DOI:
10.1039/D1RA05354J
(Paper)
RSC Adv., 2021,
11, 37932-37941
Production of liquid fuels from Kraft lignin over bimetallic Ni–Mo supported on ZIF-derived porous carbon catalyst†
Received
12th July 2021
, Accepted 1st November 2021
First published on 25th November 2021
Abstract
Non-noble bimetallic NiMo supported on zeolitic imidazolate framework-derived porous carbon (NiMo@FDC) catalyst for lignin depolymerization has been successfully developed. The synergism between Ni and Mo species in NiMo@FDC catalyst could promote the catalytic cleavage of C–O linkages in Kraft lignin. At a low reaction temperature of 240 °C and under 4 MPa H2, the lignin liquefaction yield was 98.85 wt% and minimum coke yield was 1 wt%, particularly when using 10%NiMo@FDC catalyst. Additionally, at a high reaction temperature of 300 °C and under 2 MPa H2, there was an overall yield of 86 wt% of liquid product and 42 wt% of petroleum ether soluble product. The higher heating value (HHV) increased from 27.65 MJ kg−1 to 34.11 MJ kg−1. In the cycling experiment, the bifunctional catalyst also demonstrated reversability and stability. The synergy of Ni hydrogenation sites and Mo coupled adsorption sites identified a possible mechanism path, which could offer considerable potential for lignin depolymerization.
Introduction
Recently, the over consumption of fossil fuel and global warming concerns have attracted more and more attention.1 It has become increasingly important to develop sustainable liquid fuels from renewable lignocellulose biomass feedstock.2 Particularly, lignin has been regarded as a potential candidate in producing biofuels and valuable aromatic chemicals.3 The representative natural biopolymer was made up of methoxylated phenyl-propane units, which were linked by various C–O linkages and C–C linkages in a disorderly fashion.4 Because of its rebelliousness and heterogeneity, it is rather challenging to effectively utilize this aromatic polymer.5,6 Therefore, it is of great significance to develop a novel catalytic strategy to make full use of this renewable aromatic polymer resource.
Currently, most catalytic depolymerization strategies have been developed among various lignin and lignin-derived model compounds.7,8 Thereinto, the catalytic reductive strategy using hydrogen source has been widely viewed as the more attractive method in lignin depolymerization. The degraded intermediates in catalytic depolymerization were rather sensitive to a certain extent, which were attacked to cause the unavoidable re-polymerization reaction.9 Thus, developing the bifunctional catalyst is necessary to promote lignin depolymerization and enhance the depolymerized intermediates' stabilization.
In the past decade, diverse noble metal catalysts (such as, palladium;10 rhodium;11 ruthenium;12,13 iridium;14 gold15) have been studied widely in the catalytic cleavage of C–O bonds in lignin model compounds. However, considering the high process cost, lignin depolymerization studies began to pay close attention to the non-noble metal catalysts.16 In recent studies, many molybdenum-based catalysts have been extensively studied in biomass conversion on account of their excellent catalytic performance and low costs.17 These Mo-based monometallic catalysts featured the unsaturated vacancy of active Mo adsorption sites, which could couple with the oxygen atoms of aromatic oxy-compounds.18
To pursue the desired reactivity and selectivity, the bimetallic molybdenum based catalysts have been put forward. Bimetallic Mo-based catalysts with specific activities could depend on electronic properties and geometric configurations of integrated bimetallic sites.19,20 Some transition metal species have been used as the co-catalyst in some Mo-based catalytic system. Recently, Katalin Barta and other coworkers performed that catalytic depolymerization of Kraft lignin to alkyl-phenols using various sulfide NiW catalysts on different acidic and basic supports.21 Similar work has been reported that various sulfurized CoMo catalysts represent catalytic performance in producing aromatic monomers from Kraft lignin.22 Nevertheless, the poisonousness of these sulfide catalysts might cause the non-reversible damage to nature environment, and they also likely engender the sulphur-containing compounds in the degradation products.23 Thus, it is highly desired to develop the non-sulfurized bimetallic Mo-based catalyst, which could facilitate the cleavage of C–O bonds and stabilize the intermediates.
In the process of lignin depolymerization, the catalyst supports played another crucial factor.23,24 High surface area and polyporous supports were beneficial to increase the dispersion of active species.17 One of the particular members of metal organic frameworks (MOFs), zeolitic imidazolate frameworks (ZIFs) was known as a porous coordination polymer, which has recently received more and more attention in the utilization of biomass.25 In recent years, ZIFs-derived porous carbon support also exhibited catalytic activity in lignin depolymerization, owing to its tenability, high specific area, porosity and thermo-stability.26,27 These specific properties are crucial to achieve the optimal geometry and electronic properties via incorporating some transition metal active sites.26–29
In this work, non-precious bimetallic NiMo embedded in ZIFs-derived porous carbon catalyst was prepared and studied in Kraft lignin depolymerization. The fabricated bifunctional NiMo@FDC catalyst was characterized by H2-TPR, NH3-TPD, TEM, SEM, PXRD, BET, XPS and TG analysis techniques. The depolymerized products were further analyzed by GC-MS, GC-FID, elemental analysis and 2D-HSQC-NMR techniques. On the basis of the synergy between Ni and Mo species and product analysis, the potential mechanism route was proposed to understand and explore thoroughly the structure–activity relationship on lignin depolymerization.
Materials and methods
Materials
Fe(NO3)3·9H2O, Zn(NO3)2·6H2O, N,N-2-dimethyl formamide (DMF), methanol, Ni(NO3)2·6H2O, (NH4)6Mo7O24·4H2O and these analytical grade reagents including 1,4-dioxane (AR), ethyl acetate (EA; AR), petroleum ether (PE; AR) and acetone (AR) were all purchased from Sinopharm Chemical Reagent Co., Ltd (Shanghai, China). Indulin AT™ Kraft lignin, one of the alkaline pine lignin, was bought from MeadWestvaco. The Kraft lignin consisted of 65.44 wt% C, 6.28 wt% H, 27.73 wt% O, 0.61wt% N, 1.73 wt% S. Other analytical grade reagents used in GC-FID analysis all were bought from Aladdin Chemical Reagent Co., Ltd.
Catalyst preparation
The support was firstly synthesized by sand bath process according to the reported procedure.30 As demonstrated in Fig. 1a, firstly, 1.695 g of Zn(NO3)2·6H2O, 1.97 g of 2-methylimidazole (2-MI) and 0.1 g of Fe(NO3)3·9H2O were stirred and mixed in 25 mL of DMF solvent. Then, the mixed solution was sealed in the glass bottle and sand bath heated at 60 °C for 24 h. Subsequently, the shallow brick-red sediment was washed three times with ethanol and dried in a vacuum oven at 60 °C overnight. Finally, the precursor was treated at 700 °C in a tube furnace under N2 atmosphere (10 mL min−1) for 3 h to obtain the ZIFs-derived porous carbon support. This fresh prepared support was nominated as FDC. Additionally, the NiMo@FDC catalyst was prepared by the wet impregnation method. The mixture with the equal molar ratio of (NH4)6Mo7O24·4H2O and Ni(NO3)2·6H2O was impregnated into FDC support, followed by drying at 80 °C for 12 hours. To this end, the product was grinded in a Quartz mortar. Then it was subjected to a heating treatment in a tube furnace at 500 °C under N2 atmosphere (5 mL min−1) for 2 h to obtain NiMo@FDC catalyst. Similarly, the FDC supported catalysts with different contents and various active species were prepared successively.
