
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
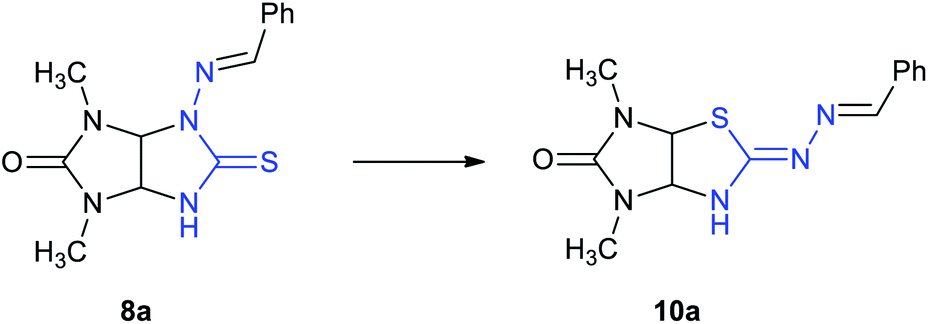
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDimroth-type N/S-interchange of N-aminothioglycolurils in the synthesis of 2-hydrazonoimidazo[4,5-d]thiazolones†
Ekaterina E. Vinogradovaa,
Galina A. Gazieva *a,
Alexei N. Izmest'eva,
Valentina A. Karnoukhovab and
Angelina N. Kravchenkoac
*a,
Alexei N. Izmest'eva,
Valentina A. Karnoukhovab and
Angelina N. Kravchenkoac
aN. D. Zelinsky Institute of Organic Chemistry, Russian Academy of Sciences, Leninsky Prosp., 47, Moscow 119991, Russian Federation. E-mail: gaz@ioc.ac.ru
bA. N. Nesmeyanov Institute of Organoelement Compounds, Russian Academy of Sciences, Vavilova Str., 28, Moscow 119991, Russian Federation
cPlekhanov Russian University of Economics, Stremyanny Lane, 36, Moscow 117997, Russian Federation
First published on 23rd August 2021
Abstract
An original method for the synthesis of 2-hydrazonoimidazo[4,5-d]thiazolone derivatives has been developed based on a one-pot acid-induced sequence of hydrazone formation from 3-thioxoperhydroimidazo[4,5-e]-1,2,4-triazinones and aromatic aldehydes, triazine ring contraction to imidazolidine one, and Dimroth-type N/S-interchange of N-aminothioglycolurils formed in situ into 2-hydrazonoimidazo[4,5-d]thiazolones. 3-Phenylacroleine derivatives are also suitable substrates for the reaction with thioxoperhydroimidazotriazinones.
Introduction
Nitrogen- and sulfur-containing heterocycles are found in many natural products and biologically active molecules.1 This diversity of compounds includes imidazolidin-2-one and hydrogenated thiazole derivatives. For example, biotin (5-[2-oxohexahydro-1H-thieno[3,4-d]imidazol-4-yl]pentanoic acid) is involved in a wide range of metabolic processes in living organisms as a cofactor for carboxylase enzymes,2 and leucogen (2-(2-ethoxy-2-oxo-1-phenylethyl)thiazolidine-4-carboxylic acid) is a veterinary vaccine used to protect cats against feline leukemia virus.3Furthermore, 2-hydrazonothiazolidin-4-one derivatives possess antifungal4 and anti-Toxoplasma gondii activities5 while the biological activity of their imidazolidine fused analogues, i.e. 2-hydrazonoimidazo[4,5-d]thiazoles, has not been studied. Among the imidazo[4,5-d]thiazole derivatives, only 3,4-dihydro-2-phenylimino-2H-imidazo[4,5-d]thiazole 1 was recently described as anticancer agent against human colorectal carcinoma (HCT-116), human prostate adenocarcinoma (PC-3), and human liver hepatocellular carcinoma (HepG-2) cell lines (Fig. 1).6 Apparently, this is due to the low availability of these compounds. Few examples of imidazo[4,5-d]thiazole derivatives are found in the literature.
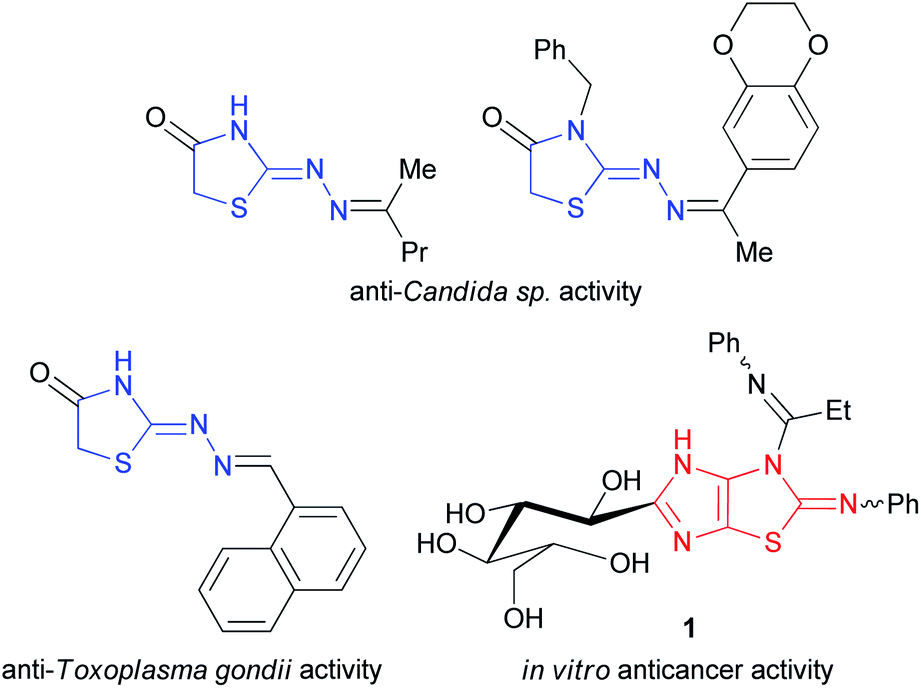 | ||
| Fig. 1 Bioactive 2-hydrazonothiazolidin-4-one and imidazo[4,5-d]thiazole derivatives. | ||
Presented at Fig. 1 compound 1 was produced from thiazole-4,5-diamine derivative with D-glucose in acetic acid in the presence of iodine used as the oxidizing agent at room temperature.6 3a,6a-Diphenylimidazo[4,5-d]thiazolediones 2 were prepared by the reaction of 1-substituted 5-hydroxy-4,5-diphenyl-1H-imidazol-2(5H)-ones with potassium thiocyanate in the presence of acetic acid in acetonitrile in the yields of 53–92%; however, only 3a,6a-diphenylimidazothiazoles are accessible by this method (Fig. 2).7 3a,6a-Bis(trifluoromethyl)imidazo[4,5-d]thiazolones 3 and 4 were synthesized by treatment of 4,5-dihydroxy-4,5-bis(trifluoromethyl)imidazolidin-2-one(thione) with NH4SCN or thiourea, respectively.8 Imidazo[4,5-d]thiazoles 5 devoid of substituents at positions 3a,6a were prepared in several stages by the cycloaddition of isothiocyanates with N-benzylsubstituted 4-methyl-5-(2-hydroxyethyl)thiazolium ylides followed by treatment with liquid ammonia and thermal desulfurization.9 Imidazothiazoles 6 (R = H, Piv) were isolated as by-products when 1,6-dimethylthioglycoluril was acylated with pivaloyl chloride and potassium tert-butoxide in the yields of 4 and 16%, respectively.10 Thus, the development of synthetic approaches to imidazo[4,5-d]thiazoles is still relevant as no general method for their synthesis has been designed.
