
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceHydroxylation of benzene to phenol over heteropoly acid H5PMo10V2O40 supported on amine-functionalized MCM-41†
Yanjun Lia,
Shichao Li *a and
Yan Kongb
*a and
Yan Kongb
aFaculty of Materials and Chemical Engineering, Yibin University, Yibin, Sichuan 644000, China. E-mail: mysimpledesign@163.com
bDepartment of Safety and Operation Management, Yibin Tianyuan Group Company Limited, Yibin 644000, China
First published on 3rd August 2021
Abstract
Supported catalysts with Keggin type heteropoly acids (H5PMo10V2O40) loaded onto amine-functionalized MCM-41 for the catalytic hydroxylation of benzene to phenol with H2O2 were prepared by a wet impregnation method. The effects of the preparation conditions on the properties and activity of the supported catalysts were fully investigated. The results showed that the catalyst retained the mesoporous structure of MCM-41 and H5PMo10V2O40 was dispersed uniformly on the surface of the amine-functionalized MCM-41. Meanwhile, the reusability and catalytic performance of the catalyst were affected by two key factors, i.e., the interaction between the heteropoly acid and the surface of MCM-41, and the hydrophobicity of the catalyst since they decide the leaching of H5PMo10V2O40 and the adsorption of benzene. The catalyst with H5PMo10V2O40 loaded onto amine-functionalized MCM-41, which was prepared using ethanol as the solvent, exhibited the highest phenol yield (20.4%), a turnover frequency value of 20.3 h−1 and good reusability. We believe this work offers an effective and facile strategy for the preparation of a new catalyst for hydroxylation of benzene to phenol.
Introduction
Phenol as one of the most indispensable organic chemical intermediates is widely used in the fields of manufacturing plastics, agrochemicals, pharmaceuticals and fine chemicals.1–4 At present, nearly 90% of the phenol in the world is produced by the traditional cumene process which was first reported by Hock.5 However, the traditional synthesis process suffers from excessive energy consumption, low atom utilization, and serious environmental problems. With the increasing demand for phenol in the market and the enhancement of people's environmental awareness, it is necessary to develop a simple and environmentally friendly alternative technology for the production of phenol.Direct hydroxylation of benzene to phenol using an oxidant or aerobic oxidation is a one-step process with economic and environmental advantages, attracting great attention in recent years.6–12 The commonly used oxidants are H2O2, O2 and N2O, etc.13–15 Among them, H2O2 shows relatively higher activity.16,17 Therefore, great efforts have been done in the last decades to develop excellent catalysts for hydroxylation of benzene to phenol using H2O2. Various catalysts, such as noble metals, molecular sieves, V-containing oxides, activated carbon, polyoxometalates (POMs), V-g-C3N4 and FeOCl have been explored.2,18–24 In particular, POMs with Keggin type structure show good catalytic performance. In the Keggin type structure, a central PO4 tetrahedron is surrounded by 12 molybdenum–oxygen octahedra and the molybdenum can be partially replaced with vanadium.25 Benefitting from its special structure, POMs have several advantages such as strong acidity, high redox property and stability, low waste generation and easy handing. Therefore, they are widely used in many oxidation reactions (such as selective oxidation methacrolein (MAL) to methacrylic acid (MAA), conversion of sugar to sugar acid, etc.).26–33
However, the direct use of POMs for hydroxylation of benzene to phenol is adverse to the catalyst recovery and reuse since it is difficult to separate POMs from reaction media. An effective strategy is to immobilize POMs onto high surface area carriers (such as activated carbon, silica, MCM-41, titania, resin, etc.).34,35 Especially, MCM-41 is usually used as a promising carrier because of its large surface area (>1000 m2 g−1), high thermal stability (ca. 900 °C) and adjustable pore size (2–8 nm).36,37 Its suitable pore size is favor of entrance of POMs cluster (the radius is about 6 A) and it as a neutral carrier can improve the stability of POMs.38,39 Therefore, POMs loaded on MCM-41 have been proven to be superior catalysts in several esterification and oxidation reactions.37,40 Unfortunately, when POMs were directly loaded onto the carriers, the weak interaction between them and the carriers results in the easy leaching of POMs. It is reported that surface modification of the carriers including deposition of basic alumina clusters,41 doping42 and amine-functionalization43 can enhance combination of POMs and the carriers. Specially, modification by amine-functionalization can firmly bond POMs with amino groups on the surface of the silicate by acid–base interaction44, which effectively prevents the leaching of POMs. Hence, loading POMs onto amine-functionalization MCM-41 should be an available way to synthesis excellent supported catalysts. Nevertheless, the potential of the catalyst with POMs loaded on amine-modified MCM-41 for hydroxylation of benzene to phenol has seldom been reported to our knowledge.
In the paper, POMs (H5PMo10V2O40) loaded on amine-functionalized MCM-41 was synthesized by wet impregnation method. The effects of the preparation conditions (such as solvent and carrier handling mode) on the properties and activity of the supported catalysts are studied by XRD, FT-IR, nitrogen adsorption, TG, UV-vis, TEM and XPS analysis. All catalysts were tested in hydroxylation of benzene to phenol. Finally, the loss of heteropolyanion and the catalytic ability of the recovered catalysts in repeating cycles were also studied.
Result and discussion
Catalysts characterization
