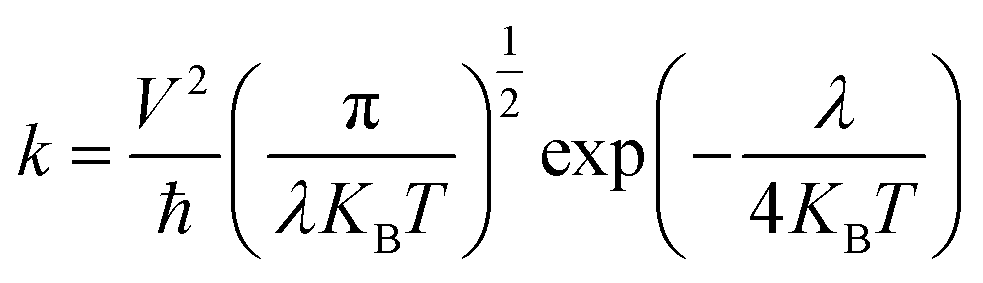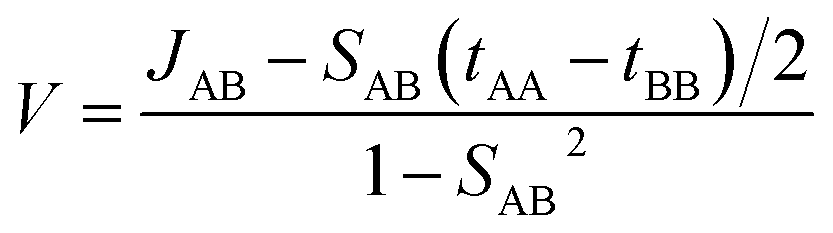DOI:
10.1039/D1RA05274H
(Paper)
RSC Adv., 2021,
11, 33703-33713
Influence of non-covalent interactions in dictating the polarity and mobility of charge carriers in a series of crystalline NDIs: a computational case study†
Received
8th July 2021
, Accepted 17th August 2021
First published on 15th October 2021
Abstract
Polycyclic aromatic compounds and their derivatives have emerged as potential molecular entities for air-stable n-type organic semiconductors. In particular, naphthalene diimide (NDI)-derived compounds stand out as one of the most promising classes of molecules that have been studied extensively. There have been a lot of debatable experimental reports on the OFET performance characteristics of some of these materials, which have not yet been resolved completely. Hence, the critical intrinsic aspect of the molecular materials during charge transport in a bulk crystalline state would be essential to categorise the potential candidates. As a case study, in this comprehensive computational approach, we investigated the structural and supramolecular organization in single crystals and the role of those aspects in the bulk carrier transport of a group of selected end-substituted NDI derivatives. A subtle alteration of the end group was observed to result in the modulation of the polarity of charge transport and the charge carrier mobility in the single crystalline state. The disparity is addressed by considering the electronic coupling of the transport states, symmetry of the frontier molecular orbitals and various non-covalent intermolecular interactions. We expect that the present study would benefit towards the rational designing of air-stable n-type organic molecular semiconductors for efficient electronic devices.
1. Introduction
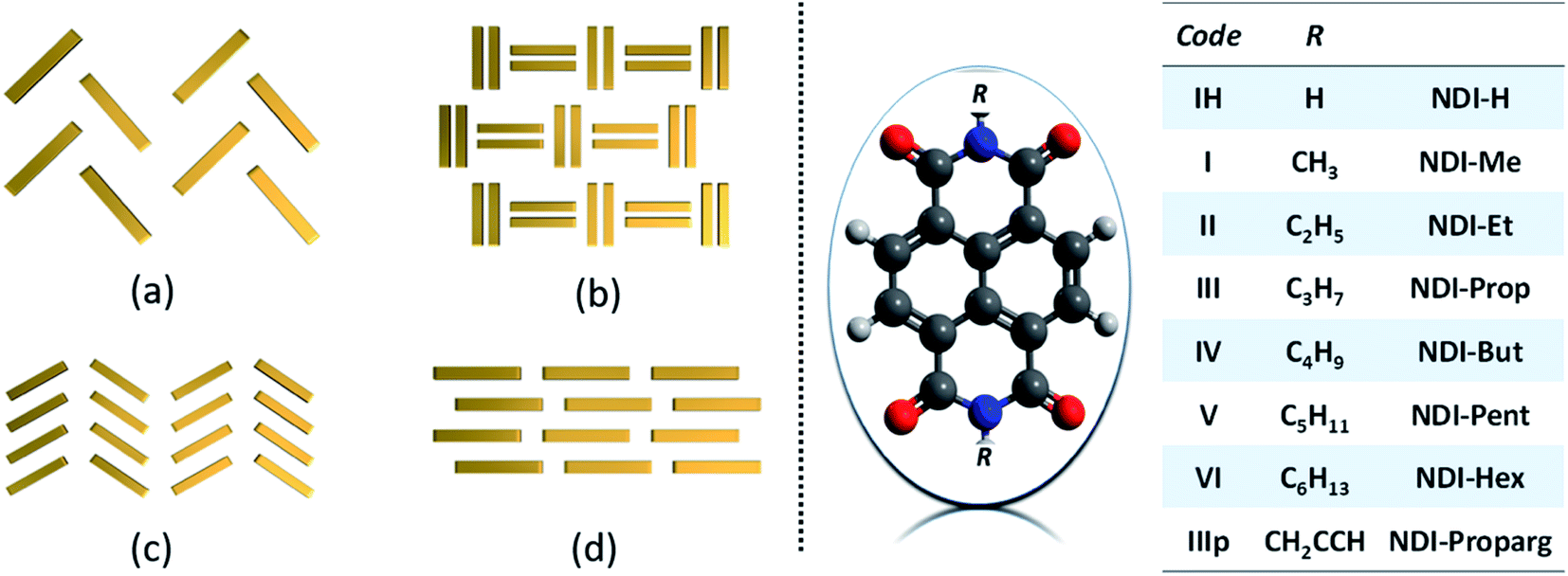
Optical and charge carrier transport properties of molecular semiconductors are exceedingly dependent on the arrangement of chromophores in the film/crystalline state and very much illustrious in their solid-state and thin-film devices.1–3 Charge transport behaviour of such molecular semiconductors is primarily governed by intermolecular interactions in lattice or molecular aggregates.4 Since a subtle alteration in intermolecular interaction yields a remarkable disparity in the optical and charge transport properties of solid semiconductor materials, it is highly desirable to have strong control over the crystal packing characteristics of molecular semiconductors in their bulk or single crystalline state.5–8 In recent years, several reports have emphasized the importance of crystallographic arrangements of molecular semiconductors for the illustration of optoelectronic properties.9–13 Due to immense quest for ambient stable organic semiconductor materials, especially n-type, polycyclic aromatic hydrocarbon-based chromospheres such as naphthalene diimides (NDIs) have been reported to be one of the most promising candidates for environmentally stable n-type semiconductors because of the enormous possibility of modulating the HOMO and LUMO energy levels by suitable substitution at the bay position and controlling the molecular packing, and thereby, the intermolecular interactions in the solid state by terminal substitution.14–17 In general, high electron affinity and good electron carrier hoping rate make these classes of molecules interesting to the organic semiconductor researchers. In order to understand the molecular stacking and intermolecular interactions, a guideline proposed by Desiraju and Gavezzotti in the case of polycyclic aromatic hydrocarbons (PAH) was followed.18 A broad categorization into herringbone, sandwich herringbone, γ motif, and β sheet is well known in small-molecular weight semiconductor research as well.6 For chromophoric PAHs containing only carbon and hydrogen, the competition of face-to-face (π–π) and edge-to-face (C–H⋯π) non-covalent interactions dictates the assembly of molecules into the above-mentioned crystallographic arrangements (Fig. 1).