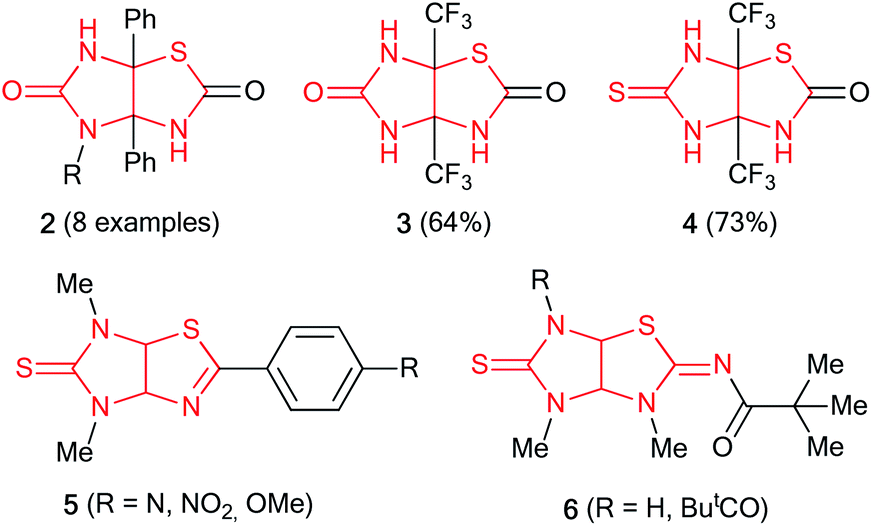 | ||
| Fig. 2 Some imidazo[4,5-d]thiazole derivatives. | ||
Amidst the imidazo[4,5-d]thiazole derivatives, compound 6 (R = H) is resulted from rearrangement of imidazolidinethione ring of thioglycoluril 7. Authors10 suppose that deprotonation of compound 7 lead to formation of thioglycoluril anion which can undergo neighboring ring opening followed by the reclosure at sulfur (Scheme 1a). This fact encouraged us to research possibility of thioglycolurils rearrangement.
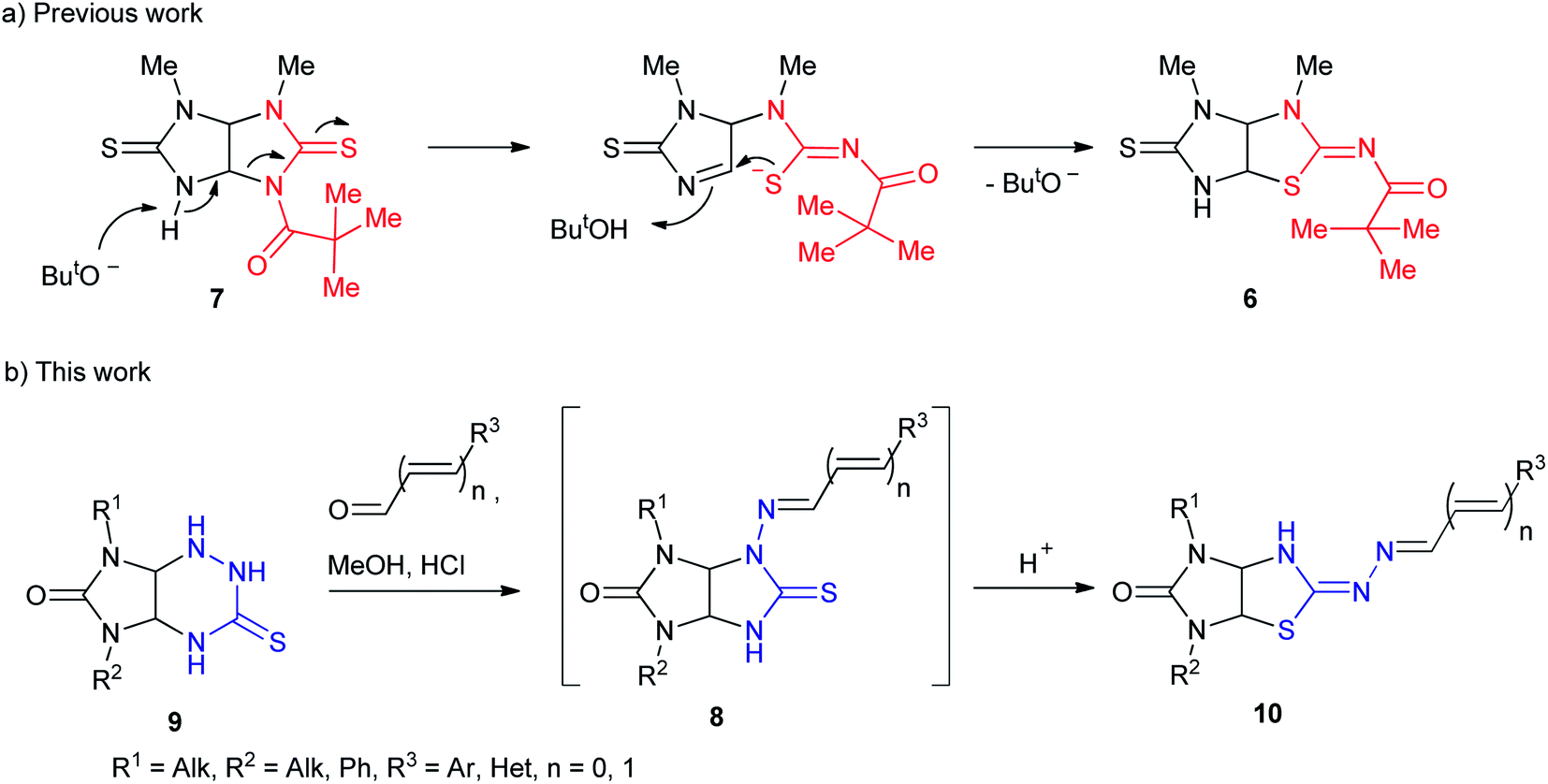 | ||
| Scheme 1 Background and general concept of this work. | ||
Recently,11 we have developed an approach to preparation of thioglycolurils 8 based on the tandem hydrazone formation and triazine ring contraction reaction of 3-thioxoperhydroimidazo[4,5-e]-1,2,4-triazin-6-ones 9 with aromatic aldehydes or 3-phenylacrylaldehyde derivatives.
Here, we propose an original method for the synthesis of 2-hydrazonoimidazo[4,5-d]thiazolone derivatives 10 based on the one-pot acid-induced sequence of hydrazone formation from thioxoperhydroimidazo[4,5-e]triazinones 9 and aromatic