DOI:
10.1039/D1RA05341H
(Paper)
RSC Adv., 2021,
11, 26672-26682
An interesting possibility of forming special hole stepping stones with high-stacking aromatic rings in proteins: three-π five-electron and four-π seven-electron resonance bindings†
Received
12th July 2021
, Accepted 30th July 2021
First published on 4th August 2021
Abstract
Long-range hole transfer of proteins plays an important role in many biological processes of living organisms. Therefore, it is highly useful to examine the possible hole stepping stones, which can facilitate hole transfer in proteins. However, the structures of stepping stones are diverse because of the complexity of the protein structures. In the present work, we proposed a series of special stepping stones, which are instantaneously formed by three and four packing aromatic side chains of amino acids to capture a hole, corresponding to three-π five-electron (π:π∴π↔π∴π:π) and four-π seven-electron (π:π∴π:π↔π:π:π∴π) resonance bindings with appropriate binding energies. The aromatic amino acids include histidine (His), phenylalanine (Phe), tyrosine (Tyr) and tryptophan (Trp). The formations of these special stepping stones can effectively reduce the local ionization potential of the high π-stacking region to efficiently capture the migration hole. The quick formations and separations of them promote the efficient hole transfer in proteins. More interestingly, we revealed that a hole cannot delocalize over infinite aromatic rings along the high π–π packing structure at the same time and the micro-surroundings of proteins can modulate the formations of π:π∴π↔π∴π:π and π:π∴π:π↔π:π:π∴π bindings. These results may contribute a new avenue to better understand the potential hole transfer pathway in proteins.
1. Introduction
The understanding of long-range electron (or hole) transfer pathways in proteins is the key to examining the catalyzing dynamics and mechanisms for many redox biological processes,1–6 including photosynthesis,7 respiration,8 biological nitrogen fixation,9 gene replication,10,11 biological denitrification12 and so on. Therefore, electron migration in proteins13 or single-chained polypeptides9,14–16 is currently the subject of intense experimental and theoretical research. Many factors control the actual electron (or hole) transport pathways in the complex biomacromolcules,16–19 including the distance between the donor and the acceptor, hydrogen bonds, weak interactions, dipole orientation, the specific side chains of amino acids and the conformation dynamic properties. These factors make the diversiform mechanisms of the electron transfer reactions according to the specific micro-surroundings in proteins.20,21 The common accepted mechanisms are the one-step superexchange process and the multi-step hopping process.22–25 The former is more suitable for the short-range electron transfer in proteins because the rate of electron transfer drops exponentially with the increase of the distance between the donor and the acceptor, which can't explain the efficient long-range electron transfer in many biological processes.21 However, the multi-step hopping mechanism can provide a reasonable explanation for electron (or hole) translocation across long distances in proteins. In multi-step hopping model, the efficient electron translocation is dependent on the presence of suitable stepping stones between the primary donor and final acceptor, which can transiently hold the transferred charge and increase the electronic coupling along the electron pathway.4,25–28 Therefore, it is crucial to uncover the rational structures of stepping stones along the hole migration pathways in proteins.
Over the past few years, there have been evidences4,25 to support the idea that many special groups can serve as stepping stones to facilitate the long-range hole transfer in proteins. The basic feature for the stepping stone is to have a low ionization potential, which can quickly be transformed between the oxidated and reduced states. Two famous examples is that the side chains of tryptophan (Trp) and tyrosine (Tyr) have lower ionization potentials among the twenty natural amino acids and can take part in electron transfer as stepping stones in many enzymes, including ribonucleotide reductase,10 DNA photolyase,29 diheme enzyme MauG,30 azurin28 and cytochrome c peroxidase.31 However, it is difficult to elucidate the details of long-range electron hole pathways in complex biological systems because there are more alternative electron pathways in the same system. Polypeptides with specific functional groups provide an excellent alternative for the big biological molecules to examine the influence of different factors on electron transfer. Yu and their co-workers have revealed that the increasing number of electron-rich alkenyl groups in polypeptides can efficiently enhance the rate of electron transfer, which substantiated that the alkenyl groups can be used as stepping stones to promote electron transfer through the peptides.17,32 In addition, the infrared reflection absorption spectroscopy and electrochemical studies by Kimura and coworkers revealed that the backbone peptide units can serve as stepping stones to promote hole transfer along a long-distance α-helix.33 Giese and coworkers have carried out a series of innovative works to confirm that the electron-rich aromatic rings can accelerate electron transfer along the polypeptide chain.4,34–36 All these valuable examinations proposed that the electron-rich groups/fragments with low ionization potentials can promote hole transfer in proteins or polypeptides.
More interestingly, some special structures or transient conformations have the potential ability to promote hole transfer in proteins because the interactions of neighbor groups can reduce the local ionization potential. Methionine (Met) has a high ionization potential,37 however, the interaction of the sulfur-atom with an adjacent amide oxygen-atom or an aromatic ring can efficiently reduce the redox potential to take part in hole migration in polypeptides.38–42 Our density functional theory (DFT) calculations have also confirmed that the side chain of Met interacts with a neighboring aromatic ring can form a S∴π three-electron hemibond with a lower ionization potential to participate in hole transfer in proteins.43 Later, Lednev and coworkers have discovered that the S∴π three-electron hemibond is produced during the fibrillation of lysozyme using Raman spectroscopy, electron paramagnetic resonance and UV-vis absorption spectroscopy.44 In addition, our recent DFT calculations have revealed that the associative interactions among two nearly parallel aromatic rings and a sulfur-containing group or two sulfur-containing groups and an inserted aromatic ring can significantly decrease the localized ionization potential by instantly forming special π∴π:S, π∴S:π or S∴π:S three-π five-electron resonance structures.45,46 More recently, Fujii group have confirmed the formations of S∴S and S∴π two-center hemibonds, S∴π:S multicenter hemibond in the [benzene-(H2S)n]+ cluster by the IR spectroscopy and DFT calculations.47 The essential features of the instant stepping stones forming during the hole transfer processes include that the each constituent moiety is electron-rich with lone pair- or π-orbitals, the formations of them can significantly decrease the ionization energy of the associative complex relative to the each moiety, the stabilization of them is moderate, which ensures the effective formation and dissociation of stepping stone to promote hole migration in proteins. In spite of the great efforts to investigate the possible relay stones, however, the accurate pathway of hole transfer in proteins remains incomprehensible owing to the complexity of proteins.
Recently, we have proposed that the stacking structure of two adjacent aromatic rings may take part in long-range hole transfer in proteins by the transient formation of the π:π three-electron hemibond, which can significantly reduce the local ionization energy of the two-aromatic-ring region relative to the isolated aromatic ring and has a moderate binding energy.48 The high π–π stacking structures consisted of three or more aromatic rings also exist in some protein crystal structures,49,50 and the structural properties of interacting pairs of aromatic rings have also been probed experimentally. In the designed synthetic helical peptides, containing repeating tetrapeptide unit with Phe residues aligned on the same face, Aravinda and his colleagues observed nearly parallel-displaced benzemers with a very low value of interplanar angle through X-ray diffraction.51 For the cases of the opposing antiparallel column of helices, close phenylalanine side chains interact with each other by forming rungs of the ladder.51 In addition, it is well known that the π-stacking conformations plays a vital role in the functionality of charge transfer for DNA and RNA.52,53 In proteins, however, the functionality of the high aromatic repeat rings formed by the aromatic side chains for the long-range hole transfer of proteins remain unaddressed. Therefore, the effects of aromatic side chains and the corresponding number in the high π-stacks on the hole transfer of proteins require to be urgently examined.
Motivated by these issues and inspired by the previous work,46–51 we are inquisitive about whether the interactions of three, four or more close aromatic rings may generate a new type of stepping stone to prompt hole transportation in proteins. Therefore, we carry out a DFT computational investigation on the electronic structures and the potential conductance of higher π-stacking structures in the present work. We systemically examine the possible influence of the number of aromatic rings in the higher π-stacking systems on the relay functionality, as well as the effect of the peptide backbone on the formations of the higher π-stacking stepping stones. The results of this study may broaden a new way to explore the electron migration pathway54 during the dynamic processes of proteins.
2. Computational details
Survey of the protein structures in the protein data bank revealed that the high π-packing structures exit in many crystal structures of proteins, as shown in Fig. S1 and S2.† It should be noted that most of the high π-packing structures are not cofacial stacked interaction, which are edge-to-face T-shape or parallel displaced interactions in the natural proteins because the latter are more stable than the former in most cases.55,56 However, the movement of proteins may modulate the conformations of the high π-clusters, which can be changed from the edge-to-face T-shape or parallel displaced conformations to the cofacial stacking conformations to achieve some special functionality of proteins. In contrast, the interactions among the electron-deficient aromatic ring and electron-rich aromatic rings favor the formations of the cofacial stacking conformation.57,58 Therefore, the migration of hole in proteins may favor the transient formations of the high π-stacking conformations for the several close aromatic rings. Furthermore, the transient formations of the high π-stacks promote the efficient transport of hole in proteins.53,54 The discrimination of the high π-stacks among the aromatic amino acids as well as their possible functionality in proteins will take a long time to identify in the future.50,59 In the present paper, we will get insight into the structural characteristics of the high π-stacks and the possible function in the hole transport in proteins.