The small-angle XRD patterns of MCM-41, MCM-41-NH2 (amine-functionalized MCM-41) and supported catalysts (HPMoV/MCM-41: impregnating MCM-41 with an ethanol solution of H5PMo10V2O40; HPMoV[ethanol]/MCM-41-NH2: impregnating MCM-41-NH2 with an ethanol solution of H5PMo10V2O40; HPMoV[water]/MCM-41-NH2: impregnating MCM-41-NH2 with a deionized water solution of H5PMo10V2O40) were shown in Fig. 1(a). For MCM-41 (Fig. 1(a)5), one intense reflection in 2θ less than 2.5° and two lower peaks in 2θ at 3.9° and 4.5° could be observed, which were attributed to (100), (110) and (200) plans with hexagonal pore arrays.45 MCM-41-NH2 (Fig. 1(a)4) and other supported catalysts (Fig. 1(a)1–3) all showed the same reflections mentioned above, indicating that hexagonal mesoporous arrangement of MCM-41 were maintained after modifying MCM-41 with 3-aminopropyltriethoxysilane (APTES) or loading HPMoV on MCM-41. However, the width and intensity of reflections of MCM-41-NH2 and supported catalysts were reduced or shifted compared with those of MCM-41, which manifested less ordered hexagonal mesoporous arrangement. An interaction between the silicon hydroxyl (Si–OH) of APTES (or heteropolyanion) and silanol group inside the wall of MCM-41 was responsibility for this phenomenon.46,47 Therefore, it was concluded that the modification by APTES or loading HPMoV onto MCM-41 had been occurred in the internal pores of MCM-41. In addition, it could be seen that these peaks became broader and weaker after loading HPMoV on MCM-41-NH2 (Fig. 1(a)1 and 2), showing that the hydrolysis of MCM-41 under acidic conditions also caused the loss of long-range order structure. The results were agreement with previous literatures.48,49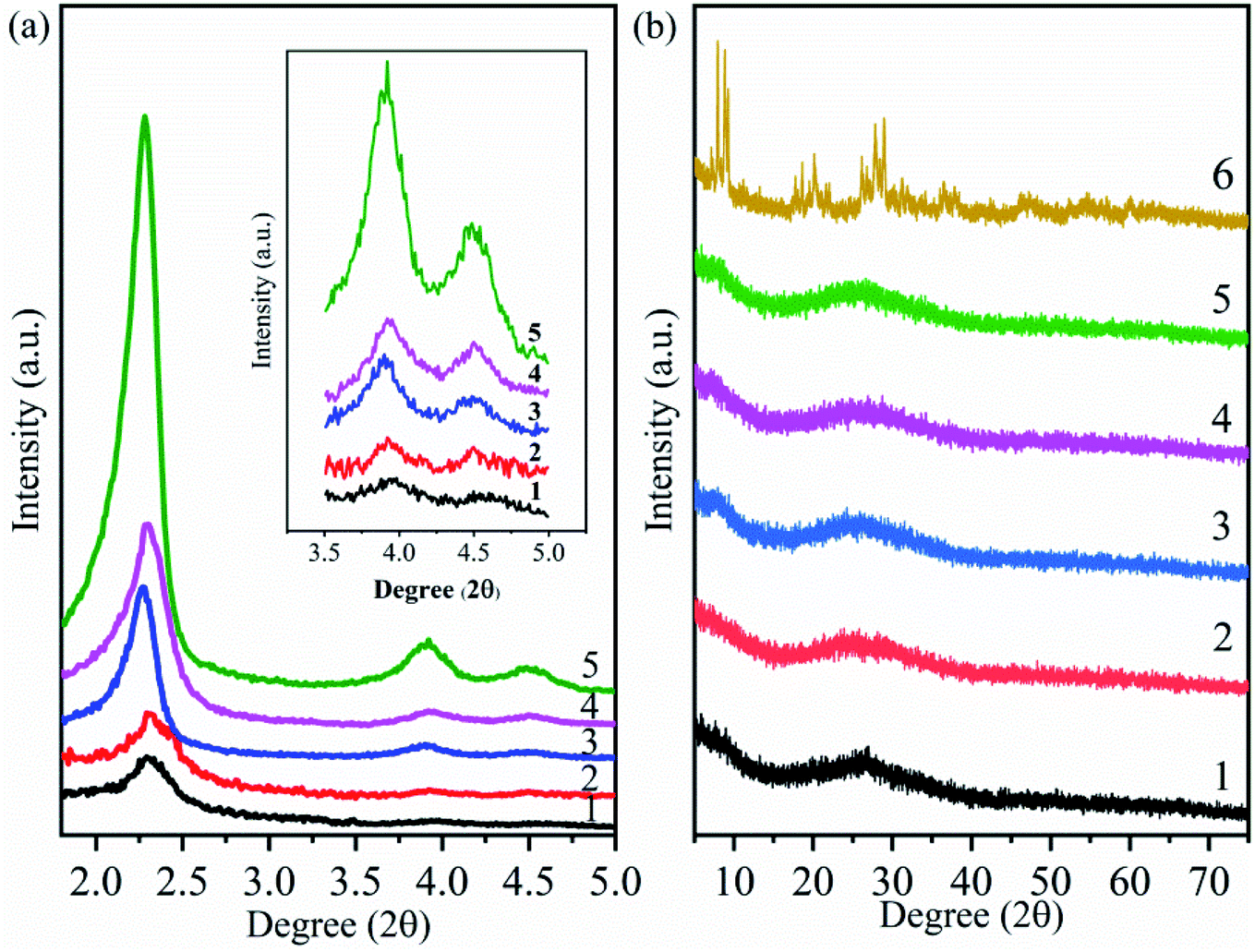 | ||
| Fig. 1 Small-angle (a) and wide-angle (b) XRD patterns of different catalysts. (1) HPMoV[ethanol]/MCM-41-NH2; (2) HPMoV[water]/MCM-41-NH2; (3) HPMoV/MCM-41; (4) MCM-41-NH2; (5) MCM-41; (6) HPMoV. | ||
Fig. 1(b) illustrated the wide-angle XRD patterns of the samples and HPMoV, respectively. The XRD pattern of HPMoV (Fig. 1(b)6) showed that a series of split peaks in 2θ range of 7°–10° (7°, 7.9°, 8.8°, 9.3°), 17°–24° (17.8°, 18.1°, 18.6°, 19.5°, 20.1°, 20.9°, 21.5°, 22.2°, 24.7°), 25°–30° (26°, 26.7°, 27.8°, 28.4°, 28.9°) and 31°–37° (31.2°, 31.9°, 35.3°, 35.9°, 36.5°, 37.6°) were belonged to the heteropoly acid in a triclinic crystal phase.38 The wide-angle XRD patterns of MCM-41, MCM-41-NH2 and HPMoV/carriers (Fig. 1(b)1–5) exhibited a broad peak caused by the amorphous silica walls of MCM-41. There were no reflection patterns corresponding to HPMoV in all supported catalysts. It indicated that HPMoV was well dispersed inside the pore of MCM-41 and MCM-41-NH2. The result showed good agreement with Juan.42 They reported that the 30 wt% POMs loaded on MCM-41 was still well dispersed.
Fig. 2a showed the FT-IR spectroscope of MCM-41. A broad vibration around 1000–1300 cm−1 was assigned to asymmetric stretching mode of Si–O–Si, while two more bands at 804 and 460 cm−1 corresponded to symmetric stretching vibration and bending vibration of Si–O–Si, respectively. A band at 1620 cm−1 was attributed to bending vibrations of H–O–H.50 In the case of MCM-41-NH2 (Fig. 2b), except for the bands of MCM-41, it could be observed the band at 1512 cm−1 corresponding to vibration of NH2. It confirmed that propylamine groups were successfully grafted onto the surface of MCM-41.51 Fig. 2f showed four characteristic absorption bands of the Keggin-type HPMoV at 1064, 961, 876 and 780 cm−1 those could be assigned to P–O stretching, terminal Mo![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretching, Mo–O–Mo asymmetric stretching of corner and edge sharing bridged oxygens, respectively.52 After loading HPMoV onto MCM-41(Fig. 2c), partial bands such as 1064 and 780 cm−1, were covered by the strong peak of Si–O–Si. Therefore, they could't be distinguishable. The bands at 961 and 876 cm−1 were found in the supported catalysts spectrum and there was no shift compared with that of HPMoV, suggesting that HPMoV actually existed in the pores of MCM-41 and the Keggin-type structure was retained after impregnation. For HPMoV[ethanol]/MCM-41-NH2 and HPMoV[water]/MCM-41-NH2 (Fig. 2d and e), the bands were similar with those of MCM-41-NH2 and HPMoV/MCM-41. This result indicated that HPMoV were successfully loaded onto amine-modified mesoporous material.
O stretching, Mo–O–Mo asymmetric stretching of corner and edge sharing bridged oxygens, respectively.52 After loading HPMoV onto MCM-41(Fig. 2c), partial bands such as 1064 and 780 cm−1, were covered by the strong peak of Si–O–Si. Therefore, they could't be distinguishable. The bands at 961 and 876 cm−1 were found in the supported catalysts spectrum and there was no shift compared with that of HPMoV, suggesting that HPMoV actually existed in the pores of MCM-41 and the Keggin-type structure was retained after impregnation. For HPMoV[ethanol]/MCM-41-NH2 and HPMoV[water]/MCM-41-NH2 (Fig. 2d and e), the bands were similar with those of MCM-41-NH2 and HPMoV/MCM-41. This result indicated that HPMoV were successfully loaded onto amine-modified mesoporous material.
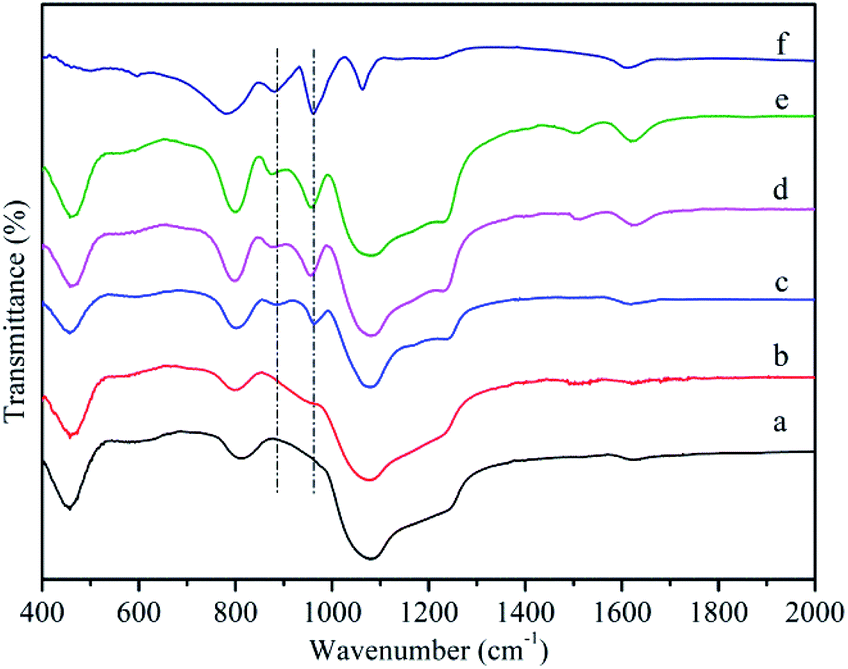 | ||
| Fig. 2 FT-IR spectra of (a) MCM-41, (b) MCM-41-NH2, (c) HPMoV/MCM-41, (d) HPMoV[ethanol]/MCM-41-NH2, (e) HPMoV[water]/MCM-41-NH2, (f) HPMoV. | ||
N2 adsorption–desorption isotherm for samples were depicted in Fig. 3 and the textural properties analysis based on nitrogen adsorption including specific surface area, pore volume and pore diameter were presented in Table 1. Isotherm curves for all samples showed a type IV isotherm, typical of a uniform order mesoporous structure, and there was capillary condensation region at a relative pressure range between 0.3 and 0.4. The reduction of the specific surface area, pore volume and pore diameter could be observed for the MCM-41-NH2 and HPMoV/MCM-41. This illustrated that HPMoV and ATPES occupied space of the mesopores, causing the decrease of the adsorption region and pore size. As further loading HPMoV onto MCM-41-NH2, more reduction of the specific surface area, pore volume and pore diameter could be seen, indicating that there was interaction between HPMoV and ATPES. This was agreement with the results of FT-IR analysis. However, the specific surface area and pore volume of HPMoV[water]/MCM-41-NH2 was higher than that of HPMoV[ethanol]/MCM-41-NH2. This fact led us to assume that the stronger polarity of water prevented the interaction between HPMoV and APTES.