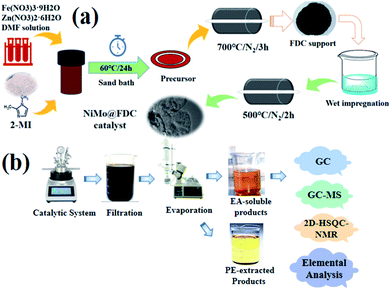 |
| | Fig. 1 (a) Illustration of NiMo@FDC catalyst preparation; (b) illustration of Kraft lignin catalytic depolymerization. | |
The relevant characterizations of catalysts were analyzed by different analysis techniques, which were described detailedly in ESI.†
Catalytic test and product analysis
The detailed reductive depolymerization was described in Fig. 1b. The catalytic reactions were carried out in a 50 mL temperature-controlled stainless steel autoclave equipped with a motor stirrer (Anhui Kemi Machinery Technology Co., Ltd, Hefei, Anhui, China). Before the typical run, the feedstock (Kraft lignin; 0.5 g) was dissolved in the desired mixed solution under the ultrasonic condition. Then, the mixture and the catalyst (0.2 g) were added into the reactor. Meanwhile, the reactor was sealed and purged three times with hydrogen to remove the remaining air. Subsequently, the autoclave was charged to the definite H2 pressure at room temperature. The reactor was then heated to the desired reaction temperature under vigorous stirring with the speed of 800 rpm. After finishing the reaction, the reactor was cooled to room temperature. The liquid products were qualitatively analyzed by GC-MS (7890A GC; 5975C MS, Agilent, Palo Alto, California, USA) and quantitatively analyzed by GC equipped with a HP-5 column (GC-2010, Shimadzu, Kyoto, Japan). The GC setup of GC/MS analysis were carried out as following, the column temperature was increased as 50 °C and maintained for 3 min, and then it was heated to the temperature of 180 °C with the ramp rate of 4 min−1, and continued to ramp up to the 260 °C with 10 °C min−1. The quantitative GC analysis were performed as below, the oven temperature was programmed from 50 °C to 250 °C with the ramp rate of 10 °C min−1 and held for 10 min. The yields of aromatic monomers in liquid products were determined and calculated through adding acetophenone as the internal standard. The residue was filtrated and then put in a drying oven at 378 K for 12 h. Additionally, during the cyclic stability test, the spent catalyst was separated after each run, and then washed with methanol and acetone several times. The dried spent sample was used for the next run directly.
The achieved mixture was successively extracted with ethyl acetate (EA) and petroleum ether (PE). All the remaining solvents were removed using a rotary evaporator (E-2000B, Yarong Instrument Company, Shanghai, China). Considering that the liquid product was all dissolve in ethyl acetate, the yield of EA-extracted product was in accordance with the yield of liquid product. The weight of low polarity liquid fuels was determined by the yield of PE-soluble product. These corresponding calculated equations were shown as below:
| | |
Yliquid products (wt%) = 2 × MEA-extracted product/MKL × 100%
| (1) |
| | |
Ysolid products (wt%) = Msolid product/MKL × 100%
| (2) |
| | |
Ygas products (wt%) = 100% − Ysolid product − Yliquid products
| (3) |
| | |
Conversion (%) = (MKL − Msolid product)/MKL × 100%
| (4) |
| | |
YPE-extracted products (wt%) = 2 × MPE-extracted product (wt)/MKL × 100%
| (5) |
| | |
Ymonomers (wt%) = Mmonomers/MKL × 100%
| (6) |
Mx: the mass of various products (x:, solid product, liquid products, EA/PE-extracted products, monomers).
Yx′: the yield of various products (x′: solid product, liquid products, gas products, PE-extracted products, monomers).
MKL: the mass of Kraft lignin;
Ms: the mass of acetophenone.
Ai: the peak area of the monomer;
As: the peak area of acetophenone.
λ: the calculated relative mass correction factor.
The elemental compositions analysis (including carbon, hydrogen, oxygen, nitrogen elements) was measured on an elemental analyzer (Elementar Vario EL cube instrument, Elementar Analysen systeme GmbH, Germany).
Two-dimensional Hetero nuclear Single Quantum Coherence (HSQC) correlation NMR (2D-HSQC-NMR) analysis was carried out on a Bruker AVANCE 500 MHz spectrometer equipped with a 5 mm double resonance broadband BBI inverse probe using a coupling constant J13C–1H of 147 Hz. Firstly, the samples were dissolved in DMSO-d6 and performed with a Bruker phase-sensitive gradient-edited HSQC pulse sequence ‘hsqcetgpsisp2’.
Results and discussion
Structural properties of catalysts
SEM/TEM. Fig. 2 shows the specific morphology and structure of the uncalcined precursor, FDC support and NiMo@FDC catalyst. As shown in Fig. 2a–c, it could be seen that the uncalcined precursor and FDC support have a similar typical hexahedral structure. Obviously, the crystal surface of the precursor was considerably smooth and the crystal size was 215–235 nm (Fig. 2a and b). After the heat treatment, zinc species in ZIFs is easily evaporated under high heat-treated temperature, thereby generating the porous carbon structures. Thus, as demonstrated in Fig. 2c, the crystal surface became less distinct and the carbon supports were more porous, and the crystal size increased slightly. When introducing the NiMo species, the facets of carbon became inconspicuous. It contained more micropores and mesopores, and the fine and uniform porosity was also observed in the fresh catalyst (Fig. 2d and e). The subtle changes of morphology structure indicated that the porosity structures of NiMo@FDC catalyst might have an advantage in anchoring and immobilizing the NiMo active sites, the NiMo nanoparticles could be founded in Fig. 2f. As demonstrated in Fig. 2g–k, the element mapping showed the actual presence of all the elements (Ni, Mo, C and few N) were distributed evenly on the surface of newly formed catalyst. The SEM images of the reactivated catalyst, the spent catalyst and the hydrothermal catalyst were also carried out in Fig. 4. Fig. 4a and b shows that the similar morphology structure of the re-activated catalyst as the fresh catalyst, which indicates that the porosity structures of NiMo@FDC catalyst could be well recovered after the regeneration process. As presented in Fig. 4c and d, the partial surfaces of spent NiMo@FDC catalyst were covered with carbon deposits, which also was consistent with the decrease of the specific surface area in spent catalyst. As shown in Fig. 4e and f, the hydrothermal catalyst presented the similar surface morphologic structure as the fresh catalyst. The corresponding elemental mapping images are showed in Fig. 4g–j, it can be observed that the Mo, Ni, C and N are all evenly distributed in the catalyst, just as with the fresh catalyst.
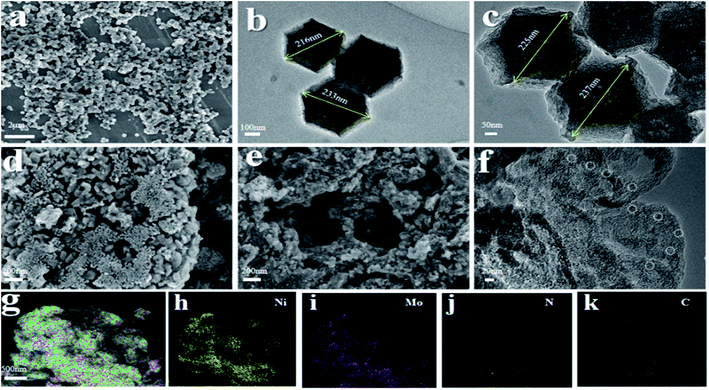 |
| | Fig. 2 SEM images (a), TEM images (b) of uncalcined precursor; TEM images (c) of ZIFs-derived carbon support; SEM images (d and e), TEM images (f), element mapping (g–k) of NiMo@FDC catalyst. | |
Textural properties. The textures and properties of various NiMo@FDC catalysts are shown in Table 1 and Fig. S1.† The surface area and pore distribution of FDC, fresh 10%NiMo@FDC and spent catalyst were detected by the nitrogen adsorption–desorption analysis. Clearly, the three samples had the coexistent micro-mesoporous textural structure. It also displayed that the pore volume and diameter all decreased after adding the Ni and Mo species. It was likely to result from the pore destruction generated by the thermal decomposition of nickel species.31 It also can be founded that the derived porous FDC support possesses the large BET surface area (505.31 m2 g−1), which is similar with the traditional activated carbon materials. Compared with the bare FDC carrier, the NiMo@FDC catalyst was equipped with a smaller special surface area (57.94 m2 g−1). This might be the partial cavity blocking caused by the incorporated NiMo species on the porous carbon support. For the spent catalyst, the dramatic dropping of SBET and pore volume and diameter was observed. It might be engendered by the aggregated carbon deposits on the surface of catalyst, which lead to the appearance of some pore blockages.