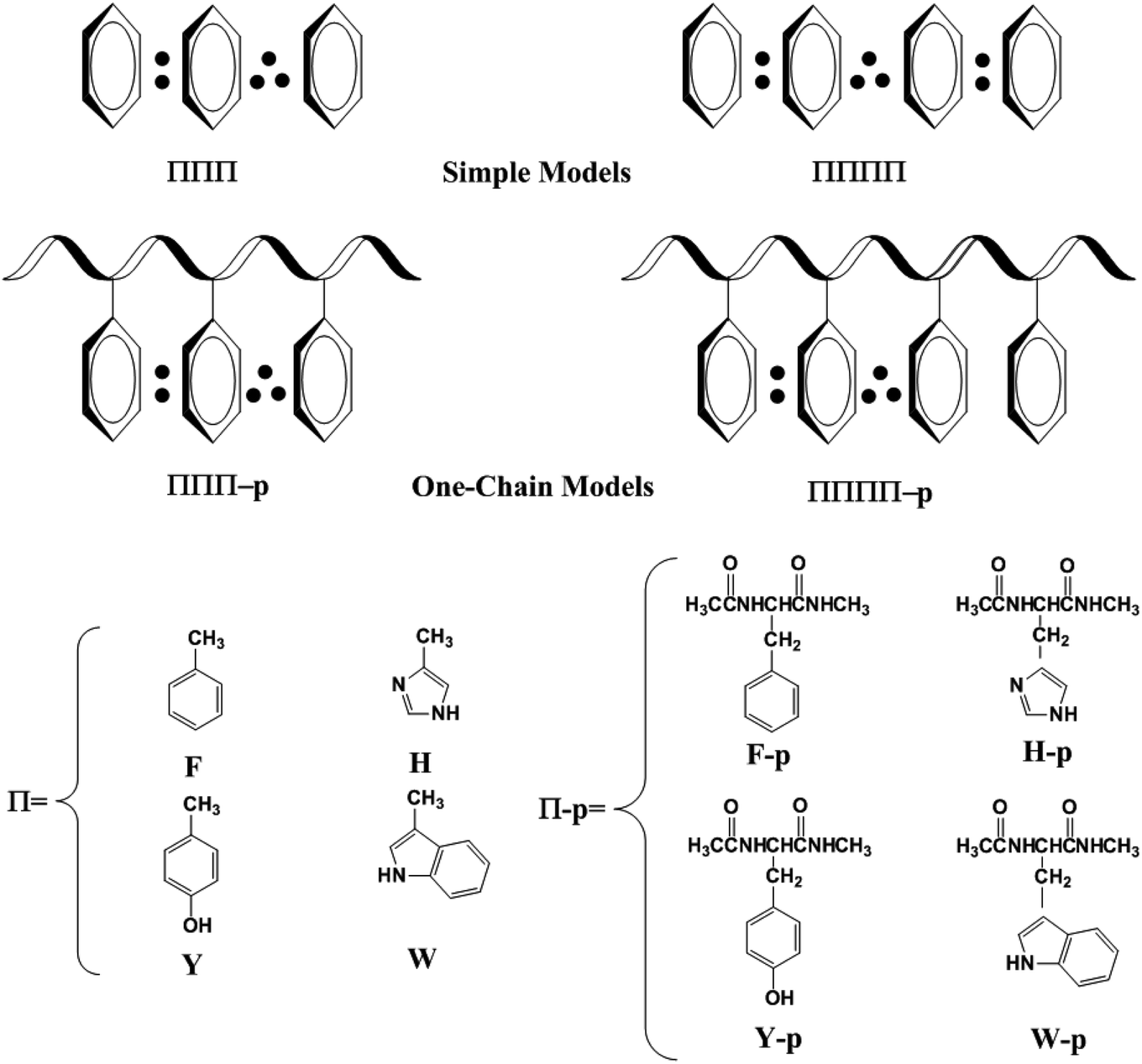
To well address this issue, we firstly used simple models instead of the aromatic amino acids, as shown in Scheme 1. The simple models only include the side chains of Phe, His, Tyr and/or Trp. F, H, Y and W are used to denote the side chains of Phe, His, Tyr and Trp, respectively. The higher π–π stacks optimized at the univalent cationic state are named ΠΠΠ and ΠΠΠΠ (Π = F, H, Y or W) for the three-π and four-π systems. We examine the interactions between the side chains of aromatic cation residues from the simple to complex models. In a one-chain model, all the aromatic residues lie close in the same peptide chain, which are named as ΠΠΠ-p or ΠΠΠΠ-p.
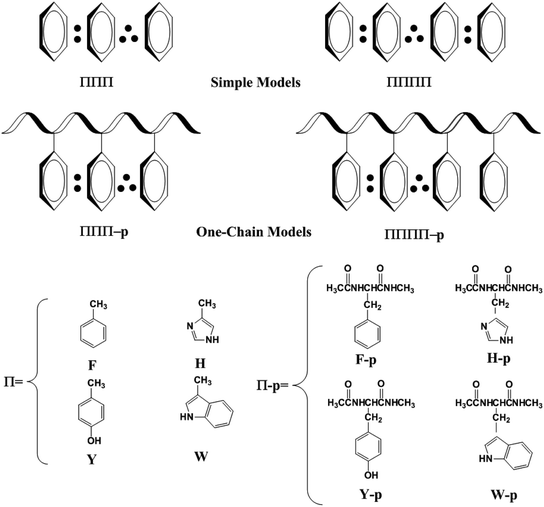 |
| | Scheme 1 Schematic representation of simple models and one-chain models for the considered three-π system and four-π system. “:” indicates the two-electron bond and “∴” denotes the three-electron bond. | |
All gas-phase calculations are carried out by using the Gaussian 09 (ref. 60) suite of programs. In an endeavor to make a thorough investigation about the electron relay functionality of three-π and four-π stacks in proteins, some proper parameters should be used to measure the interactions in three-π and four-π systems, such as the vertical ionization potential (IPV), binding energy (BE) and ultraviolet (UV) spectrum. IPV is the energy difference between a univalent cationic state and its corresponding neutral state without any change in structure according to the optimized structure at the univalent cationic state. For three-π systems, the binding energy is used to evaluate the difference between the energy of whole complex models and the sum energy of an aromatic fragment on the edge and the remaining two rings. Hence, there are two BE values for a three-π system and the lowest one is employed in this work. Homoplastically, the lowest value is adopted for a four-π system, which has three BE values, including interactions between the edge aromatic ring (two edge rings) and the remaining three aromatic rings, two aromatic rings and two aromatic rings. The BEs for the cationic high-π stacking complexes are calculated by subtracting basis set superposition errors (BSSEs)61 through the counterpoise approach.61 Time-dependent density functional response theory (TD-DFT)63 calculations are performed to determine the energy needed to excite an electron from a doubly occupied bonding orbitals to the singly occupied antibonding orbital (π/π*) of the cationic high-π stacking complexes. The corresponding electron transition spectra are calculated by the SWizard program of revision 5.0 (ref. 64) to highlight the spectroscopic features that are characteristics of the cationic high-π stacks. The M06-2X65 hybrid functional mixed with 6-31+G(d,p)66–69 basis set is utilized to optimize all geometries fully and to perform the harmonic vibrational analyses for confirming minima (all real frequencies). All these parameters are gained by single-point calculations at the M06-2X/6-311++G(d,p) level of theory. In addition, to confirm the reliability of the M06-2X hybrid functional for the high π-stacking structures, the long-range (LR)-corrected CAM-B3LYP70 and the LR- and dispersion-corrected ωB97X-D71 functionals are employed to optimize the structures of YYY, FFF and YYYY by using the same basis set. The calculation results reveal that it is suitable to use the M06-2X functional to examine the relay function of the high π-stacking structures in proteins and the details are shown in ESI.† It has been reported that average dielectric constant of proteins is about 4 and the interface of two proteins may have a high dielectric constant.72,73 Therefore, dielectric continuum theories74,75 are used to explore the effect of protein polarizable environment on the relay functionality of the high π–π packs. For all the structures, single-point calculations are carried out in three different continuum solvents, including diethyl ether (ε = 4.335),76,77 dichloroethane (ε = 10.36) and water (ε = 78.36) at the M06/6-311+G(d,p) level of theory by means of the conductor-like polarizable continuum model (CPCM).78,79 Other significant data including BEs, IPVs, electron transition spectra, structural parameters, orbital characteristics and correlations among several quantities for all structures are listed in the ESI.†
3. Results and discussion
3.1 Three-π five-electron bindings
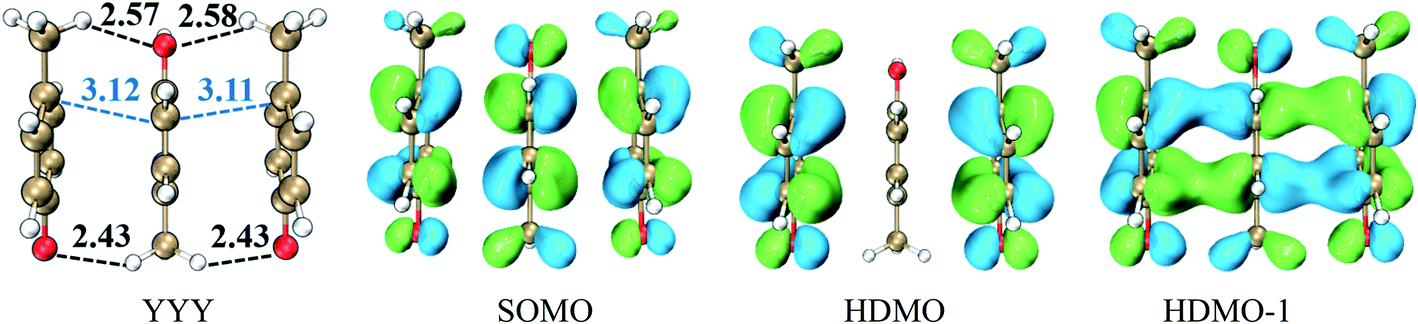
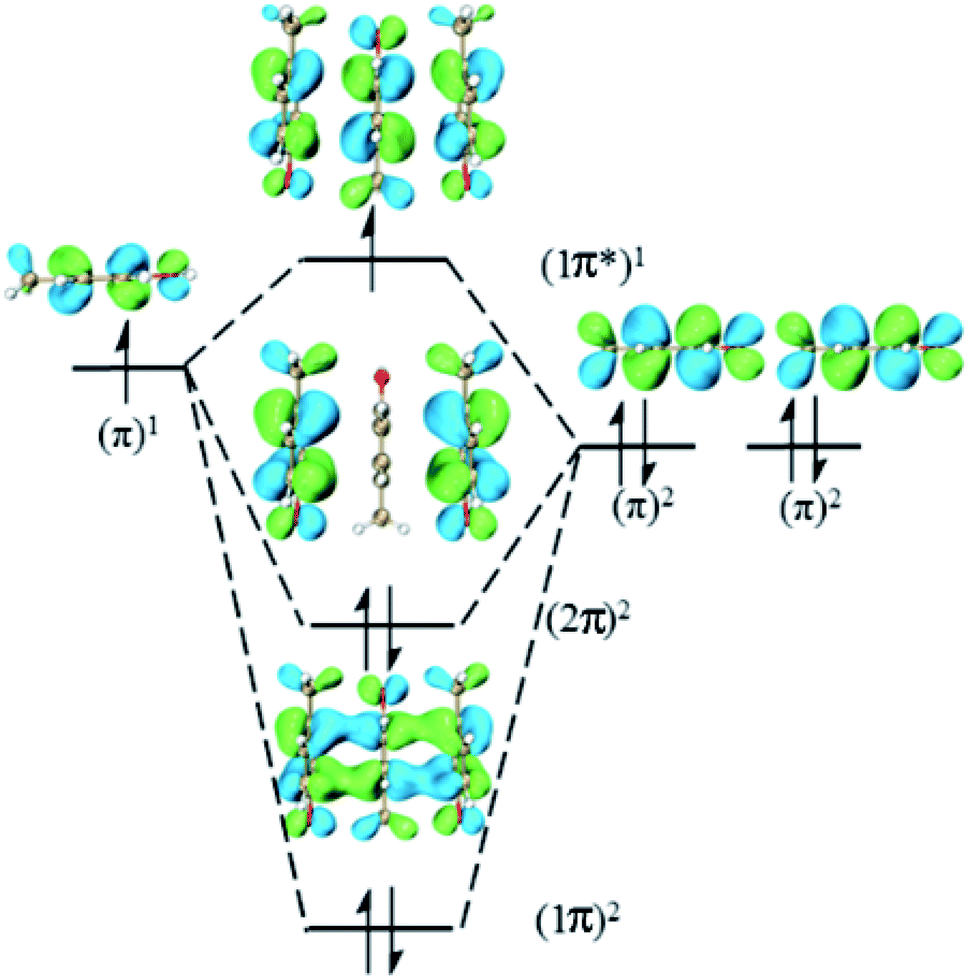