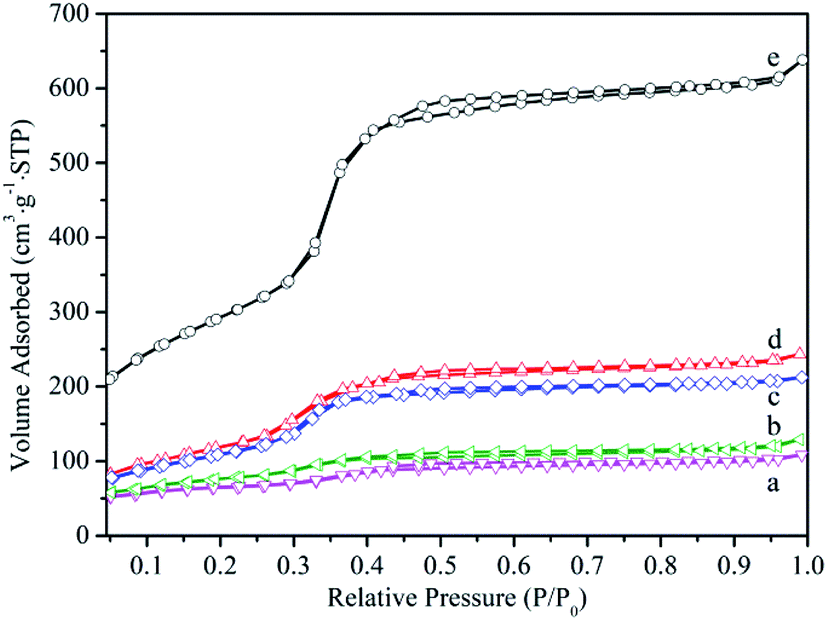 | ||
| Fig. 3 Nitrogen adsorption–desorption isotherm of (a) HPMoV[ethanol]/MCM-41-NH2, (b) HPMoV[water]/MCM-41-NH2, (c) HPMoV/MCM-41, (d) MCM-41-NH2, (e) MCM-41. | ||
| Samples | SBET (m2 g−1) | Vpore (ml g−1) | Dpore (nm) |
|---|---|---|---|
| MCM-41 | 1074 | 0.94 | 3.12 |
| MCM-41-NH2 | 467 | 0.35 | 2.74 |
| HPMoV/MCM-41 | 406 | 0.28 | 2.91 |
| HPMoV[ethanol]/MCM-41-NH2 | 267 | 0.16 | 2.66 |
| HPMoV[water]/MCM-41-NH2 | 302 | 0.18 | 2.71 |
The TEM image of MCM-41 was presented in Fig. 4a. The image showed the long-range order channel of hexagonal mesoporous material. After modifying MCM-41 with APTES (Fig. 4b), the long strips still could be seen distinctly, which proved the structural integrity of MCM-41. It confirmed that the weakening of the crystal structure of MCM-41-NH2 shown in XRD was mainly due to the interaction between species rather than hydrolysis of MCM-41. Fig. 4c and d showed that long-range order was partially turned amorphous, which proved that siloxane linkage of MCM-41 was easily hydrolyzed under acidic conditions, leading to the partial collapsed of mesoporous structure. However, in general a good ordered mesoporous structure was maintained for the supported catalysts. It could be inferred that the effect of loading HPMoV onto MCM-41 or MCM-41-NH2 under present preparation conditions on the destruction of mesoporous structure could be ignored.
 | ||
| Fig. 4 TEM image of (a) MCM-41, (b) MCM-41-NH2, (c) HPMoV/MCM-41, (d) HPMoV[ethanol]/MCM-41-NH2. | ||
Moreover, several darker spots (HPMoV nanoparticles) on nanoscale were evidently visible in the TEM images of HPMoV/MCM-41 and HPMoV[ethanol]/MCM-41-NH2. As shown in Fig. 4c and d, these darker spots were evenly dispersed in the interior of the channel of the carriers and there was no sign of bulk HPMoV outside the edge of MCM-41 and MCM-41-NH2. It was concluded that HPMoV were well dispersed and located inside the pores of MCM-41 and MCM-41-NH2. This showed good agreement with XRD and nitrogen adsorption results. The element mapping of HPMoV/MCM-41 and HPMoV[ethanol]/MCM-41-NH2 were shown in Fig. S1 and S2.† The element was homogeneously distributed on the surface of the MCM-41 and MCM-41-NH2, which further evidenced the good dispersion of HPMoV.
The fresh and used HPMoV[ethanol]/MCM-41-NH2 were tested by XPS spectra to confirm the active species in the reaction, respectively. Fig. 5 presented the chemical states of the constituting elements on the surface of the catalyst. The peak values were numerically fitted using Gaussian components to evaluate the respective valence state. As shown in the survey XPS spectrum (Fig. 5a), the photoelectron peak was detected for O, V, Mo, N, C, Si, P, which constituted the catalyst. Fig. 5b showed that the N 1s and Mo 3p5/2 spectra of the catalyst were fitted. N 1s spectrum was divided into two peaks at 399.6 eV and 401.8 eV which were related to C–N and NH4+.52,53 Fig. 5c showed the high resolution of P 2p which was deconvoluted to two peaks at 133.7 eV and 134.5 eV belonged to 2p3/2 and 2p1/2 of P5+, respectively.54 The peak of Mo 3p5/2 was assigned to the Mo6+ species. The assignment of the Mo 3d spectra for the catalyst was shown in Fig. 5d. These were decomposed into two independent peaks. The peaks at 232.8 eV and 235.8 eV were related to 3d5/2 and 3d3/2 of Mo6+, respectively.54 The high resolution of V 2p was shown in Fig. 5e. This was divided into two hands between 515 and 520 eV. The characteristic binding energy at 516.4 eV and 517.6 eV was belonged to V5+ and V4+ species, respectively.55 For the P, N and Mo species, no change was observed between the fresh catalyst and used catalyst. However, it was found that the molar ratio of V4+/V5+ (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1.4) for the used catalyst was higher than that of the fresh catalyst (1:3.7) according to quantitative analysis. That is to say that V5+ reduced to V4+ in the reaction. XPS spectra of HPMoV/MCM-41 were presented in Fig. S3.† As shown in the survey XPS spectrum, the HPMoV/MCM-41 contained Si, O, P, Mo, V as the constructing elements. The change of the molar ratio of different elements before and after reaction was similar with that for HPMoV[ethanol]/MCM-41-NH2. Based on the results of XPS, the V in the HPMoV should be the active species and thus the HPMoV was the reaction species during the reaction. This is consistent with the precious reports.22,56
:1.4) for the used catalyst was higher than that of the fresh catalyst (1:3.7) according to quantitative analysis. That is to say that V5+ reduced to V4+ in the reaction. XPS spectra of HPMoV/MCM-41 were presented in Fig. S3.† As shown in the survey XPS spectrum, the HPMoV/MCM-41 contained Si, O, P, Mo, V as the constructing elements. The change of the molar ratio of different elements before and after reaction was similar with that for HPMoV[ethanol]/MCM-41-NH2. Based on the results of XPS, the V in the HPMoV should be the active species and thus the HPMoV was the reaction species during the reaction. This is consistent with the precious reports.22,56