Table 1 Textures and properties of various catalysts
| Catalyst |
Nitrogen adsorption–desorption |
| SBETa (m2 g−1) |
Vpb (cm3 g−1) |
dpc (nm) |
| Calculated from nitrogen adsorption isotherms at 77 K using BET equation. Calculated from nitrogen adsorption isotherms at 77 K at P/Po = 0.99. Calculated from nitrogen adsorption isotherms at 77 K using BJH model. |
| FDC |
505.31 |
0.27 |
21.33 |
| 10%NiMo@FDC-fresh |
57.94 |
0.23 |
20.16 |
| 10%NiMo@FDC-spent |
10.69 |
0.02 |
12.21 |
XPS. The chemical valence of active metal sites was analyzed by XPS analysis. The Mo 3d and Ni 2p spectra of the best performing 10%NiMo@FDC catalyst are exhibited in Fig. 3a and b. Firstly, Fig. 3a shows the peak of Mo 3d spectrum, the peak of Mo 3d5/2 at 232.7 eV and the peak of Mo 3d3/2 at 235.9 eV. The result indicates that Mo element in the fresh catalyst mainly exists in the form of Mo6+ species. Then, Fig. 3b shows the peak of Ni 2p spectrum, the two peaks show the presence of Ni 2p3/2 and Ni 2p1/2, and each of them are the binding energies of 856.4 eV and 873.8 eV. The result suggests the characteristic features of Ni2+ species.32
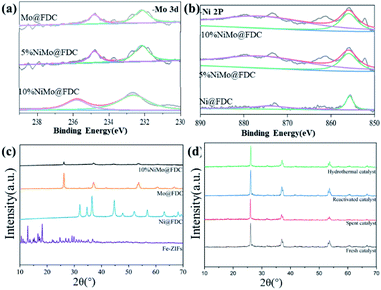 |
| | Fig. 3 Characterization of NiMo@FDC catalysts. Mo-3d profile (a); Ni–2P profile (b) in XPS analysis; PXRD patterns of various of catalysts (c and d). | |
PXRD. The PXRD patterns of fresh NiMo@FDC catalysts are demonstrated in Fig. 3c, as compared to the uncalcined precursor, other fresh catalysts displayed different diffraction patterns. The XRD spectra of monometallic Mo@FDC catalyst was exhibited at 37.12°, 53.72°, 60.56° corresponding to (210) (022) and (031) of MoO2 phase; the peak of 2θ = 27.3° was attributed to MoO3 phase, and the rest were assigned to MoC1−x phase.33,34 The monometallic Ni@FDC catalyst showed the diffraction peak of 2θ values of 44.60°, 47.84°, 52.06° assigned to (111), (200), (220) crystal planes of Ni2C.35 By contrast, the peak of 2θ values located at 27.3°, 44.60°, 37.12°, and 53.72° were all detected in the bimetallic NiMo@FDC catalyst. This patterns indicate the coexistence of MoO2, MoO3, MoC1−x and Ni2C species, whereas the derived porous carbon does not show any phase features in PXRD analysis. Fig. 3d also shows the PXRD patterns of spent catalyst (after 6 h) and hydrothermal catalyst. Compared to the standard simulated PXRD analysis of the fresh catalyst, the reactivated catalyst and the hydrothermal catalyst had the same peaks of 2θ, and it doesn't make any difference in the intensity. This also demonstrated that it has almost the invariable phase states among the cooccurrence of MoO2, MoO3, MoC1−x and Ni2C species, when the catalyst was conducted after the regeneration procedure and the hydrothermal treatment. By comparison, the spent catalyst shows the similar phase state, but the intensity of phase state both have a slight reduction. This difference might be attributed to the transformation of molybdenum species to the molybdenum oxycarbohydride phase. Nonetheless, no diffraction peaks of sulfurized compounds were observed in the spent catalyst.
TG. TG analysis of 10%NiMo@FDC catalyst is shown in Fig. S2a.† It is clear to observe two sections of the weight loss in the fresh catalyst. The first weight loss of 1.8 wt% was founded might be attributable to the removal of dimethyl formamide solvents and the residual 2-methylimidazole organic ligand. The second sharp decrease of weight loss is 7.7 wt%, which might be caused by the fewer dissociation of organic linker species. The weight of final residue was more than 90 wt%. The above results demonstrate that NiMo@FDC catalyst has good thermal stability, and its overall structure is probably not damaged when used below 700 °C. Additionally, TG-DTA of the spent catalyst is shown in Fig. S2b,† the dramatic increase of weight loss was observed under the lower treatment temperature, indicating the reduction of thermal stability and specific surface area in spent catalyst.
H2-TPR. Table 2 and Fig. 4 demonstrate the profiles of H2-TPR analysis and NH3-TPD analysis of different catalysts. Firstly, the hydrogenation ability and the interaction between NiMo active sites with the FDC support was detected in H2-TPR analysis. It can be seen that the monometallic Mo@FDC catalyst was reduced into metallic molybdenum at 641 K, which is attributed to the reduction of MoO2 to metallic Mo (Fig. 4a). However, the reduction peak of bimetallic NiMo@FDC catalyst moved towards higher temperature ranging from 400 °C to 600 °C (Fig. 4a). This reflected the interaction of NiMo active sites and FDC support. Simultaneously, with the loading of bimetallic NiMo active sites, the amount of hydrogen consumption increased from 1282.4 μmolH2 gcat−1 to 8463.2 μmolH2 gcat−1. Additionally, the reduction peaks of all catalysts were lower than bulk MoO3 species. These observations suggest that the addition of Ni species can engender the magnanimous improvement of hydrogenation ability, which might be conductive to lignin depolymerization.
Table 2 Acidity and active sites of fresh catalysts
| Catalyst |
Total acid sitesa (mmolNH3 g−1) |
Peak position (°C) |
Active sitesb (μmolH2 gcat−1) |
| θ1 |
θ2 |
| Measured by NH3-TPD analysis. Measured by H2-TPR analysis. |
| Mo@FDC |
0.80 |
361.9 |
— |
689.7 |
| 5%NiMo@FDC |
0.92 |
362.1 |
— |
1282.4 |
| 10%NiMo@FDC |
1.15 |
358.5 |
447.2 |
8463.2 |
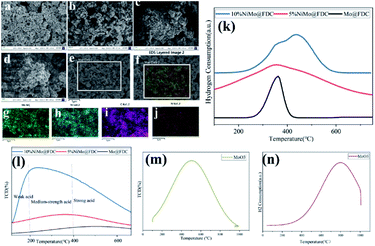 |
| | Fig. 4 SEM images of the re-activated NiMo@FDC catalyst (a and b); the spent catalyst (c and d); the hydrothermal catalyst (e) and the corresponding EDS-mapping (f–j); H2-TPR analysis of various catalysts (k) and bulk MoO3 (n); NH3-TPD analysis of various catalysts (l) and bulk MoO3 (m). | |
NH3-TPD. The acid sites on the surface of the catalyst were measured by NH3-TPD analysis. As presented in Fig. 4c, the acid sites were made up of three kinds of acids (that is, the weak acid, the medium-strength acid and strong acid). The total acid content of monometallic Mo@FDC catalyst was 0.80 mmolNH3 g−1. After loading Ni and Mo species simultaneously, the total acid amount of 10%NiMo@FDC catalyst was enhanced to 1.15 mmolNH3 g−1. Fig. 4c also shows that the strength of medium-strength acid increases and the strength of strong acid weakens. These results demonstrate that the interaction between Ni and Mo might be beneficial to improve Mo species' absorption ability. Meanwhile, the temperature of acid sites' peaks was lower than bulk MoO3 species, it also suggests the mutual effect between NiMo active sites and porous carbon carrier. Briefly, these changes in fresh NiMo@FDC catalyst might play an active role in catalytic cleavage of lignin.