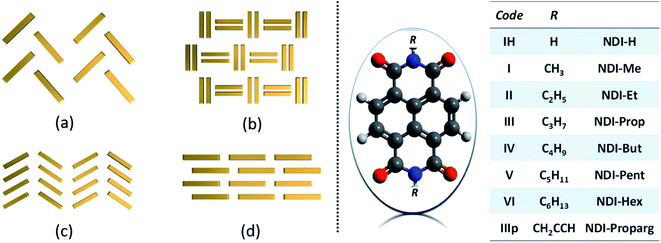 |
| | Fig. 1 Schematic of four major types of molecular packing (left panel) seen in polycyclic aromatic hydrocarbons: (a) herringbone; (b) sandwich herringbone; (c) γ motif and (d) β sheet and the chemical structure of NDI derivatives considered for the present investigation (right panel). | |
Though pendant alkyl chain groups are considered to be the least significant component while computing the single molecular carrier transport features, it has been found to be extremely crucial in dictating the charge transport efficiency by controlling the supramolecular stacks.11,19–23 In this comprehensive computational approach, we have investigated the influence of structural factors by varying the alkyl chain length of the N-substitution and their supramolecular organization in single crystals on the bulk carrier transport properties of a group of selected N-substituted NDI derivatives. In general, the functionalization of the NDI chromospheres is made by two different approaches, namely, (a) end substitution on the imide nitrogen atom and (b) substitution on the NDI core unit. End substitution maintains the planarity of the NDI aromatic core, whereas core substitution leads to a varying degree of distortion in the core due to the steric hindrance present in the bay region. Though core substitution involves synthetic challenges, introducing electron-withdrawing groups into the NDI core unit has been proven to be a good strategy to lower the LUMO energy level.24
In the present investigation, in order to maintain uniform structural topographies to compare the transport characteristics, we have chosen a group of end-substituted NDI materials with known single-crystal structural data. Such a choice enables the core units to maintain the planarity in the optimized geometry (see ESI†). The chemical structures are depicted in Fig. 1. Molecular semiconductors are distinguished from insulating ones by their frontier orbitals' sizable spatial extent. This dictates the carrier transport characteristics in the crystalline lattice or in the condensed state. In hole transport, the orbital of relevance is the highest occupied molecular orbital (HOMO), whereas, for electron transport, it is the lowest unoccupied molecular orbital (LUMO). In insulating molecules, these orbitals are small, localized on just a few atoms. As a result, the spatial overlap between frontier orbitals of neighbouring molecules is expected to be minimal and so the likelihood of charge transfer between them is vanishing. Hence, the spatial orbital distribution and the energy level (with respect to the vacuum state) are crucial parameters to begin the discussion.
2. Results and discussion
2.1 Single molecular transport parameters
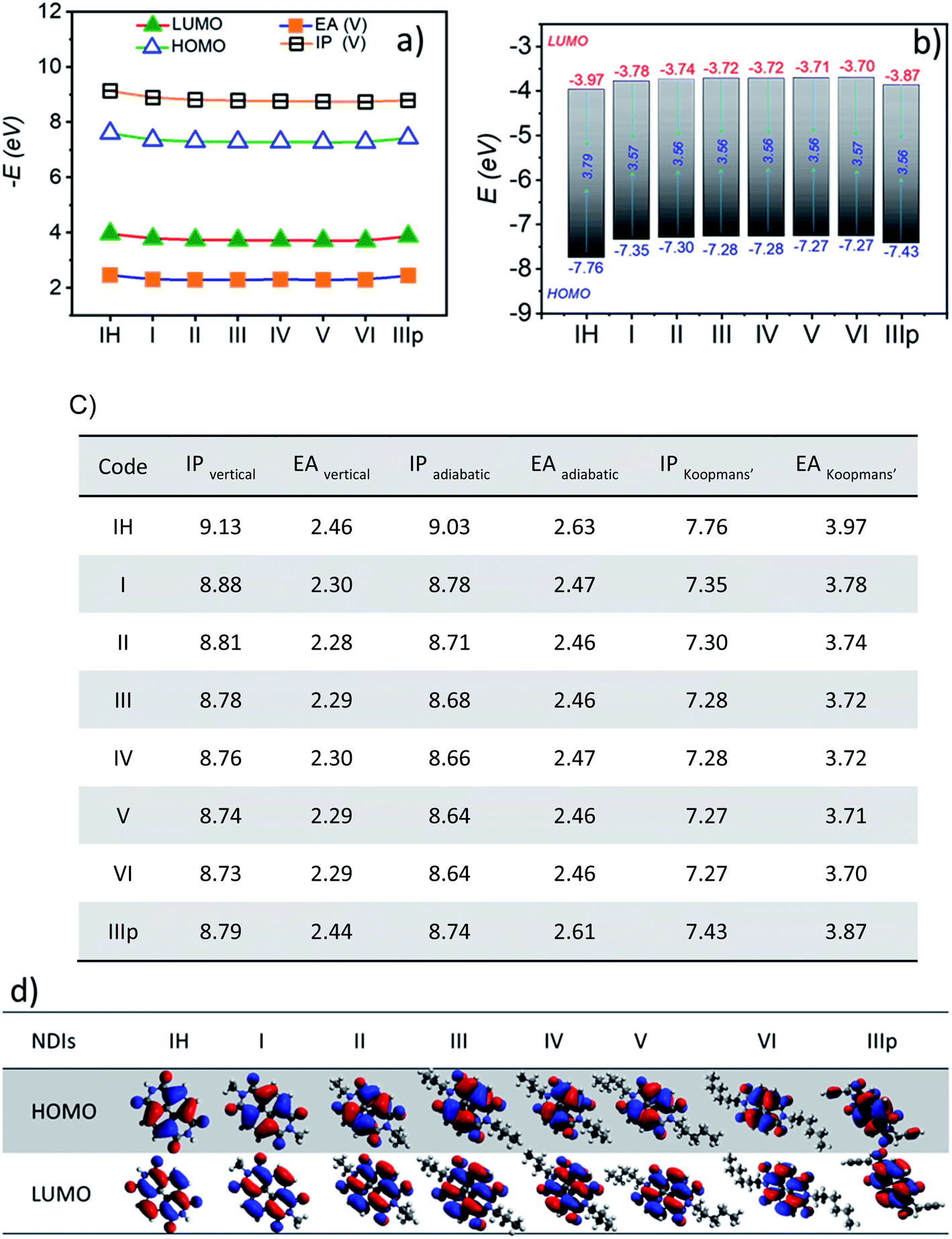
Computed frontier molecular orbital energy levels of all the derivatives are summarized in Fig. 2b and diagrammatically represented in Fig. 2d. The HOMO–LUMO energy levels of all the derivatives IH to VI are closely indexed as expected (Fig. 2a). The HOMO and LUMO orbitals of alkyl substituted NDIs embrace nodes along the long molecular axis and, thus, on the imide N atoms as well (Fig. 2d). Hence, end substituents are not expected to induce any significant alteration of the electronic properties of the individual NDI molecules at their monomeric state. All of them have a very similar HOMO–LUMO energy gap (∼3.6 eV) as well. At the same time, in the case of two other derivatives, namely, IH and IIIp, the energy levels have a slight offset of ∼200 meV and ∼150 meV respectively (both HOMO and LUMO) in comparison to the other derivatives. This difference in the case of IH is attributed to the absence of the +I effect of the alkyl chain connected to the N-centre of the NDI ring resulting in the lowering of HOMO and LUMO energy levels, whereas the same observation in the case of IIIp is attributed to the contribution of the propargyl group with two sp hybridised carbon centres to the HOMO and LUMO wave functions (Fig. 2d). The HOMO and LUMO of all the NDI derivatives are predominantly π-type and distributed mostly over the NDI core. In the case of IIIp, the alkyne (CH2–C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) CH) pendant group has a significant contribution to the HOMO, whereas HOMO−1 locates entirely on the end group (Fig. S1†). Besides the frontier molecular energy levels, other molecular parameters such as ionization potential (IP) and electron affinity (EA) are also indispensable parameters in elucidating the charge-injection process and ambient stability of these molecules and determining the polarity of charge carrier transport. It is known that low ionization potentials (IPs) and low electron affinities (EAs) are usually the characteristics of hole-transporting materials (p-type), whereas materials with high EA and high IP function as electron-transporting materials (n-type). Computed (i) adiabatic ionization potential (IPa) and electron affinity (EAa) and (ii) vertical ionization potential (IPv) and electron affinity (EAv) of the studied derivatives are summarized in Fig. 2c and Table S1†.
CH) pendant group has a significant contribution to the HOMO, whereas HOMO−1 locates entirely on the end group (Fig. S1†). Besides the frontier molecular energy levels, other molecular parameters such as ionization potential (IP) and electron affinity (EA) are also indispensable parameters in elucidating the charge-injection process and ambient stability of these molecules and determining the polarity of charge carrier transport. It is known that low ionization potentials (IPs) and low electron affinities (EAs) are usually the characteristics of hole-transporting materials (p-type), whereas materials with high EA and high IP function as electron-transporting materials (n-type). Computed (i) adiabatic ionization potential (IPa) and electron affinity (EAa) and (ii) vertical ionization potential (IPv) and electron affinity (EAv) of the studied derivatives are summarized in Fig. 2c and Table S1†.