aldehydes or 3-phenylacrylaldehyde derivatives, triazine ring contraction to imidazolidine one, and rearrangement, i.e. Dimroth-type N/S-interchange, of thioglycolurils 8 formed in situ into 2-hydrazonoimidazo[4,5-d]thiazolones (Scheme 1b).
Results and discussion
At first, N-aminothioglycoluril derivatives 8a–o were prepared according to earlier developed procedure starting from 3-thioxoperhydroimidazo[4,5-e]-1,2,4-triazines 9a–c and aromatic aldehydes or 3-phenylacrylaldehyde derivatives (Scheme 2).11 The yields of thioglycolurils 8a–j, l–o were close to described values.11 New compound 8k was synthesized in 35% yield. Introduction of phenyl substituent at the N(5) atom of starting compound 9c led to the significant decrease of the reaction product yield compared to the yields of 8a,f.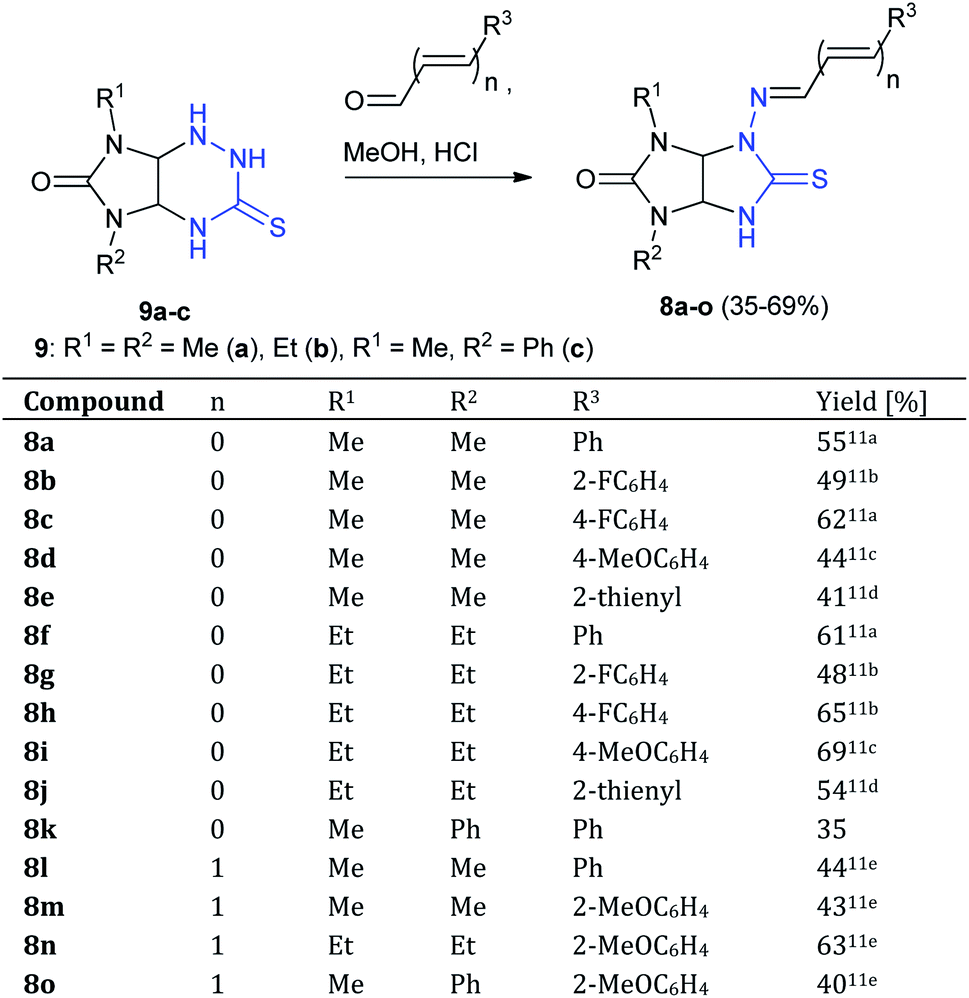 | ||
| Scheme 2 Synthesis of thioglycolurils 8a–o. | ||
To optimize the procedure for the rearrangement of thioglycolurils 8, compound 8a was chosen as a model substrate. The solvent, base/acid, reaction time, and temperature were varied. The representative results were summarized in Table 1.
