown are an average of three trials. Analysis of the data for competitive titrations of 2H/D with Cl− reveals a normal DEIE of 1.014 ± 0.002, calculated from monitoring the C2 13C NMR signal. The Cab and C1 13C NMR signals have the largest percent error in the calculated DEIE and show no statistically significant DEIE (i.e., DEIE = 1) for Cl− binding; however, because there is only one DEIE in the system, these positions must not be sensitive enough to the vibrational energy of the C–H/D bond in the free host and the host–guest complex to reveal the normal DEIE.
| 13C NMR Signal | DEIE (R2) | |
|---|---|---|
| Carbon | Cl− | Br− |
| Cab | 0.983 ± 0.017 (0.997) | 1.006 ± 0.010 (0.999) |
| C1 | 1.006 ± 0.007 (0.999) | 1.009 ± 0.018 (0.997) |
| C1 | 1.014 ± 0.002 (1.00) | 0.990 ± 0.046 (0.981) |
Notably, our experimental DEIE value for Cl− binding with 2H/D in 10% DMSO-d6/CD3CN is smaller than the computed value of 1.020 for Cl− binding with 1H/D in DMSO-d6.12 Our previously published computational study revealed that the DEIE of Cl− binding resulted from distorted urea N–H⋯Cl− hydrogen bonding geometry affecting the vibrational frequency of the C–H/D bond in the host–guest complex. Replacing the NO2 functional group in 1H/D (σp = 0.78) with a CF3 functional group (σp = 0.54) in 2H/D decreases the polarization of the C–H/D bond and subsequently makes it a slightly poorer hydrogen bond donor. In addition, the DEIE of Cl− binding in this current study is in a less polar solvent system (10% DMSO/CH3CN, ε ∼ 42) compared to the previous study (DMSO, ε = 47). We hypothesize that the decreased polarization of the C–H/D bond and the lower solvent polarity either relieve the distorted N–H⋯Cl− hydrogen bonding geometry or decrease their influence on the vibrational frequency of the C–H/D bond in the host–guest complex. To deconvolute and better understand the role of both C–H/D hydrogen bond donor polarity and solvent on the DEIE of Cl− binding in these receptors, a systematic study of these two variables would be required, similar to those previously reported, which we intend to pursue in future work.14,27,28
Analysis of the data for competitive titrations of 2H/D with Br− revealed no DEIE at any of the tracked 13C NMR signals; however, each calculated DEIE has a relatively large percent error (0.99–4.64%, compared to 0.20% for the DEIE of Cl− binding), which could potentially obscure small DEIEs. We attribute these large percent errors to a limitation in the Perrin method that assumes that the hosts are fully bound by guest at saturation. This limitation can potentially decrease the precision of this method for weakly bound guests with low Ka, such as Br− which has a Ka of 173 ± 9 M−1 with 2H in 10% DMSO-d6/CD3CN at 25 °C.14
Using the combined data from 11 experiments, we were unable to determine a DEIE for HS− binding. The C1 13C NMR signal appeared to be the most sensitive to the change in vibrational energy of the C–H/D bond in the free host and the host–guest complex; however, in over half these trials, data from the C1 13C NMR signal showed a poor linear fit (R2 < 0.99). In addition, we were unable to triplicate any DEIE from the data which showed a good linear fit (R2 > 0.99). We hypothesize that the high nucleophilicity and air and water sensitivity of HS− made it incompatible with the long experiment times needed for 13C NMR spectroscopy titrations. In addition it is important to note that HS− is the only protic guest investigated in these studies, and it is also possible that vibrational coupling between the S–H motif and the receptor may further complicate the measurement of these small EIEs. Such coupling between S–H and other motifs has been implicated previously in the IR inactivity of S–H stretching modes in many metal-sulfhydryl complexes.29
In conclusion, deuterium equilibrium isotope effects (DEIE) can be used to elucidate non-covalent driving forces behind anion binding in our arylethynyl bisurea receptors. We endeavored to use DEIE studies to further investigate a preference of polarized C–H hydrogen bond donors for HS− over Cl− and Br− which we reported previously.14 In this current work, we highlight a convenient method to selectively and completely deuterate the aryl C–H hydrogen bond donor in our supramolecular anion receptors. We then found a DEIE of 1.014 ± 0.002 for Cl− binding with 2H/D. This DEIE was smaller than the computed DEIE of Cl− binding with 1H/D which features a more polarized C–H hydrogen bond donor and in a more polar solvent. Finally, we reveal challenges in using the Perrin method and 13C NMR spectroscopy titrations in determining small and precise EIE for weakly binding or highly reactive guests.
From this work, we have identified several areas that need further research. The first is to study how solvent and hydrogen bond donor polarity affect EIE of guest binding. A computational study from Paneth and coworkers suggest that both these variables can be used to influence 35/37Cl EIE in supramolecular hosts.13 We also were unable to determine a DEIE of HS− binding in our receptors, likely due to its high reactivity. A new method to determine small, precise EIE of reactive species such as HS− is needed in order to learn more about the supramolecular chemistry of this biologically relevant anion and to develop new strategies for selectively binding HS− over other competing anions.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
Research reported in this publication was supported by National Institutes of Health (R01-GM087398 to D. W. J./M. M. H.), the NSF (CHE-2107602 to M. D. P.), and the UO Knight Campus Undergraduate Scholars program (scholarship to R. A. N.). This work was also supported by the Bradshaw and Holzapfel Research Professorship in Transformational Science and Mathematics to D. W. J. We also thank Professor Charles L. Perrin for helpful discussions.Notes and references
- H.-J. Schneider, Int. J. Mol. Sci., 2015, 16, 6694 CrossRef CAS PubMed.
