
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceStudy on the synthesis and thermal stability of silicone resins reinforced by Si–O–Ph cross-linking†
Huadong Zhanga,
Zhongyi Yana,
Zhizhou Yang *a,
Jinshui Yaoa,
Qiuhong Mub,
Dan Pengb and
Hui Zhaoac
*a,
Jinshui Yaoa,
Qiuhong Mub,
Dan Pengb and
Hui Zhaoac
aSchool of Materials Science and Engineering, Qilu University of Technology (Shandong Academy of Sciences), Jinan 250353, China. E-mail: yzz@qlu.edu.cn
bShandong Provincial Key Laboratory of Special Silicone-Containing Materials, Advanced Materials Institute, Qilu University of Technology (Shandong Academy of Sciences), Jinan 250014, China
cSchool of Chemical Engineering, Sichuan University, Chengdu 610065, China
First published on 20th September 2021
Abstract
A novel silicone resin (SR-OH) containing phenolic hydroxyl (Ph–OH) groups was designed and synthesized via co-hydrolysis/condensation and catalytic hydrogenation. During the curing process, the cross-linking degree of the resin was further increased by the Si–O–Ph bonds formed by the reaction of the Ph–OH and terminal Si–OH groups. Thermogravimetric analysis (TGA) showed that the cured resin product exhibited excellent thermal and thermo-oxidative stability, which was much higher than that of a typical methyl phenyl silicone resin (SR-Ph). The temperature at which 5% weight loss occurs (Td5) was up to 606 °C (nitrogen) and 542 °C (air), and its char yield at 800 °C was 91.1% and 85.3% in nitrogen and air, respectively. The significant improvement in thermal stability was mainly attributed to the formation of Si–O–Ph bonds which not only increases the cross-linking degree of the resin but also significantly prevents degradation by the ‘back-biting’ and oxidative cleavage.
1. Introduction
In recent decades, the wide application of resin-based materials has contributed to the rapid development of aerospace, electronics and other technological industries. Thermosetting resins exhibit unique advantages in mechanical and electrical properties, thermal stability, and strength-to-mass ratio due to their inherent molecular structure and composition.1–5 With the advancement and development of technology, thermosetting resin materials play an important role in materials used in extreme environmental conditions. For example, high-temperature resistant resins are used as coatings,6 adhesives7,8 and matrix components9 to protect aircraft fuselage against thermal shock caused by atmospheric friction, and to ensure long-term and stable operation of engines and electronic equipment at high-temperature. Among thermosetting polymeric materials, polyimide,10–12 bismaleimide,13–15 phthalonitrile16,17 and silicone resins have become important paragons in the field of high-temperature resistance due to their excellent thermal stability. Since the bond energy of the Si–O bond (460.5 kJ mol−1) is much higher than that of the C–C (304.0 kJ mol−1) and C–O (358.0 kJ mol−1) bonds, silicone resins show improved thermal stability compared to other resins mentioned above with C–C and C–O bonds as the framework.18,19 In addition, the unique semi-inorganic molecular structure of silicone resins also gives it excellent electrical insulation, weather resistance and good hydrophobicity, which makes it suitable for use in aerospace, electronic technologies, thermal protection and ablation resistance. However, the development in these areas puts forward a higher demand for heat-resistant silicone resins. To be suitable for harsher environments and expand their applications, increasing efforts have been made to further improve the thermal stability of silicone resins.The addition of inorganic fillers (such as SiO2,20 Al2O3,21,22 graphene23 and carbon nanotubes24) is a simple and practical method to improve the thermal stability of silicone resins. However, the agglomeration of inorganic particles in the resin matrix may lead to inhomogeneous dispersion and phase separation. In addition, the methyl group in silicone resins has been replaced by a phenyl group to improve its thermal stability.19,25,26 The introduction of functional groups into the aromatic ring that participates in the cross-linking reaction has been a successful method to improve the degree of cross-linking. Huang et al.27 introduced trifluorovinyl ether (TFVE) groups into the silane monomer and enhanced the cross-linking through the [2 + 2] ring polymerization of TFVE to improve the thermal stability of the silicone resin. Although increasing the content of TFVE could effectively increase Td5 and Td10, the final char yield (800 °C) decreased significantly.
The degradation of silicone resins is mainly attributed to the breaking and rearrangement of Si–O–Si bonds and the oxidative degradation of the side groups at high temperatures. The ‘back-biting’ reaction initiated by terminal Si–OH can cause the silicone resin to undergo ‘unlocking’ degradation and produce low molecular weight rings.28,29 For silicone resins containing phenyl, the terminal Si–OH also promotes the breakage of the Si–Ph bond, leading to branching of the main chain.30 The amount of Si–OH groups in silicone resin can be effectively reduced by using mono-functional siloxane25,31 (such as hexamethyldisiloxane and 1,1,3,3-tetramethyl divinyl disiloxane) as an end-capping agent. In addition, POSS32,33 and polysilazanes34,35 have been reported to react with Si–OH to reduce the ‘back-biting’ effect. However, the improvement in the heat resistance of the silicone resin by these methods has been met with limited success.
There is a current need for the design and synthesis of novel silicone resins with a high-temperature resistant threshold. According to previous reports, Si–O–C groups and phenol groups can directly undergo transesterification under thermal conditions. Li36 and Yun37 prepared silicone/phenolic resins containing a Si–O–Ph structure through the transesterification reaction of methyltrimethoxysilane or methyltriethoxysilane oligomers with the phenolic resin. The results showed that the formation of Si–O–Ph groups effectively protected the phenolic hydroxyl group from oxidation, improving its stability to thermal oxidation. Gao et al.38 reported that at temperatures higher than 200 °C, the Si–O–CH2–CH3 in the benzoxazine containing silicon formed a reversible Si–O–Ph bond with the phenol group increasing the cross-linking degree of the resin and thus its thermal stability. However, these methods are only applicable to the preparation of hybrid resin materials, and excessive organic components in the hybrid resin limit its thermal threshold. Taking into account the chemical properties of Si–O–C and Si–OH, we envisioned that Si–O–Ph structures may also be formed by condensation of Si–OH with Ph–OH groups. Therefore, the introduction of Ph–OH groups into the silicone resin may be a feasible solution to improve its thermal stability. In our work, a silicone resin (SR-OH) containing Ph–OH groups was successfully synthesized via co-hydrolysis/condensation and catalytic hydrogenation. As additional active points, Ph–OH groups interacted with terminal Si–OH groups to give Si–O–Ph cross-linked polymeric material. The formation of the Si–O–Ph structure is effective in increasing the degree of resin cross-linking and reducing the content of terminal hydroxyl groups. The curing behavior of the resin was studied, and the effect of the installation of Ph–OH groups on the thermal properties of the resin was characterized in detail.