|
||||
|---|---|---|---|---|
| Entry | Solvent (base/acid) | T, °C | Time, h | Yield of 10a, %b |
| a Reaction conditions: stirring the mixture of thioglycoluril 8a (1.0 mmol) and base (1.0 mmol) in methanol (20 ml) either stirring the suspension of thioglycoluril 8a (1.0 mmol) in acid (or in the mixture of MeOH with acid) (20 ml) for 2–3 h.b Isolated yield.c d = 1.170, C = 34.18%.d Compound 11a was formed in the 62% yield. | ||||
| 1 | MeOH (NaOMe) | rt | 2 | 0 |
| 2 | MeOH (KOH) | rt | 2 | 0 |
| 3 | HClc | rt | 2 | 47 |
| 4 | MeOH![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :HClc (1:10) :HClc (1:10) |
rt | 2 | 70 |
| 5 | MeOH:HClc (1:10) |
60 | 2 | 18 |
| 6 | MeOH:HClc (1:1) |
rt | 2 | 80 |
| 7 | MeOH:HClc (1:1) |
rt | 3 | 78 |
| 8 | AcOH | rt | 5 | 0 |
| 9 | AcOH | 60 | 2 | 0 |
| 10 | AcOH | 110 | 2 | 0 |
| 11 | HCOOH | 60 | 2 | 0d |
The Dimroth rearrangement was carried out upon treatment with bases, acids, heat, or, rarely, light.12 Therefore, we started our research by testing sodium methylate and potassium hydroxide (Table 1, entries 1, 2). However, no rearrangement products were obtained under these conditions. The rearrangement of thioglycoluril 8a was observed in concentrated HCl at room temperature (entry 3). Subsequently, we screened the acid reaction conditions.
Addition of methanol that dissolved starting compound 8a to hydrochloric acid (in a volume ratio of 1:10) provided an increase of the yield of the desired product 10a to 70% (entry 4). Heating the reaction mixture at 60 °C for the same time (2 h) reduced the yield of the rearrangement product by almost four times (entry 5). Increasing the share of methanol (1:1) in the reaction mixture at room temperature enhanced the yield of 10a to 80% (entry 6). Prolongation of the reaction time to 3 h didn't lead to improvement of the yield of target compound 10a (entry 7). When the reaction was carried out in an organic acid (acetic and formic acids) compound 10a was not formed at all (entries 8–11). Starting thioglycoluril 8a or known N-{4,6-dimethyl-5-oxo-2-thioxohexahydroimidazo[4,5-d]imidazol-1(2H)-yl}formamide (11a) were isolated (Fig. 3).13 Thus, the best result was achieved performing rearrangement in the mixture of equal volumes of methanol and concentrated hydrochloric acid at room temperature for 2 h (method A).
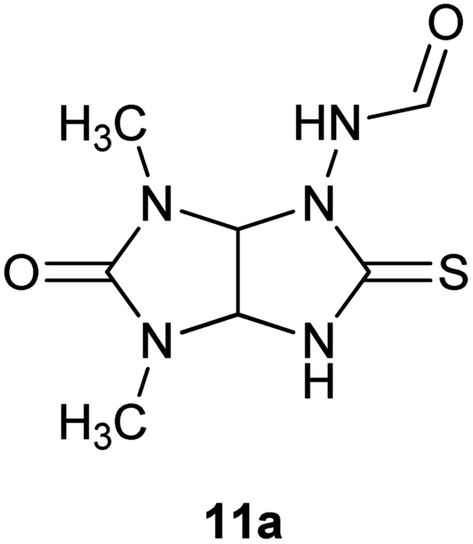 | ||
| Fig. 3 Structure of N-{4,6-dimethyl-5-oxo-2-thioxohexahydroimidazo[4,5-d]imidazol-1(2H)-yl}formamide (11a). | ||
The scope of the Dimroth-type N/S-interchange of 1,3-disubstituted 4-[(arylmethylidene)amino]thioglycolurils 8a–k was subsequently investigated under the optimized conditions. It was found that apart from model substrate 8a, 1,3-diethyl- and 3-methyl-1-phenylsubstituted thioglycolurils containing 4-benzylideneamino fragment (8f,k), 1,3-dimethyl- and 1,3-diethylsubstituted compounds with electron withdrawing (8b,c,g,h) and electron donating substituents in arylmehtylideneamino moieties at position 4 (8d,i), as well as with 4-[(thiophene-2-ylmethylidene)amino] fragment (8e,j) underwent rearrangement to afford the desired products 10a–k in 50–86% yields (Scheme 3).