Catalytic upgrading of Kraft lignin
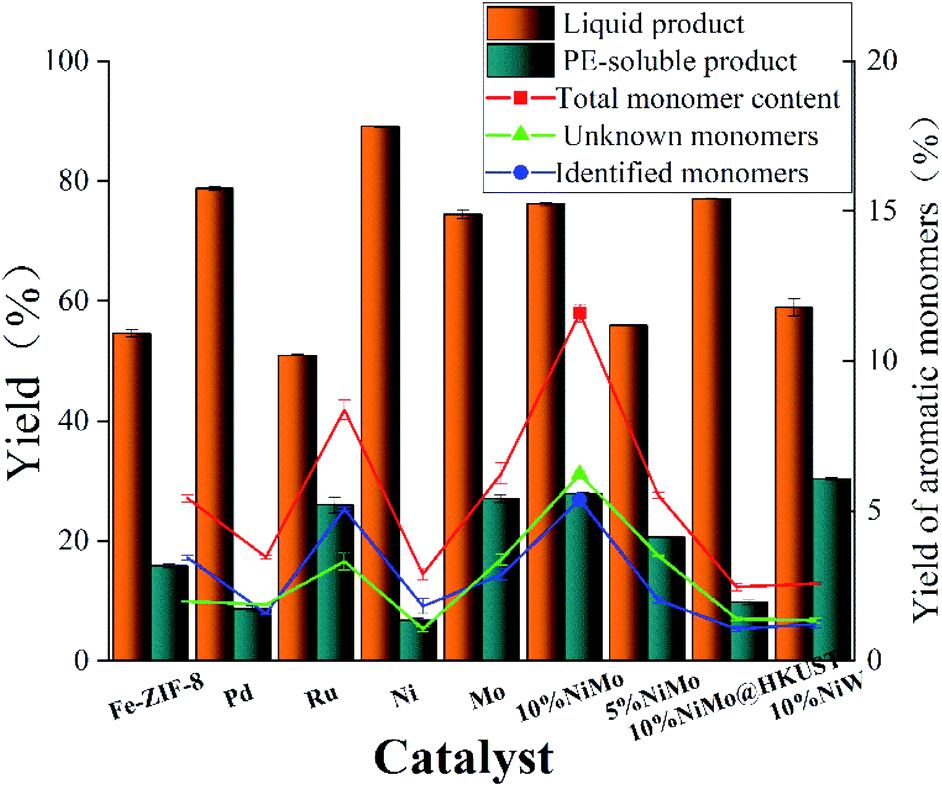
The reductive depolymerization performance of IndulinAT Kraft lignin was evaluated using various supported catalysts (including various FDC supported catalysts and HKUST (1-(2-methyl-4-(2-oxopyrrolidin-1-yl) phenyl)-3-morpholino- 5,6-dihydropyridin-2(1H)-one) derived porous carbon supported NiMo catalyst). The catalytic reaction was conducted at 300 °C, 2 MPa H2 and 3 h. As shown in Fig. 5, it is not surprising that trace amounts of liquid product and PE-soluble product were obtained using the individual FDC support. It manifests that FDC support has poor reactivity in Kraft lignin depolymerization. Moreover, the higher liquefaction ratio (89.1%) was obtained by monometallic Ni@FDC catalyst, compared to monometallic Mo@FDC catalyst (74.45%). However, the latter (27.02%) was superior to the former (6.79%) in the yield of PE-soluble product. It indicates that the sole Ni-based catalyst is inclined to liquify Kraft lignin, and the individual Mo-based catalyst could promote catalytic cleavage by stabilizing the intermediates. As expected, when using NiMo@FDC catalysts, quite appreciable volume of production in liquid product (76.2%) and PE-extracted product (28.3%) were obtained. But when using HKUST-derived porous carbon supported NiMo catalyst and other FDC supported precious metals (Pd and Ru) catalysts, the yields of liquid product and PE-extracted product all were much lower than that of NiMo@FDC catalysts. Tell its tale, 58% of liquid product was obtained over NiW@FDC catalyst, which was lower than NiMo@FDC catalyst. Besides, as the loading enhanced from 5 to 10 wt%, the yields of liquid product and PE-soluble product were increasing linearly from 55.95% to 76.2% and from 20.6% to 28.3%. Based on the above results, 10% NiMo@FDC catalyst was chosen to study in the following researches.
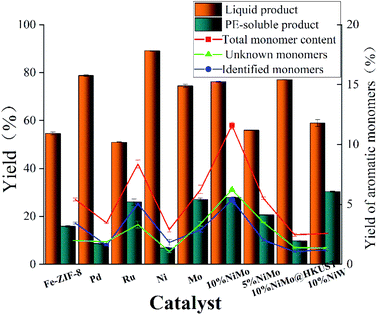 |
| | Fig. 5 The catalytic performance in Kraft lignin depolymerization of various composite catalysts. Reaction conditions: 0.5 g of Kraft lignin, 0.2 g of catalyst, 25 mL of 1,4-dioxane and 5 mL of methanol, 300 °C, 3 h, 2 MPa H2. The 10% NiMo* is the HKUST-derived porous carbon supported NiMo catalyst, others are M@FDC catalysts (M = Pd, Ru, Ni, Mo, 5%NiMo, 10%NiMo, 10%NiW). | |
The effect of the reaction condition
The effect of the reaction condition was investigated among reaction temperature, reaction time, reaction solution and initial hydrogen pressure in Fig. 6. The influence of different reaction temperatures (240–300 °C) over NiMo@FDC catalyst is showed in Fig. 6a. It is obvious that the yield of PE-soluble products is rising linearly from 9.95% to 28.3% with the increasement of reaction temperature. Nevertheless, the yield of liquid product had a slight decline from 86.45% to 76.2%. These results indicate that increasing temperature is beneficial to the production of PE-soluble product, and this also may lead to the intermediates' condensation reaction at a higher reaction temperature. The influence of the reaction time parameter in lignin depolymerization was demonstrated in Fig. 6b. With the increase of reaction time, the yield had the linear increasing of PE-extracted product (28.3–43%) as a function of liquid product yield raised from 70.7% to 86%. Additionally, the less solid product was detected at a longer reaction time. It was attributed to the presence of soft-coking carbonization (MoOxCyHz phase) between active Mo species and the carbon residue, which is also confirmed in our previous studies.20 On the basis of these results, the extension of reaction time was favorable for lignin depolymerization.
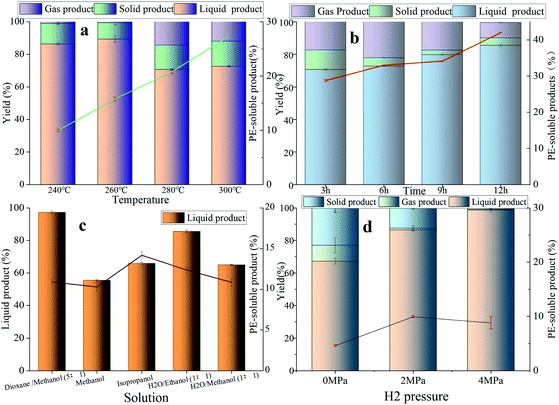 |
| | Fig. 6 The influence of reaction temperature (a); the impact of reaction time (b); the role of reaction solvent (c); and the effect of initial hydrogen pressure over NiMo@FDC catalysts (d). | |
In order to better investigate the effect of the organic phase in Kraft lignin depolymerization, the catalytic studies were performed in different reaction solvents. The catalytic depolymerization experiments were carried out under a mild reaction condition (260 °C, 2 MPa H2 and 3 h) in Fig. 6c. Although the yields of PE-soluble product for all the reaction solvents were similar, the yield of EA-soluble product were diverse. Obviously, 55.59% of EA-soluble product and 65.96% of EA-soluble product were obtained in the monophasic methanol (30 mL) and isopropanol (30 mL), respectively. It is worth noting that the EA-soluble products' outcomes in 15 mL H2O/15 mL methanol was only 65.05%. It also can be seen that the yield of liquid product in 15 mL H2O/15 mL methanol is further increased to 85.62%. To sum up, these are lower than the yield of EA-soluble product in 25 mL 1,4-dioxane/5 mL methanol (97.26%). Thus, the mixed solution of 25 mL of 1,4-dioxane and 5 mL of methanol was regarded as the optimal reaction solvent in this study.
In addition to the above reaction parameters, initial hydrogen pressure also was the vital indicator to evaluate the optimal conditions in lignin depolymerization (Fig. 6d). Meanwhile, in order to evaluate the catalytic performance under a lower reaction temperature, catalytic studies using different hydrogen pressures were operated at 240 °C. When initial H2 pressure was increased from 0 MPa to 4 MPa, the yield of EA-extracted liquid product kept pace with growth, ranging from 67.25% to 98.85%. Minimally of 1wt% solid product was obtained at 240 °C and 4MPaH2. Apparently, the high hydrogen pressure had the positive advantage in lignin hydroliquefaction, thereby generating less solid product. However, the coupling hydrodeoxygenation reaction of degraded intermediates was inhibited when the H2 pressure increased from 2 MPa to 4 MPa. This resulted in the low yield of PE-extracted product and aromatic monomers. As a result, the initial H2 pressure of 2 MPa was selected to conduct other experiments in this study.