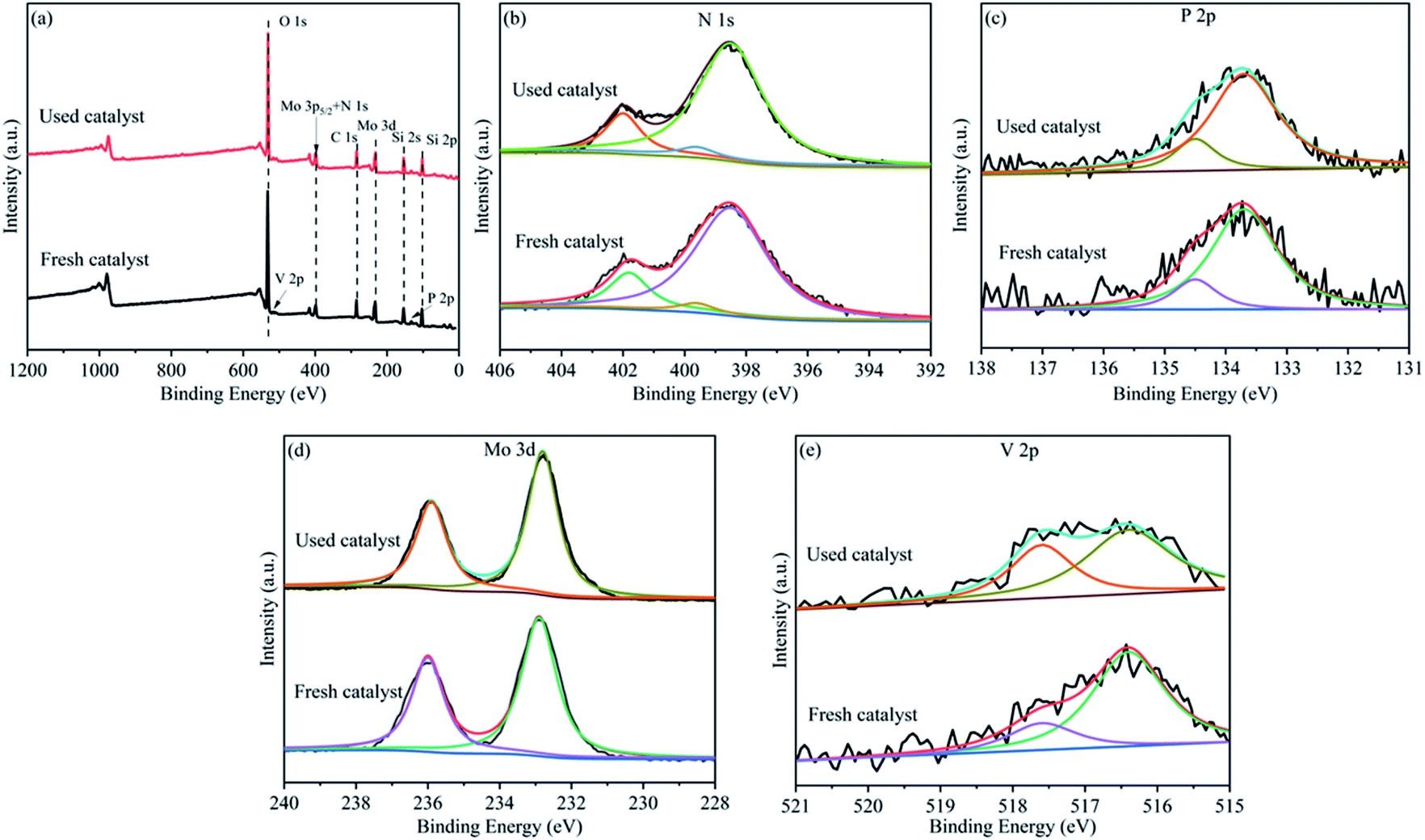 | ||
| Fig. 5 XPS spectra of (a) survey spectra, (b) N 1s, (c) P 2p, (d) Mo 3d, (e) V 2p, for the HPMoV[ethanol]/MCM-41-NH2 catalyst. | ||
Catalytic activity in benzene hydroxylation
The catalytic performance of various catalysts for hydroxylation of benzene to phenol using H2O2 was tested and the results were illustrated in Table 2. For MCM-41 and MCM-41-NH2, slight phenol was observed by carefully analyzing the liquid mixture, which showed that MCM-41 and MCM-41-NH2 had no catalytic activity. The formation of the phenol was because that acetic acid was oxidized by H2O2 to form acetic acid peroxide, and then acetic acid peroxide proceeded the reaction. When HPMoV was use as the catalyst (entry 3), a conversion of benzene of 22.2% and 97.4% of phenol selective were achieved. The amount of HPMoV equaled to that of HPMoV in HPMoV/support was also used (entry 4) in the reaction to assess catalytic activity for the same amount of HPMoV. The slightly decrease of the catalytic performance was detected due to the fewer catalysts. In fact, this reaction proceeded in homogeneous system due to the high solubility of HPMoV in polar solvents, which was difficult to recycle the catalyst. After loading HPMoV on MCM-41 (entry 5), a heterogeneous catalyst was prepared and the catalytic performance decreased. However, the high yield of phenol could also be achieved after loading HPMoV onto MCM-41-NH2 (entry 6 and 7). Particularly, the catalytic activity of HPMoV[ethanol]/MCM-41-NH2 was comparable to that of homogeneous HPMoV. According to data shown in Table 2 (entry 5 and 6), the conversion of H5PMo10V2O40/MCM-41 (17.2% ± 0.4) was lower in compared with H5PMo10V2O40/MCM-41-NH2 (21% ± 0.3). There were three reasons for this phenomenon. Firstly, the amount of HPMoV in HPMoV/MCM-41 (15 wt%) was lower than that in HPMoV[ethanol]/MCM-41-NH2 (17.9 wt%), which led to the less HPMoV on MCM-41 taken part in the reaction by comparing to HPMoV[ethanol]/MCM-41-NH2. Secondly, the leaching of HPMoV from MCM-41 was easy. This could be confirmed by the released heteropolyanions in the solution discussion later (Fig. 8). The leaching of HPMoV decreased the amount of HPMoV on the carriers, which was disadvantageous for the reaction process. Moreover, hydrophobic of catalysts surface also affected the adsorption behavior of benzene. In hydroxylation of benzene reaction system, acetonitrile or acetic acid was usually used for solving the problem of immiscibility between benzene and hydrogen peroxide. Hydrophobic property of the catalysts would facilitate the accessibility of hydrophobic reactants (benzene) to the catalyst surface, and then elevated catalytic efficiency.57 The ratio of the amount of adsorbed water to the adsorbed hydrocarbon could represent the relative degree of hydrophobicity of the catalysts.58 The test on the adsorption of catalysts (50 mg) in an equal mole mixture of hydrophobic benzene (15 μmol) and hydrophilic phenol (15 μmol) was conducted to investigate the hydrophobic capacity of the samples. After achieving sorption equilibrium, the molar ratio of the remaining benzene to phenol in the solution was 0.85, 0.96 and 0.96 for the HPMoV/MCM-41, HPMoV[ethanol]/MCM-41-NH2 and HPMoV[water]/MCM-41-NH2, respectively. It showed that benzene was more likely to attach to the catalysts with amine-functionalized support due to hydrophobic nature of C–H bond of ATPES, which reduced the diffusion resistance of benzene. Therefore, the relative high hydrophobicity of the prepared catalysts may be beneficial to the enhancement of the catalytic activity.| Entry | Catalysts | XBenzene (%) | Sphenol (%) | CBc (%) |
|---|---|---|---|---|
| a Reaction conditions: benzene (3 ml), H2O2 (9 ml), solvent of acetonitrile and acetic acid (30 ml, volume ratio 1:1), catalysts (0.2 g), temperature (70 °C), reaction time (9 h).b Reaction conditions: benzene (3 ml), H2O2 (9 ml), solvent of acetonitrile and acetic acid (30 ml, volume ratio 1:1), catalysts (0.036 g), temperature (70 °C), reaction time (9 h).c CB: carbon balance; Table S1 in the ESI shows the calculation details for the carbon balance. |
||||
| 1 | MCM-41a | 0.25 ± 0.03 | 100 | 99.6 |
| 2 | MCM-41-NH2a | 0.32 ± 0.03 | 100 | 99.7 |
| 3 | HPMoVa | 22.5 ± 0.3 | 97.1 ± 0.3 | 99.6 |
| 4 | HPMoVb | 20.7 ± 0.4 | 97.2 ± 0.3 | 99.3 |
| 5 | HPMoV/MCM-41a | 17.2 ± 0.4 | 96.5 ± 0.3 | 99.7 |
| 6 | HPMoV[ethanol]/MCM-41-NH2a | 21 ± 0.3 | 97.3 ± 0.4 | 99.5 |
| 7 | HPMoV[water]/MCM-41-NH2a | 19.6 ± 0.3 | 96.3 ± 0.4 | 99.4 |
The comparison of the catalytic activity of HPMoV[ethanol]/MCM-41-NH2 for benzene hydroxylation using H2O2 with some previously reported heterogeneous V-based catalysts was presented in Table 3. The synthesized HPMoV[ethanol]/MCM-41-NH2 showed the highest catalytic performance in comparison to others catalysts. Therefore, the supported catalyst in this study may be one of the appropriated catalysts for the hydroxylation of benzene to phenol.