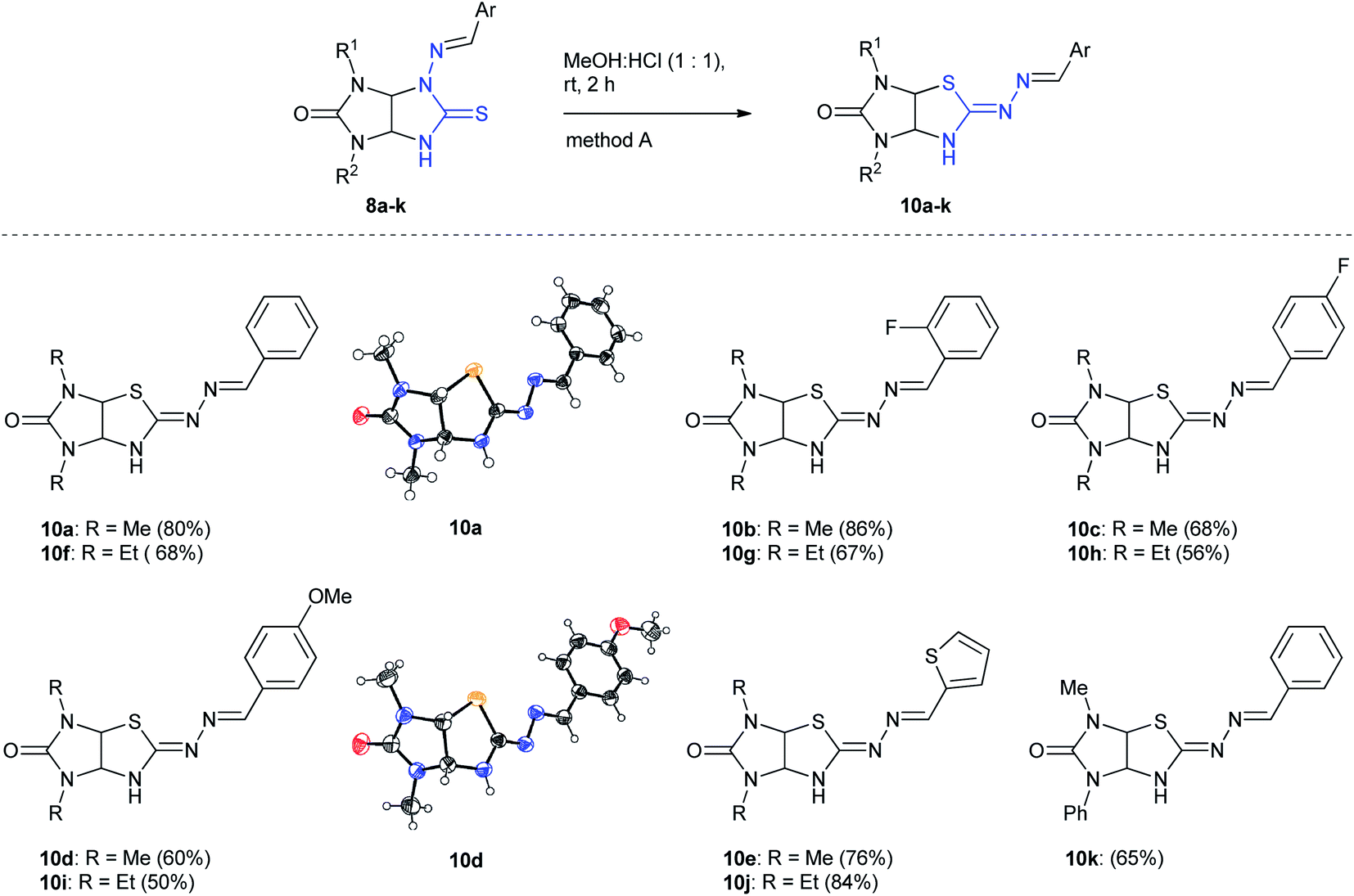 | ||
| Scheme 3 Synthesis of imidazothiazoles 10a–k by method A. | ||
Next, we studied behavior of [(E)-((E)-3-phenylallylidene)amino]thioglycolurils 8l–o under these conditions. The rearrangement of compounds 8l–o also proceeded successfully to give imidazothiazoles 10l–o in the yields of 53–80% (Scheme 4).
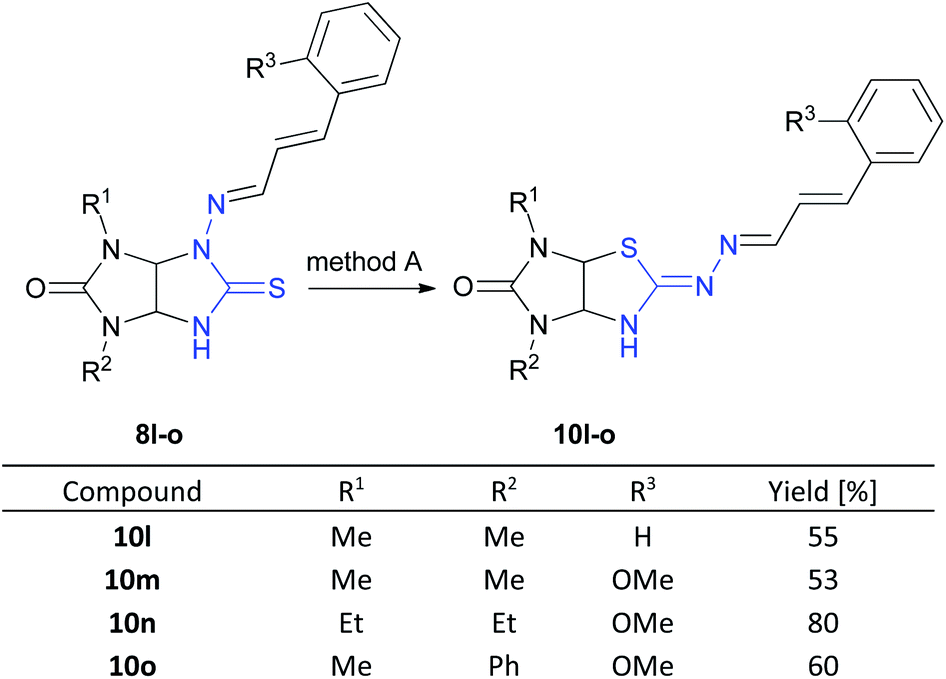 | ||
| Scheme 4 Synthesis of imidazothiazoles 10l–o (method A: MeOH:HCl (1:1), rt, 2 h). | ||
Finally, we tried to carry out these reactions in one-pot version starting from imidazotriazines 9a–c and aldehydes because both the synthesis of thioglycolurils 8 and their rearrangement into imidazothiazole derivatives 10 proceeded in methanol with hydrochloric acid (Scheme 5, method B). A mixture of imidazotriazine 9, corresponding aldehyde and methanol in the presence of hydrochloric acid (1 drop, 0.03 mL, 0.33 mmol) was refluxed with stirring for 1.5 h. Then the reaction mixture was cooled to room temperature, diluted with hydrochloric acid (in a volume ratio of 1:1), and stirred for 2 h. The yields of imidazothiazoles 10a–o obtained by one-pot method were higher (36–63%) as compared to yields of 23–45% obtained by two step method (Schemes 2–4) based on starting imidazotriazine 9.