Product analysis
GC-MS qualitative analysis. To comprehend the definite chemical structure of aromatic monomers in the degraded liquid products, the EA-soluble product was investigated by GC-MS-FID chromatography. The main products were detected and identified including Guaiacol, Veratrole, 4-methylguaiacol, 4-ethylguaiacol, eugenol, 4-propyl guaiacol, vanillin, 3,4,5-trimethoxytoluene, isoeugenol, aceto-vanillone, 2,6-di-tert-butyl p-phenol, 3,4-dimethoxy acetophenone, benzyl phenyl ether and homovanillic acid. All of these chemical structure and name of major compounds were shown in Table S1.†
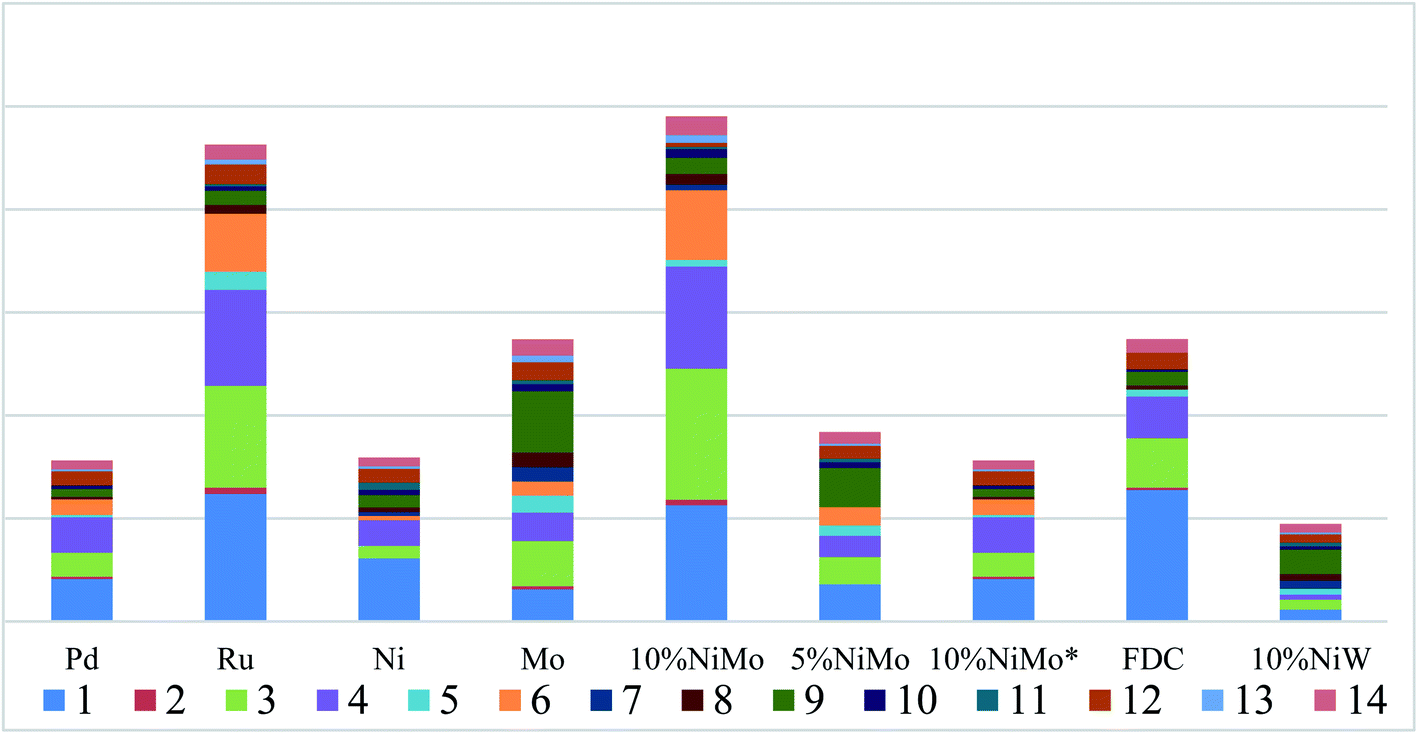
GC-FID quantitative analysis. Apart from the qualitative identification in aromatic monomers by GC-MS-FID technique, the aromatic products were analyzed quantificationally using GC-FID measurements in Fig. 5 and 7. Firstly, Fig. 5 shows the yield of aromatic monomers, including the total monomers content, the identified aromatic product and the unknown aromatic product. There is clear difference in the yield distribution under the respective catalysts' main catalytic activity in lignin depolymerization. As expected, the total yield of aromatic monomers (12.1%) of NiMo@FDC catalyst was more than other supported catalysts. In combination with the above chemical structure of aromatic monomers in GC-MS analysis, Fig. 7 also shows the distribution of identified aromatic monomers in GC-FID analysis. The content of guaiacol, 4-methylguaiacol, 4-ethylguaiacol, and 4-propylguaiacol were higher than other identified aromatic monomers when using 10%NiMo@FDC catalyst. These results suggest that the NiMo@FDC catalyst has an active effect in converting Kraft lignin into the lower molecular weight of aromatic products.
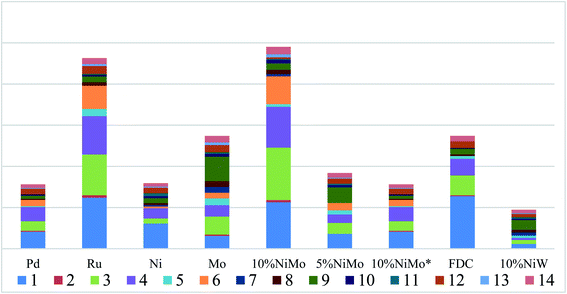 |
| | Fig. 7 The distribution of identified aromatic monomers in GC-FID analysis over various catalysts. Reaction conditions: 0.5 g of Kraft lignin, 0.2 g of catalyst, 25 mL of 1,4-dioxane and 5 mL of methanol, 300 °C, 3 h, 2 MPa H2. | |
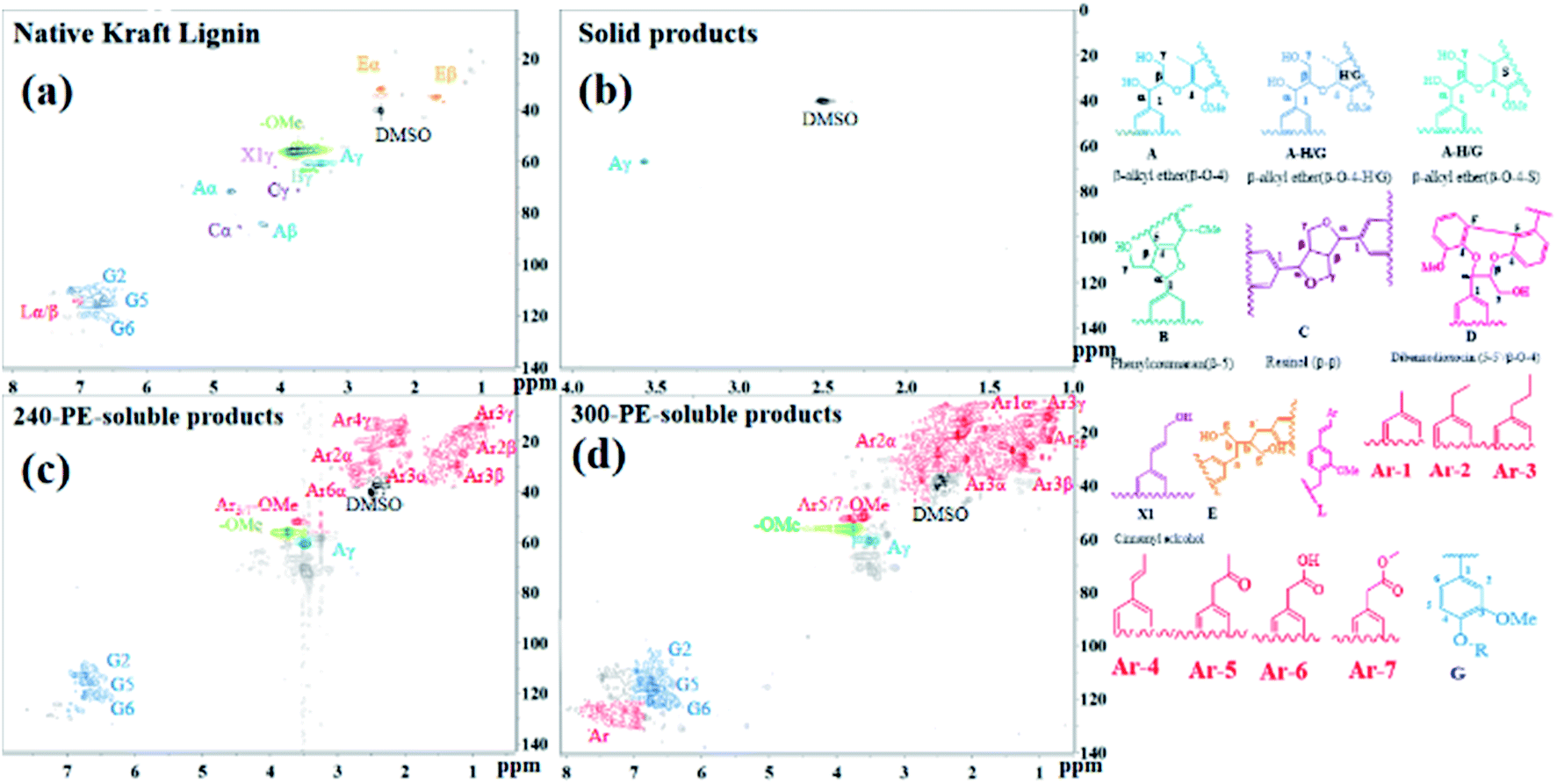
2D-HSQC-NMR analysis. 2D HSQC NMR analysis of native Kraft lignin, PE-soluble product and solid product were recorded in Fig. 8 to further study Kraft lignin depolymerization. As presented in Fig. 8a, the NMR spectrum of native Kraft lignin in DMSO-d6 shows the prominent linkages. These respectively are β-O-4 linkage (Aα, δC/δH = 71.0/4.8 ppm); β-O-4 linkage (Aβ, δC/δH = 84.0/4.3 ppm, this is associated with G-type and S-type units); β-O-4 linkage (Aγ, δC/δH = 60.0/3.3–3.7 ppm); β-5 linkage (Bγ, δC/δH = 62.7/3.7 ppm); β–β linkage (C, Cα-δC/δH = 84.8/4.6 ppm; Cβ-δC/δH = 53.5/3.1 ppm; Cγ-δC/δH = 70.7/3.8 and 4.2 ppm); G-type linkages (G2-δC/δH = 109–122/6.7–7.1 ppm; G5-δC/δH = 110–117/6.4–7.1 ppm; and G6-δC/δH = 102–105/6.8–7.3 ppm), and other linkages of 5–5′(D), E, X1, L and the typical methoxyl group in the oxygenated alkyl region.9,36,37 The NMR spectrum of PE-soluble product obtained after lignin depolymerization was demonstrated in Fig. 7c and d, it is distinct that apart from the linkages of Aγ, G2, G5, G6 and –OMe, other signals almost have vanished away. In addition, the newly emerging saturated structures (including Ar1, Ar2, Ar3, Ar4, Ar5, Ar6 and Ar7) also observed clearly. This also was supported by the result of