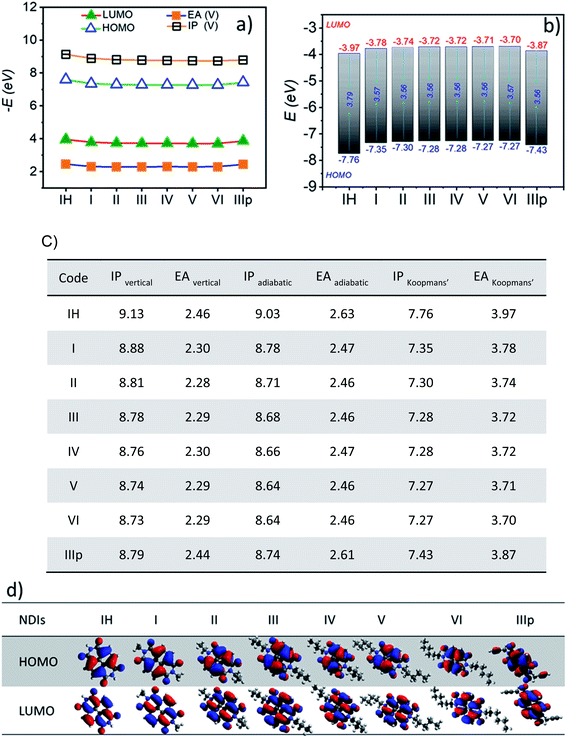 |
| | Fig. 2 a) Frontier molecular orbital energy, vertical ionization potential (IPvertical), and electron affinity (EAV) of NDIs (the lines indicate guide to the eye); (b) HOMO and LUMO energy levels (EHOMO and ELUMO) and the energy gap (ΔEH–L); (c) vertical, adiabatic and Koopmans' ionization potential (IP) and electron affinity (EA) values of NDI monomers and (d) Frontier molecular orbital diagrams (HOMO and LUMO) of NDIs. MOs are generated at an isosurface value of 0.03 from the optimized geometry. (All energies are expressed in eV). | |
As expected, all the molecules possess high electron affinity indicative of the intrinsic n-type character. Nevertheless, the polarity of carrier transport depends a lot on the supramolecular features besides their molecular parameters. For the series of chosen molecules, very similar frontier energy levels, IE, and EA values were obtained, and hence, these factors would not be a determinant strand in the charge transport behaviour of the aggregated/solid state. Since the bulk charge transport properties of molecular semiconductors are highly sensitive to the relative orientations of individual molecules with respect to the neighbouring ones in the lattice and their 3D molecular packing, a detailed analysis of their supramolecular interactions in solid state has been carried out. To investigate such supramolecular features, the reported single-crystal structures of all the molecules were taken from the CCDC database.
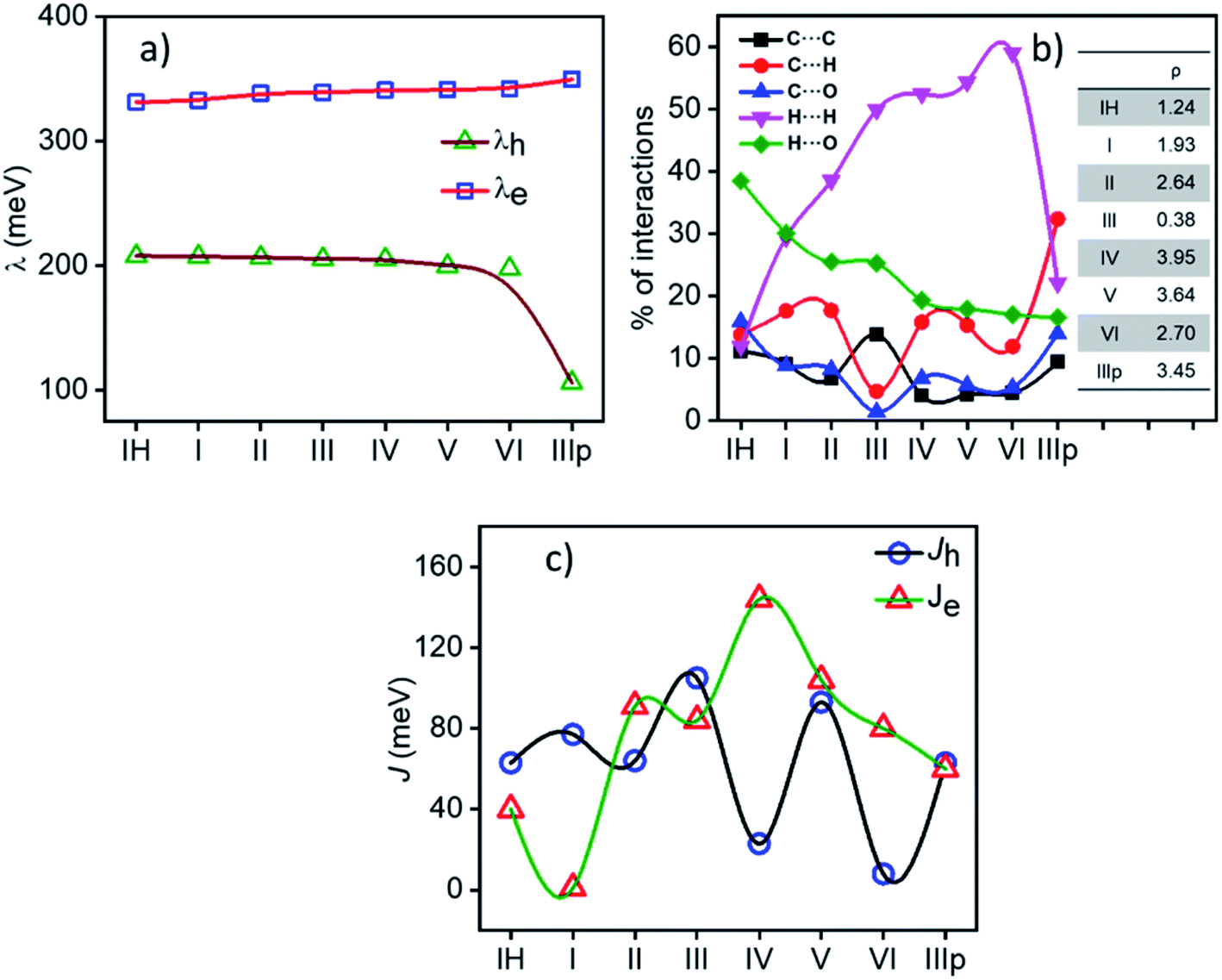
2.1.1 Reorganization energy. Reorganization energy (λ) is an important molecular parameter to be addressed carefully to rationalize the charge transport behaviour of molecular semiconductors under the umbrella of Marcus–Hush formalism. In most simplified means, reorganization energy (inner sphere) accounts for the energy associated with the coordinate change in a process of electron or hole transfer from its neutral state to the corresponding charged state (see the schematic representation in Fig. S2†) or, concisely, during the charge transport process, energy expense due to the geometric reorganization can be effectively defined by the parameter λ. In general, λ influences the rate of electron transfer ke inversely as per the Marcus–Hush formalism. For the NDI derivatives under consideration, the effect of substitution on the planar NDA (1,4,5,8-naphthalenetetracarboxylic dianhydride) was found to result in an increase in inner sphere electron reorganization energy λe (λe for the planar NDA unit is 197.2 meV) and the computed reorganization energy values of all the NDIs are in the range of 207.4–106.1 meV for holes (λh) and 331.4–349.7 meV for electrons (λe). In brief, λe for all the derivatives are found to be larger than their λh (Fig. 3a). Interestingly, we observed a linear dependence of λh and λe on the alkyl chain length substitution varying from IH (n = 0) to VI (n = 6) except for the abnormally low λh (106.1 meV) for IIIp. λh decreases with the increase in alkyl chain length, whereas λe increases from 332.7 meV for I to 342.1 meV for VI with the increase in alkyl chain length. The internal reorganisation energy parameter is predisposed more towards the π-character of the IIIp derivative.