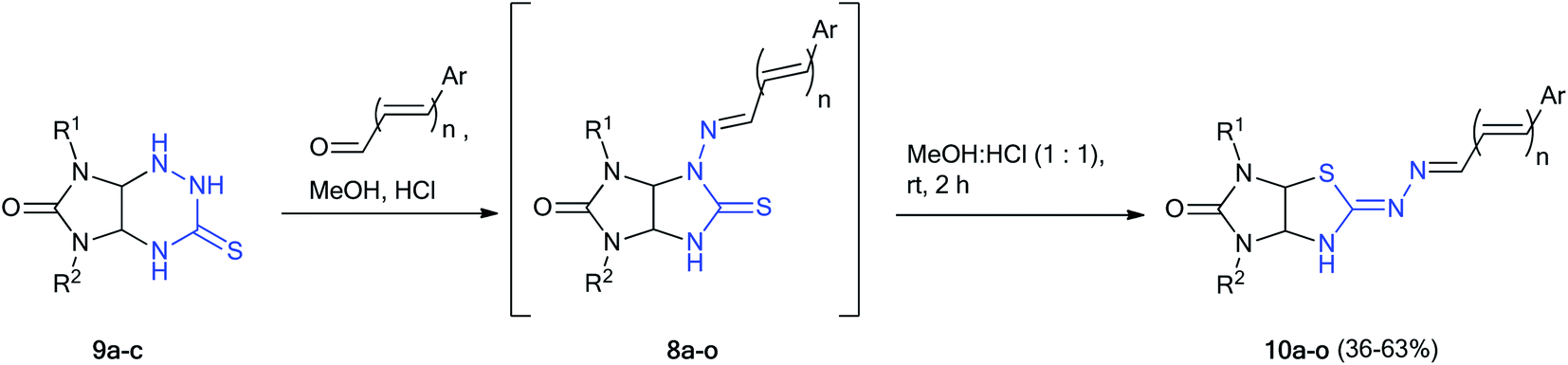 | ||
| Scheme 5 One-pot synthesis of imidazothiazoles 10a–o by method B. | ||
The structures of new compounds 10a–o were established by IR, 1H and 13C NMR and HRMS analysis. The IR spectra of imidazothiazoles 10a–o showed intense absorption bands at 1682–1727 cm−1 that are characteristic of carbonyl group. Characteristic of C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N bonds absorption bands were observed at 1599–1636 cm−1. In the 1H NMR spectra of imidazothiazoles 10a–o as compared to the spectra of 8a–o, there are upfield shifts of the signals of NH-, NCH-groups, aromatic protons, one of the bridged CH-proton, and one of the NMe-groups (Fig. 4). Protons of CH2-groups of N-ethyl fragments are diastereotopic and resonate as four separate complicated signals which partially overlap. The structures of 10a and 10d were also confirmed using single-crystal X-ray diffraction analysis. Bond lengths and angles in the structures are within normal ranges that was confirmed by the Mogul geometry check.14 The configurations of CN and NCH double bonds were Z and E, respectively. The hydrogen atoms at the C(3a) and C(6a) atoms are in the cis arrangement similar to that in thioglycolurils 8.11 So, both compounds 10 and thioglycolurils 8 are formed as a racemate. Other diastereomer with the trans arrangement of hydrogen atoms or other substituents at the bridged C(3a) and C(6a) atoms has never been observed.10,11,15
N bonds absorption bands were observed at 1599–1636 cm−1. In the 1H NMR spectra of imidazothiazoles 10a–o as compared to the spectra of 8a–o, there are upfield shifts of the signals of NH-, NCH-groups, aromatic protons, one of the bridged CH-proton, and one of the NMe-groups (Fig. 4). Protons of CH2-groups of N-ethyl fragments are diastereotopic and resonate as four separate complicated signals which partially overlap. The structures of 10a and 10d were also confirmed using single-crystal X-ray diffraction analysis. Bond lengths and angles in the structures are within normal ranges that was confirmed by the Mogul geometry check.14 The configurations of CN and NCH double bonds were Z and E, respectively. The hydrogen atoms at the C(3a) and C(6a) atoms are in the cis arrangement similar to that in thioglycolurils 8.11 So, both compounds 10 and thioglycolurils 8 are formed as a racemate. Other diastereomer with the trans arrangement of hydrogen atoms or other substituents at the bridged C(3a) and C(6a) atoms has never been observed.10,11,15
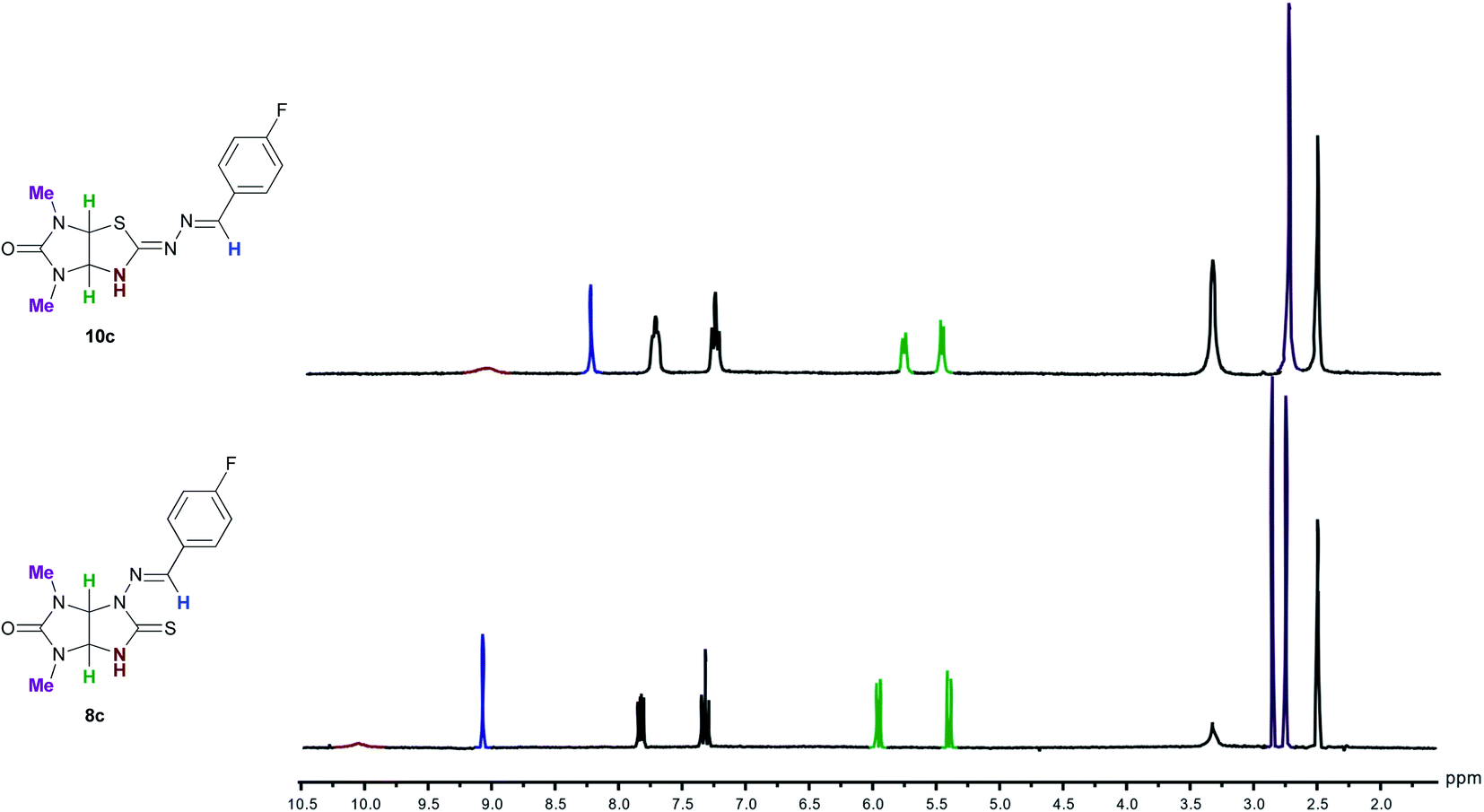 | ||
| Fig. 4 Comparison of the 1H NMR spectra of thioglycoluril 8c and imidazothiazole 10c in the region 2.0–10.5 ppm in DMSO-d6. | ||