| Catalyst | Catalyst amount (mg) | Reaction temperature (°C) | Yield of phenol (%) | Selectivity of phenol (%) | TOFa (h−1) | Ref. |
|---|---|---|---|---|---|---|
| a Based on vanadium mol%, TOF: mole of phenol/(mole of V × reaction time in hour scale).b 1 ml benzene, molar ratio of H2O2:benzene (3.5:1), 6 ml acetonitrile and acetic acid (1:1), 8 h.c 1 ml benzene, molar ratio of H2O2:benzene (3.5:1), 3 ml acetonitrile and acetic acid (1:1), 9 h.d 1 ml benzene, molar ratio of H2O2:benzene (3:1), 3 ml acetonitrile and acetic acid (1:1), 8 h.e 1 ml Benzene, molar ratio of H2O2:benzene (2.7:1), 6 ml acetonitrile and acetic acid (5:1), 6 h.f 0.9 ml benzene, molar ratio of H2O2:benzene (3:1), 6 ml acetonitrile and acetic acid (1:1), 4 h.g 1 ml benzene, molar ratio of H2O2:benzene (3:1), 25 ml acetonitrile and acetic acid (1:1), 5 h.h 1 ml benzene, molar ratio of H2O2:benzene (2:1), 6 ml acetonitrile, 2 h.i 0.3 ml benzene, molar ratio of H2O2:benzene (5.2:1), 5 ml acetonitrile, 4 h.j 4.4 ml benzene, molar ratio of H2O2:benzene (1:1), 20 ml acetonitrile, 8 h.k 1 ml benzene, molar ratio of H2O2:benzene (4:1), 5 ml acetonitrile and acetic acid (1:1), 9 h.l 0.5 ml benzene, molar ratio of H2O2:benzene (2:1), 5 ml acetic acid, 3 h.m 3 ml benzene, molar ratio of H2O2:benzene (3:1), 30 ml acetonitrile and acetic acid (1:1), 9 h. |
||||||
| PMo10V2/pg-C3N4b | 100 | 60 | 25.7 | 99.7 | 7.8 | 16 |
| PMoV2/DMA16-CMPSc | 100 | 65 | 21.9 | 99.3 | 17.4 | 59 |
| PMoV2/titaniad | 150 | 60 | 27.3 | 99.1 | 7.8 | 60 |
| PMoV2@SiO2e | 150 | 60 | 21.6 | 100 | 6.7 | 61 |
| [Dmim]2.5PMoV2f | 100 | 70 | 26.5 | 100 | 7.4 | 62 |
| Py1-PMoV2g | 200 | 80 | 20.5 | 98 | 2.1 | 63 |
| [(CH3)4N]4PMo11VO40h | 200 | 60 | 12.4 | 85.7 | 6.2 | 4 |
| VOx/SBA-16i | 10 | 60 | 13.8 | 97.5 | 12.5 | 64 |
| V2O5/Al2O3j | 1180 | 65 | 22.4 | 100 | 0.5 | 65 |
| PMoV2-IL-Al-MCM-41k | 50 | 60 | 14.8 | 100 | 20 | 66 |
| CsPMoV2l | 30 | 65 | 19.2 | 96.3 | 11.9 | 67 |
| HPMoV[ethanol]/MCM-41-NH2m | 200 | 70 | 20.4 | 97.3 | 20.3 | This work |
To check out the durability of the supported catalysts in hydroxylation of benzene to phenol, the catalytic recyclability of HPMoV/MCM-41, HPMoV[ethanol]/MCM-41-NH2 and HPMoV[water]/MCM-41-NH2 was measured using a five-run test under the same conditions employed previously. After each catalytic run, all tested samples was recovered by centrifugation, and then dried at 100 °C overnight. The recovered catalysts were used in the next cycle run without further treatment. The catalytic performance of the three catalysts for each run was shown in Fig. 6. The benzene conversion decreased as the number of reused. This decrease was possibly caused by two reasons, i.e. the change and leaching of active phase during the reaction.
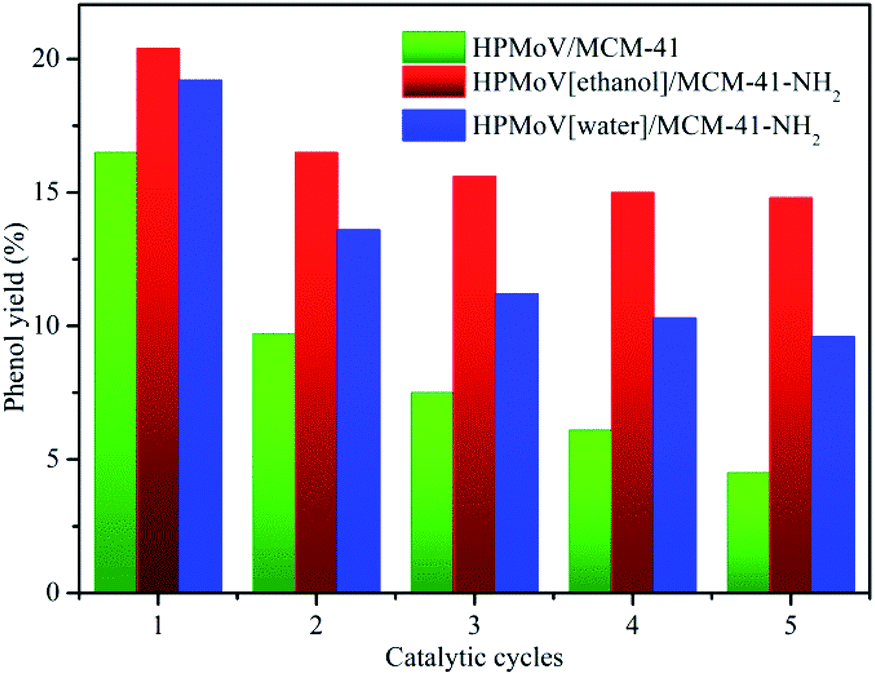 | ||
| Fig. 6 The catalytic recycle of the supported catalysts. | ||
The TG-DTA of HPMoV and HPMoV[ethanol]/MCM-41-NH2 was analyzed to illustrate the thermo stability of the catalysts, shown as in Fig. 7. Three weight losing steps could be observed in TG curves of HPMoV. Two endothermic peaks and one exothermic peak were shown from DTA curve at about 66, 290 and 430 °C, respectively.68 The mass loss in the range of 30–330 °C was related to physically absorbed water and constitution water, which corresponded to the two endothermic peaks in DTA curve. This amount of mass loss was 12.4%, and 14H2O per Keggin unit could be calculated form the mass loss. The mass loss from 330 to 500 °C corresponded to decomposition of HPMoV to oxides, and the exothermic peak in DTA curve reflected this change of heteropoly acids structure. Similarly, the weight transformation and thermal effects were observed in TG-DTA of HPMoV[ethanol]/MCM-41-NH2. The result showed that HPMoV[ethanol]/MCM-41-NH2 had the same strong thermal stability as HPMoV.