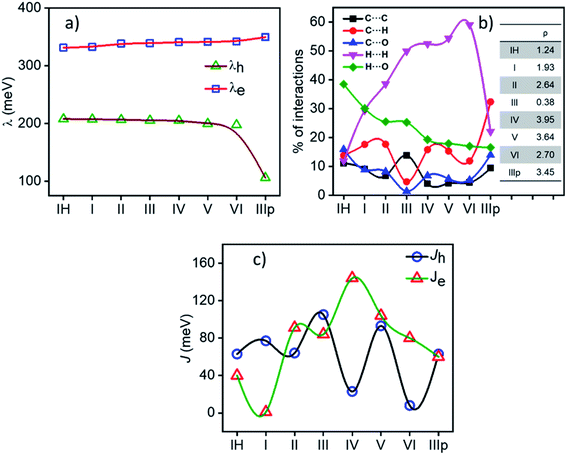 |
| | Fig. 3 (a) Variation in internal reorganization energy of the NDIs under consideration. (b) Percentage of non-covalent interactions obtained from the Hirshfeld surface analysis (values of ρ[(% C⋯H)/(% C⋯C)] are shown in the inset). (c) Hole and electron charge transfer integrals along the M–D1 pair of NDIs. | |
2.2 Single-crystal molecular arrangement and transport properties of the aggregated state
The electronic couplings among the adjacent molecular units in the single crystals of organic semiconductors are known to be dependent primarily on the molecular packing. Before discussing the charge transport properties in the aggregates, various supramolecular interactions that dictate the crystallographic arrangements in the set of molecules under consideration need to be elaborated. The crystal structural data of all the NDI derivatives were collected from the Cambridge Crystallographic Data Centre as reported for IH (CCDC 189606), I (CCDC 1029338), II (CCDC 1029339), III (1029340), IV (819749), V (238148), VI (671518), and IIIp (100718). Detailed lattice parameters are provided in Table 1
Table 1 Crystal structure data of the studied NDIs. (a, b, and c are in Å and α, β, and γ are in degree)
| |
CCDC |
Crystal system |
Shape |
a |
b |
c |
α |
β |
γ |
| IH |
189606 |
Triclinic |
Plate |
7.867 |
5.305 |
12.574 |
90 |
72.73 |
90 |
| I |
1029338 |
Monoclinic |
Needle/flexible plastic |
4.621 |
8.019 |
17.024 |
90 |
93.99 |
90 |
| II |
1029339 |
Monoclinic |
Needle/flexible elastic |
4.844 |
7.736 |
18.315 |
90 |
90.12 |
90 |
| III |
1029340 |
Orthorhombic |
Needle |
6.962 |
17.242 |
27.580 |
90 |
90 |
90 |
| IV |
819749 |
Triclinic |
Prismatic |
5.223 |
7.840 |
11.132 |
103.72 |
94.28 |
93.86 |
| V |
238148 |
Monoclinic |
Acicular |
5.028 |
8.107 |
24.026 |
90 |
90.79 |
90 |
| VI |
671518 |
Triclinic |
Plate |
4.898 |
8.284 |
14.524 |
96.33 |
98.10 |
93.59 |
| IIIp |
100718 |
Monoclinic |
Needle |
9.746 |
6.310 |
12.867 |
90 |
111.78 |
90 |
Various non-covalent intermolecular interactions present in the crystals were computed and are summarized. Fig. 3b describes the percentage contributions for C⋯C, C⋯H, C⋯O, H⋯H, and H⋯O interactions of all the considered NDIs. The percentage contributions of these weak intermolecular interactions dictate the molecular packing. A broad classification is possible by analysing these intermolecular interactions. NDIs in which the edge-to-face C⋯H interactions are dominant ended up with a herringbone molecular stack, whereas for crystals having large face-to-face C⋯H interactions they lead to β motif packing models. A ratio (ρ) of % C⋯H/% C⋯C is known to be a guiding factor to predict the crystallographic arrangements.25
Among the studied materials, IIIp, IV and V having dominant % C⋯H (edge-to-face) interaction, show herringbone molecular packing in the single crystal. However, III has the highest % C⋯C face-to-face interaction and thus arranges in columnar stacks (β motif). Other derivatives, namely,. IH, I, II, and VI with comparable % C⋯H and % C⋯C interactions fall into the γ motif category. Detailed crystal structure analysis and pie diagrams to summarize all the non-covalent interactions in supramolecular stacks for the individual materials are provided in the ESI (Fig. S3–S6†) and are summarized in Fig. S7.†
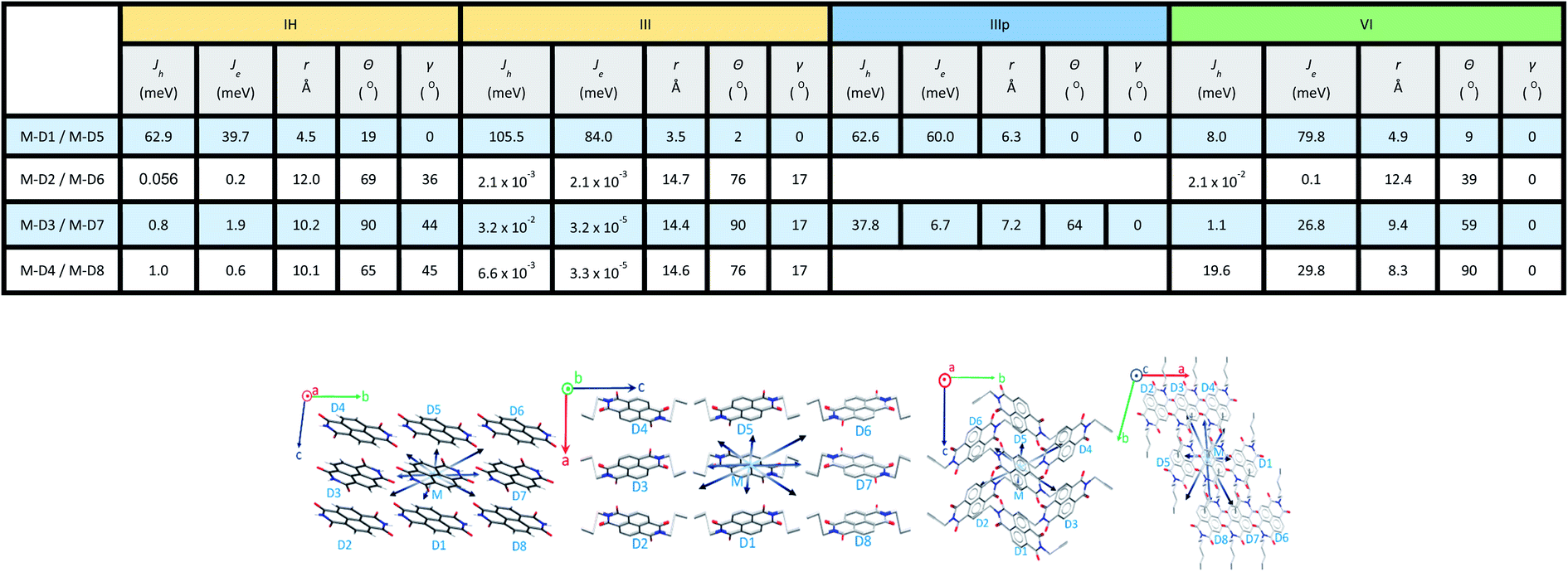
2.2.1 Charge transfer integral. Intermolecular electronic coupling is quantified in terms of electron and hole transfer integral (Jh and Je) that is described as follows. Charge transfer integral (J) of a crystalline organic semiconductor pair is known to be highly sensitive to the relative orientations of the charge transport centres in the crystal lattice, which reflects the electronic couplings among the adjacent molecular unit.11,26–30 In this study, to calculate the charge transfer integral between the pairs of stacks, we used the dimer projection (DIPRO) method proposed by Valeev et al. and Baumeier et al.27,31 The effect of electronic polarization (site energy correction) is considered while calculating the charge transfer integral values as proposed by Valeev et al.27 A detailed discussion of this method is provided in the ESI.†Charge carrier transport characteristics among the adjacent NDI molecules in the crystalline state are computed using the distinct molecular pairs around the identified origin molecule (M) along all the directions towards the neighbouring pairs (D1, D2, D3, D4, D5, etc.). The co-facial dimeric pair is denoted as D1. Other interacting neighbouring molecules are successively labelled as represented in Fig. 4. For all the NDI derivatives, charge transfer integrals (both the Je and Jh) are highest along the cofacial dimeric pair (M–D1) direction compared to the other interacting dimers. Though the NDI substituted molecules are known for electron transport characteristics, in the case of molecule IH, I and III the highest hole transfer integral is higher than Je (see the discussion henceforth). Similarly, in the case of molecule IIIp, the highest Jh and Je are comparable indicating an ambipolar transport ability of the crystalline form of the molecule, while for the rest of the molecules (II, IV, V, and VI) the electron transfer integral dominates over the hole. They are categorized into three groups based on the polarity of charge transport. Here, r is the centroid-to-centroid distance between the hoping dimers, theta (θ) is the angle between any dimer of our interest (vector connecting two centroids) and the reference axis (crystallographic a, b or c). Gamma (γ) is the projection angle of any dimer of our interest to the reference plane.