A plausible rearrangement mechanism is depicted in Scheme 6. Probably, rearrangement is a result of imidazolidinethione ring opening of compound 8 upon treatment with acid and recyclization of the thiazolidine ring involving other nucleophilic center, sulfur atom. The reaction proceeds towards the product with a bulk substituent localized on an exocyclic heteroatom.
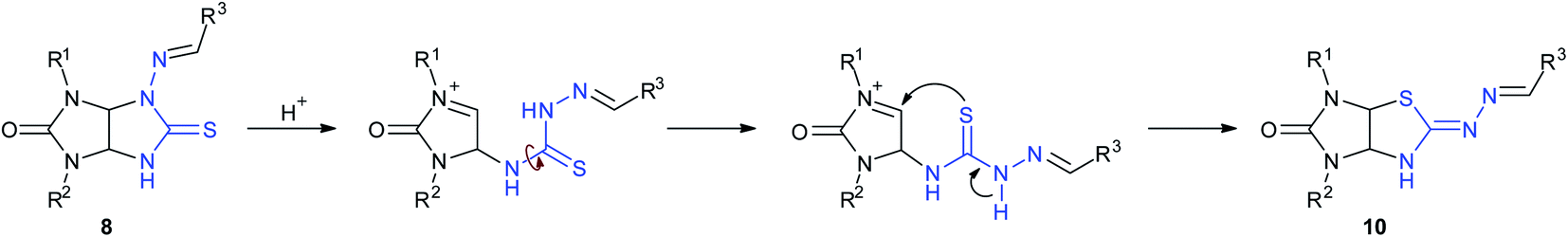 | ||
| Scheme 6 Plausible rearrangement mechanism. | ||
As we shown earlier,11c formation of thioglycolurils 8 accompanied by side process: instead of a recyclization step, the elimination of aldehyde thiosemicarbazone can occur. Apparently, the process takes place also during the rearrangement under study that result in decrease of the product yields.
Conclusion
We have developed an original method for the synthesis of earlier unavailable 2-hydrazonoimidazo[4,5-d]thiazolone derivatives based on the one-pot acid-induced sequence of hydrazone formation from 3-thioxoperhydroimidazo[4,5-e]-1,2,4-triazinones and aromatic aldehydes or 3-phenylacroleine derivatives, triazine ring contraction to imidazolidine one, and Dimroth-type N/S-interchange of N-aminoimidazolidine-2-thione ring into 2-hydrazonothiazolidine fragment. The synthesized structures represent a new class of promising bioactive heterocyclic compounds. Investigations of antiproliferative activity of the prepared products against cancer cell lines are in progress.Conflicts of interest
There are no conflicts to declare.Acknowledgements
High resolution mass spectra were recorded in the Department of Structural Studies of N.D. Zelinsky Institute of Organic Chemistry, Moscow. Crystal structure determination for compound 10a was performed in the Department of Structural Studies of N.D. Zelinsky Institute of Organic Chemistry, Moscow. X-ray diffraction experiment of 10d was performed with financial support from the Ministry of Science and Higher Education of the Russian Federation using equipment at the Center for Molecular Composition Studies of INEOS RAS.Notes and references
- For selected reviews, see: (a) E. K. Davison and J. Sperry, J. Nat. Prod., 2017, 80, 3060 CrossRef CAS PubMed; (b) L. Konnert, F. Lamaty, J. Martinez and E. Colacino, Chem. Rev., 2017, 117, 13757 CrossRef CAS PubMed; (c) N. A. Meanwell, Adv. Heterocycl. Chem., 2017, 123, 245 CrossRef CAS; (d) S. Pathania, R. K. Narang and R. K. Rawal, Eur. J. Med. Chem., 2019, 180, 486 CrossRef CAS PubMed; (e) N. N. Makhova, L. I. Belen’kii, G. A. Gazieva, I. L. Dalinger, L. S. Konstantinova, V. V. Kuznetsov, A. N. Kravchenko, M. M. Krayushkin, O. A. Rakitin, A. M. Starosotnikov, L. L. Fershtat, S. A. Shevelev, V. Z. Shirinian and V. N. Yarovenko, Russ. Chem. Rev., 2020, 89, 55 CrossRef CAS; (f) L. I. Belen’kii, G. A. Gazieva, Y. B. Evdokimenkova and N. O. Soboleva, Adv. Heterocycl. Chem., 2020, 132, 385 CrossRef.