2. Experimental
2.1 Materials
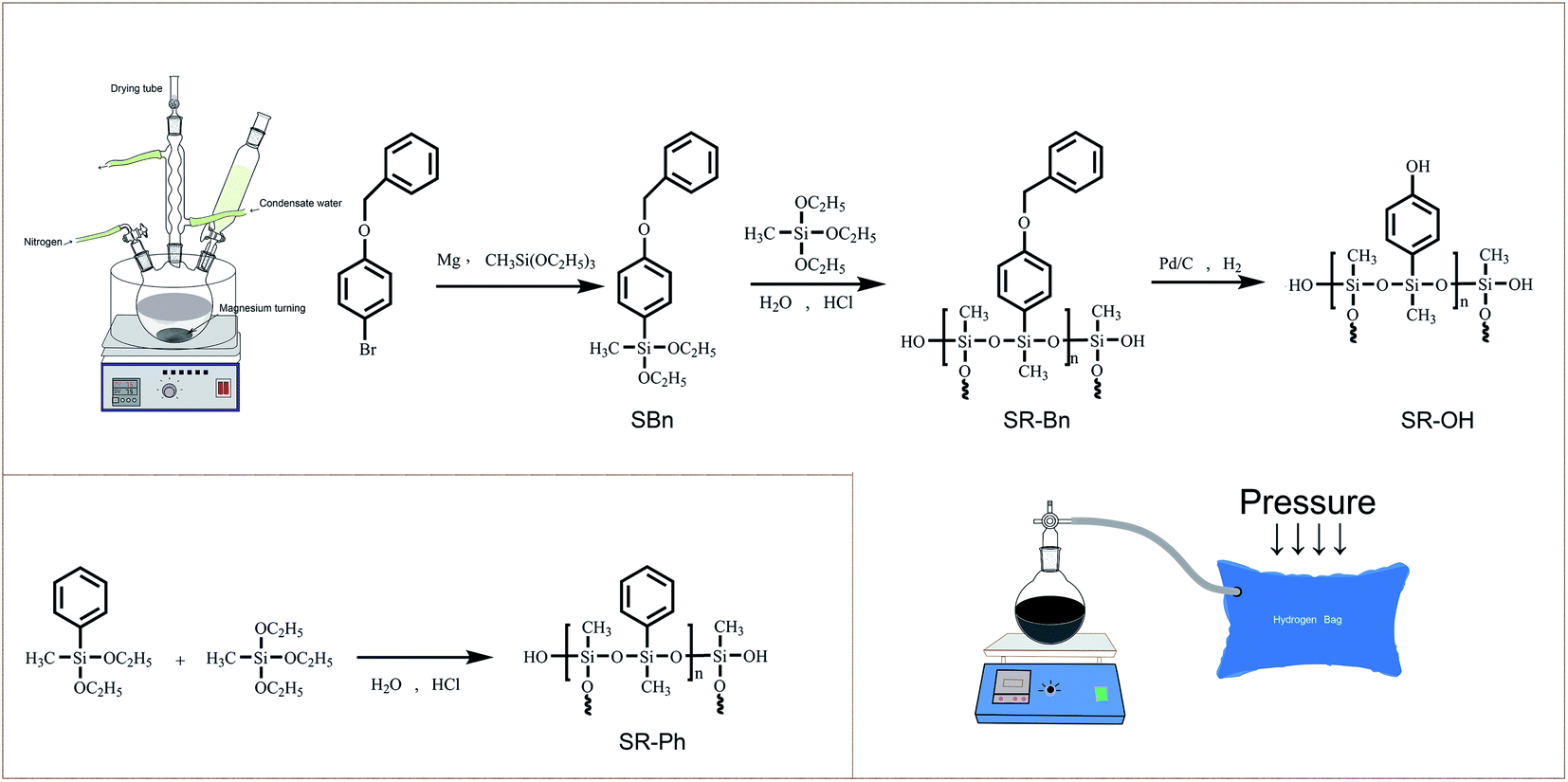
1-Bromo-4-(phenylmethoxy)benzene was purchased from Jiuding Chemistry Co., Ltd (Shanghai, China). Methyltriethoxysilane (MTES) and palladium on activated carbon (10% Pd) were obtained from Shanghai Macklin Biochemical Co., Ltd. Diethoxymethylphenylsilane (DEMPS) was purchased from Shanghai Aladdin Biochemical Technology Co., Ltd (Shanghai, China). Tetrahydrofuran (THF) was dried and distilled with sodium-diphenylketone for use. Other reaction reagents were commercial available. Unless otherwise specified, all chemical reagents were used without further purification.2.2 Synthesis of 1-(phenylmethoxy)-4-(diethoxymethylsilyl)-benzene (SBn)
Magnesium (4.43 g, 0.18 mol), methyltriethoxysilane (32.52 g, 0.18 mol), THF (80 mL) and a small amount of iodine crystals were added to a 250 mL three-neck flask equipped with magnetic agitator, condenser and dropping funnel. The mixture solution was heated to 75 °C under a nitrogen atmosphere. Followed THF (120 mL) solution of 1-bromo-4-(phenylmethoxy)-benzene (40.00 g, 0.15 mol) was added dropwise into reaction system over 2 h. After 12 h of stirring, THF was removed and n-hexane (400 mL) was added. The resulting mixture was filtered to obtain a filtrate. Then the solvent was removed and crude product was purified by vacuum distillation to afford a colorless liquid (26.89 g, 54.21%). FT-IR (KBr, cm−1): 1502, 1562, 1596 (aromatic rings), 1246 (Ar–O–C), 1081, 1105 (Si–O). 1H NMR (400 MHz, CDCl3, ppm): 0.36 (s, 3H, Si–CH3), 1.26 (t, 6H, O–CH2–CH3), 3.83 (m, 4H, Si–O–CH2–), 5.10 (s, 2H, O–CH2–Ph), 7.00–7.60 (9H, H of aromatic rings). 13C NMR (400 MHz, CDCl3, ppm): −3.88 (Si–CH3), 18.52 (O–CH2–CH3), 58.53 (Si–O–CH2–), 69.74 (O–CH2–Ph), 114.61, 126.37, 127.55, 128.04, 128.67, 135.80, 137.10, 160.56 (12C, C of aromatic rings). HR-MS (ESI, m/z): calculated for [C18H24O3Si + H]+ 317.1568, found 317.1566.2.3 Synthesis of the silicone resin containing benzyl groups (SR-Bn)
The silicone resin containing benzyl (SR-Bn) was synthesized by co-hydrolysis of SBn and MTES. Firstly, SBn (4.75 g, 15 mmol) and MTES (2.67 g, 15 mmol) were dissolved in THF (30 mL), then hydrochloric acid solution (6.75 g, pH = 1) was added while stirring, and the reaction was carried out at room temperature for 24 hours and 60 °C for another 24 hours. Subsequently, THF was removed and the product was extracted with ethyl acetate, washed with water, and dried with anhydrous calcium chloride for 24 hours. Finally, ethyl acetate was removed and the viscous silicone precursor was obtained.A typical methyl phenyl silicone resin (SR-Ph) was synthesized from DEMPS and MTES in a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 molar ratio, using the same co-hydrolysis/condensation method as SR-Bn.
:1 molar ratio, using the same co-hydrolysis/condensation method as SR-Bn.
2.4 Synthesis of the silicone resin containing phenol groups (SR-OH)
A THF (150 mL) solution containing SR-Bn (5.00 g) and 10% palladium on carbon (300 mg) was placed in a 500 mL round-bottom flask equipped with tee valve and connected to a hydrogen gas bag. This was placed under an atmosphere of hydrogen and allowed to react with stirring for 72 h at room temperature. The catalyst (Pd/C) was removed by filtration, and the solvent was removed by rotary evaporator to afford a yellowish viscous silicone resin.2.5 Curing of the silicone resins
The synthesized silicone resins were cured by direct heating. Each silicone resin was transferred into a mold and cured at 90 °C for 2 hours, followed by cured at 120 °C, 150 °C, 180 °C, 210 °C, 240 °C, 270 °C, 300 °C and 350 °C for 1 hour respectively to obtain the cured silicone resin material.2.6 Thermal treatment of SR-OH cured at 350 °C
The SR-OH samples cured at 350 °C were transferred into an alumina crucible and placed in a tubular furnace, where they underwent thermal treatment at different temperatures (400 °C, 500 °C, 550 °C, 600 °C, 700 °C and 800 °C) for 1 h under nitrogen and air.2.7 Characterization
Fourier transform infrared (FT-IR) spectra were measured using a Nicolet iS20 spectrometer with KBr pellets at a resolution of 2 cm−1 from 4000 to 400 cm−1.Proton nuclear magnetic resonance (1H NMR) and 29Si nuclear magnetic resonance (29Si NMR) spectra were recorded on a Bruker AVANCE II 400 spectrometer using CDCl3 and (CD3)2SO as solvents and (CH3)4Si as internal reference. 13C nuclear magnetic resonance (13C NMR) spectrum was obtained on a Bruker 400M spectrometer using CDCl3 as solvent. Solid-state 29Si NMR spectra were obtained on an Agilent 600 M spectrometer.
High resolution mass spectrum (HR-MS) was measured on a Q Exactive Focus mass spectrometer (Thermo Fisher Scientific, USA) using acetonitrile as solvent.
The molecular weight of the resin was recorded using a gel permeation chromatography (GPC) (WH2-07, WYATT, USA) at a flow rate of 1.00 mL min−1 with DMF as the solvent.
Different scanning calorimetry (DSC) curves were obtained on a DSC Q10 instrument (TA Instruments, USA) with a heating rate of 10 °C min−1 under a nitrogen flow rate of 50 mL min−1.
Thermogravimetric analysis (TGA) was performed using an HCT-4 synchronous thermal analyzer (Hengjiu, China) at a heating rate of 10 °C min−1 under a gas flow rate of 50 mL min−1.