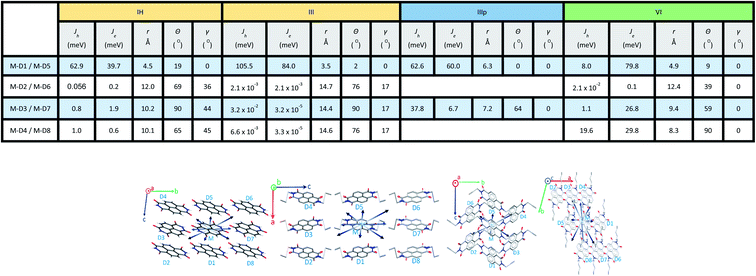 |
| | Fig. 4 Illustration of various charge hoping (transport) pathways of the representative NDIs (IH, III, IIIp, and VI) with the corresponding hole (Jh) and electron (Je) transfer integrals (in meV). | |
To gain more insights into the diverse transfer integral characteristics of a group of very similar NDI derivatives, we investigated (i) the orbital interaction in terms of FMO splitting energy (ΔE) and (ii) the symmetry of the frontier molecular orbitals of the hoping dimers. In line with the tight binding model, the interaction of monomer HOMOs (LUMOs) leads to a splitting of HOMOs (LUMOs) in dimeric pairs. Thus, the hole charge transfer integral is related to the energy difference between the HOMO and HOMO−1 orbitals taken from the closed-shell configuration of a dimer, whereas the electron charge transfer integral is related to that of LUMO and LUMO+1. The frontier molecular orbital energies of IH, IV, VI and IIIp along with the HOMO and LUMO energy splitting values are tabulated in Table 2. The same for other derivatives are listed in Table S3.†
Table 2 Frontier Molecular orbital energies (HOMO, HOMO−1, and LUMO, LUMO+1) of dimeric pairs of IH, IIIp and VI along with the energy splitting values (energy unit is eV)
| |
Monomer |
Dimers |
ELUMO+1 |
ELUMO |
EHOMO |
EHOMO−1 |
ΔE(LUMO+1)–LUMO |
ΔEHOMO–(HOMO−1) |
| HOMO |
LUMO |
| IH |
−7.759 |
−3.973 |
M–D1 |
−4.030 |
−4.125 |
−7.623 |
−7.740 |
0.095 |
0.117 |
| M–D4 |
−3.915 |
−3.980 |
−7.557 |
−7.588 |
0.065 |
0.031 |
| IIIp |
7.428 |
−3.871 |
M–D1 |
−3.892 |
−4.014 |
−7.454 |
−7.573 |
0.122 |
0.119 |
| M–D2 |
−3.742 |
−3.960 |
−7.289 |
−7.541 |
0.218 |
0.252 |
| VI |
−7.428 |
−3.704 |
M–D1 |
−3.570 |
−3.721 |
−6.177 |
−6.178 |
0.151 |
0.001 |
| M–D2 |
−3.618 |
−3.619 |
−6.134 |
−6.136 |
0.001 |
0.002 |
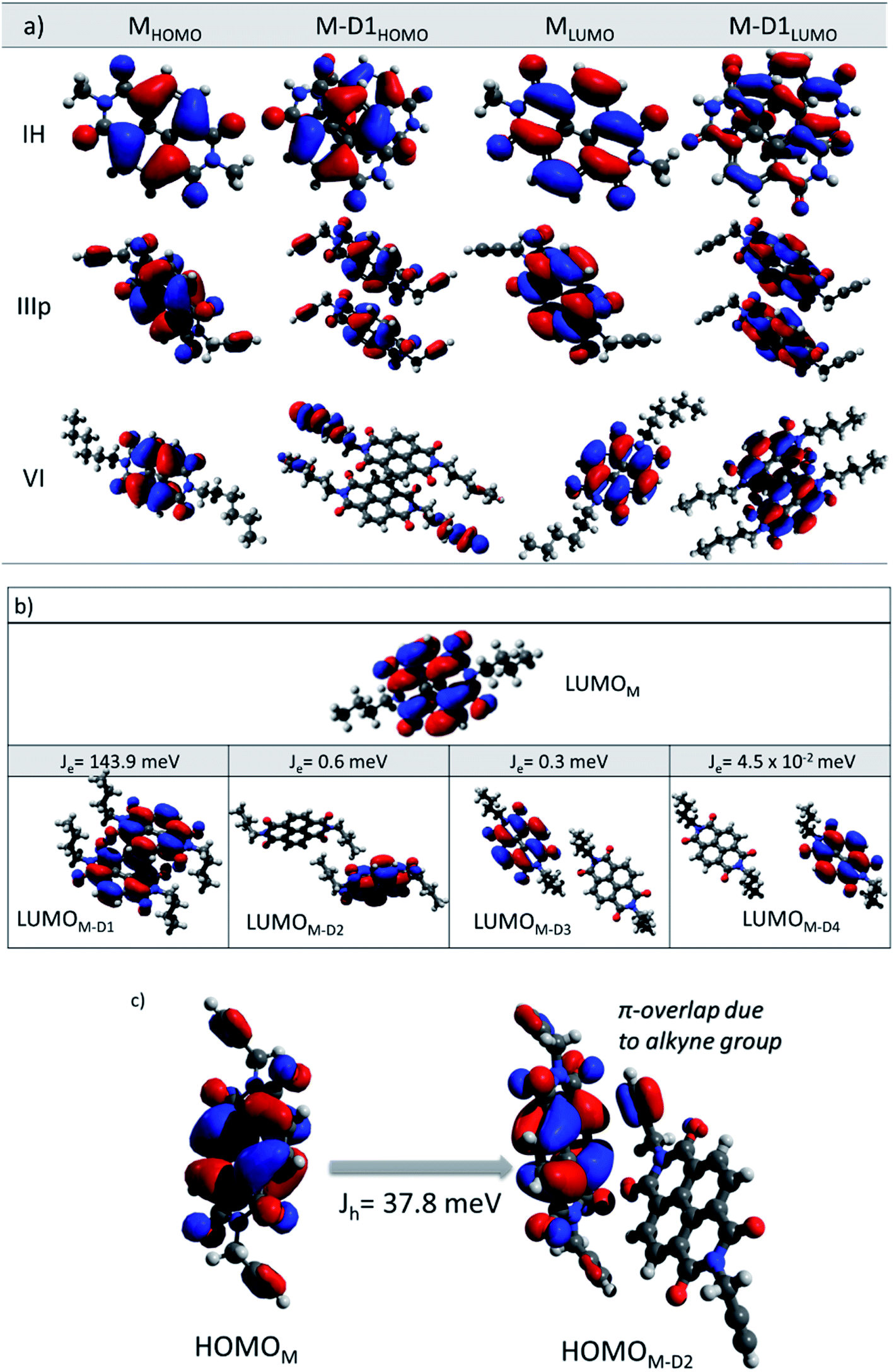
For the category 1 derivatives (IH, I and III having Jh > Je along the M–D1 direction), a strong constructive overlap between HOMOs is responsible for the predominant hole transport characteristics. For example, in the case of IH, the monomeric HOMO and LUMO energy values are −7.759 and −3.973 eV respectively. In the M–D1 pair HOMO gets stabilized (−7.623 eV) compared to the monomeric HOMO energy level (−7.759 eV), whereas LUMO gets destabilized (−4.125 eV) compared to the monomeric LUMO level (−3.973 eV). Moreover, the splitting of HOMO energy in the dimer is found to be as high as 0.117 eV. This large HOMO energy splitting (ΔEHOMO–(HOMO−1)) is an indicative of constructive overlap between HOMOs, and thus, the preferential hole transport along the M–D1 direction. For category 2 (example IIIp), the dimeric pair (M–D1) orbital symmetry is evidently constructive for both the HOMOs and LUMOs. Hence, an ambipolar transport can be expected and the electron (60.0 meV) and the hole (62.6 meV) transfer integral values support the claim. The same can be understood from the fact that both HOMOs and LUMOs are equally split in the dimer (ΔEHOMO–(HOMO−1) = 0.122 eV and ΔELUMO–(LUMO−1) = 0.119 eV). Both the HOMO (−7.454 eV) and LUMO (−4.014 eV) energy levels of the dimer get stabilized in comparison to the monomeric HOMO (−7.428 eV) and LUMO (−3.871 eV) energy levels. In contrast, in the case of category 3 NDIs (example VI), the constructive symmetry overlap (of the M–D1 pair) is observed to be large for the LUMOs in comparison to that of the HOMOs. The dimeric LUMO energy value is stabilized (−3.721 eV) in comparison to the monomeric LUMO energy level (−3.704 eV), whereas dimeric HOMO (−6.177 eV) energy is destabilized compared to the monomeric HOMO energy (−7.428 eV). This asserts the constructive overlap between the LUMOs and destructive overlap between HOMOs. As can be seen from the energy splitting value, the ΔEHOMO–(HOMO−1) = 0.001 eV is much less than ΔELUMO–(LUMO−1) = 0.151 eV. Hence, exclusive electron transport is observed in the case of this dimeric configuration. It can be correlated for the other derivatives of the same series as well.