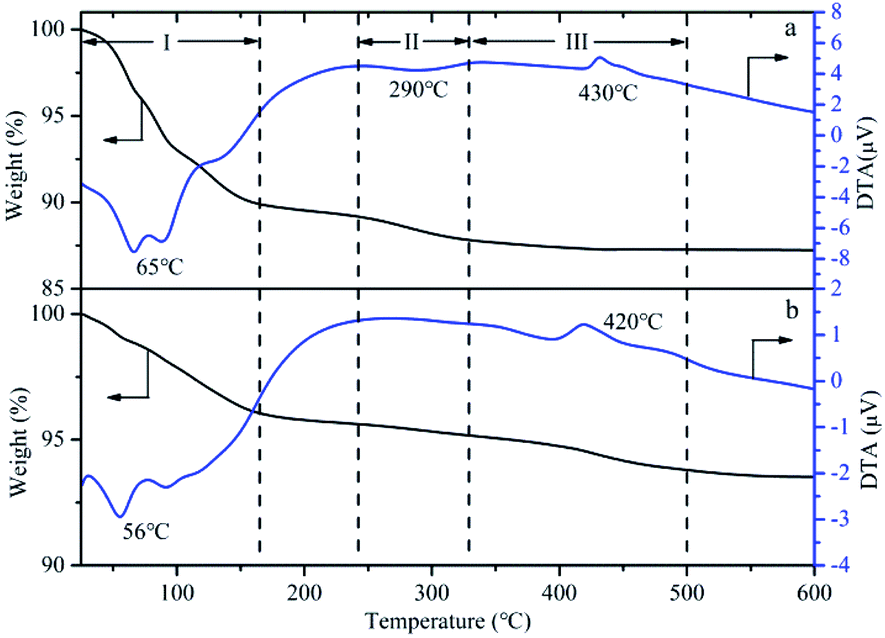 | ||
| Fig. 7 TG-DTA curve of (a) HPMoV and (b) HPMoV[ethanol]/MCM-41-NH2. | ||
The released heteropolyanions in the solution after reaction could be detected using UV-vis spectroscopy to inspect the leaching of HPMoV. The absorption band at 308 nm was assigned to the charge transfer oxygen (O2−) ligand to octahedral coordinated metal (V5+) in Keggin-type heteropolyanion.69 The intensity of this absorption peak was direct proportion to the content of the catalysts in the solution. Fig. 8 showed the relationship between the content of released heteropolyanion and number of catalytic run for HPMoV/MCM-41, HPMoV[ethanol]/MCM-41-NH2 and HPMoV[water]/MCM-41-NH2. For all samples, the amount of released heteropolyanion decreased rapidly at the first three catalytic run, and then declined gradually, which was agreement with the change rules of catalytic activity. Therefore, based on the above analysis, we could infer that the decrease of yield of phenol was attributed to the loss of heteropolyanion during reaction rather than the distortion of the catalyst phase.
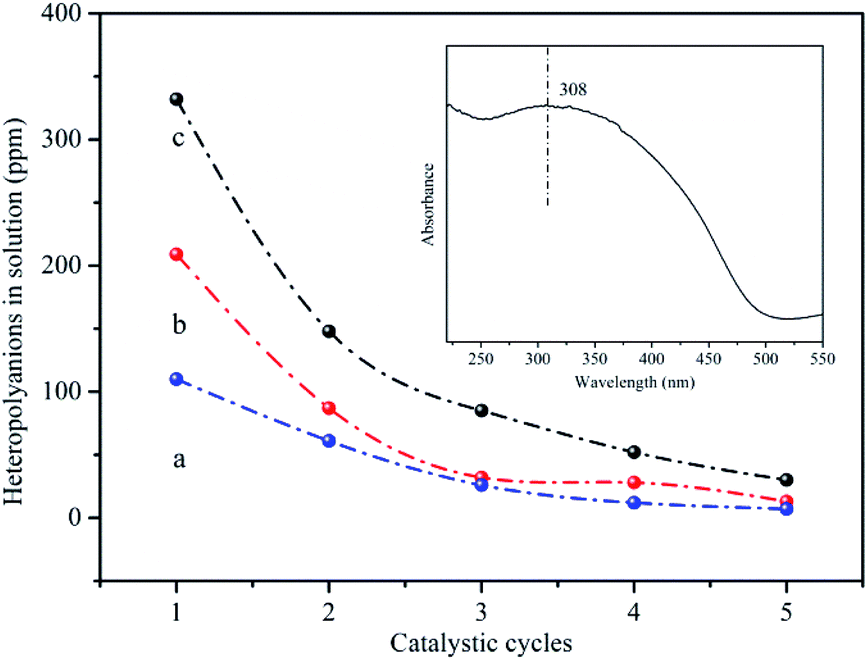 | ||
| Fig. 8 The content of heteropolyanion in the reaction solution (a) HPMoV[ethanol]/MCM-41-NH2; (b) HPMoV[water]/MCM-41-NH2; (c) HPMoV/MCM-41. | ||
It could be seen from Fig. 6 and 8 that the catalytic performance depended on the number of HPMoV. For HPMoV/MCM-41, the heteropolyanions physically adsorb on the surface of MCM-41 by van der Waals force. This binding force was weak and heteropolyanions easily leached into the reaction solution, and then the activity reduced as the losing of HPMoV. Inversely, in the case of HPMoV[ethanol]/MCM-41-NH2 and HPMoV[water]/MCM-41-NH2, the active species (HPMoV) was loaded on amino-functionalized MCM-41 by the interaction between the ammonium group and heteropolyanion. The reduction of the loss of HPMoV was obvious and the performance of the catalysts could maintain relatively high level. The leached heteropolyanion for the two catalysts could be portion physically adsorbed heteropolyanion on the surface of the carriers. The amount of released heteropolyanions in HPMoV[ethanol]/MCM-41-NH2 was lower than that in HPMoV[water]/MCM-41-NH2 due to the difference on polarity of solvent in impregnation method. The stronger polarity of water possibly prevented heteropolyanion from binding to ammonium group compared with ethanol. It could be deduced from N2 adsorption–desorption result in which the relative higher specific surface area and pore volume for HPMoV[water]/MCM-41-NH2 could be observed. Hence, the catalytic activity of HPMoV[ethanol]/MCM-41-NH2 was higher than that of HPMoV[water]/MCM-41-NH2 for each run.
Among all prepared supported catalysts, employed HPMoV[ethanol]/MCM-41-NH2 catalyst manifested high initially yield of phenol (20.4%) and a TOF of 20.3 (TOF = mole of phenol/(mole of V × reaction time in hour scale)). After the fifth run, there was a moderately decrease in the benzene conversion (15%), showing good survivability in the harsh reaction conditions and remarkable catalytic performance.
It could be seen from the cycle experiment that the leaching of HPMoV during the reaction obviously restrained the performance of the catalysts. It is necessary to pay more attention focused on the incorporation method of the amine groups into the silica framework and enhancement of the organic ligands capacity to solve the leaching of HPMoV problem. For the present method, organosiloxanes was covalently bonded on the inorganic walls of the calcined MCM-41. The post-synthesis treatment had several drawbacks. First, the attachment of the functional groups on the channel wall would decrease pore size and pore volume, and even led to pore blockage. Secondly, the loading amount of the functional groups was limited to a low-level because of scarcity of surface silanols. Moreover, grafting was significantly hindered due to the restricted access for the loading species and poor diffusion behavior in the limited channel of the material.70 The co-condensation method shows a good application prospect. Various functional groups including aliphatic hydrocarbon, phenyl, amine, sulfonic groups have been incorporated on the silica skeleton.71–74 This method can offer a higher and more uniform surface distribution of functional groups and a better control over the surface properties of the materials,70,75,76 which is not only favor to HPMoV grafting but also increase the interaction between the HPMoV and amine groups. The further work using co-condensation method to prepare HPMoV/support catalysts is under way. In addition, the leaching of HPMoV can be overcome by partially exchanging protons of the HPAs with large cations, such as Cs+, K+ and Rb+ etc., forming water-insolubility compounds.
Experimental
Materials
All chemicals were used without further purification. Trimethylammonium hydroxide (TMAOH), sulfuric acid, 3-aminopropyltriethoxysilane (APTES), benzene, hydrogen peroxide (30%), ethyl alcohol were supplied by Macklin Co., Ltd. Acetic acid, cetyltrimethylammonium bromide (CTAB), sodium molybdate (Na2MoO4), sodium hydrogen phosphate (Na2HPO4), sodium metavanadate (NaVO3), hydrochloric acid (37%), ethyl ether were purchased from Sinopharm Chemical Reagent Co., Ltd.Preparation of the catalysts
The heterogeneous properties of the catalyst were demonstrated according to the method reported by Sheldon.79 A reaction was carried out at 70 °C and then the catalyst was withdrawn at the reaction temperature after 15 min. The filtrate was maintained at the reaction temperature under stirred for 2 h. No further reaction was observed in the experiment, which demonstrated the heterogeneous character of the catalyst.