2.8 TGA-FTIR analysis
TGA-FTIR technique was used to identify the composition of gaseous products during degradation. A powder sample of approximately 5 mg was added to a suitable ceramic crucible and placed in a specific location on the NETZSCH STA 449 F5 Jupiter Thermogravimetric analyzer. The temperature was gradually increased from 50 °C to 800 °C at a heating rate of 10 °C min−1 under a nitrogen flow rate of 50 mL min−1. The volatile decomposition products generated in the process of thermal gravimetric loss were introduced into the optical path of the Nicolet iS10 FT-IR spectrometer through the pipeline and gas pool by purging gas. Continuous FT-IR spectra with a detection range from 4000 to 400 cm−1 were obtained.3. Results and discussion
3.1 Structural characterization of the monomer and silicone resins
The synthetic routes of the silane monomer and the three silicone resins are shown in Scheme 1. The structures were preliminarily characterized by FT-IR spectra, as shown in Fig. 1. The SR-Bn was synthesized by the co-hydrolysis/condensation reaction of SBn and MTES in a 1:1 molar ratio. The molecular weight of SR-Bn was obtained by GPC, and the results showed that its number-averaged molecular weight and weight-averaged molecular weight were 1882 and 2012 g mol−1, respectively. According to the FT-IR spectrum of SR-Bn, the disappearance of the characteristic peak for Si–O–CH2–CH3 group at 940 cm−1 and the appearance of the characteristic absorption peak for Si–OH group around 3400 cm−1 revealed the occurrence of hydrolysis reaction. The broad peak at 1000–1100 cm−1 corresponds to the Si–O–Si group, indicating condensation between the Si–OH groups. SR-OH was synthesized by catalytic hydrogenation of SR-Bn. In the FT-IR spectrum of SR-OH, the absorption peak around 3400 cm−1 was significantly more pronounced compared with that in SR-Bn. In addition, the characteristic peak of the Ar–O–C functional group was absent at 1246 cm−1. These changes confirmed the deprotection of the benzyl group and the presence of the phenolic hydroxyl group. The characteristic absorption peaks from Si–OH at 3400 cm−1, Si–O–Si at 1000–1100 cm−1, benzene ring at 1500–1600 cm−1, and Si–CH3 at 1428 cm−1 and 1260 cm−1 were also found in the FT-IR spectrum of SR-Ph, which indicated that the hydrolysis and condensation reactions had occurred.
 | ||
| Scheme 1 Synthetic routes of the silane monomer (SBn) and three silicone resins (SR-Bn, SR-OH and SR-Ph). | ||
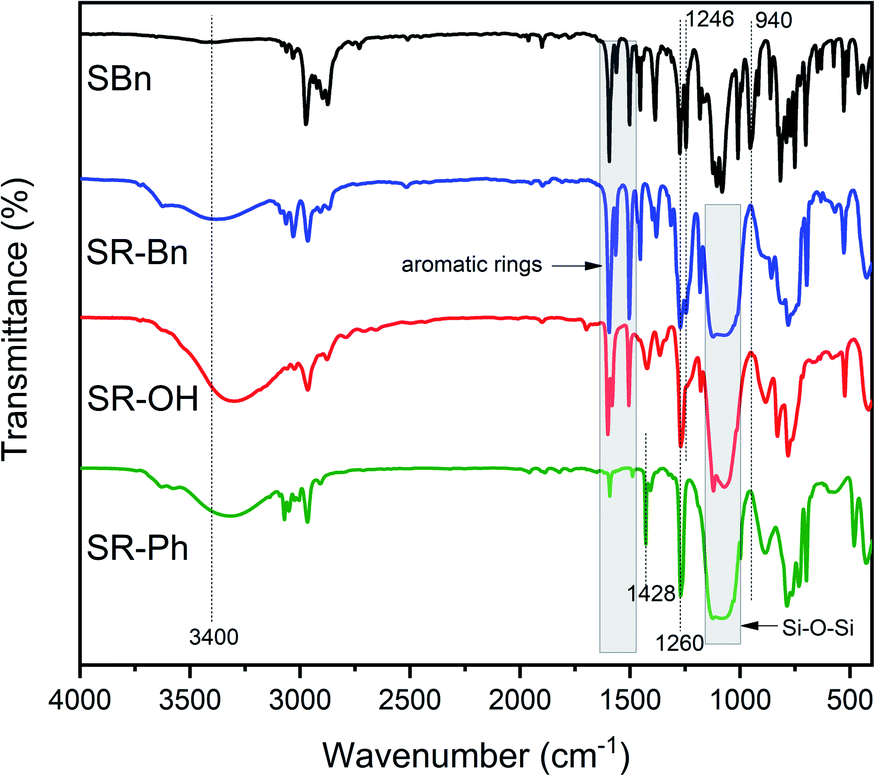 | ||
| Fig. 1 FT-IR spectra of SBn, SR-Bn, SR-OH and SR-Ph. | ||
The structures of the silane monomer and three silicone resins were further confirmed by 1H NMR, as shown in Fig. 2. In the 1H NMR spectrum of SBn, the peaks from 7.01 ppm to 7.61 ppm correspond to the C–H protons on the aromatic rings. The methylene peak in Ar–O–CH2– appears at 5.10 ppm. The resonance signals of methylene and methyl in Si–O–CH2–CH3 arise at 3.83 ppm and 1.26 ppm, respectively. The signal at 0.36 ppm belongs to the methyl group directly attached to the silicon atom. From the 1H NMR spectrum of SR-Bn, the absence of resonance signals at 1.26 ppm and 3.83 ppm indicates that the ethoxy groups in both monomers have been hydrolyzed completely. According to the 1H NMR spectrum of the SR-OH, the methylene peak of the Ar–O–CH2 group was absent at 5.10 ppm, indicating deprotection of the benzyl group after catalytic hydrogenation. In addition, the resonance peak of the phenolic hydroxyl appeared at 9.5 ppm. In the spectrum of SR-Ph, the proton peaks on both the benzene ring (7.00–8.00 ppm) and Si–CH3 (around 0.36 ppm) are present. Furthermore, the actual ratio of R and Me calculated by 1H NMR data were close to the theoretical values (Table 1), which further indicated successful synthesis of the monomer and the three silicone resins.