In order to rationalize the directional dependency of Jh and Je (asymmetric charge transport), some of the salient features of the orbital symmetry analysis as described above is applied for the other dimeric pairs as well and is compiled below taking a few examples. As shown in Fig. 5b, the maximum Je for IV is 143.9 meV along the direction of the parallel M–D1 dimer, which is because of the maximum constructive overlapping of the LUMO orbitals in a co-facially stacked dimeric form. However, relatively small effective LUMO coupling leads to lower electron transfer integral for the M–D2 (0.6 meV), M–D3 (0.3 meV) and M–D4 (4.5 × 10−2 meV) dimers as compared to the M–D1 pair. Fig. 5b represents the LUMO orbitals for M–D1, M–D2, M–D3 and M–D4 dimers of IV. For the M–D4 pair, there is hardly any effective orbital overlap between the LUMOs, which rationalizes much smaller electronic couplings and J values in the case of M–D4 pair. In the case of VI, the reason for almost exclusive electron transfer for the M–D1 pair can be justified by the following FMOs symmetry analysis of the M–D1 dimer (Fig. 5a). The HOMO for the M–D1 pair is centred on the alkyl chain and there is no contribution from the NDI core unit, as expected, the hole transfer integral would be minimal (Jh = 8.0 meV) along this particular pair. However, the symmetry and the NDI-centred LUMOs ensure the effective electronic coupling for the same dimer, which results in ten times higher electron transfer integral (Je = 79.8 meV).
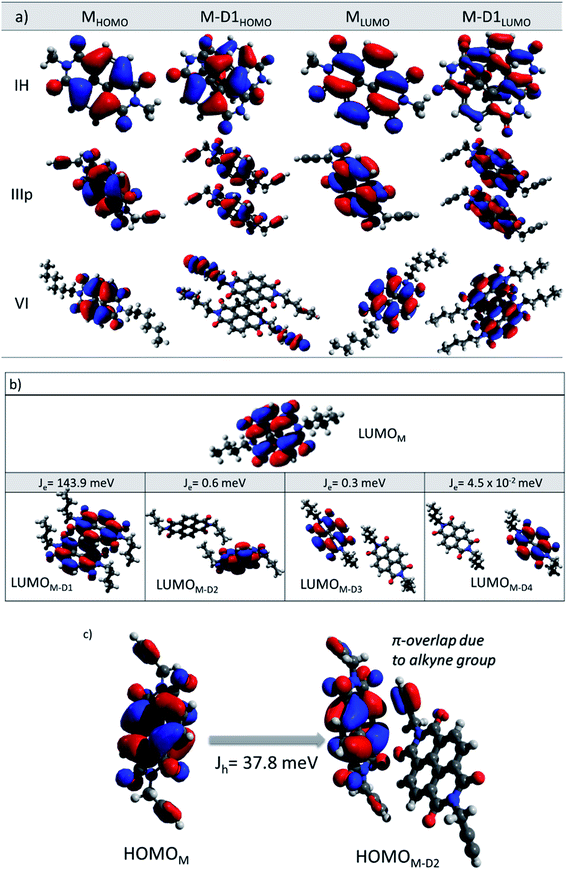 |
| | Fig. 5 (a) Frontier molecular orbital diagrams (HOMO and LUMO) of monomers and M–D1 dimers of IH, IIIp and VI. (b) LUMO orbital of monomers and M–D1, M–D2, M–D3 and M–D4 dimers of IV. (c) HOMO orbital of monomer and M–D2 dimers of IIIp frontier molecular orbitals are generated at an isosurface value of 0.03. | |
Apart from the M–D1 hopping channel, charge transfer integral along other directions are less prominent for almost all the NDI derivatives except for IIIp. For IIIp, due to the presence of an alkyne end group, it was found to facilitate the FMO coupling along with the M–D2 pair mediated by the pi-cloud of the alkyne group. A considerable constructive overlap between HOMOs for the M–D2 pair (Fig. 5c) ensures considerable Jh along the M–D2 direction also. Briefly, for the studied NDIs, symmetry matching of the FMOs was found to be crucial for effective electronic coupling, and thereby, constructive transfer integral along the specific direction. This would impart a large spatial asymmetry in charge transport for the molecules with unidirectionally overlapped FMOs. For the better transport of holes, the symmetry of HOMOs should match for constructive overlap. Whereas favourable electron transport is achieved with the same thumb rule for the LUMOs.
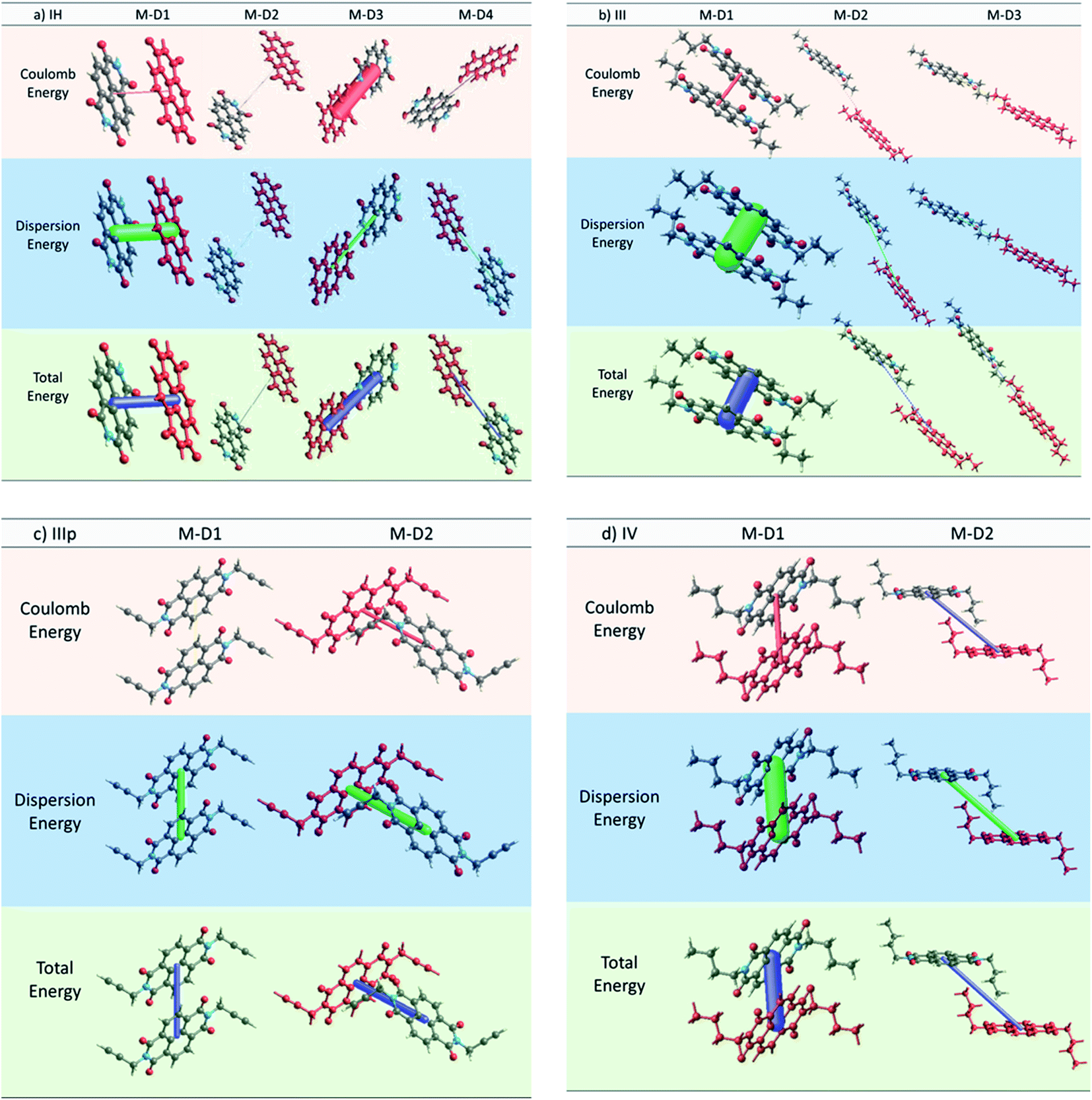
2.2.2 Interaction energies and its relation to the charge transfer integral. In recent times, decomposition of the intermolecular interactions between two molecules into physically meaningful energy contributions (e.g. electrostatics, polarization, dispersion, exchange repulsion) is rapidly becoming a popular means of rationalizing the morphological and transport properties of organic semiconductors.32–37 Here, we calculated pairwise interaction energy for different dimers for all the NDIs using the method proposed by Turner et al.37 As discussed earlier for all the NDI derivatives, we observed that the M–D1 pair has the highest charge transfer integral. To better understand this fact, we critically examined the electrostatics, polarization, dispersion, and exchange repulsion energy values obtained from the pairwise framework energy calculation for individual dimers. For IH, the M–D1 pair has the highest dispersion energy of −74.8 kJ mol−1 (Table 3). This particular pair also has the highest charge transfer integral. Although the M–D3 pair has a significant coulomb attractive energy (−69.2 kJ mol−1), we observed that the charge transfer integral is significantly less for latter pair compared to that of the former one. This suggests that dispersion interaction has a superior role in the charge hoping process than the electrostatic interaction. Similarly, for IV, the M–D1 pair, which has the highest electron transfer integral (143.9 meV), shows an exceedingly high value of dispersion energy (−96.4 kJ mol−1) (Table 3). In the case of IIIp, both the non-identical interacting dimer pairs have comparable dispersion energy (M–D1: Edispersion = −39.3 kJ mol−1, M–D2: Edispersion = −44.5 kJ mol−1) though repulsion energy is higher for M–D2 (29.5 kJ mol−1) than for M–D1 (9 kJ mol−1). Significant dispersion energy along the M–D2 also validates our observation regarding the considerable transfer integral along that direction (Fig. 5c and 6). Subsequent observations made for the dimers of the other derivatives explicitly led us to draw a clear correlation between the long range dispersive interaction energy and charge transfer integral of the NDI derivatives (Fig. S16 and Table S4†). Thus we propose that high magnitude of the dispersive interaction leads to efficient charge hopping between the dimers.