GC result, as described in the section of GC analysis, the more productive products are guaiacol, 4-methylguaiacol, 4-ethylguaiacol, 4-propylguaiacol. And there's the big difference that the more G-type linkages and some aromatic structures were detected in the 300-PE-soluble product than that of 240-PE-soluble product. This is agreement of the catalytic results in this study, which shows that the steadily increased PE-extracted product yield with the reaction temperature. Additionally, the NMR spectrum of solid product in Fig. 7b shows the sole linkages of Aγ in the oxygenated aliphatic hydrocarbon region. These observed results indicate that the original β-O-4 linkages in native Kraft lignin disappeared via the cleavage of C–O linkages, it could lead to the presence of the newly-presented G-type linkages and other aromatic monomers.
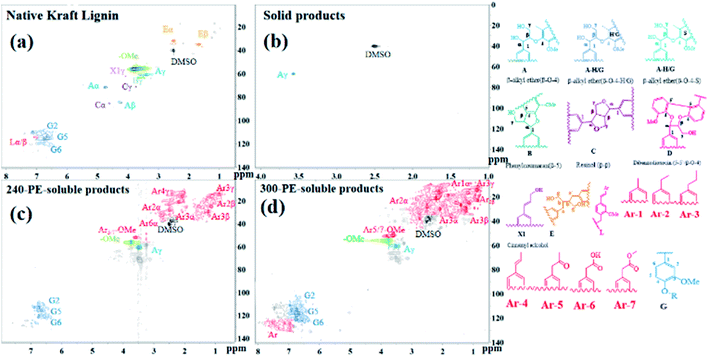 |
| | Fig. 8 1H–13C HSQC NMR spectra of lignin and PE-soluble products obtained after catalytic depolymerization using 10%NiMo@FDC catalyst at 300 °C, 2 MPa H2 and 12 h; (a) Native Kraft Lignin, (b) solid product; (c and d) PE-soluble products. | |
Element analysis. Table 3 shows the elemental analysis among the Kraft lignin, liquid product and PE-soluble product. Compared to the Kraft lignin, it can be discovered that the content of carbon (C), hydrogen (H) elements increased in the liquid product and PE-soluble product, but the elemental content of oxygen (O) decreased. These observations showed that the fresh NiMo@FDC catalyst is beneficial to the hydrogenation and hydrodeoxygenation reaction in lignin depolymerization. The relevant higher heating value (HHV) in the liquid product (32.18 MJ kg−1) and PE-soluble product (34.11 MJ kg−1) were higher than native Kraft lignin (27.65 MJ kg−1). By comparison, the HHV of PE-soluble product was slightly higher than that of liquid product, which implies that the degraded liquid fuels may potentially become the additives for petroleum products.
Table 3 Elemental analysis of the Kraft lignin, liquid product and PE-soluble product
| Sample |
Elemental contentb (wt%) |
HHVc (MJ kg−1) |
| C |
H |
O |
N |
S |
| Reaction condition: 0.5 g Kraft lignin, 0.2 g 10%NiMo@FDC catalyst, 300 °C, 2 MPa H2, 12 h, 800rmp. Measured by elemental analysis. The higher heating value (HHV)was calculated by DIN 51900 standard,38 which was showed as follow: HHV (MJ kg−1) = (34C + 124.3H + 6.3N + 19.3S − 9.8O)/100. |
| Kraft lignin |
65.44 |
6.28 |
27.73 |
0.61 |
1.73 |
27.65 |
| Liquid producta |
70.71 |
7.88 |
19.79 |
0.44 |
1.32 |
32.18 |
| PE-soluble producta |
69.46 |
9.84 |
20.41 |
0.37 |
1.28 |
34.11 |
The stability test of the catalyst
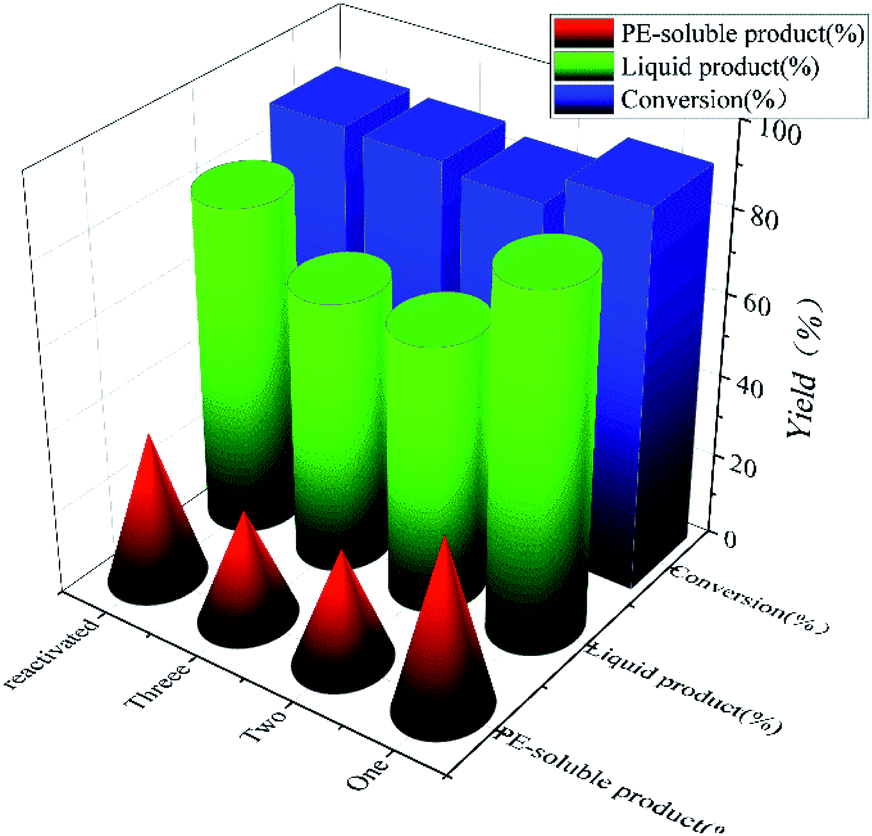
To test the recyclability of NiMo@FDC catalyst, it was used in Kraft lignin depolymerization without any further treatment at 300 °C, 2 MPa H2 and 12 h. In Fig. 9, while the conversion rate remained nearly at 88%, the yield of liquid product and PE-soluble product decreased marginally (42–28%) after the third runs. When the spent catalyst was activated at 500 °C in a nitrogen atmosphere, the catalytic activity of reactive NiMo@FDC catalyst could recover as the fresh catalyst. It demonstrates the high durability and stability of NiMo@FDC catalyst, as well as its suitability for lignin depolymerization.