This type consists of three nearly parallel aromatic rings. Here, we illustrate the properties of three-π five-electron resonance structures by an example, which consists of three p-methylphenols. Fig. 1 depicts the structure of YYY with the corresponding singly occupied molecular orbital (SOMO), the highest doubly occupied MO (HDMO) and the second HDMO (HDMO−1). The shortest distances between the three adjoining rings are 3.11 and 3.12 Å, respectively, which are shorter than that (3.33 Å) of corresponding two-body Y∴Y hemibond previously reported by Sun.48 In addition, two intermolecular hydrogen-bonds (H-bonds) are formed between hydroxyl oxygen atoms (O-atoms) and methyl hydrogen atoms (H-atoms) to link the adjacent rings. The short distances between the neighboring rings indicate the strong interactions among the three aromatic pieces. In this case, the plot of SOMO delocalizes over the three pieces with an antibonding feature (1π*). Conversely, HDMO−1 (1π) and HDMO (2π) are bonding to delocalize over the three pieces. The linear interactions of SOMO, HDMO and HDMO−1 yield a new five-electron orbital to connect the three nearly parallel rings, named as a π:π∴π↔π∴π:π resonance binding, in which the hole can delocalize over the three rings through the π–π interactions. The corresponding forming process of π:π∴π↔π∴π:π resonance binding is described in Scheme 2. When a hole moves towards a parallel three-aromatic-ring domain, the highest occupied molecular orbitals (HOMOs) of three isolated monomers (three Ys) combines each other to produce three new MOs, two bonding orbitals (1π and 2π) and an antibonding orbital (1π*). 1π, 2π and 1π* for YYY are HDMO−1, HOMO and SOMO, respectively, as mentioned above. Notably, the interaction of the three new MOs brings out an alliance which not only can draw three fragments close but also effectively reduce the local ionization potential of the three-piece domain. The IPV of YYY is 7.02 eV, which is lower than that (7.90 eV) of p-methylphenol and also lower than that (7.12 eV) of the two-body Y∴Y hemibond.48 In reality, this IPV of YYY is even lower than that of Tyr (7.90 eV) at the M06-2X/6-311++G(d,p)//M06-2X/6-31+G(d,p) level of theory, which are proposed as an efficient ET relay station.28,48 This result indicates that the generation of π:π∴π↔π∴π:π bonding can stabilize a hole in proteins. The value (12.23 kcal mol−1) of BE for YYY are modest, which assures that the formation and separation of π:π∴π↔π∴π:π bonding may be quickly occurred in proteins. Therefore, the lower IPV and the appropriate BE favor the YYY stack to take part in hole transfer in proteins by the transient formation of π:π∴π↔π∴π:π binding. The approaching of the three near parallel aromatic rings can reduce the ionization potential of the three-aromatic-ring domain to capture a hole by the formation of π:π∴π↔π∴π:π bonding and the separation of the three close pieces then increases the local ionization potential, which promote hole to move to the next stepping stone. Consequently, it is rational to deduce that the formation of π:π∴π↔π∴π:π bonding could facilitate long-range hole transfer in proteins as an effective stepping stone.
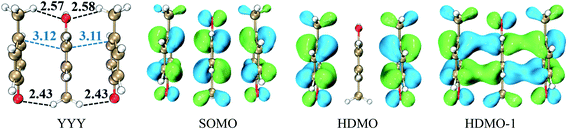 |
| | Fig. 1 Interaction of three aromatic rings and the corresponding singly occupied molecular orbital (SOMO), the highest doubly occupied molecular orbital (HDMO) and the second highest doubly occupied molecular orbital (HDMO−1). The numbers are the shortest distances in Å for the neighboring aromatic rings obtained at the M06-2X/6-31+G(d,p) level. | |
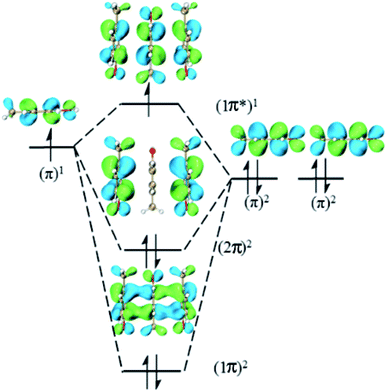 |
| | Scheme 2 Exhibition of forming π:π∴π↔π∴π:π five-electron bindings via the highest occupied molecular orbitals of three isolated p-methylphenols. | |
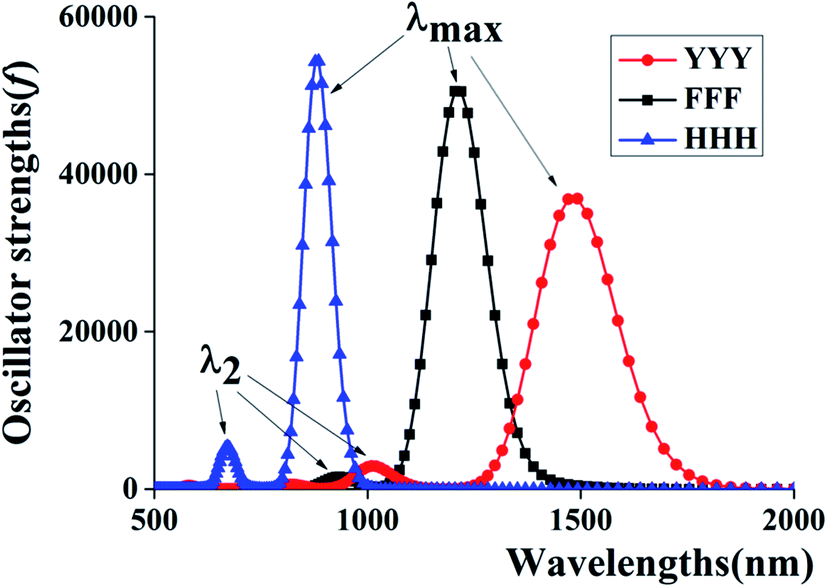
Additionally, to provide more features of the π:π∴π↔π∴π:π bonding, the TD-DFT calculations are also carried out to analyze the absorption spectra of YYY, as shown in Fig. 2. There are two distinct absorption peaks in the absorption curve, corresponding to electron transitions from the two doubly occupied MOs to the SOMO. The λmax for YYY is 1482.33 nm in correspondence with electron transition from HDMO to SOMO. Another absorption peak (λ2) at 1013.00 nm stands for electron transition from HDMO−1 to SOMO. The spectra identification is an index of forming π:π∴π↔π∴π:π bonding. The appearance and disappearance of the two absorption peaks indicate the formation and dissociation of the π:π∴π↔π∴π:π bonding, which may provide a signal to identify the relay functionality of the three-π stacking cluster.