Table 3 Pairwise interaction energy for different dimers of IH, III, IIIp and IV (all energies are expressed in kJ mol−1)
| IH |
r (Å) |
ECoulomb |
Epolarization |
Edispersion |
Erepulsion |
Etotal |
| M–D1 |
4.5 |
−5.3 |
−3.1 |
−74.8 |
40.8 |
−41.7 |
| M–D2 |
12.0 |
−1.4 |
−0.1 |
−1.1 |
0 |
−2.5 |
| M–D3 |
10.2 |
−69.2 |
−18.4 |
−14.2 |
58.1 |
−48.2 |
| M–D4 |
10.1 |
−4.9 |
−0.6 |
−5 |
0.1 |
−9.8 |
| III |
r (Å) |
ECoulomb |
Epolarization |
Edispersion |
Erepulsion |
Etotal |
| M–D1 |
3.48 |
−17.5 |
−4.0 |
−119.2 |
59.5 |
−79.6 |
| M–D2 |
15.02 |
−0.5 |
−0.2 |
−8.9 |
3.4 |
−5.9 |
| M–D3 |
14.51 |
0.2 |
−1.1 |
−7.4 |
2.1 |
−5.6 |
| IIIp |
r (Å) |
ECoulomb |
Epolarization |
Edispersion |
Erepulsion |
Etotal |
| M–D1 |
6.3 |
0.9 |
−1.8 |
−39.3 |
9.0 |
−28.4 |
| M–D2 |
7.2 |
−16.0 |
−4.8 |
−44.5 |
29.5 |
−35.6 |
| IV |
r (Å) |
ECoulomb |
Epolarization |
Edispersion |
Erepulsion |
Etotal |
| M–D1 |
5.2 |
−22.9 |
−6.3 |
−96.4 |
57.4 |
−68.7 |
| M–D2 |
11.1 |
−4.3 |
−0.5 |
−40.9 |
20.1 |
−25.3 |
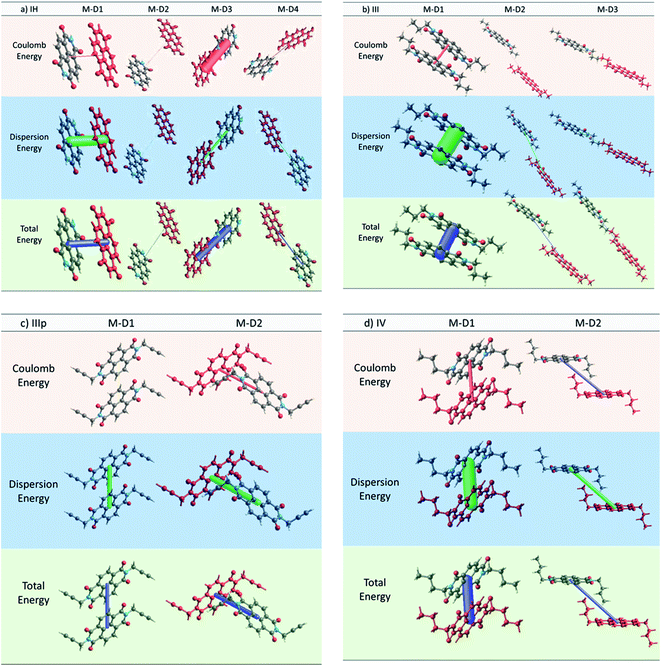 |
| | Fig. 6 Crystal explorer based energy frameworks of different dimers of (a) IH, (b) III, (c) IIIp and (d) IV with 100 energy scale factor and zero energy threshold. | |
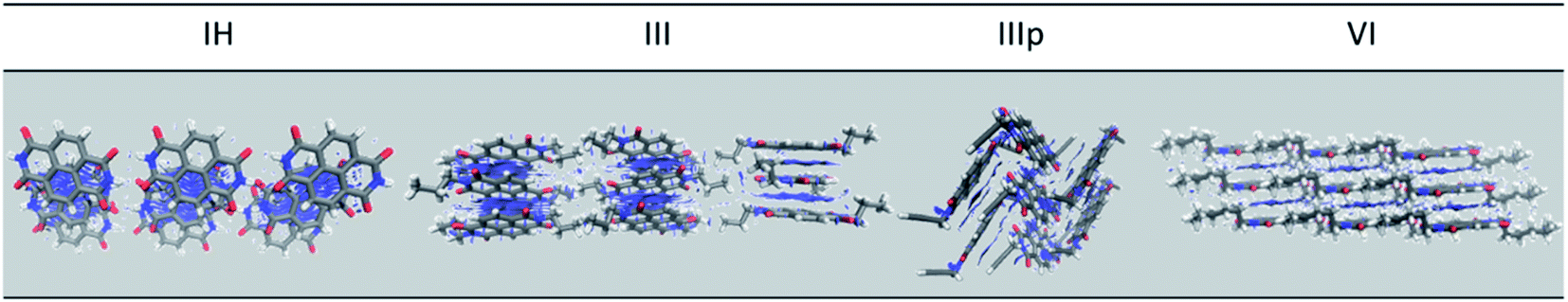
Furthermore, the role of non-covalent intermolecular interaction in the electronic coupling of NDIs is also explored. The non-covalent interactions are so crucial in organic solids in dictating the charge transport properties through band narrowing or widening, which impacts the transport levels immensely.38 To get a qualitative idea about the extent of non-covalent interactions, we performed density overlap region indicator (DORI) analysis.39,40 The NCI plots obtained from the DORI analysis for IH, III, IIIp and VI are shown in Fig. 7. The same for the other derivatives are shown in Fig. S17.† From the non-covalent interaction plot, we could draw a direct correlation between the extent of non-covalent interactions and magnitude of electronic coupling that can be clearly understood from the charge transfer integral (J) values of the respective dimers. It can be seen from the figure that there are high degrees of NCI along the M–D1 direction of IH (Fig. 7). This particular dimer (M–D1) also has the highest transfer integral. The M–D1 dimeric pair shows hole and electron transfer integrals of 62.9 and 39.7 meV respectively. The other dimers have a significantly lower NCI and so as their charge transfer integral values. Similar observation is seen in the case of other derivatives also. Moreover, the NCI plot shows that there are very less non-covalent interactions due to the alkyl group (I, II, III, IV, V, and VI) itself. However, it does influence the stacking pattern of the overall chromophore, thereby influencing the extent of interactions between the NDI cores (Fig. S17†). Moreover, we observed a significant NCI due to the alkyne group (IIIp). The pi cloud of the alkyne group has considerable interactions with the pi-cloud of the adjacent NDI aromatic core. The alkyne group of the central IIIp molecule interacts with the core NDI unit of the D2 unit. This observation strengthens our hypothesis that the presence of additional pi clouds on the N-substituted pendant group can introduce an additional transport channel in the bulk (Fig. 7).