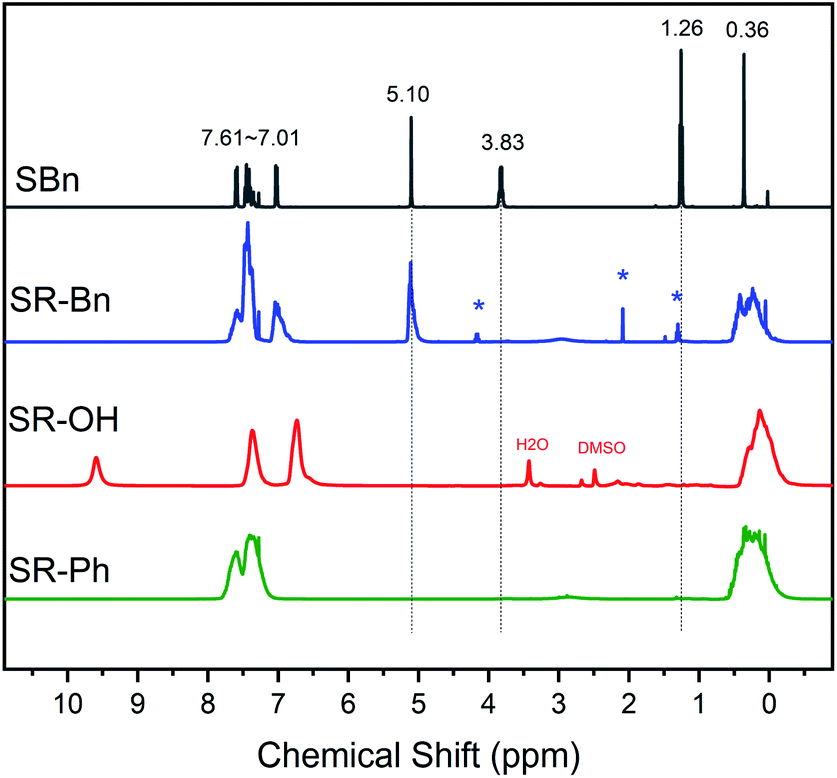 | ||
| Fig. 2 1H NMR spectra of SBn, SR-Bn, SR-OH and SR-Ph. (SBn, SR-Bn and SR-Ph: CDCl3 as solvent; SR-OH: DMSO as solvent; *: ethyl acetate). | ||
3.2 Curing behavior of silicone resins
Scheme 2 illustrates the curing process of SR-OH. To study the structural changes of SR-OH during the curing process, the FT-IR spectra of the samples cured at different temperatures were analyzed and are shown in Fig. 3. Upon increasing the cured temperature from 90 °C to 150 °C, the intensity of the peak at 3400 cm−1 decreased, indicating further cross-linking between the terminal Si–OH. A new peak at 936 cm−1 corresponding to Si–O–Ph bond appeared at 150 °C, indicating the condensation reaction of Si–OH and Ph–OH groups at this temperature. The temperature was increased from 150 °C to 350 °C and the reaction was monitored by FT-IR. The intensity of the peak at 3400 cm−1 further decreased, with a gradual increase of a peak at 936 cm−1 corresponding to Si–O–Ph, indicating the increased amount of formation of Si–O–Ph bonds. In order to further confirm the formation of the Si–O–Ph structure, the solid-state 29Si NMR spectra of the samples cured at 270 °C and 350 °C were obtained. As shown in Fig. 4, the peak at −36.37 ppm corresponded to the PhCH3Si(O–Si)2 structure, and the peak at −69.31 ppm can be attributed to the Si–O–Ph and CH3Si(O–Si)3 structures, based on analyses from previous literature reports.36,37 Compared with the sample cured at 270 °C, the peak at −69.31 ppm for the sample cured at 350 °C was stronger, further indicating that the higher curing temperature increased the amount of formation of Si–O–Ph bonds.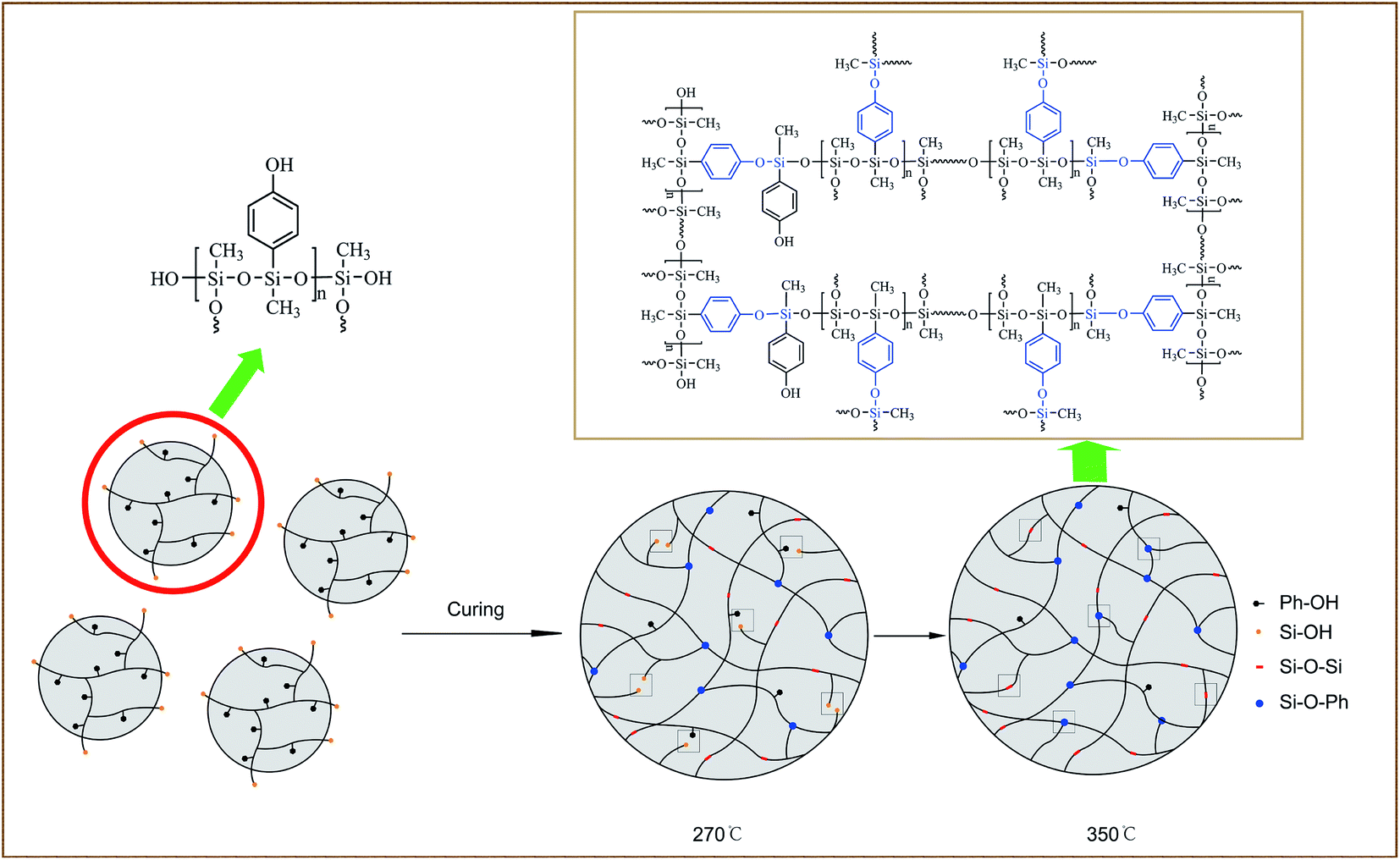 | ||
| Scheme 2 The curing process of the silicone resin containing phenolic hydroxyl groups. | ||
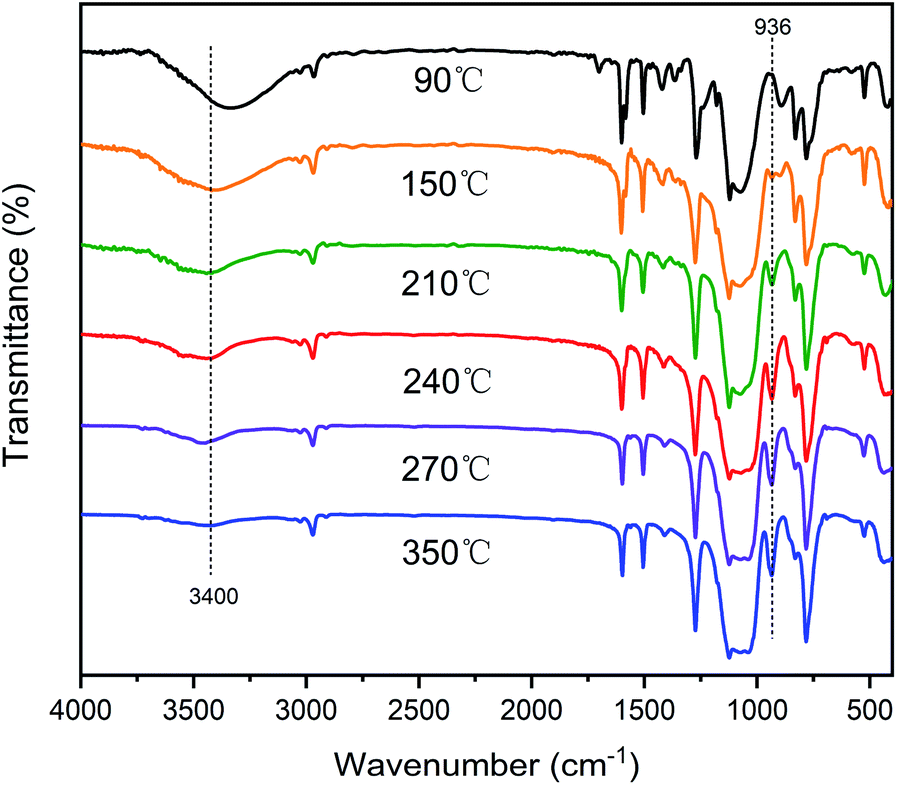 | ||
| Fig. 3 FT-IR spectra of the SR-OH at different temperatures during the curing process. | ||
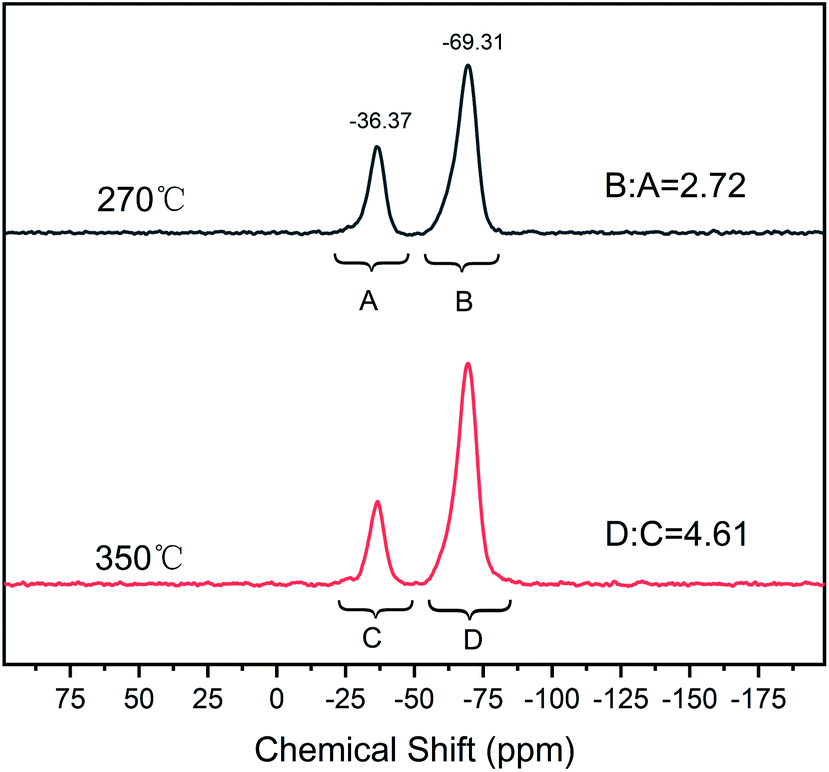 | ||
| Fig. 4 Solid-state 29Si NMR spectra of SR-OH samples cured at 270 °C and 350 °C. | ||
In addition, DSC curves of the three resins were obtained and the results are shown in Fig. 5. Compared with the other two silicone resins, the SR-OH resin shows a stronger exothermic peak. During the curing process, the initial exothermic temperature of SR-OH was about 150 °C, which is consistent with the temperature of initial formation of Si–O–Ph bond in the FT-IR spectra (Fig. 3).