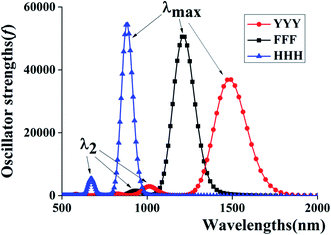 |
| | Fig. 2 Two characteristic peaks of HDMO → SOMO (λmax) and HDMO−1 → SOMO (λ2) for the absorption spectra of YYY, FFF and HHH. | |
Similar analyses can be applied for the other three-π systems, including FFF, HHH, YFY, YHY, YWY, FYY, WYY, HYY, FYF, FHF, FFY, HFH, HYH, FYH, FHY, FWY, YFH and WHW. The relative information about their structures and frontier orbitals are given in the ESI.† Among these structures, the shortest distances between the neighboring fragments (dmin) are in the range of 2.94–3.25 Å, and their BE values coverage from 7.33 kcal mol−1 for FYY to 16.93 kcal mol−1 for WHW (Table 1), backing for the formation of the π:π∴π↔π∴π:π bindings. More interesting, the IPVs for them are in the range of 6.52–7.57 eV, which are sharply lower than that of the corresponding isolated monomer, as shown in Table 1. The values of IPVs for the 19 three-π systems are comparable to the IP values of Trp (7.35 eV) and Tyr (7.90 eV) at the M06-2X/6-311++G(d,p)//M06-2X/6-31+G(d,p) level, two effective relay residues in proteins.28 Therefore, the stacking interactions among three close aromatic side chains can significantly decrease the ionization potential of the three-π packing region relative to the every monomer, implying that the three-π pack has a potential functionality to promote hole transport by transient forming the π:π∴π↔π∴π:π binding in protein.
Table 1 Shortest distance between the neighboring aromatic rings (dmin, in Å), binding energy (BE, in kcal mol−1), vertical ionization potential (IPV, in eV) and decreasing value of IPV (ΔIP, in eV) compared to the corresponding monomer for all the multi-π stacking systems
| Species |
F |
H |
Y |
W |
YYY |
FFF |
HHH |
YFY |
YHY |
| dmin |
— |
— |
— |
— |
3.11/3.12 |
3.18/3.19 |
2.94/2.94 |
3.25/3.12 |
2.97/3.11 |
| BE |
— |
— |
— |
— |
12.23 |
11.57 |
15.13 |
12.25 |
13.98 |
| IPV |
8.64 |
8.11 |
7.90 |
7.35 |
7.02 |
7.57 |
6.52 |
7.21 |
6.82 |
| ΔIP |
— |
— |
— |
— |
0.88 |
1.07 |
1.59 |
0.69 |
1.08 |
| Species |
YWY |
FYY |
WYY |
HYY |
FYF |
FHF |
FFY |
HFH |
HYH |
| dmin |
3.15/3.10 |
3.22/3.09 |
3.07/3.12 |
2.97/3.20 |
3.16/3.11 |
3.12/3.12 |
3.22/3.09 |
3.00/3.01 |
3.00/3.05 |
| BE |
12.77 |
8.68 |
13.33 |
12.16 |
10.91 |
11.76 |
7.33 |
10.29 |
11.42 |
| IPV |
6.55 |
7.06 |
6.86 |
6.79 |
7.11 |
6.99 |
7.31 |
7.28 |
7.06 |
| ΔIP |
0.80 |
0.84 |
0.49 |
1.11 |
0.79 |
1.12 |
0.59 |
0.83 |
0.84 |
| Species |
FHY |
FYH |
FWY |
YFH |
WHW |
FFFF |
YYYY |
HHHH |
YFHY |
| dmin |
3.12/3.07 |
3.21/2.96 |
3.21/3.18 |
3.04/3.04 |
2.97/2.98 |
3.19 |
3.16 |
3.03 |
3.18 |
| BE |
11.75 |
9.23 |
10.90 |
10.57 |
16.93 |
8.03 |
10.42 |
9.56 |
9.46 |
| IPV |
6.98 |
7.15 |
6.53 |
7.16 |
6.62 |
7.42 |
6.74 |
6.37 |
6.82 |
| ΔIP |
0.92 |
0.75 |
0.82 |
0.74 |
0.73 |
1.22 |
1.16 |
1.74 |
1.08 |
| Species |
FYYY |
FYFY |
YFFY |
FFFFF |
YYYYY |
HHHHH |
FFFFFF |
YYYYYY |
| dmin |
3.26 |
3.23 |
3.18 |
3.23 |
3.13 |
2.98 |
3.22 |
3.13 |
| BE |
7.04 |
8.28 |
9.46 |
6.33 |
6.92 |
6.48 |
5.38 |
7.25 |
| IPV |
6.86 |
6.95 |
7.23 |
7.37 |
6.69 |
6.39 |
7.30 |
6.59 |
| ΔIP |
1.04 |
0.95 |
0.67 |
1.27 |
1.21 |
1.72 |
1.34 |
1.31 |
| Species |
YYY-p |
FYY-p |
HYY-p |
YHY-p |
FHF-p |
FHY-p |
YYYY-p |
FFFF-p |
YFYY-p |
| dmin |
3.07/3.03 |
3.14/3.03 |
2.97/3.05 |
2.99/3.08 |
3.12/3.19 |
3.18/3.02 |
3.09 |
3.06 |
3.07 |
| IPV |
6.87 |
7.02 |
6.75 |
6.89 |
6.99 |
6.92 |
6.75 |
7.35 |
6.93 |
| ΔIP |
0.95 |
0.80 |
1.07 |
0.93 |
1.06 |
0.90 |
1.07 |
0.70 |
1.01 |
In addition, several interesting trends can be gained by the careful analyses of the data in Table 1. For the three-π systems consisting of the same aromatic rings, the decreasing order of IPV is FFF (7.57 eV) > YYY (7.02 eV) > HHH (6.52 eV), which is opposite to the order of BEs of them, as shown in Table 1. The BEs of FFF, YYY and HHH are 11.57, 12.23 and 15.13 kcal mol−1 (Table 1), respectively. The decreasing values of (ΔIPs) of the three-π systems relative to the monomers are 1.07, 0.88 and 1.59 eV for FFF, YYY and HHH, respectively. This changes in BE, IP and ΔIPs indicate that the larger BE results in the lower IP and the larger ΔIP for the three-same-π systems. YYY is a special case because the BE value (12.23 kcal mol−1) of YYY is larger than that (11.57 kcal mol−1) of FFF and the ΔIP values is in reverse order. This maybe attribute to the fact that the additional intermolecular H-bonds forming between the neighboring pieces in YYY enhance the BE value relative to FFF and the H-binding interactions can't reduce the IPV of the systems, which comes from the π–π interactions. Therefore, despite the higher BE for YYY, the ΔIP value of YYY is less than that of FFF. Additionally, a further analysis reveals that there is a distinct linear correlation among the BE values and the decreasing values of IPVs (ΔIPs) for the three-π models except the cases with the formations of strong intermolecular H-bonds, including YYY, YFY, YWY, WYY and WHW, as exhibited in Fig. 3. The correlation of BE and ΔIP substantiates that the strong π–π binding strength can efficiently reduce the IP value of the three-π cluster by the formation of three-π five-electron binding.
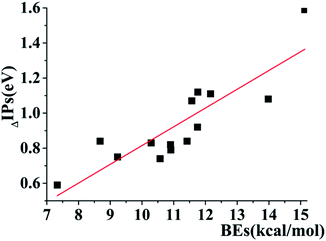 |
| | Fig. 3 The relationship among the binding energies (BEs) and the decreasing values of vertical ionization potentials (ΔIPV) for three-π models except for YYY, YFY, YWY, WYY and WHW. It is clear that the higher the binding strengths of the three-π stacks are, the larger the values of ΔIP are. | |
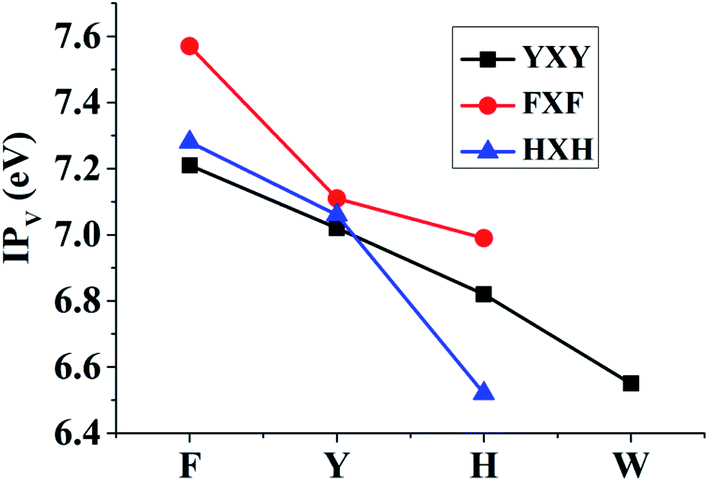
The corresponding TD-DFT calculations also confirm the formations of the π:π∴π↔π∴π:π bondings with two characteristic peaks for YYY, FFF and HHH, as shown in Fig. 2. The two characteristic peaks indicate the electron transitions from HDMO (λmax) and HDMO−1 (λ2) to SOMO. The interactions of SOMO, HDMO and HDMO−1 produce the π:π∴π↔π∴π:π bondings, as mentioned above. The λmaxs for YYY, FFF and HHH are 1482.33, 1211.92 and 881.3 nm, respectively, indicating λmax shifts blue with the decrease of the ring size in the three-π systems. Additionally, when the two side aromatic rings are the same, the IP value of three-π system decreases with the diminishing of the IPV of the middle aromatic ring, such as YFY (7.21 eV) > YYY (7.02 eV) > YWY (6.55 eV) (Table 1). However, the IPV of YHY with H as the middle ring is lower than that of the case with Y as the middle ring, YHY (6.82 eV) < YYY (7.02 eV). Although the IPV value (7.90 eV) of Y is lower than that (8.11 eV) of H, the surface area of the five-number ring of H is less than that of the six-number ring with an O-atom of Y and the small area causes the strong π–π packing interactions with the two side aromatic rings in YHY, which results in the IPV value of YHY (6.82 eV) is lower than that of YYY (7.02 eV). Therefore, the decreasing order of IPVs for the cases of YXY is YFY < YYY < YHY < YWY. The same trend in IPV can be found for FXF and HXH (X changes from F, Y to H), as shown in Fig. 4.