- (a) A. León-Del-Río, J. Inherit. Metab. Dis., 2019, 42, 647 CrossRef; (b) R. Ahmed, E. Spikings, S. Zhou, A. Thompsett and T. Zhang, J. Immunol. Methods, 2014, 406, 143 CrossRef CAS PubMed.
- S. Langhammer, U. Fiebig, R. Kurth and J. Denner, Intervirology, 2011, 54, 78 CrossRef CAS PubMed.
- (a) C. De Monte, S. Carradori, B. Bizzarri, A. Bolasco, F. Caprara, A. Mollica, D. Rivanera, E. Mari, A. Zicari, A. Akdemir and D. Secci, Eur. J. Med. Chem., 2016, 107, 82 CrossRef CAS PubMed; (b) S. Carradori, B. Bizzarri, M. D'Ascenzio, C. De Monte, R. Grande, D. Rivanera, A. Zicari, E. Mari, M. Sabatino, A. Patsilinakos, R. Ragno and D. Secci, Eur. J. Med. Chem., 2017, 140, 274 CrossRef PubMed.
- M. D'Ascenzio, B. Bizzarri, C. De Monte, S. Carradori, A. Bolasco, D. Secci, D. Rivanera, N. Faulhaber, C. Bordón and L. Jones-Brando, Eur. J. Med. Chem., 2014, 86, 17 CrossRef PubMed.
- A. A. Ghoneim and A. G. A. Hassan, Polycycl. Aromat. Comp., 2021 DOI:10.1080/10406638.2020.1866035.
- A. N. Kravchenko, M. M. Antonova, V. V. Baranov and Yu. V. Nelyubina, Synlett, 2015, 26, 2521 CrossRef CAS.
- (a) L. V. Saloutina, A. Ya. Zapevalov, M. I. Kodess, I. N. Ganebnykh, V. I. Saloutin and O. N. Chupakhin, J. Fluorine Chem., 2018, 212, 144 CrossRef CAS; (b) L. V. Saloutina, A. Ya. Zapevalov, M. I. Kodess, P. A. Slepukhin, I. N. Ganebnykh, V. I. Saloutin and O. N. Chupakhin, J. Fluorine Chem., 2019, 227, 109362 CrossRef CAS.
- A. Takamizava and S. Matsumoto, Chem. Pharm. Bull., 1973, 21, 1300 CrossRef.
- P. A. Duspara, C. F. Matta, S. I. Jenkins and P. H. M. Harrison, Org. Lett., 2001, 3, 495 CrossRef CAS PubMed.
- (a) G. A. Gazieva, P. A. Poluboyarov, L. D. Popov, N. G. Kolotyrkina, A. N. Kravchenko and N. N. Makhova, Synthesis, 2012, 44, 3366 CrossRef CAS; (b) G. A. Gazieva, T. V. Nechaeva, N. N. Kostikova, N. V. Sigay, S. A. Serkov and S. V. Popkov, Russ. Chem. Bull., 2018, 67, 1059 CrossRef CAS; (c) G. A. Gazieva, L. V. Anikina, S. A. Pukhov, T. B. Karpova, Yu. V. Nelyubina and A. N. Kravchenko, Mol. Diversity, 2016, 20, 837 CrossRef CAS PubMed; (d) G. A. Gazieva, T. B. Karpova, L. D. Popov, Yu. V. Nelyubina and A. N. Kravchenko, J. Heterocycl. Chem., 2015, 52, 1390 CrossRef CAS; (e) G. A. Gazieva, L. V. Anikina, T. V. Nechaeva, S. A. Pukhov, T. B. Karpova, S. V. Popkov, Yu. V. Nelyubina, N. G. Kolotyrkina and A. N. Kravchenko, Eur. J. Med. Chem., 2017, 140, 141 CrossRef CAS PubMed.
- (a) V. A. Mamedov, N. A. Zhukova and M. S. Kadyrova, Chem. Heterocycl. Compd., 2021, 57, 342 CrossRef CAS PubMed; (b) V. F. Ferreira, T. de B. da Silva, F. P. Pauli, P. G. Ferreira, L. da S. M. Forezi, C. G. de S. Lima and F. de C. da Silva, Curr. Org. Chem., 2020, 24, 1999 CrossRef CAS; (c) A. Krajczyk and J. Boryski, Curr. Org. Chem., 2017, 21, 2515 CrossRef CAS; (d) E. S. H. El Ashry, S. Nadeem, M. R. Shah and Y. El Kilany, Adv. Heterocycl. Chem., 2010, 101, 161 CrossRef CAS.
- G. A. Gazieva, T. B. Karpova, T. V. Nechaeva, Yu. V. Nelyubina, I. E. Zanin and A. N. Kravchenko, Synlett, 2017, 28, 858 CrossRef CAS.
- I. J. Bruno, J. C. Cole, M. Kessler, J. Luo, W. D. S. Motherwell, L. H. Purkis, B. R. Smith, R. Taylor, R. I. Cooper, S. E. Harris and A. G. Orpen, J. Chem. Inf. Comput. Sci., 2004, 44, 2133 CrossRef CAS PubMed.
- S. Y. Panshina, O. V. Ponomarenko, A. A. Bakibaev and V. S. Malkov, J. Struct. Chem., 2020, 61, 1315 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d1ra05568b |
| This journal is © The Royal Society of Chemistry 2021 |