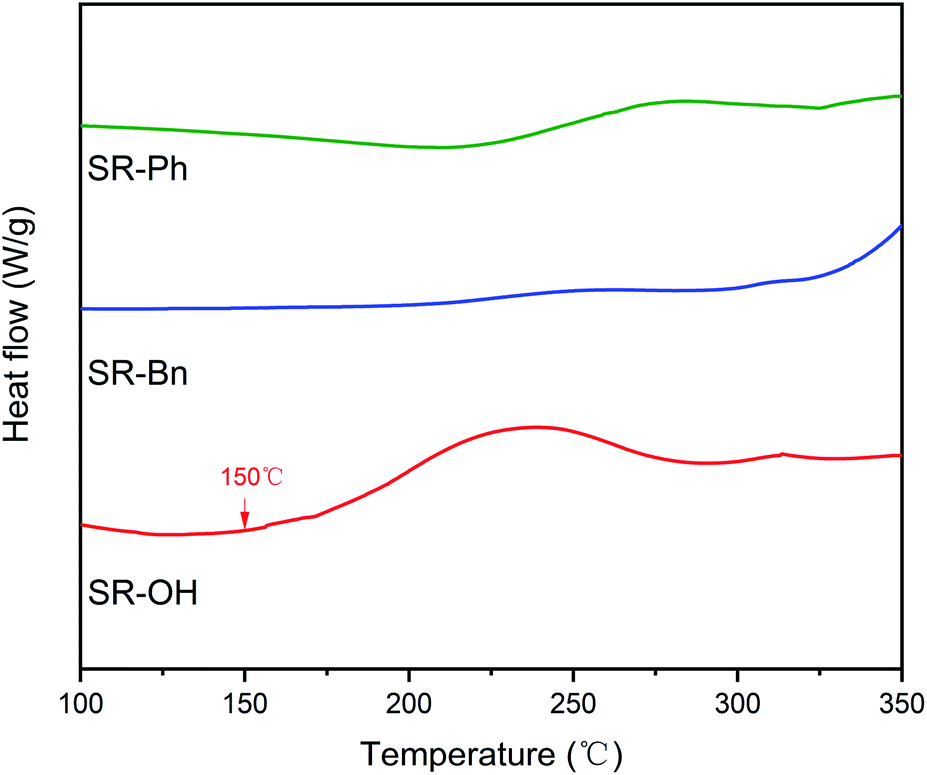 | ||
| Fig. 5 DSC curves of the three silicone resins. | ||
3.3 Thermal performance of silicone resins
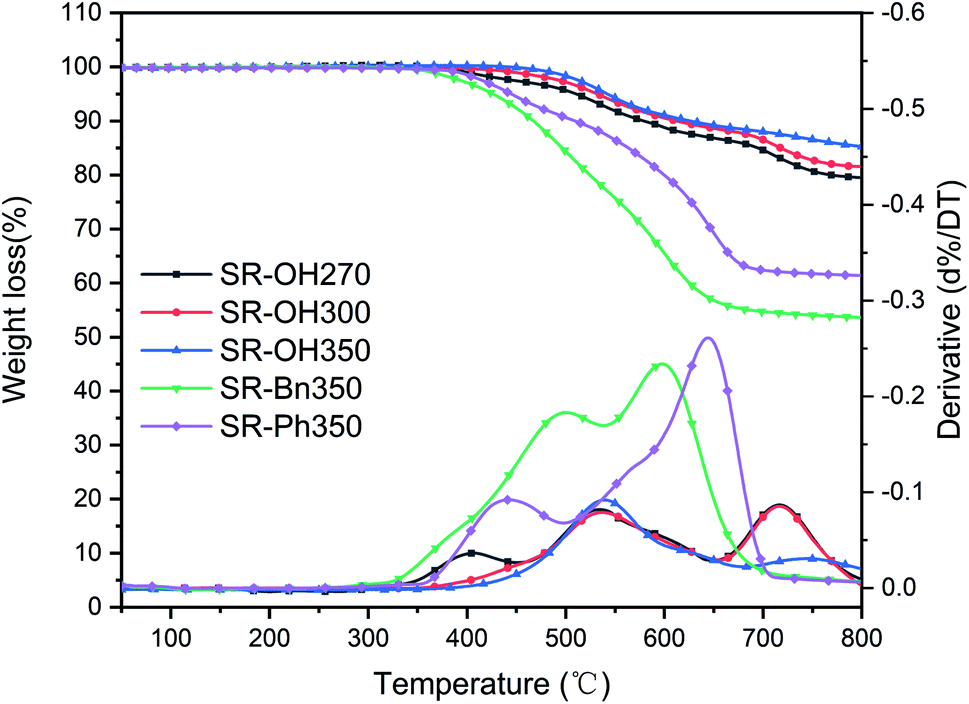
The TGA and DTG curves of different silicone resins in nitrogen are shown in Fig. 6, and further characterization is provided in Table 2. SR-OH270, SR-OH300 and SR-OH350 resin products were obtained at 270 °C, 300 °C and 350 °C, respectively. According to Fig. 6 and Table 2, the 5% weight-loss temperature (Td5), 10% weight-loss temperature (Td10) and char yield at 800 °C (Y800) of the SR-OH increased with curing temperature. Moreover, the DTG curves indicate that as the curing temperature increased, the degradation peak at around 400 °C gradually disappeared (Fig. 6). In order to study the degradation of the resin at this stage, TGA-FTIR was used to analyze the degradation products of SR-OH cured at different temperatures. Fig. 7 shows the FT-IR spectra of the thermal decomposition volatiles of SR-OH270, SR-OH300 and SR-OH350 samples in nitrogen. The results show that the thermal decomposition volatile of the three samples at temperatures lower than 500 °C was mainly water. This indicated that the difference in the thermal stability of SR-OH cured at different temperatures at this stage was mainly due to further dehydration and condensation of residual –OH at high temperatures. The high curing temperature could reduce the residual amount of –OH in the silicone resin so that its thermal stability at this stage was significantly improved with the increase of the curing temperature. Previous studies have shown that Si–OH groups in the silicone resin could cause its degradation through a series of reactions, such as in the ‘back-biting’ reaction.28,29 But for SR-Bn and SR-Ph, even after curing at 350 °C, their thermal stability was also much lower than that of SR-OH, indicating that high temperature alone cannot eliminate Si–OH groups in the silicone resin. Therefore, we attribute the improved thermal stability observed in SR-OH mainly due to the phenolic hydroxyl groups. The phenol groups reacted with residual Si–OH groups at high temperatures to form Si–O–Ph bonds, eliminating the effect of the Si–OH groups on resin degradation and increased the degree of cross-linking. After curing at 350 °C, the Td5 and char yield of SR-OH increased to 606 °C and 91.1%, respectively, showing excellent thermal stability. This is much higher than most silicone resins that have been previously reported, indicating that this type of silicone resin has potential applications in high-temperature-resistant materials and ceramic precursors.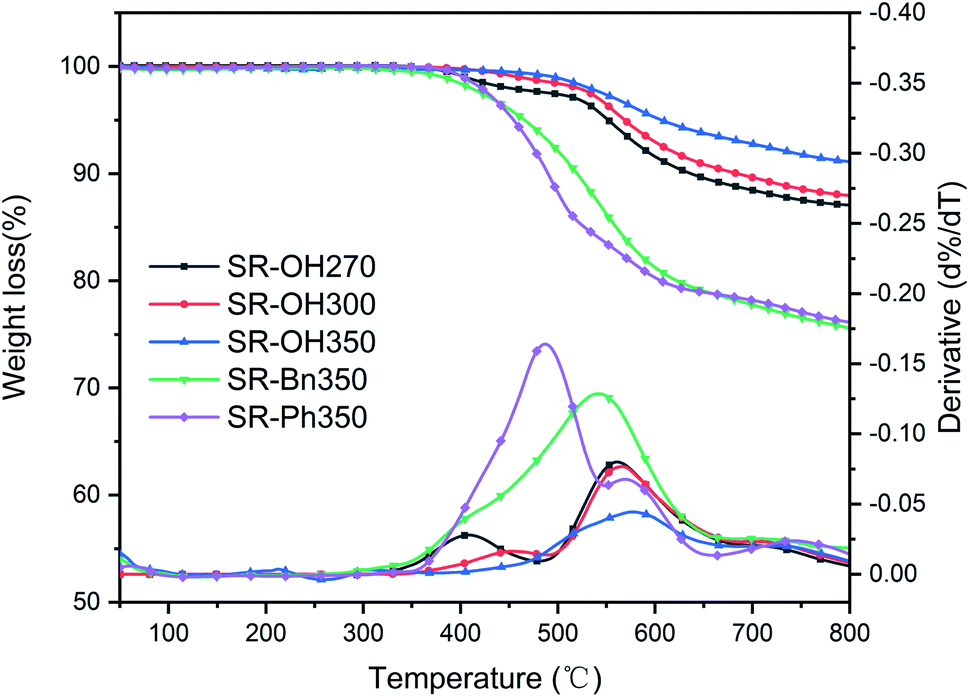 | ||