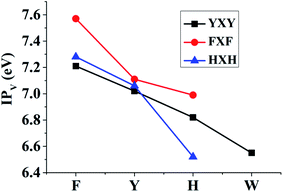 |
| | Fig. 4 The changing tendencies of IPs for the YXY, FXY and HXH with X changing from F, Y, H to W. | |
3.2 Four-π seven-electron bindings
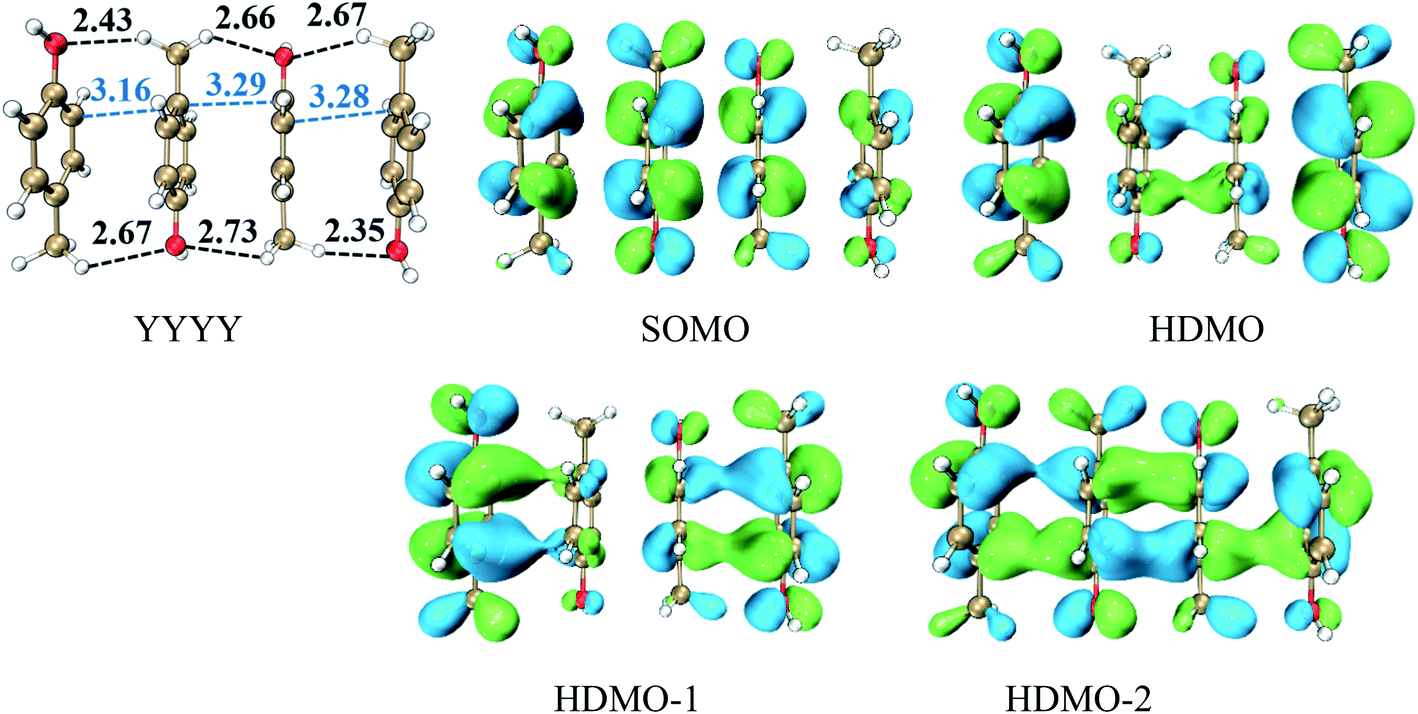
Analogously, we mainly explore the relay function of four nearly parallel aromatic rings in this section. A representative example is the π–π packing interactions among the side chains of four Tyr residues at the one-electron oxidation state, named as YYYY, as shown in Fig. 5. The shortest distances between adjacent p-methylphenol planes are 3.16 Å, 3.29 Å and 3.28 Å, respectively, which are shorter than the distance between two-body Y∴Y hemibond (3.33 Å) in the previous work.48 Additionally, there are two intermolecular H-bonds forming between two adjacent pieces, as shown in Fig. 5. The BE value of YYYY is 10.42 kcal mol−1. These data cast light on the strong interactions among the four Y pieces. In this case, its SOMO delocalizes over the four aromatic rings with an antibonding characteristic, which indicates the hole is distributed over the four bodies. In addition, HDMO, HDMO−1 and HDMO−2 are all delocalized over the four pieces with the bonding features. Furthermore, the plot of HDMO exhibits a bonding link between the two middle aromatic rings, the plot of HDMO−1 shows the bonding links between the two terminal aromatic rings symmetrically and the plot of HDMO−2 shows the bonding characteristic to connect the four pieces. Then, the combination of SOMO, HDMO, HDMO−1 and HDMO−2 produces a special π:π∴π:π↔π:π:π∴π resonance binding, as shown in Fig. S8.† More significantly, the IPV value of YYYY is 6.74 eV, which is lower than the IPV of the monomer (7.90 eV) and even lower than that (7.02 eV) of YYY, as mentioned above. This result indicates that the IP of the π–π packing system decreases with the increase of the number of aromatic rings. More importantly, the features of the YYYY imply that the high π–π packing interactions can modulate the hole migration pathway by reducing the IP of the local domain and then encourage long-range hole transfer in proteins.
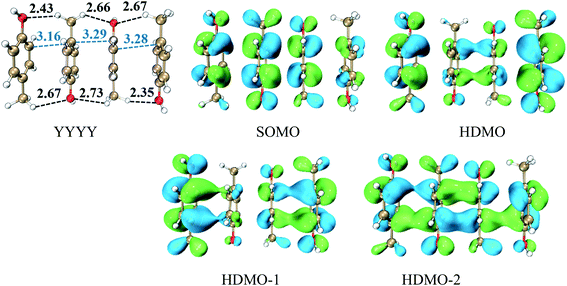 |
| | Fig. 5 Structure of YYYY and the corresponding singly occupied molecular orbitals (SOMO) and three doubly occupied molecular orbitals (HDMOs) obtained at the M06-2X/6-31+G(d,p) level. | |
Similar analyses can be available for the other four-π systems, including FFFF, HHHH, YFHY, FYYY, YFYF and YFFY (the details are in the ESI†). The IPV values of these four-π systems are in the scope of 6.37–7.42 eV (Table 1), which are all lower than these of the corresponding monomers and are compared with the IP value of the side chain of Trp (7.35 eV), an effective relay station in proteins.28 The binding energies of the four-π seven-electron bindings are in the range of 7.04–10.42 kcal mol−1 (Table 1). The YYYY, FFFF and HHHH systems all consist of four duplicated aromatic rings. The electron hole in FFFF is delocalized spatially wider than in HHHH because the size of F is large than that of H, which results in that the BE (8.03 kcal mol−1) of FFFF is less than that of HHHH (9.56 kcal mol−1). However, YYYY (10.42 kcal mol−1) goes counter to this rule because of the existence of the intermolecular H-bonds.
To further get the reliable evidence for four-π seven-electron bindings, the electron transition spectra of YYYY, FFFF and HHHH are achieved through carrying out the TD-M06-2X/6-311++G(d,p) calculations, as diagrammed in Fig. 6. There are three distinct absorption peaks in every absorption curve, corresponding to main electron transitions from the three doubly occupied MOs to the SOMO [(1π)2 → (1π*)1, (2π)2 → (1π*)1 and (3π)2 → (1π*)1]. For instance, the λmax for YYYY is 1634.25 nm with a strong oscillator strength (f = 0.23), in correspondence with an electron migration from HDMO to SOMO. The other two absorption peaks at 1052.48 nm and 852.2 nm with two weaker oscillator strengths stand for electron transitions from HDMO−1 to SOMO and HDMO−2 to SOMO, respectively. It's worth noting that a larger size of the aromatic ring can bring out a red shift in the λmax, as well as a longer distance between the two side fragments in the four-π systems. For example, the increasing order of λmax values is HHHH (1044.76 nm) < FFFF (1542.29 nm) < YYYY (1634.25 nm), which follows the increasing order of distance, HHHH (9.45 Å) < FFFF (9.65 Å) < YYYY(9.73 Å).