Characterization
Powder X-ray diffraction (XRD) was performed on a X-ray diffractometer (Bruker AXS D8 Advance X-ray powder diffractometer). The sample was recorded at 2θ = 1–10° with Cu Kα radiation (λ = 1.5418 Å) and 2θ = 5–60°, respectively. FT-IR spectra were recorded on Fourier transform infrared spectrophotometer (Nicolet-380, Thermal Electron Corporation, US). The sample was pelletized with KBr and the FTIR spectra for the prepared samples were obtained ranging from 400–4000 cm−1. N2 adsorption–desorption isotherms were obtained using Micromeritics Model ASAP 2020 instrument at 77 K. All samples were degassed at 150 °C under vacuum for 6 h. Specific surface areas were calculated with Brunauer–Emmett–Teller (BET) equation, and pore size distribution and total pore volume was calculated based Barret–Joyner–Halenda (BJH) method. The micro topography of catalysts was observed by field emission electron microscope (TEM, JEM-2100F, JEOL, Japan). Samples were analyzed by a conducting thermogravimetry/differential thermal analyzer (TG/DTA, STA-449-FS, Netzsch, German). Sample sizes of approximately 20 mg were heated from room temperature to 600 °C at 10 °C min−1 under 10 ml˙min−1 flow rate in an air atmosphere. Ultraviolet-visible (UV-vis) spectra (UV3600, Shimadzu, Japan) were obtained with a PE Lambda 950 spectrometer. XPS spectra were taken with a Thermo Scientific K-Alpha electron spectrometer using monochromatic Al Kα X-ray excitation source to study the changes of chemical valence states of element on the surface of the catalysts. ICP results were detected by an inductively coupled plasma mass spectrometry (NEXION 300X, PerkinElmer, US) to quantify the amount of HPMoV in the supported carriers. The amount of N was tested by Elemental Analyzer (ONH836, LECO Corporation, US).Catalytic test
Hydroxylation of benzene to phenol was carried out in a 50 ml three-neck round bottom flask equipped with a magnetic stirring bar, and a reflux condenser was connected with it. Typically, 0.2 g catalyst (the amount of catalyst base on benzene was 7.5 wt%), 3 ml benzene and 30 ml acetonitrile and acetic acid solvent (1:1) were added into the flask. The suspension was heated to 60 °C under stirring and then 9 ml hydrogen peroxide (molar ratio of H2O2/benzene was about 3) was added dropwise to the flask using constant pressure drop funnel in 1 h. The mixture was stirred constantly for 8 h. After the reaction stopped, the catalyst was separated from the solution, and the liquid product was collected to be analyzed. The liquid products were analyzed by a gas chromatograph equipped with a PEG-20M capillary column and a flame ionization detector. The reactants and products were confirmed by comparing with the retention time of the standard samples. Moreover, the reaction mixture was also further qualitative analyzed by GC/MS. They were retrieved by mass spectral data system, and then according to the standard spectrum analysis, the relative abundance of the base peak is compared to determine the product. The products were phenol, hydroquinone and benzoquinone based on above analysis. Toluene was used as an internal standard to quantify conversions based on benzene and selectivity. The phenol selectivity was calculated as the molar ratio of the produced phenol to the converted benzene, while the phenol yield was calculated as the molar ratio of the produced phenol to the initial benzene. Hydroquinone and benzoquinone selectivity was calculated as the molar ratio of the produced hydroquinone and benzoquinone to the converted benzene, respectively. The experiment was repeated three times at least for every catalyst including MCM-41 and MCM-41-NH2.
Conclusions
V-substituted polyoxometalates loaded onto MCM-41 and amine-modified MCM-41 were successfully synthesized as a heterogeneous catalyst for hydroxylation of benzene to phenol by using H2O2. The prepared supported catalysts were characterized by XRD, FT-IR, BET, TEM, TG-DTA, UV-vis and XPS techniques. The results showed that the catalyst maintained mesoporous structure of MCM-41 and H5PMo10V2O40 were dispersed uniformly on the surface of amine-modified MCM-41. The catalytic activity of the supported catalysts was related to the heteropolyanion number and hydrophobicity of the catalysts. Strong interaction between heteropolyanions and amine group could reduce the leaching of HPMoV, and hydrophobicity of the catalysts was favor to the adsorption of benzene. HPMoV[ethanol]/MCM-41-NH2 showed highest capable of selective oxidation of benzene to phenol (20.4%) and good stability against leaching. According to the results in this work, a strategy of a new catalyst prepared for the hydroxylation of benzene was suggested that immobilized the active species by strong interaction between heteropolyanion and the carriers, and improvement hydrophobicity of the catalysts should be needed for efficiently catalytic reaction.Author contributions
Y. Li and S. Li designed the experiments and wrote the paper. Y. Kong supervised the study. All of the authors discussed the results and commented on the manuscript.Conflicts of interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.Acknowledgements
We are grateful from Yibin University, where the work was done.References
- C. Gabriele and P. Siglinda, Catal. Today, 2008, 143, 145–150 Search PubMed
.
- L. Balducci, D. Bianchi, R. Bortolo, R. D’Aloisio, M. Ricci, R. Tassinari and R. Ungarelli, Angew. Chem., Int. Ed., 2003, 115, 5087–5090 CrossRef
- J.-S. Choi, T.-H. Kim, K.-Y. Choo, J.-S. Sung, M. B. Saidutta, S.-O. Ryu, S.-D. Song, B. Ramachandra and Y.-W. Rhee, Appl. Catal., A, 2005, 290, 1–8 CrossRef CAS
- K. Lemke, H. Ehrich, U. Lohse, H. Berndt and K. Jähnisch, Appl. Catal., A, 2003, 243, 41–51 CrossRef CAS
- H. Hock and S. Lang, Ber. Dtsch. Chem. Ges. B, 1944, 77, 257–264 CrossRef
- X. Ye, Y. Cui and X. Wang, ChemSusChem, 2014, 7, 738–742 CrossRef CAS PubMed
- G. Luo, X. Lv, X. Wang, S. Yan, X. Gao, J. Xu, H. Ma, Y. Jiao, F. Li and J. Chen, RSC Adv., 2015, 5, 94164–94170 RSC
- Y. Morimoto, S. Bunno, N. Fyjieda, H. Sugimoto and S. Itoh, J. Am. Chem. Soc., 2015, 137, 5867–5870 CrossRef CAS PubMed
- V. Sanny, R. B. N. Baig, N. N. Mallikarjuna and S. V. Rajender, ACS Sustain. Chem. Eng., 2017, 5, 3637–3640 CrossRef PubMed
- R. M. Peter and R. S. Sinha, J. Mol. Catal. A: Chem., 2015, 398, 149–157 CrossRef
- T. Tomokazu, Z. A. Andre, H. Yutaka, M. Kaoru, O. Takashi, S. Yoshihito, Y. Kazunari, S. Hiroyasu and K. Masahito, Angew. Chem., Int. Ed., 2017, 56, 7779–7782 CrossRef PubMed
- W. Shan, S. Li, X. Cai, J. Zhu, Y. Zhou and J. Wang, ChemCatChem, 2019, 11, 1076–1085 CrossRef CAS
- H. Xin, A. Koekkoek, Q. Yang, R. van Santen, C. Li and E. J. M. Hensen, Chem. Commun., 2009, 7590–7592 RSC