| Fig. 6 TG and DTG curves of the three cured silicone resins in nitrogen. | ||
| Nitrogen | Air | |||||
|---|---|---|---|---|---|---|
| Td5/°C | Td10/°C | Char yield at 800 °C/% | Td5/°C | Td10/°C | Char yield at 800 °C/% | |
| SR-Ph350 | 455 | 490 | 76.1 | 445 | 510 | 61.4 |
| SR-Bn350 | 465 | 520 | 75.6 | 426 | 467 | 53.6 |
| SR-OH270 | 552 | 637 | 87.1 | 511 | 579 | 79.5 |
| SR-OH300 | 569 | 684 | 88.0 | 532 | 612 | 81.6 |
| SR-OH350 | 606 | — | 91.1 | 542 | 627 | 85.3 |
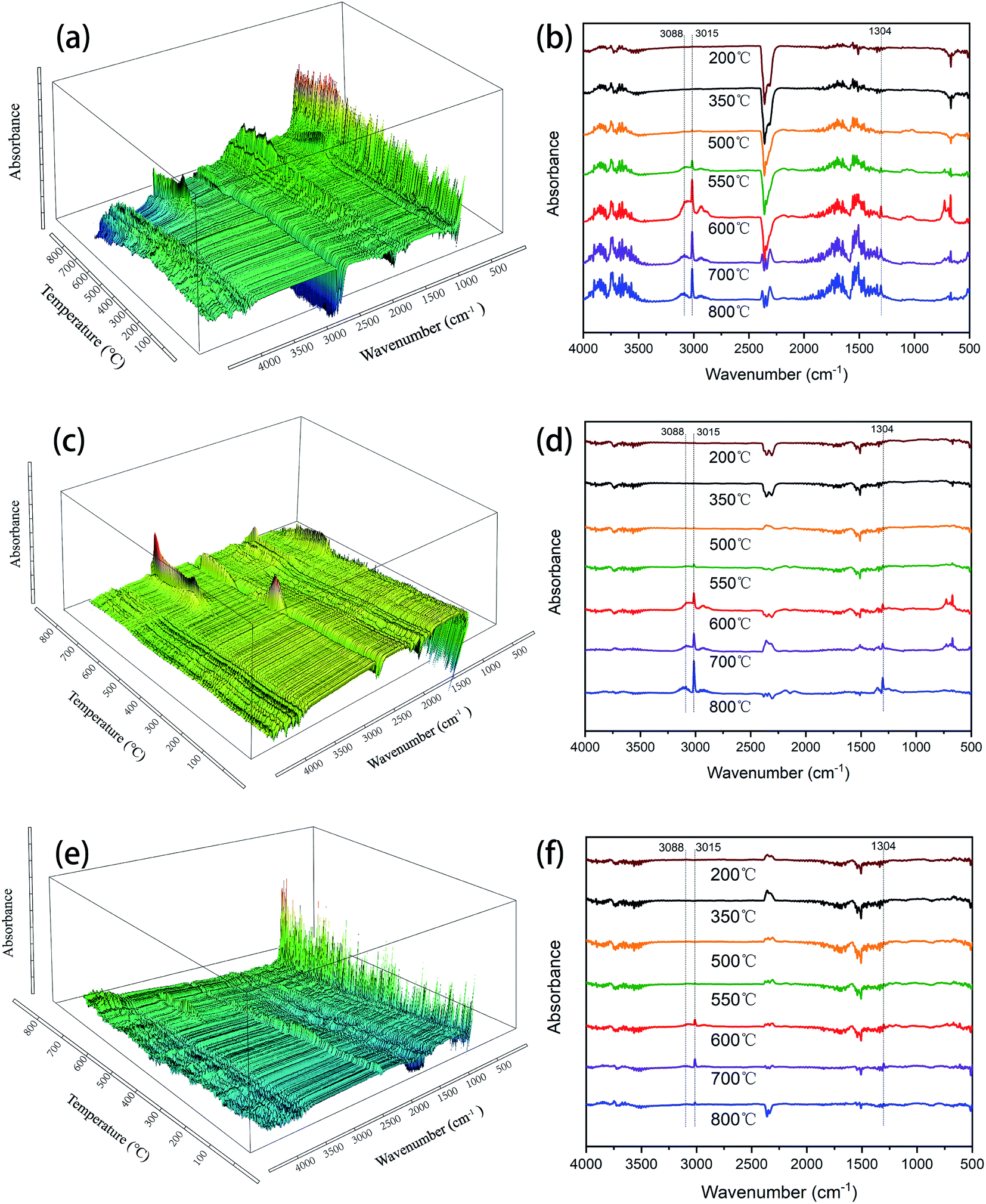 | ||
| Fig. 7 FT-IR spectra of degradation products for (a and b) SR-OH270, (c and d) SR-OH300 and (e and f) SR-OH350 from TGA. | ||
In order to further investigate the degradation process, the SR-OH resins treated at different temperatures were analyzed by FT-IR (Fig. 8). The results showed that the spectrum of SR-OH500 was similar to the cured sample, indicating that there were no significant changes in the structure of SR-OH at this stage. This was consistent with the discussion in the previous section. The characteristic peak of Si–O–Ph at 936 cm−1 decreased when the temperature reached 550 °C. Meanwhile, the characteristic peak at around 3088 cm−1 corresponding to the aromatic rings appeared in the FT-IR spectra of the thermal decomposition volatiles at 550 °C. Both results demonstrate cleavage of the Si–O–Ph at this temperature. When the temperature reached 600 °C, the characteristic peak of Si–O–Ph disappeared. Correspondingly, the absorption peak intensity at 3088 cm−1 reached the maxima as shown in Fig. 7. New characteristic peaks at 3015 cm−1 and 1304 cm−1 belonging to methane were also present between 500 °C and 800 °C, which indicated the cleavage of Si–CH3 at this stage.39,40 Additionally, the amount of the thermal decomposition volatiles decreased significantly with increasing curing temperature. In the degradation process, no obvious characteristic peaks of the compounds with Si–O–Si structure were detected in the FT-IR spectra of the thermal decomposition volatiles. This indicated that the Si–O–Si structure was less damaged in the degradation process of SR-OH, maintaining a high char yield at 800 °C. According to the above analysis, a reasonable explanation is that the consumption of Si–OH makes it difficult for the resin to undergo a ‘back-biting’ reaction at high temperature, which avoids the branching and fracture of Si–O–Si main chain. The relatively stable and complete Si–O–Si network limits the fracture and departure of side groups to a certain extent. These results demonstrate that the formation of Si–O–Ph could significantly improve the thermal stability of the silicone resin.