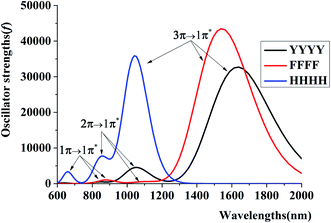 |
| | Fig. 6 Three characteristic peaks [(1π)2 → (1π*)1, (2π)2 → (1π*)1 and (3π)2 → (1π*)1] of the π:π∴π:π↔π:π:π∴π resonance binding for FFFF, HHHH and YYYY. It is shown that the maximal absorbing spectrum (λmax) exhibits an obvious red shift with the increasing the size of the four aromatic rings. | |
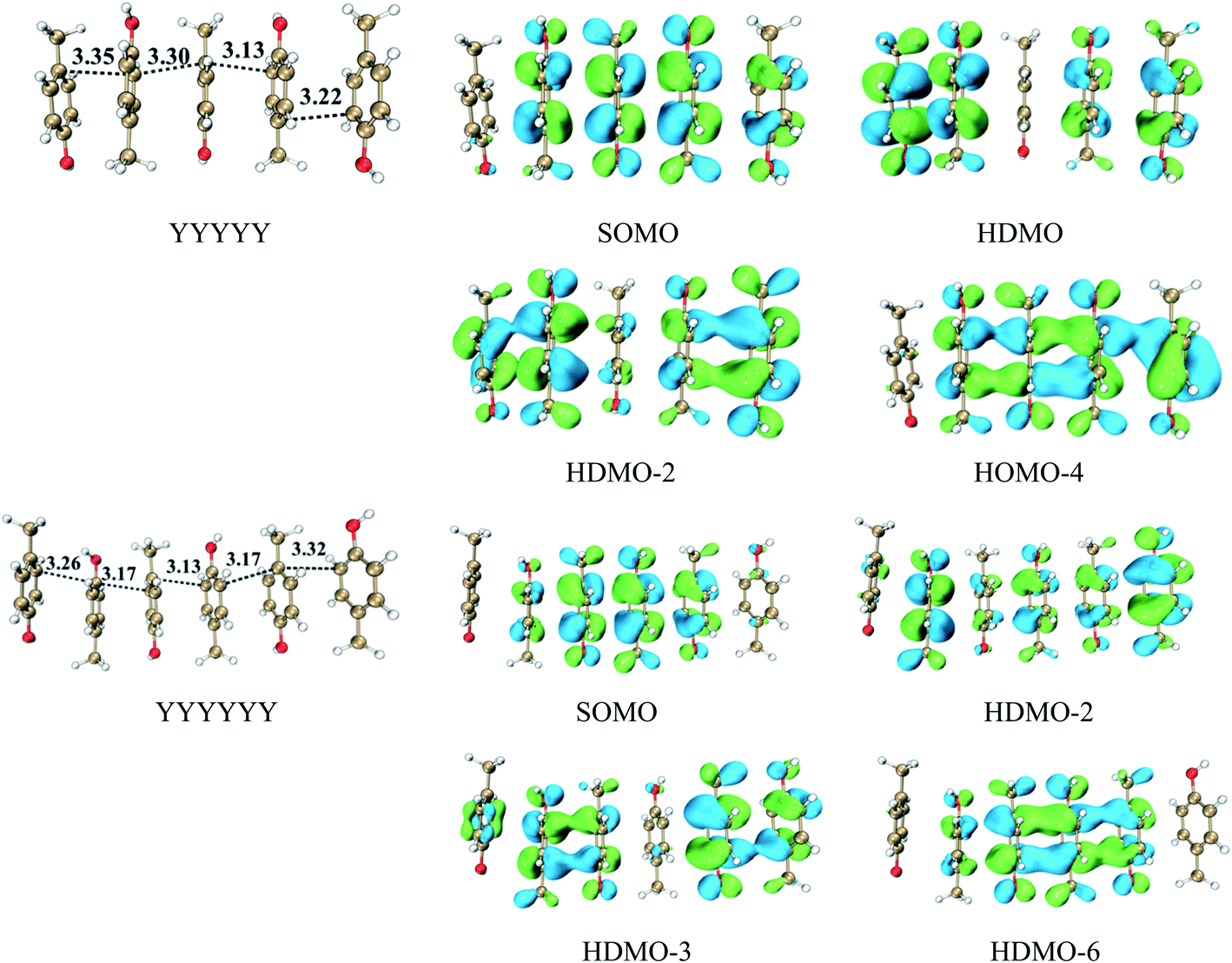
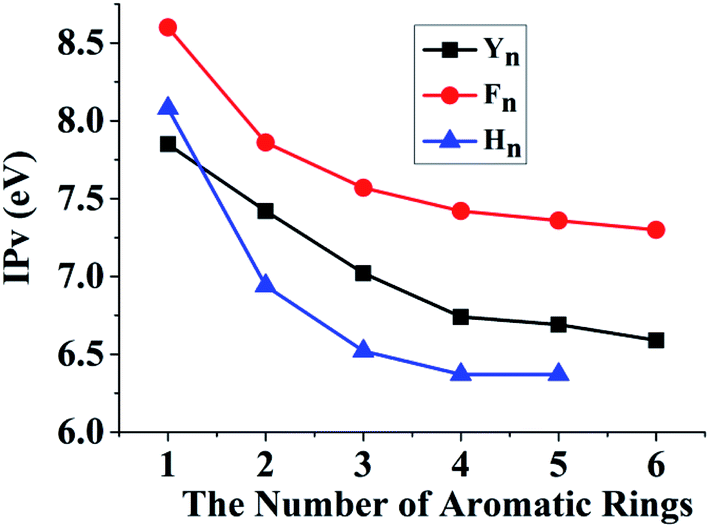
We have revealed that the IP of the same-π packing system (Xn) decreases with the increasing number of the same aromatic rings due to the formations of the π∴π hemibond, the π∴π:π↔π:π∴π binding and π:π∴π:π↔π:π:π∴π bindings for n = 2, 3, 4, respectively. This result also confirms that a larger scope for hole delocalization can enhance the stabilization of hole. Therefore, it is seemingly reasonable to infer that the hole can delocalize over more aromatic rings at the same time in a higher π–π packing system. So we continue to examine the π–π packing systems containing five and six aromatic rings. Fig. 7 shows the structures of YYYYY and YYYYYY with the corresponding π–π packing orbitals. It is surprising to reveal that SOMO delocalizes over only four aromatic pieces in YYYYY and YYYYYY, which indicates that a hole cannot delocalize over more than four aromatic rings simultaneously. The distance from the terminal aromatic ring without spin density to the adjacent ring is 3.35 Å, which is larger than that of the other neighboring rings (3.30, 3.13 or 3.22 Å) in YYYYY. The corresponding BE is 6.88 kcal mol−1, which is significant lower than that (10.42 kcal mol−1) of YYYY. More importantly, the IPV of YYYYY are 6.69 eV, nearly consistent with the IPV (6.74 eV) of YYYY. These results indicate that the IPV and BE of the Yn system decrease with the increase the number of Y in the π–π packing structure when ‘n’ is not more than 4. The similar analysis can be applied for the other π–π packing systems (Xn, X = F and H, n = 2, 3, 4) with the increasing of the aromatic rings, as shown in Fig. 8. The corresponding orbital analyses show that the maximum number of the aromatic rings participating hole resonance structure along the π–π packing is four at the same time. These results indicate that the IPV value of the π–π packing system nearly remains constant with the increase of the same aromatic rings when the number of the aromatic rings is more than four.
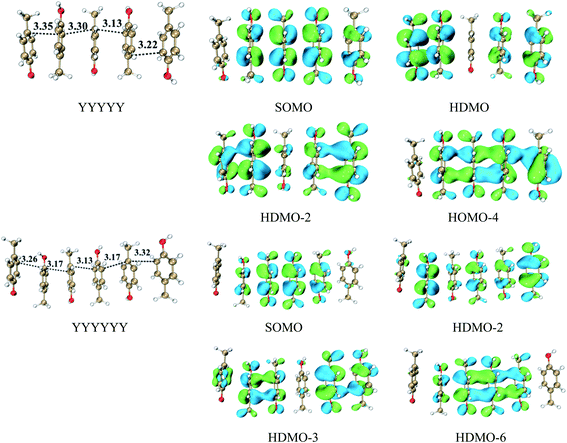 |
| | Fig. 7 Interaction of five and six aromatic rings and the corresponding MOs. | |
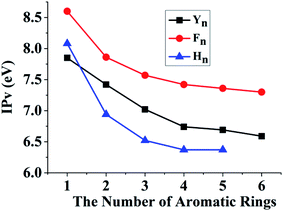 |
| | Fig. 8 The changing trend of the vertical ionization potentials (IPs) with the increasing number of the same aromatic rings in the high π–π packing systems. Y, F and H are the side chains of Tyr, Phe and His, respectively. | |
3.3 Influence of peptide chains on the formation of three-π five-electron and four-π seven-electron bindings
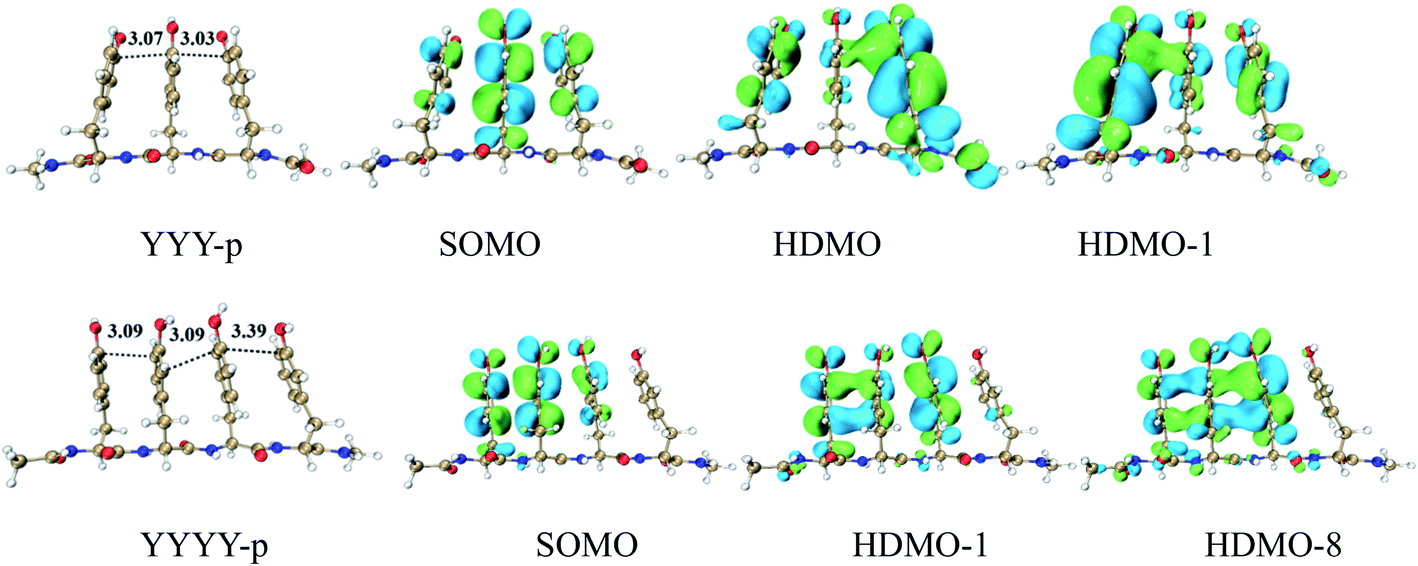
DFT calculations provide a systematic analysis of the cooperative interactions between three close aromatic residues on the same peptide chain (one-chain model) at the cation state, as shown in Fig. 9 and S12.† It should be noted that the presence of peptide chain can draw the three aromatic rings closer. For the case of YYY-p, the shortest distances between the aromatic side chains are only 3.03 and 3.07 Å respectively, which are shorter than those (3.11 and 3.12 Å) in the corresponding simple model of YYY. The distributions and interactions of the corresponding frontier MOs also favor the formation of the π:π∴π↔π∴π:π binding. As displayed in Fig. 9, SOMO, HDMO, and HDMO−1 are not only distributed on the three side chains but also partially delocalize over the peptide chain. The IPV of YYY-p is 6.87 eV, which is lower than that (7.02 eV) of YYY. This result reflects that the presence of peptide chain can reduce IPV of three-π pack and facilitate the formation of π:π∴π↔π∴π:π bindings. The similar results can be found for the other one-chain three-π stacks, as shown in Fig. S12.† In addition, the π:π∴π↔π∴π:π bindings can be formed in the cases of the three aromatic residues residing at the different peptide chains and the details are shown in Fig. S13.†
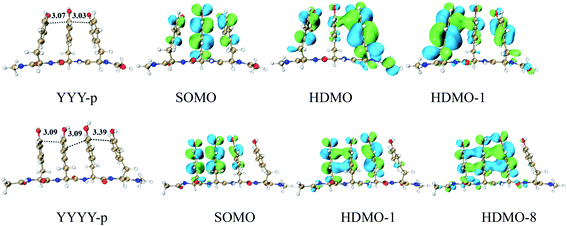 |
| | Fig. 9 Three and four Tyrs in a main chain, named YYY-p and YYYY-p, with the distributions of SOMO, HDMO and HDMO−1/8. | |
Furthermore, four adjacent Tyr residues in the same peptide chain (YYYY-p) are examined, as shown in Fig. 9. The plot of SOMO of YYYY-p unexpectedly distributes over only three neighboring aromatic side chains, indicating that the hole can be hold by three aromatic rings and cannot delocalize over four aromatic rings at the same time in this case. Additionally, the two doubly occupied molecular orbitals, HDMO and HDMO−8, mainly delocalize over the same three aromatic side chains, as shown in Fig. 9. Therefore, the interactions of SOMO, HDMO and HDMO−8 produce the π∴π:π↔π:π∴π binding, which isn't the expected four-π seven-electron binding. The corresponding geometrical information also confirms that the terminal aromatic ring without the distribution of SOMO is long away from the neighboring ring relative to the other side aromatic ring. The corresponding nearest distance is 3.39 Å, which is larger than that (3.09/3.09 Å) of the other neighboring rings. These results are different from the corresponding simple model of YYYY, in which the interactions of the four aromatic rings form the π:π∴π:π↔π:π:π∴π binding, as mentioned above. Therefore, the presence of the peptide chain can modulate the distribution of hole and the motif of the multi-electron resonance structure. It is a remarkable fact that the IPV value of YYYY-p is 6.75 eV, which is lower than that of YYY-p (6.87 eV). The lower IPV value indicates that YYYY-p can capture a hole as the efficient stepping stone by forming the π∴π:π↔π:π∴π binding. The similar analyses can be applied for the other one-chain four-π systems, including FFFF-p and YFYY-p.