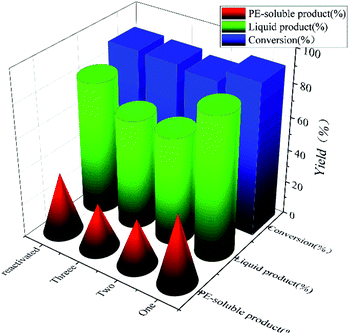 |
| | Fig. 9 Cycle experiments of 10%NiMo@FDC catalyst. Reaction conditions: 0.5 g of Kraft lignin, 0.2 g of catalyst, 25 mL of 1,4-dioxane and 5 mL of methanol, 300 °C, 12 h, 2 MPa H2. | |
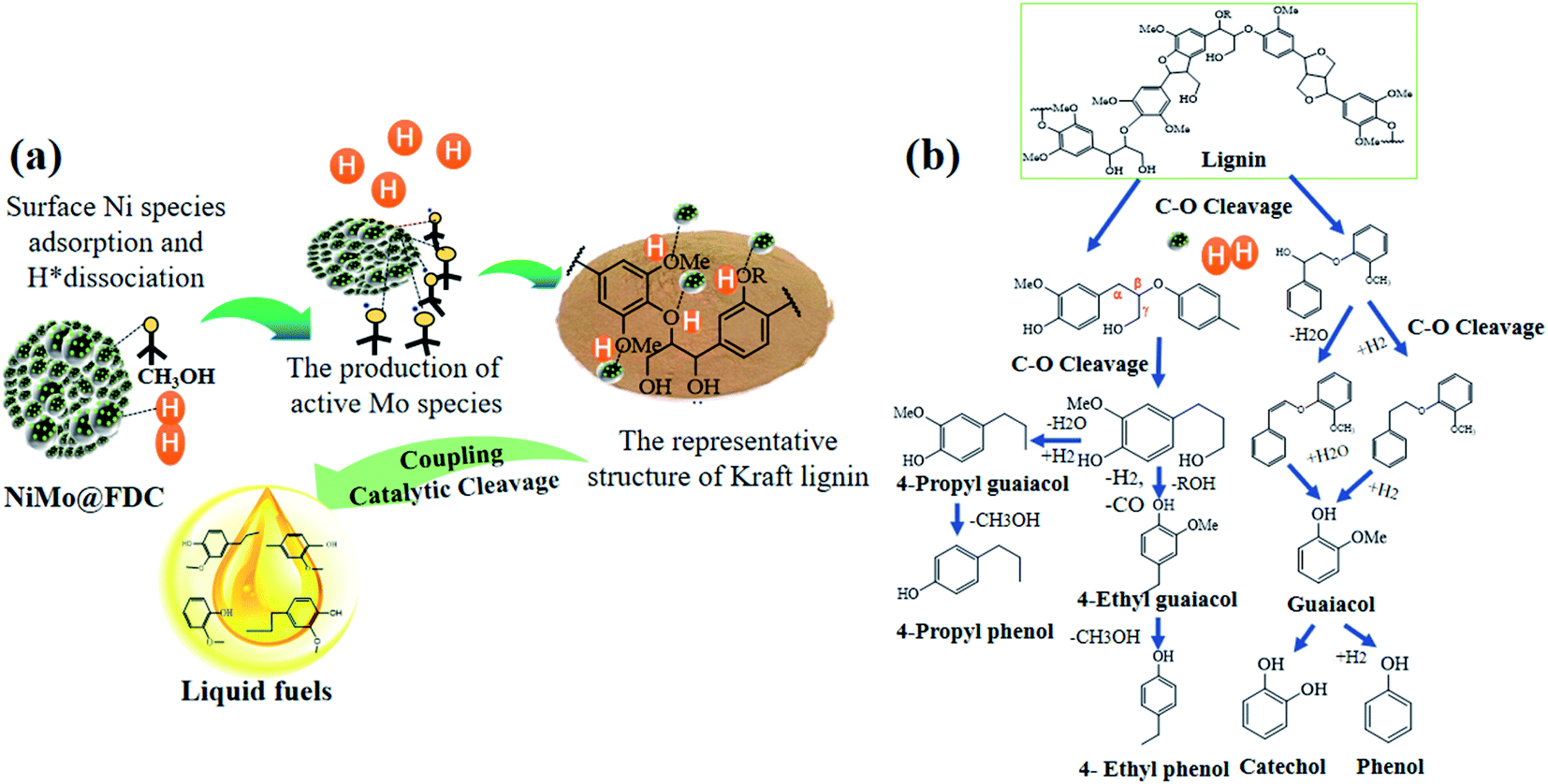
Reaction route and mechanism
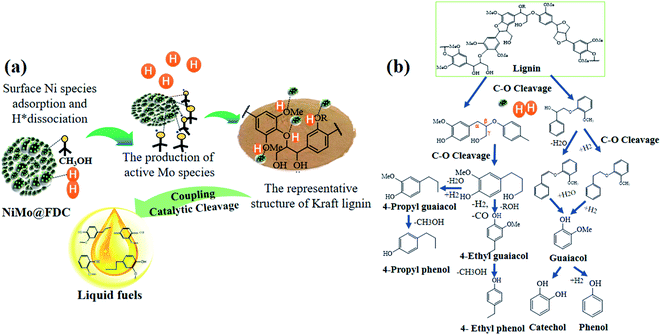
It is relatively challenging to catalytic depolymerize Kraft lignin into aromatic monomers, especially the cleavage of C–O ether linkages. To gain preliminary insights into lignin depolymerization mechanism, the possible catalytic cleavage illustration was described in Fig. 10. As depicted in Fig. 10a, for the bimetallic NiMo@FDC catalyst, the simultaneous impregnation of nickel nitrate and ammonium molybdate might lead to the solid commixture of nickel and molybdenum species. As has mentioned above, the molybdenum and nickel existed in the form of +6 and +2 oxidation valences, respectively (Fig. 3; the result of XPS analysis). Moreover, the Mo species possess the stronger bonding ability than Ni species. Additionally, Ni species have the more reducibility than Mo species, and Mo species have the specific oxophilicities (Fig. 4; the results of H2-TPR and NH3-TPD analysis). This is supported by the excellent hydrogenation capability of Ni species, which results in the irreversible chemical desorption of hydrogen.
 |
| | Fig. 10 Schematic diagram of possible catalytic depolymerization mechanism route. | |
With the above results and the corresponding product analysis combined, the catalytic cleavage reaction might be induced by the synergy of Ni and Mo species. As presented, the hydrogen molecules were possibly adsorbed on Ni active sites' surface, resulting in the formation of active H species. At the same time, the methoxy group in methanol could activate saturated Mo species, resulting in active Mo active sites with unsaturated coordination. The Ni hydrogenation performance and Mo adsorption activity were strengthened by the subtle interactions on two metal active sites and the mutual effect between the FDC support and NiMo active sites. Thus, a considerable proportion of C–O bonds were absorbed on the unsaturated vacancy of Mo active sites. Many dissociated active H species likely spilled over to Mo sites to attack the newly coupled intermediates, resulting in the cleavage of C–O linkages. Finally, as presented in Fig. 10b, the different targeted aromatic monomers in liquid products were achieved by the coupling catalytic hydrogenation and hydrodeoxygenation process.
Conclusions
In summary, the no-noble NiMo supported ZIFs derived porous carbon catalyst demonstrated its catalytic activity in converting Kraft lignin to liquid fuels. The synergistic effect of Ni and Mo species is essential for improving C–O linkages cleavage. At a low reaction temperature of 240 °C and 4 MPa H2, a high liquefaction yield (98.85 wt%) was achieved and a low solid product yield (1 wt%). Meanwhile, the yield of PE-extracted product (42 wt%) was obtained at 300 °C and 2 MPa H2. The high heating value was increased from 27.65 MJ kg−1 to 34.11 MJ kg−1. Besides, the NiMo@FDC catalyst showed the recycling and regeneration ability during the cycle experiment. The product analysis (GC, GC-MS and 2D-HSQC-NMR) and other catalyst characterization were used to depict the potentially coupled cleavage mechanism. The collaborative system of Ni hydrogenation sites and Mo coupled adsorption sites could bring new possibilities for future bio-fuel and petroleum additives from native lignin.