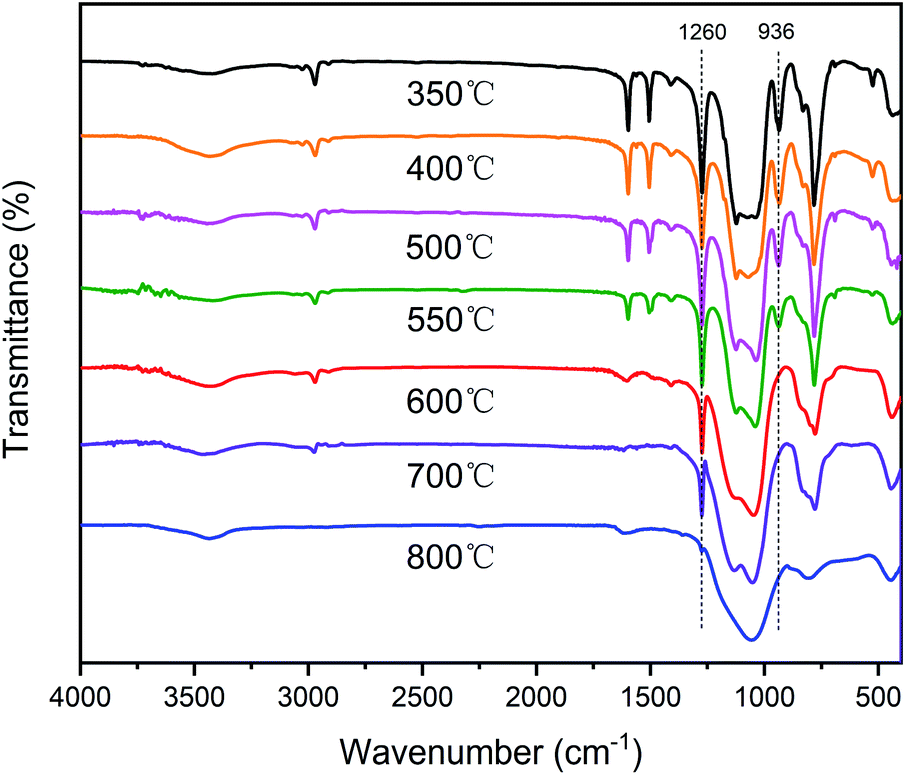 | ||
| Fig. 8 FT-IR spectra of SR-OH350 and its degradation residues at 400 °C, 500 °C, 550 °C, 600 °C, 700 °C and 800 °C in nitrogen. | ||
The TGA and DTG curves of different silicone resins in air are shown in Fig. 9, and further information is provided in Table 2. As the curing temperature increased from 270 °C to 350 °C, the Td5 of SR-OH increased from 511 °C to 542 °C and Td10 increased from 579 °C to 627 °C, showing excellent thermo-oxidative stability. In order to study the degradation process of SR-OH, the SR-OH350 resin was heated at different temperatures in air, and the FT-IR spectra of its residues are shown in Fig. 10. At temperatures below 500 °C, the FT-IR spectra of the residues for SR-OH350 in nitrogen (Fig. 8) and air (Fig. 10) atmosphere are similar, indicating that the degradation of SR-OH was not affected by the presence of oxygen below 500 °C. At this temperature, it was difficult for oxygen to diffuse and dissolve into the resin due to the relatively complete cross-linked structure. Therefore, the degradation observed during the second stage (450–650 °C) was attributed to the cleavage of all Si–O–Ph bonds and a small amount of Si–CH3, rather than oxidative degradation. After heating SR-OH350 to 550 °C, the FT-IR spectrum shows almost complete cleavage of the Si–O–Ph bond. As the cross-linking structure was ruptured, oxygen diffused into the resin and the substituents in the resin began to undergo oxidative degradation. The observed DTG curve peaks in the range of 650–800 °C are mainly caused by oxidative degradation. As the curing temperature increased, the degradation peak at this stage was significantly reduced. This could be attributed to the lack of diffused oxygen by the high-cross-linked structure and reduced content of residual –OH groups in the resin, thus significantly preventing oxidative degradation. When the heating temperature rose to 700 °C, the characteristic peak of Si–CH3 at 1260 cm−1 was absent (Fig. 10), indicating that most of the Si–CH3 cleaved at 600–700 °C. These results suggest that the thermal degradation of SR-OH350 in the third stage was mainly due to the oxidation of Si–CH3. In summary, the formation of Si–O–Ph bonds plays an important role in improving the thermo-oxidative stability of silicone resins. Therefore, the thermo-oxidative stability of the other two resins (SR-Bn350 and SR-Ph350) is far inferior to that of SR-OH350. As shown in Table 2, the Y800 of SR-OH350 in air was up to 85.3%, which is suitable for use in ablative resistant materials with potential applications in flame retardants and thermal insulation.
 | ||
| Fig. 9 TG-DTG curves of the three cured silicone resins in air. | ||
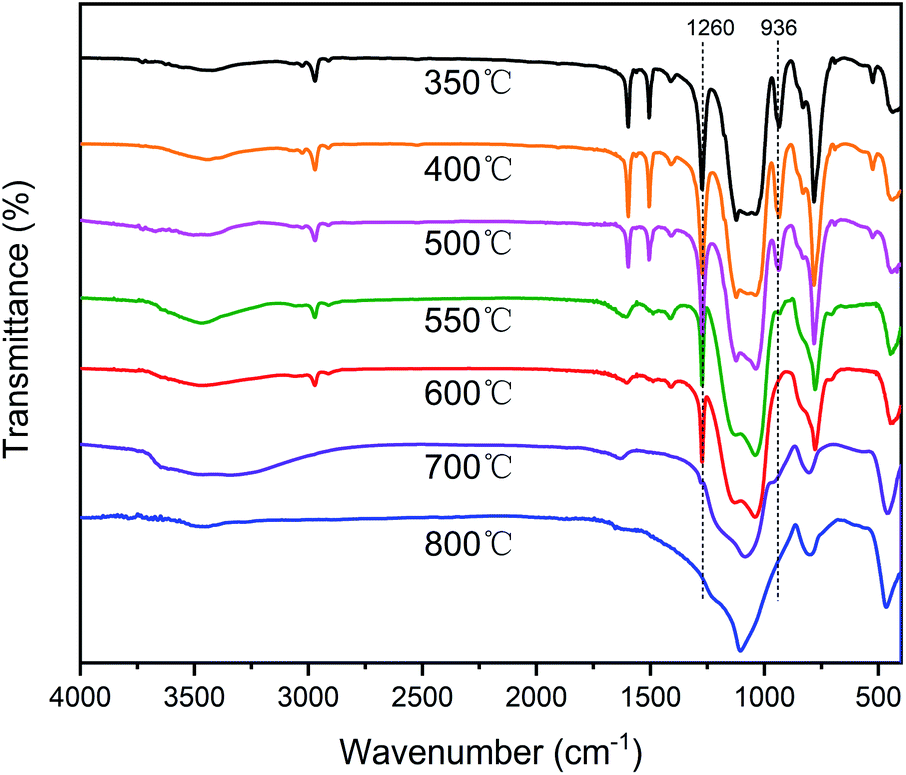 | ||
| Fig. 10 FT-IR spectra of SR-OH350 and its degradation residues at 400 °C, 500 °C, 550 °C, 600 °C, 700 °C and 800 °C in air. | ||
4. Conclusion
In summary, a high temperature resistant silicone resin was successfully synthesized by introducing phenolic hydroxyl groups into the molecular chain. The resin exhibited excellent thermal stability due to the formation of Si–O–Ph bonds during the curing process. After the curation reached completion at 350 °C, the Td5 and Y800 of SR-OH were as high as 606 °C and 91.1% in nitrogen. Even in the presence of oxygen, the Y800 of SR-OH at the same curing condition remained high (85.3%), which is only 5.8% lower than in an inert atmosphere. The high char yield of this resin makes it suitable for potential applications as a matrix component in high-performance flame-retardant materials and ablation-resistant materials. This study provides a novel strategy for significantly improving the thermal and thermo-oxidative stability of silicone resins.Conflicts of interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.Acknowledgements
This work is supported by the National Natural Science Foundation of China (51503107); Key R & D project of Shandong Province (grant No. 2019JZZY010350, No. 2019JZZY010316); Innovation Pilot Project of the Integration of Science, Education and Industry of Qilu University of Technology (Shandong Academy of Sciences) (2020KJC-ZD19); Natural Science Foundation of Shandong Province (ZR2020LFG005); Sichuan Provincial Science and Technology Innovation Talents (2018RZ0113).References
- C. Y. Bo, X. H. Yang, L. H. Hu, M. Zhang, P. Y. Jia and Y. H. Zhou, Polym. Compos., 2019, 40, 2539–2547 CrossRef CAS.