3.4 Influence of protein polarizable environment on the relay functionality of the high π–π packs
Because the average dielectric constant of proteins is about 4 and some special positions in proteins or the interface of two proteins may have a high dielectric constant,62,63 the effect of protein polarizable environment on the relay functionality of the high π–π packs were examined in the continuum solvents of diethyl ether (ε = 4.335), dichloroethane (ε = 10.36) and water (ε = 78.36) at the M06/6-311++G(d,p) level by means of the conductor-like polarizable continuum model (CPCM). The corresponding calculations reveal that the IPV values of all the high π–π packing structures decrease with the increase of the dielectric constant of continuum solvents. For example, the IPV values of YYY are 7.02, 5.83, 5.61 and 5.48 eV for the continuum polarizable environment of gas (ε = 0.00), diethyl ether (ε = 4.335), dichloroethane (ε = 10.36) and water (ε = 78.36), respectively. The other three-π and four-π packs have the similar trend for the correction between IPV and the polarizable environment, as shown in Table S1.† These results also reflect that the higher polarizable environment can facilitate the high π–π packs to take part in the hole transfer in proteins, which modulates the relay functionality of the high π–π packs.
4. Conclusion
The present electronic structure calculations elucidate that the probability of formations of π:π∴π↔π∴π:π and π:π∴π:π↔π:π:π∴π resonance bondings, which may serve as relay stones of hole transfer in proteins. Two main reasons can support these structures to participate in long-range hole transfer of proteins. One is the interactions of these electron-riched groups can lower the local IPV to capture a hole. The other is that the BE values are in the range of 7.33–16.93 kcal mol−1 for the three-π five-electron bindings and 5.48–10.42 kcal mol−1 for four-π seven-electron bindings, which are even more stronger than a hydrogen bond, indicating that the multi-π multi-electron bindings may produce in a short time during the hole transfer processes in proteins. The movement of protein may cause the formation and dissociation of the multi-π multi-electron binding to promote hole migration due to the low IP of the multi-π cluster and the modest BE of multi-π multi-electron binding. In addition, our DFT calculations reveal that the hole can't delocalize over infinite aromatic rings along the high π–π packing structures, which can delocalize over four aromatic rings for the simple models and three aromatic rings for the one-chain models. This result reflects that the micro-surroundings of proteins can modulate the existing form of hole. The discovery of the π∴π:π↔π:π∴π and π:π∴π:π↔π:π:π∴π resonance bindings may broaden the avenues to explore the microscopic hole transfer pathways in proteins. Furthermore, it will be highly valuable to explore the properties and the advanced applications of π∴π:π↔π:π∴π and π:π∴π:π↔π:π:π∴π bindings by both more experimental and theoretical approaches in the future.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
This work was financially supported by NSFC of China (21573026, 21773138), sponsored by Natural Science Foundation of Chongqing, China (cstc2018jcyjAX0643) and the graduate research and innovation foundation of Chongqing (CYB18045).
References
- C. C. Page, C. C. Moser, X. Chen and P. L. Dutton, Nature, 1999, 402, 47 CrossRef CAS PubMed.
- J. R. Winkler and H. B. Gray, J. Am. Chem. Soc., 2014, 136, 2930 CrossRef CAS.
- J. Huang, J. Zarzycki, M. R. Gunner, W. W. Parson, J. F. Kern, J. Yano, D. C. Ducat and D. M. Kramer, J. Am. Chem. Soc., 2020, 142, 10459 CrossRef CAS.
- M. Cordes and B. Giese, Chem. Soc. Rev., 2009, 38, 892 RSC.
- S. S. Skourtis, D. H. Waldeck and D. N. Beratan, Annu. Rev. Phys. Chem., 2010, 61, 461 CrossRef CAS PubMed.
- H. B. Gray and J. R. Winkler, Q. Rev. Biophys., 1999, 36, 341 CrossRef PubMed.
- S. J. Mora, E. Odella, G. F. Moore, D. Gust, T. A. Moore and A. L. Moore, Acc. Chem. Res., 2018, 51, 445 CrossRef CAS PubMed.
- P. Saura and V. R. I. Kaila, J. Am. Chem. Soc., 2019, 141, 5710 CrossRef CAS PubMed.
- O. S. Wenger, Acc. Chem. Res., 2013, 46, 1517 CrossRef CAS.
- J. Stubbe, D. G. Nocera, C. S. Yee and M. C. Y. Chang, Chem. Rev., 2003, 103, 2167 CrossRef CAS.
- X. Chen, G. Ma, W. Sun, H. Dai, D. Xiao, Y. Zhang, X. Qin, Y. Liu and Y. Bu, J. Am. Chem. Soc., 2014, 136, 4515 CrossRef CAS PubMed.
- X. Qin, L. Deng, C. Hu, L. Li and X. Chen, Chem.–Eur. J., 2017, 23, 14900 CrossRef CAS.
- H. B. Gray and J. R. Winkler, Biochim. Biophys. Acta, Bioenerg., 2010, 1797, 1563 CrossRef CAS PubMed.
- J. Yu, J. R. Horsley and A. D. Abell, Acc. Chem. Res., 2018, 51, 2237 CrossRef PubMed.
- S. Saen-Oon, M. F. Lucas and V. Guallar, Phys. Chem. Chem. Phys., 2013, 15, 15271 RSC.
- N. Amdursky, ChemPlusChem, 2015, 80, 1075 CrossRef CAS.
- L. Berstis, G. T. Beckham and M. F. Crowley, J. Chem. Phys., 2015, 143, 225102 CrossRef PubMed.
- V. L. Davidson, Acc. Chem. Res., 2008, 41, 730 CrossRef CAS.
- C. M. Paquete and R. O. Louro, Acc. Chem. Res., 2014, 47, 56 CrossRef CAS PubMed.
- S. Hammes-Schiffer and A. A. Stuchebrukhov, Chem. Rev., 2010, 110, 6939 CrossRef CAS PubMed.
- J. L. Dempsey, J. R. Winkler and H. B. Gray, Chem. Rev., 2010, 110, 7024 CrossRef CAS.
- R. A. Malak, Z. N. Gao, J. F. Wishart and S. S. Isied, J. Am. Chem. Soc., 2004, 126, 13888 CrossRef CAS PubMed.
- J. Yu, O. Zvarec, D. M. Huang, M. A. Bissett, D. B. Scanlon, J. G. Shapter and A. D. Abell, Chem. Commun., 2012, 48, 1132 RSC.
- M. Choi, S. Shin and V. L. Davidson, Biochemistry, 2012, 51, 6942 CrossRef CAS.