Conflicts of interest
The authors declare that there are no conflicts interest.
Acknowledgements
This work was supported by “Transformational Technologies for Clean Energy and Demonstration”, Strategic Priority Research Program of the Chinese Academy of Sciences, Grant No. XDA 21060101 and Key Research and Development Projects in Anhui Province No. 202004a06020053, and the National Key Technology R&D Program of China (No. 2018YFB1501601).
References
- X. Zhang, W. Tang, Q. Zhang, Y. Li, L. Chen, Y. Xu, C. Wang and L. Ma, Fuel, 2018, 215, 825–834 CrossRef CAS.
- C. Liu, S. Wu, H. Zhang and R. Xiao, Fuel Process. Technol., 2019, 191, 181–201 CrossRef CAS.
- I. Hita, P. J. Deuss, G. Bonura, F. Frusteri and H. J. Heeres, Fuel Process. Technol., 2018, 179, 143–153 CrossRef CAS.
- M. B. Figueirêdo, H. J. Heeres and P. J. Deuss, Sustainable Energy Fuels, 2020, 4, 265–276 RSC.
- L. S. Yanding Li, H. Kim, A. H. Motagamwala, J. K. Mobley, F. Yue, Y. Tobimatsu, D. Havkin-Frenkel, F. Chen, R. A. Dixon, J. S. Luterbacher, J. A. Dumesic and J. Ralph, Sci. Adv., 2018, 4, eaau2968 CrossRef PubMed.
- M. B. Figueirêdo, P. J. Deuss, R. H. Venderbosch and H. J. Heeres, Biomass Bioenergy, 2020, 134, 105484 CrossRef.
- R. Datta, A. Kelkar, D. Baraniya, A. Molaei, A. Moulick, R. Meena and P. Formanek, Sustainability, 2017, 9, 1163 CrossRef.
- R. Shu, R. Li, B. Lin, B. Luo and Z. Tian, Fuel, 2020, 265, 116962 CrossRef CAS.
- H. Li, X. Zheng, H. Zhang, X. Li and J. Long, ACS Sustainable Chem. Eng., 2020, 8, 15685–15695 CrossRef CAS.
- T. Xie, J.-P. Cao, C. Zhu, X.-Y. Zhao, M. Zhao, Y.-P. Zhao and X.-Y. Wei, Fuel Process. Technol., 2019, 188, 190–196 CrossRef CAS.
- L. Jin, W. Li, Q. Liu, J. Wang, Y. Zhu, Z. Xu, X. Wei and Q. Zhang, Fuel Process. Technol., 2018, 178, 62–70 CrossRef CAS.
- W. F. Wei Huo, W. Li, M. Zhang, H.-m. Chang and H. Jameel, Catal. Lett., 2014, 144, 1159–1163 CrossRef.
- X. Zhang, H. Yan, L. Zhu, T. Li and S. Wang, Adv. Sustainable Syst., 2020 Search PubMed.
- H. Wu, W. Li, Z. Xu, Z. Du and Q. Liu, J. Univ. Sci. Technol. China, 2017, 47, 377–384 CAS.
- X. Yang, Y. Liang, X. Zhao, Y. Song, L. Hu, X. Wang, Z. Wang and J. Qiu, RSC Adv., 2014, 4, 31932–31936 RSC.
- L. Zhang, N. Shang, S. Gao, J. Wang, T. Meng, C. Du, T. Shen, J. Huang, Q. Wu, H. Wang, Y. Qiao, C. Wang, Y. Gao and Z. Wang, ACS Catal., 2020, 10, 8672–8682 CrossRef CAS.
- M. Shetty, K. Murugappan, W. H. Green and Y. Román-Leshkov, ACS Sustainable Chem. Eng., 2017, 5, 5293–5301 CrossRef CAS.
- Y. Wang, Z. Tang, M. Chen, J. Zhang, J. Shi, C. Wang, Z. Yang and J. Wang, Energy Convers. Manage., 2020, 222, 113227 CrossRef CAS.
- I. T. Ghampson, G. Pecchi, J. L. G. Fierro, A. Videla and N. Escalona, Appl. Catal. B: Environ, 2017, 208, 60–74 CrossRef CAS.
- W. L. Ge Guo, X. Dou, A. T. Ogunbiyi, T. Ahmed, B. Zhang and M. Wu, Bioresour. Technol., 2021, 321, 124443 CrossRef.
- C. R. Kumar, N. Anand, A. Kloekhorst, C. Cannilla, G. Bonura, F. Frusteri, K. Barta and H. J. Heeres, Green Chem., 2015, 17, 4921–4930 RSC.
- A. Narani, R. K. Chowdari, C. Cannilla, G. Bonura, F. Frusteri, H. J. Heeres and K. Barta, Green Chem., 2015, 17, 5046–5057 RSC.
- Q. Chen, C. Cai, X. Zhang, Q. Zhang, L. Chen, Y. Li, C. Wang and L. Ma, ACS Sustainable Chem. Eng., 2020, 8, 9335–9345 CrossRef CAS.
- M. Liu, J. Zhang, L. Zheng, G. Fan, L. Yang and F. Li, ACS Sustainable Chem. Eng., 2020, 8, 6075–6089 CrossRef CAS.
- V. I. N. Isaeva, O. M. Kustov and M. Leonid, Catalysts, 2018, 8, 368 CrossRef.
- T. Li, H. Lin, X. Ouyang, X. Qiu and Z. Wan, ACS Catal., 2019, 5828–5836, DOI:10.1021/acscatal.9b01452.
- A. Zanon and F. Verpoort, Coord. Chem. Rev., 2017, 353, 201–222 CrossRef CAS.
- S. Zhang, A. Han, Y. Zhai, J. Zhang, W. C. Cheong, D. Wang and Y. Li, Chem. Commun., 2017, 53, 9490–9493 RSC.
- L. W. Jiao, Y. Jiang, H. L. Xu and Q., Adv. Mater., 2018, 30, e1703663 CrossRef PubMed.
- H. Zhang, S. Hwang, M. Wang, Z. Feng, S. Karakalos, L. Luo, Z. Qiao, X. Xie, C. Wang, D. Su, Y. Shao and G. Wu, J. Am. Chem. Soc., 2017, 139, 14143–14149 CrossRef CAS PubMed.
- J. Liu, J. He, L. Wang, R. Li, P. Chen, X. Rao, L. Deng, L. Rong and J. Lei, Sci. Rep., 2016, 6, 23667 CrossRef CAS.
- D. Mukherjee, R. Singuru, P. Venkataswamy, D. Damma and B. M. Reddy, Acs Omega, 2019, 4, 4770–4778 CrossRef CAS.
- Z. Cai, F. Wang, X. Zhang, R. Ahishakiye, Y. Xie and Y. Shen, Mol. Catal., 2017, 441, 28–34 CrossRef CAS.
- X. Ma, R. Ma, W. Hao, M. Chen, F. Yan, K. Cui, Y. Tian and Y. Li, ACS Catal., 2015, 5, 4803–4813 CrossRef CAS.
- R. K. Chowdari, S. Agarwal and H. J. Heeres, ACS Sustain Chem Eng, 2019, 7, 2044–2055 CrossRef CAS.
- H. W. Ma, H. W. Li, W. J. Zhao, L. X. Li, S. J. Liu, J. X. Long and X. H. Li, Green Chem., 2019, 21, 658–668 RSC.
- Z. Cai, J. Long, Y. Li, L. Ye, B. Yin, L. J. France, J. Dong, L. Zheng, H. He and S. Liu, Chem, 2019, 5, 2365–2377 CAS.
- V. Ranaware, D. Verma, R. Insyani, A. Riaz, S. M. Kim and J. Kim, Green Chem., 2019, 21, 1021–1042 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1ra05354j |
|
| This journal is © The Royal Society of Chemistry 2021 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *ab,
Tauseef Ahmeda,
DuoDuo Huc,
Ru Cuic,
Baikai Zhanga and
Xia Zhanga
*ab,
Tauseef Ahmeda,
DuoDuo Huc,
Ru Cuic,
Baikai Zhanga and
Xia Zhanga