- X. Wu, X. Yang, R. Yu, X. J. Zhao, Y. Zhang and W. Huang, J. Mater. Chem. A, 2018, 6, 10184–10188 RSC.
- J. J. Hu, Y. H. Hu, S. F. Deng, J. L. Zhou, N. Jiang, Y. Zhu and M. Sun, High Perform. Polym., 2020, 32, 1112–1121 CrossRef CAS.
- D. Zhang, C. Wang, Q. H. Wang and T. M. Wang, Tribol. Int., 2019, 140, 10 Search PubMed.
- Z. H. Lu, W. L. Feng, X. L. Kang, J. L. Wang, H. Xu, Y. P. Wang, B. Y. Liu, X. M. Fang and T. Ding, Polym. Adv. Technol., 2019, 30, 2686–2694 CrossRef CAS.
- M. N. Uddin, L. Le, R. Nair and R. Asmatulu, J. Eng. Mater. Technol., 2019, 141, 7 CrossRef.
- M. D. Banea and L. F. M. da Silva, Materialwiss. Werkstofftech., 2010, 41, 325–335 CrossRef CAS.
- E. A. S. Marques, D. N. M. Magalhaes and L. F. M. da Silva, Materialwiss. Werkstofftech., 2011, 42, 471–477 CrossRef CAS.
- W. Chen, M. Ji and S. Y. Yang, Chin. J. Polym. Sci., 2016, 34, 933–948 CrossRef CAS.
- M. Yoonessi, Y. Shi, D. A. Scheiman, M. Lebron-Colon, D. M. Tigelaar, R. A. Weiss and M. A. Meador, ACS Nano, 2012, 6, 7644–7655 CrossRef CAS PubMed.
- D. J. Liaw, K. L. Wang, Y. C. Huang, K. R. Lee, J. Y. Lai and C. S. Ha, Prog. Polym. Sci., 2012, 37, 907–974 CrossRef CAS.
- Y. Liu, X. Z. Xu, S. Mo, L. Zhai, M. H. He and L. Fan, Chem. J. Chin. Univ., 2019, 40, 187–194 CAS.
- W. Q. Jin, L. Yuan, G. Z. Liang and A. J. Gu, ACS Appl. Mater. Interfaces, 2014, 6, 14931–14944 CrossRef CAS PubMed.
- Y. J. Zhu, L. Yuan, G. Z. Liang and A. J. Gu, Polym. Degrad. Stab., 2015, 118, 33–44 CrossRef CAS.
- X. H. Xiong, P. Chen, J. X. Zhang, Q. Yu and B. C. Wang, Thermochim. Acta, 2011, 514, 44–50 CrossRef CAS.
- L. P. Sheng, J. C. Zeng, S. L. Xing, C. P. Yin, J. S. Yang, Y. D. Yang and J. Y. Xiao, High Perform. Polym., 2017, 29, 13–25 CrossRef CAS.
- S. N. Bai, X. Y. Sun, Z. J. Zhang, X. G. Chen, X. Y. Yu and Q. X. Zhang, ChemistrySelect, 2020, 5, 265–269 CrossRef CAS.
- Y. Kaneko, H. Toyodome, M. Shoiriki and N. Iyi, Int. J. Polym. Sci., 2012, 2012, 14 Search PubMed.
- W. Zhou, H. Yang, X. Guo and J. Lu, Polym. Degrad. Stab., 2006, 91, 1471–1475 CrossRef CAS.
- C. Wu, M. Q. Huang, D. H. Luo, Y. Z. Jiang and M. Yan, J. Alloys Compd., 2018, 741, 35–43 CrossRef CAS.
- Y. Liu, Z. X. Chen, Y. S. Qin, Y. Q. Shen, Y. Zhou, D. Wang, J. X. Hu and W. C. Feng, J. Electron. Mater., 2020, 49, 4379–4384 CrossRef CAS.
- Y. Y. Yao, G. Q. Lu, D. Boroyevich and K. D. T. Ngo, Polymer, 2014, 55, 4232–4240 CrossRef CAS.
- J. Lin, H. Y. Zhang, M. Y. Tang, W. Y. Tu and X. B. Zhang, J. Mater. Eng. Perform., 2015, 24, 920–929 CrossRef CAS.
- T. Shimizu, R. Kishi, K. Kobashi, T. Morimoto, T. Okazaki, T. Yamada and K. Hata, Compos. Commun., 2020, 22, 5 Search PubMed.
- X. Q. Xu, C. A. Wu, B. H. Zhang and H. Dong, J. Appl. Polym. Sci., 2013, 128, 4189–4200 CrossRef CAS.
- C. Chou and M. H. Yang, J. Therm. Anal., 1993, 40, 657–667 CrossRef CAS.
- R. Huang, J. S. Yao, Q. H. Mu, D. Peng, H. Zhao and Z. Z. Yang, Polymers, 2020, 12, 12 Search PubMed.
- N. Grassie, I. G. Macfarlane and K. F. Francey, Eur. Polym. J., 1979, 15, 415–422 CrossRef CAS.
- Y. M. Han, J. Y. Zhang, L. Shi, S. C. Qi, J. Cheng and R. G. Jin, Polym. Degrad. Stab., 2008, 93, 242–251 CrossRef CAS.
- N. Grassie, K. F. Francey and I. G. Macfarlane, Polym. Degrad. Stab., 1980, 2, 67–83 CrossRef CAS.
- J. Y. Ji, X. Ge, W. J. Liang, X. Y. Pang, R. L. Liu, S. Y. Wen, J. Q. Sun, X. J. Chen and J. F. Ge, Coatings, 2019, 9, 12 CrossRef.
- Y. Y. Xu, J. Long, R. Z. Zhang, Y. Z. Du, S. J. Guan, Y. Y. Wang, L. Y. Huang, H. W. Wei, L. Liu and Y. D. Huang, Polym. Degrad. Stab., 2020, 174, 9 CrossRef.
- H. D. Liu, G. M. Zhu and C. S. Zhang, Compos. Pt. B-Eng., 2020, 190, 12 Search PubMed.
- Y. Zhang, Y. Huang, X. Liu and Y. Yu, J. Appl. Polym. Sci., 2003, 89, 1702–1707 CrossRef CAS.
- J. T. Sun, Y. D. Huang, H. L. Cao and G. F. Gong, Polym. Degrad. Stab., 2004, 85, 725–731 CrossRef CAS.
- S. Li, Y. Han, F. H. Chen, Z. H. Luo, H. Li and T. Zhao, Polym. Degrad. Stab., 2016, 124, 68–76 CrossRef CAS.
- J. Yun, L. X. Chen, X. F. Zhang, H. Zhao, Z. Y. Wen and D. F. Zhu, J. Mater. Sci., 2018, 53, 14185–14203 CrossRef CAS.
- S. Gao, Y. Liu, S. Y. Feng and Z. J. Lu, J. Mater. Chem. A, 2019, 7, 17498–17504 RSC.
- Z. Yang, L. Feng, S. Diao, S. Feng and C. Zhang, Thermochim. Acta, 2011, 521, 170–175 CrossRef CAS.
- G. Camino, S. M. Lomakin and M. Lageard, Polymer, 2002, 43, 2011–2015 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1ra05524k |
| This journal is © The Royal Society of Chemistry 2021 |