- X. Chen, L. Zhang, L. Zhang, W. Sun, Z. Zhang, H. Liu, Y. Bu and R. I. Cukier, J. Phys. Chem. Lett., 2010, 1, 1637 CrossRef CAS.
- R. D. Teo, R. Wang, E. R. Smithwick, A. Migliore, M. J. Therien and D. N. Beratan, Proc. Natl. Acad. Sci. U.S.A., 2019, 116, 15811 CrossRef CAS PubMed.
- X. Song, F. Zhang and Y. Bu, J. Comput. Chem., 2019, 40, 988 CrossRef CAS.
- C. Shih, A. K. Museth, M. Abrahamsson, A. M. Blanco-Rodriguez, A. J. Di Bilio, J. Sudhamsu, B. R. Crane, K. L. Ronayne, M. Towrie Jr, A. Vlcek, J. H. Richards, J. R. Winkler and H. B. Gray, Science, 2008, 320, 1760 CrossRef CAS PubMed.
- A. Lukacs, A. P. Eker, M. Byrdin, K. Brettel and M. H. Vos, J. Am. Chem. Soc., 2008, 130, 14394 CrossRef CAS.
- N. A. Tarboush, L. M. R. Jensen, E. T. Yukl, J. Geng, A. Liu, C. M. Wilmot and V. L. Davidson, Proc. Natl. Acad. Sci. U.S.A., 2011, 108, 16956 CrossRef.
- J. L. Seifert, T. D. Pfister, J. M. Nocek, Y. Lu and B. M. Hoffman, J. Am. Chem. Soc., 2005, 127, 5750 CrossRef CAS.
- J. R. Horsley, J. Yu, K. E. Moore, J. G. Shapter and A. D. Abell, J. Am. Chem. Soc., 2014, 136, 12479 CrossRef CAS PubMed.
- Y. Arikuma, H. Nakayama, T. Morita and S. Kimura, Angew. Chem., Int. Ed., 2010, 49, 1800 CrossRef CAS.
- B. Giese, M. Wang, J. Gao, M. Stoltz, P. Muller and M. Graber, J. Org. Chem., 2009, 74, 3621 CrossRef CAS PubMed.
- M. Cordes, A. Kottgen, C. Jasper, O. Jacques, H. Boudebous and B. Giese, Angew. Chem., Int. Ed., 2008, 47, 3461 CrossRef CAS PubMed.
- J. A. Gao, P. Muller, M. Wang, S. Eckhardt, M. Lauz, K. M. Fromm and B. Giese, Angew. Chem., Int. Ed., 2011, 50, 1926 CrossRef CAS.
- P. Brunelle, C. Schneich and A. Rauk, Can. J. Chem., 2006, 84, 893 CrossRef CAS.
- M. Wang, J. Gao, P. Muller and B. Giese, Angew. Chem., Int. Ed., 2009, 48, 4232 CrossRef CAS.
- R. S. Glass, G. L. Hug, C. Schoneich, G. S. Wilson, L. Kuznetsova, T. M. Lee, M. Ammam, E. Lorance, T. Nauser, G. S. Nichol and T. Yamamoto, J. Am. Chem. Soc., 2009, 131, 13791 CrossRef CAS.
- R. S. Glass, C. Schoneich, G. S. Wilson, T. Nauser, T. Yamamoto, E. Lorance, G. S. Nichol and M. Ammam, Org. Lett., 2011, 13, 2837 CrossRef CAS PubMed.
- W. J. Chung, M. Ammam, N. E. Gruhn, G. S. Nichol, W. P. Singh, G. S. Wilson and R. S. Glass, Org. Lett., 2009, 11, 397 CrossRef CAS.
- N. P. A. Monney, T. Bally, G. S. Bhagavathy and R. S. Glass, Org. Lett., 2013, 15, 4932 CrossRef CAS PubMed.
- X. Chen, Y. Tao, J. Li, H. Dai, W. Sun, X. Huang and Z. Wei, J. Phys. Chem. C, 2012, 116, 19682 CrossRef CAS.
- T. Quinones-Ruiz, M. F. Rosario-Alomar, K. Ruiz-Esteves, M. Shanmugasundaram, V. Grigoryants, C. Scholes, J. Lopez-Garriga and I. K. Lednev, J. Am. Chem. Soc., 2017, 139, 9755 CrossRef CAS PubMed.
- W. Sun, H. Ren, Y. Tao, D. Xiao, X. Qin, L. Deng, M. Shao, J. Gao and X. Chen, J. Phys. Chem. C, 2015, 119, 9149 CrossRef CAS.
- W. Sun, M. Shao, H. Ren, D. Xiao, X. Qin, L. Deng, X. Chen and J. L. Gao, J. Phys. Chem. C, 2015, 119, 6998 CrossRef CAS.
- D. Wang, K. Hattori and A. Fujii, Chem. Sci., 2019, 10, 7260 RSC.
- W. Sun, H. Dai, Y. Tao, D. Xiao, Y. Zhang, Z. Wei and X. Chen, J. Phys. Chem. C, 2013, 117, 18325 CrossRef CAS.
- G. M. Chourasia, G. M. Sastry and S. Narahari, Int. J. Biol. Macromol., 2011, 48, 540 CrossRef PubMed.
- R. Thakuria, N. K. Nath and B. K. Saha, Cryst. Growth Des., 2019, 19, 523 CrossRef CAS.
- A. Subrayashastry, S. Narayanaswamy, D. Chittaranjan and S. Arumugam, J. Am. Chem. Soc., 2003, 125, 5308 CrossRef.
- D. Porath, A. Bezryadin, S. de Vries and C. Dekker, Nature, 2000, 403, 635 CrossRef CAS PubMed.
- D. N. Beratan, C. Liu, A. Migliore, N. F. Polizzi, S. S. Skourtis, P. Zhang and Y. Zhang, Acc. Chem. Res., 2014, 48, 474 CrossRef.
- J. M. Bollinger, Science, 2008, 320, 1730 CrossRef CAS.
- M. Mas-Torrent and C. Rovira, Chem. Rev., 2011, 111, 4833 CrossRef CAS.
- K. D. Schladetzky and T. S. Haque, J. Org. Chem., 1995, 60, 4108 CrossRef CAS.
- G. B. McGaughey, M. Gagne and A. K. Rappe, J. Biol. Chem., 1998, 273, 15458 CrossRef CAS PubMed.
- C. A. Hunter and J. K. M. Sanders, J. Am. Chem. Soc., 1990, 112, 5525 CrossRef CAS.
- M. Kang, P. Zhang, H. Cui and S. M. Loverde, Macromolecules, 2016, 49, 994 CrossRef CAS PubMed.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery Jr, J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, T. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, O. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian 09, Revision D.01, Gaussian, Inc, Wallingford, CT, 2013 Search PubMed; M. J. Frisch, G. W. Trucks, H. B. Schlegel, et al., Gaussian 09, revision D.01, Gaussian Inc.: Wallingford, CT, 2013 Search PubMed.
- F. B. Vanduijneveldt, J. G. C. M. Vanduijneveldtvanderijdt and J. H. Vanlenthe, Chem. Rev., 1994, 94, 1873 CrossRef CAS.
- S. F. Boys and F. Bernardi, Mol. Phys., 2002, 100, 65 CrossRef.
- M. E. Casida, C. Jamorski, K. C. Casida and D. R. Salahub, J. Chem. Phys., 1998, 108, 4439 CrossRef CAS.
- S. I. Gorelsky and A. B. P. Lever, J. Organomet. Chem., 2001, 635, 187 CrossRef CAS.
- Y. Zhao and D. G. Truhlar, Theor. Chem. Acc., 2008, 120, 215 Search PubMed.
- A. D. Mclean and G. S. Chandler, J. Chem. Phys., 1980, 72, 5639 CrossRef CAS.
- R. Krishnan, J. S. Binkley, R. Seeger and J. A. Pople, J. Chem. Phys., 1980, 72, 650 CrossRef CAS.
- T. Clark, J. Chandrasekhar, G. W. Spitznagel and P. V. Schleyer, J. Comput. Chem., 1983, 4, 294 CrossRef CAS.
- M. J. Frisch, J. A. Pople and J. S. Binkley, J. Chem. Phys., 1984, 80, 3265 CrossRef CAS.
- T. Yanai, D. P. Tew and N. C. Handy, Chem. Phys. Lett., 2004, 393, 51 CrossRef CAS.
- J.-D. Chai and M. Head-Gordon, Phys. Chem. Chem. Phys., 2008, 10, 6615 RSC.
- P. E. Smith, R. M. Brunne, A. E. Mark and W. F. van Gunsteren, J. Phys. Chem., 1993, 97, 2009 CrossRef CAS.
- T. Simonson and C. L. Brooks III, J. Am. Chem. Soc., 1996, 118, 8452 CrossRef CAS.
- C. J. Cramer and D. G. Truhlar, Chem. Rev., 1999, 99, 2161 CrossRef CAS.
- J. Tomasi and M. Persico, Chem. Rev., 1994, 94, 2027 CrossRef CAS.
- S. Ghosh, A. Dey, Y. Sun, C. P. Scholes and E. I. Solomon, J. Am. Chem. Soc., 2009, 131, 277 CrossRef CAS.
- S. Miertus, E. Scrocco and J. Tomasi, Chem. Phys., 1981, 55, 117 CrossRef CAS.
- V. Barone and M. Cossi, J. Phys. Chem. A, 1998, 102, 1995 CrossRef CAS.
- M. Cossi, N. Rega, G. Scalmani and V. Barone, J. Comput. Chem., 2003, 24, 669 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: Details for the choice of methods, the data of structures, front molecular orbitals, influence of protein polarizable environment, electron transition spectra for all structures. See DOI: 10.1039/d1ra05341h |
| ‡ Li and Sun contributes equally to this work. |
|
| This journal is © The Royal Society of Chemistry 2021 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *ab
*ab