- H. Han, T. Jiang, T. Wu, D. Yang and B. Han, ChemCatChem, 2015, 7, 3526–3532 CrossRef CAS
- B. Liu, S. Wu, Y. Liu, Y. Liu, W. Cai, Q. Sun and Y. Li, Appl. Organomet. Chem., 2019, 33, e4662 CrossRef
- H. Wang, M. Zhao, Q. Zhao, Y. Yang, C. Wang and Y. Wang, Ind. Eng. Chem. Res., 2017, 56, 2711–2721 CrossRef CAS
- T. Jiang, W. Wang and B. Han, New J. Chem., 2013, 37, 1654–1664 RSC
- S.-i. Niwa, M. Eswaramoorthy, J. Nair, A. Raj, N. Itoh, H. Shoji, T. Namba and F. Mizukami, Science, 2002, 295, 105–107 CrossRef CAS PubMed
- Y.-W. Chen and Y.-H. Lu, Ind. Eng. Chem. Res., 1999, 38, 1893–1903 CrossRef CAS
- Y. Zhou, Z. Ma, J. Tang, N. Yan, Y. Du, S. Xi, K. Wang, W. Zhang, H. Wen and J. Wang, Nat. Commun., 2018, 9, 2931 CrossRef PubMed
- J. Xu, H. Liu, R. Yang, G. Li and C. Hu, Chin. J. Catal., 2012, 33, 1622–1630 CrossRef CAS
- J. Zhang, Y. Tang, G. Li and C. Hu, Appl. Catal., A, 2005, 278, 251–261 CrossRef CAS
- J. Yang, G. Sun, Y. Gao, H. Zhao, P. Tang, J. Tan, A.-H. Lu and D. Ma, Energy Environ. Sci., 2013, 6, 793–798 RSC
- E. E. Ahmed, E. Ghada, Z. Y. Fatma, M. A.-S. Ahmed and K. Søren, ACS Catal., 2018, 8, 10668–10675 CrossRef
- N. Mizuno and M. Misono, Chem. Rev., 1998, 98, 199–218 CrossRef CAS PubMed
- L. Zhou, S. Zhang, Z. Li, J. Scott, Z. Zhang, R. Liu and J. Yun, RSC Adv., 2019, 5, 34065–34075 RSC
- S. Illies and B. Kraushaar-Czarnetzki, Ind. Eng. Chem. Res., 2016, 55, 8509–8518 CrossRef CAS
- X. Liu, H. Li, H. Pan, H. Zhang, S. Huang, K. Yang, W. Xue and S. Yang, J. Energy Chem., 2016, 25, 523–530 CrossRef
- J. Albert, D. Lüders, A. Bösmann, D. M. Guldi and P. Wasserscheid, Green Chem., 2014, 16, 226–237 RSC
- J. Tian, J. Wang, S. Zhao, C. Jiang, X. Zhang and X. Wang, Cellulose, 2010, 17, 587–594 CrossRef CAS
- P. Lanzafame, D. M. Temi, S. Perathoner, A. N. Spadaro and G. Centi, Catal. Today, 2012, 179, 178–184 CrossRef CAS
- X. Luo, H. Wu, C. Li, Z. Li, H. Li, H. Zhang, Y. Li, Y. Su and S. Yang, Front. Chem., 2020, 8, 580146 CrossRef CAS PubMed
- R. Palkovits, K. Tajvidi, A. M. Ruppert and J. Procelewska, Chem. Commun., 2011, 47, 576–578 RSC
- A. Sinnema, R. J. J. Jansen, K. Pamin and H. van bekkum, Catal. Lett., 1995, 30, 241–252 Search PubMed
- M. Kanno, T. Yasukawa, W. Ninomiya, K. Ooyachi and Y. Kamiya, J. Catal., 2010, 273, 1–8 CrossRef CAS
- B. R. Jermy and A. Pandurangan, Appl. Catal., A, 2005, 295, 185–192 CrossRef CAS
- J. C. Juan, J. Zhang and M. A. Yarmo, J. Mol. Catal. A: Chem., 2007, 267, 265–271 CrossRef CAS
- E. Wang, C. Hu and L. Xu, Introduction to Polyoxometalate, Chemical Industry Press, Beijing, 1998 Search PubMed
- A. A. Concellón, P. Vázquez, M. Blanco and C. Cáceres, J. Colloid Interface Sci., 1998, 204, 256–267 CrossRef PubMed
- A. Popa, V. Sasca and J. Halasz, Appl. Surf. Sci., 2008, 255, 1830–1835 CrossRef CAS
- X. L. Sheng, Y. M. Zhou, Y. W. Zhang, M. W. Xue and Y. Z. Duan, Chem. Eng. J., 2012, 179, 295–301 CrossRef CAS
- X. Sheng, J. Kong, Y. Zhou, Y. Zhang, Z. Zhang and S. Zhou, Microporous Mesoporous Mater., 2014, 187, 7–13 CrossRef CAS
- A. N. Kharat, S. Moosavikia, B. T. Jahromi and A. Badiei, J. Mol. Catal. A: Chem., 2011, 348, 14–19 CrossRef CAS
- M. Kamada, H. Kominami and Y. Kera, J. Colloid Interface Sci., 1996, 182, 297–300 CrossRef CAS
- C. T. Kresge, M. E. Leonowicz, W. J. Roth, J. C. Vartuli and J. S. Beck, Nature, 1992, 359, 710–712 CrossRef CAS
- A. Ghanbari-Siahkali, A. Philippou, J. Dwyer and M. W. Anderson, Appl. Catal., A, 2000, 192, 57–69 CrossRef CAS
- G. Ma, J. Zhang, L. Chen, T. Liu, L. Yu, X. Liu and C. Lu, Appl. Catal., A, 2014, 4, 41341–41347 CAS
- L. Y. Chen, S. Jaenicke and G. K. Chuah, Microporous Mater., 1997, 12, 323–330 CrossRef CAS
- S. Kawi and S. C. Shen, Mater. Lett., 2000, 42, 108–112 CrossRef CAS
- D. Kumar and C. C. Landry, Microporous Mesoporous Mater., 2007, 98, 309–316 CrossRef CAS
- G. Sun, Y. Chang, S. Li, Q. Li, R. Xu, J. Gu and E. Wang, Dalton Trans., 2009, 23, 4481–4487 RSC
- Y.-L. Cao, L. Wang, B.-H. Xu and S.-J. Zhang, Chem. Eng. J., 2018, 334, 1657–1667 CrossRef CAS
- S. Yuan, J. Gu, Y. Zheng, W. Jiang, B. Liang and S. O. Pehkonen, J. Mater. Chem. A, 2015, 3, 4620–4636 RSC
- I. Tamiolakis, I. N. Lykakis, A. P. Katsoulidis, M. Stratakis and G. S. Armatas, Chem. Mater., 2011, 23, 4204–4211 CrossRef CAS
- L. Zhou, L. Wang, S. Zhang, R. Yan and Y. Diao, J. Catal., 2015, 329, 431–440 CrossRef CAS
- K. Nomiya, Y. Nemoto, T. Hasegawa and S. Matsuoka, J. Mol. Catal. A: Chem., 2000, 152, 55–68 CrossRef CAS
- M. Kimura, T. Nakato and T. Okuhara, Appl. Catal., A, 1997, 165, 227–240 CrossRef CAS
- T. Okuhara, Chem. Rev., 2002, 102, 3641–3666 CrossRef CAS PubMed
- H. Wang, L. Fang, Y. Yang, L. Zhang and Y. Wang, Catal. Sci. Technol., 2016, 6, 8005–8015 RSC
- H. Wang, C. Wang, M. Zhao, Y. Yang, L. Fang and Y. Wang, Chem. Eng. Sci., 2018, 177, 399–409 CrossRef CAS
- Y. Li, Z. Wang, R. Chen, Y. Wang, W. Xing, J. Wang and J. Huang, Catal. Commun., 2014, 55, 34–37 CrossRef CAS
- Y. Leng, J. Wang, D. Zhu, L. Shen, P. Zhao and M. Zhang, Chem. Eng. J., 2011, 173, 620–626 CrossRef CAS
- Y. Leng, H. Ge, C. Zhou and J. Wang, Chem. Eng. J., 2008, 145, 335–339 CrossRef CAS
- Y. Zhu, Y. Dong, L. Zhao and F. Yuan, J. Mol. Catal. A: Chem., 2010, 315, 205–212 CrossRef CAS
- J. K. Joseph, S. Singhal, S. L. Jain, R. Sivakumaran, B. Kumar and B. Sain, Catal. Today, 2009, 141, 211–214 CrossRef CAS
- M. Taheri, M. Ghiaci, A. Moheb and A. Shchukarev, Appl. Organomet. Chem., 2019, 33, e5012 CrossRef
- S. Feng, L. Zhang, Y. Ren, B. Yue, L. Ye, Y. Wang, X. Chen and H. He, Acta Chim. Sin., 2012, 70, 2316–2322 CrossRef CAS
- B. K. Hodnett and J. B. Moffat, J. Catal., 1984, 88, 253–263 CrossRef CAS
- G. Ranga Rao and T. Rajkumar, Catal. Lett., 2008, 120, 261–273 CrossRef
- A. Walcarius, C. Despas and J. Bessière, Microporous Mesoporous Mater., 1998, 23, 309–313 CrossRef CAS
- A. S. Maria Chong, X. S. Zhao, A. T. Kustedjo and S. Z. Qiao, Microporous Mesoporous Mater., 2004, 72, 33–42 CrossRef
- X.-M. Wang, X.-Z. Du, C.-L. Li and X. Cao, Appl. Surf. Sci., 2008, 254, 3753–3757 CrossRef CAS
- L. Zhu, X. Liu, T. Chen, Z. Xu, W. Yan and H. Zhang, Appl. Surf. Sci., 2012, 258, 7126–7134 CrossRef CAS
- J. A. Melero, G. D. Stucky, R. van Grieken and G. Morales, J. Mater. Chem. A, 2002, 12, 1664–1670 RSC
- V. Zeleňák, M. Skřínska, F. R. Siperstein and A. Patti, Appl. Surf. Sci., 2019, 476, 886–896 CrossRef
- T. Deschner, Y. Liang and R. Anwander, J. Phys. Chem. C, 2010, 114, 22603–22609 CrossRef CAS
- A. Corma, A. Martínez and V. Martínez-Soria, J. Catal., 1997, 169, 480–489 CrossRef CAS
- G. A. Tsigdinos and C. J. Hallada, Inorg. Chem., 1968, 7, 437–441 CrossRef CAS
- G. J. Meuzelaar, L. Maat and R. A. Sheldon, Catal. Lett., 1998, 56, 49–51 CrossRef CAS
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1ra04269f |
| This journal is © The Royal Society of Chemistry 2021 |