- M. M. G. Antonisse and D. N. Reinhoudt, Chem. Commun., 1998, 443 RSC.
- M. M. J. Smulders, S. Zarra and J. R. Nitschke, J. Am. Chem. Soc., 2013, 135, 7039 CrossRef CAS PubMed.
- K. Świderek and P. Paneth, Chem. Rev., 2013, 113, 7851 CrossRef.
- Z. R. Laughrey, T. G. Upton and B. C. Gibb, Chem. Commun., 2006, 970 RSC.
- D. Rechavi, A. Scarso and J. Rebek, J. Am. Chem. Soc., 2004, 126, 7738 CrossRef CAS.
- Y.-L. Zhao, K. N. Houk, D. Rechavi, A. Scarso and J. Rebek, J. Am. Chem. Soc., 2004, 126, 11428 CrossRef CAS PubMed.
- T. Haino, K. Fukuta, H. Iwamoto and S. Iwata, Chem.–Eur. J., 2009, 15, 13286 CrossRef CAS.
- D. Wade, Chem.-Biol. Interact., 1999, 117, 191 CrossRef CAS PubMed.
- J. S. Mugridge, R. G. Bergman and K. N. Raymond, J. Am. Chem. Soc., 2012, 134, 2057 CrossRef CAS PubMed.
- C. Zhao, R. M. Parrish, M. D. Smith, P. J. Pellechia, C. D. Sherrill and K. D. Shimizu, J. Am. Chem. Soc., 2012, 134, 14306 CrossRef CAS.
- B. W. Tresca, A. C. Brueckner, M. M. Haley, P. H.-Y. Cheong and D. W. Johnson, J. Am. Chem. Soc., 2017, 139, 3962 CrossRef CAS.
- A. Paneth and P. Paneth, J. Phys. Chem. B, 2021, 125, 1874 CrossRef CAS PubMed.
- H. A. Fargher, N. Lau, H. C. Richardson, P. H.-Y. Cheong, M. M. Haley, M. D. Pluth and D. W. Johnson, J. Am. Chem. Soc., 2020, 142, 8243 CrossRef CAS PubMed.
- R. Wang, Physiol. Rev., 2012, 92, 791 CrossRef CAS PubMed.
- B. K. Czyzewski and D.-N. Wang, Nature, 2012, 483, 494 CrossRef CAS.
- D. Sippel, M. Rohde, J. Netzer, C. Trncik, J. Gies, K. Grunau, I. Djurdjevic, L. Decamps, S. L. A. Andrade and O. Einsle, Science, 2018, 359, 1484 CrossRef CAS PubMed.
- M. D. Hartle, R. J. Hansen, B. W. Tresca, S. S. Prakel, L. N. Zakharov, M. M. Haley, M. D. Pluth and D. W. Johnson, Angew. Chem., Int. Ed., 2016, 55, 11480 CrossRef CAS PubMed.
- N. Lau, L. N. Zakharov and M. D. Pluth, Chem. Commun., 2018, 54, 2337 RSC.
- J. Vázquez and V. Sindelar, Chem. Commun., 2018, 54, 5859 RSC.
- H. A. Fargher, N. Lau, L. N. Zakharov, M. M. Haley, D. W. Johnson and M. D. Pluth, Chem. Sci., 2018, 10, 67 RSC.
- F. W. Wassmundt and W. F. Kiesman, J. Org. Chem., 1995, 60, 1713 CrossRef CAS.
- D. M. Lindsay, W. Dohle, A. E. Jensen, F. Kopp and P. Knochel, Org. Lett., 2002, 4, 1819 CrossRef CAS.
- C. N. Carroll, O. B. Berryman, C. A. Johnson, L. N. Zakharov, M. M. Haley and D. W. Johnson, Chem. Commun., 2009, 252 Search PubMed.
- C. L. Perrin and M. A. Fabian, Anal. Chem., 1996, 68, 2127 CrossRef CAS PubMed.
- C. L. Perrin and P. Karri, J. Am. Chem. Soc., 2010, 132, 12145 CrossRef CAS PubMed.
- B. W. Tresca, R. J. Hansen, C. V. Chau, B. P. Hay, L. N. Zakharov, M. M. Haley and D. W. Johnson, J. Am. Chem. Soc., 2015, 137, 14959 CrossRef CAS PubMed.
- T. J. Sherbow, H. A. Fargher, M. M. Haley, M. D. Pluth and D. W. Johnson, J. Org. Chem., 2020, 85, 12367 CrossRef CAS.
- M. D. Pluth and Z. J. Tonzetich, Chem. Soc. Rev., 2020, 49, 4070 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Synthetic methods and characterization, copies of NMR spectra, competitive titration details and representative data. See DOI: 10.1039/d1ra05711a |
| This journal is © The Royal Society of Chemistry 2021 |