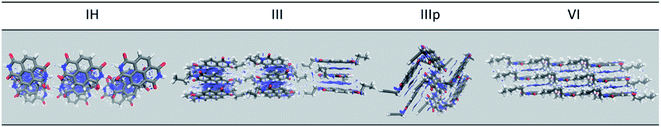 |
| | Fig. 7 NCI plot of IH, III, IIIp and VI (all plots are visualized at an isosurface value of 0.6). | |
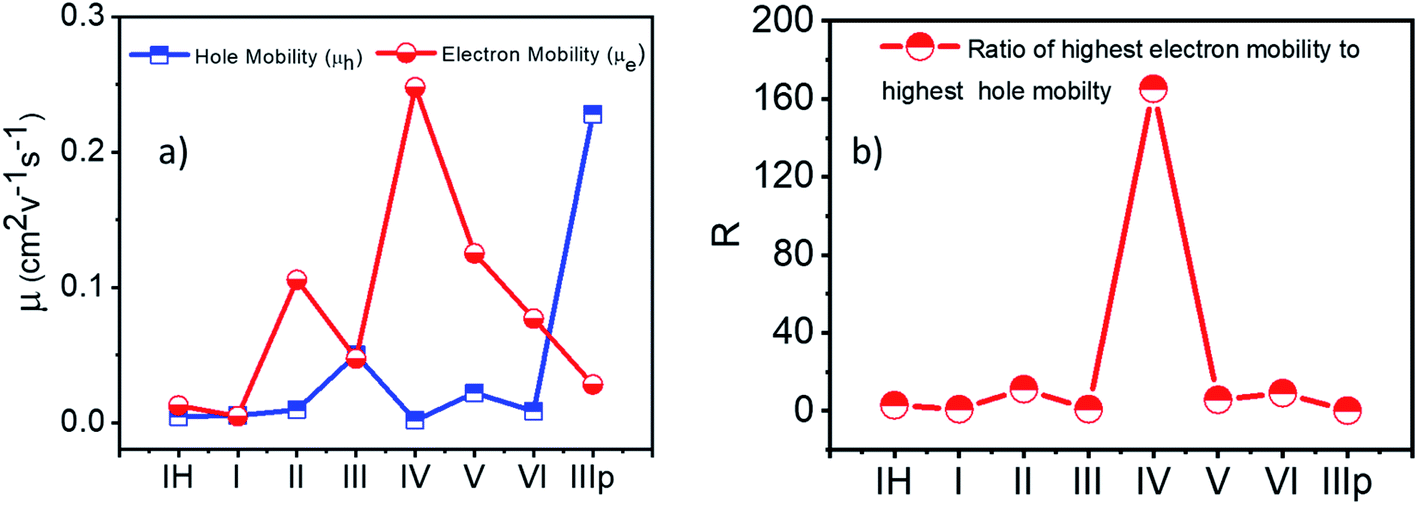
2.2.3 Charge carrier mobility. Finally, collating the computed transfer integral values and the reorganization energies of the NDIs, spatially resolved electron and hole carrier mobilities (μe and μh respectively) were calculated. The essential parameter considered is the charge transfer rate constant (k), which, in turn, depends mainly on the reorganization energy (λ) and the transfer integral (J) as well as the center-to-center distance (r) between the interacting molecules (Tables S5–S12†). To achieve a high carrier mobility, low reorganization energy and high transfer integral values are required for the material. At room temperature, the charge transports in the disordered organic crystals are reported to be anisotropic in nature, which is because of the enhancement of fluctuations in the intermolecular transfer integrals. Based on the above-calculated reorganization energy and transfer integral values, and geometrical parameters (r, θ, and γ), anisotropic carrier mobilities of the molecules were investigated by the method established by Deng et al.41 This method has been applied to compute the anisotropic carrier mobility of organic semiconducting molecules.42–45 A detailed representation of the spatially resolved carrier mobility is summarized in the ESI (Fig. S18†). Fig. 7 shows the highest anisotropic mobility values of the studied NDIs. For category 3 derivatives II, IV, V, and VI, the electron mobility was found to be higher than the hole mobility, thereby showing the intrinsic n-type charge transfer characteristics. Material IV exhibits the highest electron mobility among the series of molecules (0.25 cm2 V−1 s−1) with a ratio of electron mobility to hole mobility (R) around 160 (Fig. 8b). Observation of a higher electron mobility in IV implies an efficient charge-hopping capability between the stacks, due to the high electron transfer integral value observed for the M–D1 dimer of IV. In the case of IIIp, hole mobility value is higher than that of electron mobility. This can be attributed to two factors: (i) λh ≪ λe and (ii) Jh > Je. Furthermore, IIIp is found to have the largest hole mobility in comparison to the other derivatives in the series, which implies exclusive p-type charge transfer characteristics of IIIp. The detailed analysis procedures of angular anisotropic electron mobility of NDI crystals are illustrated in the ESI (Fig. S18†).
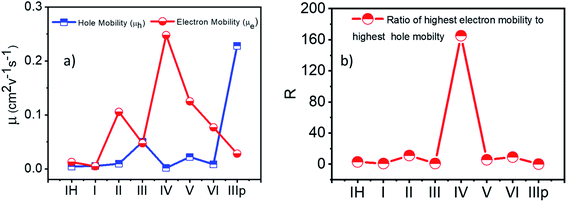 |
| | Fig. 8 (a) Highest anisotropic hole and electron mobility. (b) Ratio of the highest electron mobility to the highest hole mobility. | |
3. Computational methods
All the electronic structure theory calculations presented in the manuscript are carried out using the Gaussian16 program unless stated otherwise. The monomer geometries are initially optimized using the B3LYP-D3/6-31+G* level of theory. Internal reorganization energy (λ) was computed using the “four point energy” approach at the B3LYP-D3/6-31+G* level of theory.
Geometry optimization and single point energy calculations of all the neutral structures were carried out using B3LYP-D3/6-31+G* level of theory whereas that of all the charged species were done at uB3LYP-D3/6-31+G* level of theory. Details of the four-point energy method used are described in the ESI (Section 2†).
The charge transfer integral is mathematically written as follows:
where
ψA and
ψB represent the wave functions of two different quantum states A and B, which are orthogonal and
Ĥ represents the Hamiltonian operator. Here
J measures the extent of electronic coupling between states. The larger the value of
J, the larger the probability that a charge in state A will move to state B and
vice versa. In this study, to calculate the charge transfer integral between the stacks of the active material, we used the dimer projection, in short, the DIPRO method proposed by Baumeier
et al.30 The effect of electronic polarization was considered to estimate the charge transfer integral values.
Charge transfer integral is directly related to the rate of electron transfer according to the Marcus-Hush theory. At room temperature, according to the Marcus–Hush hopping mechanism, the charge hopping rate (k) is given as follows:
| |
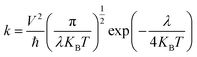 | (2) |
where ℏ is the Planck constant,
KB is the Boltzmann constant,
T is the temperature, and
λ is the reorganization energy.
V is the electronic coupling expressed as follows:
| |
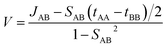 | (3) |
where,
To calculate the transfer integral, we first chose a reference crystallographic plane and a reference axis. The reference axis must lie on the reference plane. Usually, the reference axis is the pi-stacking direction. Then, we chose different interacting hoping dimers and calculated the charge transfer integral using the aforementioned method. For transfer integral calculations, we used the B3LYP-D3/6-31+G* level of theory.
All the transfer integral calculations can be reproduced using the open source code available on GitHub.46
4. Conclusion
In summary, computational analysis of charge transport features of a series of NDI derivatives has revealed several critical factors that control the bulk carrier transport ability of ‘so-called’ n-type semiconductor materials. Interestingly, some of the derivatives discussed are predominantly p-type merely because of slight end group variance. Both the structural and supramolecular factors that dictate the carrier mobility and the polarity of transport are elaborated in detail. A quick prediction of the polarity of transport could be obtained by the charge transfer integral values and a decent correlation is drawn using this assumption. Despite very similar frontier molecular orbital energies, ionisation potential and electron affinity (vertical), the carrier mobility of individual crystalline state was found to be largely dependent on the interaction energies and we found that the dispersion energy plays a major role in dictating the amplitude of carrier mobility and charge transport of NDIs. To the best of our knowledge, among the studied materials, the experimental mobility values are reported only for VI. Our data suggest that some of our studied material, especially IV, can be good candidates for the potential application in OFET devices. Moreover, our result suggests that the end-substitution of an additional pi-stacking group can be a useful strategy for the development of new active materials for better charge transport. Further experimental and computational investigations on these directions are in progress.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
K. J. K. acknowledges IISER K and I. G. acknowledges UGC for the fellowships. This work was supported by IGSTC 2+2 Project LABELONIK. The authors acknowledge Dr Debangshu Chaudhuri for the fruitful discussion. We thank IISER Kolkata for the supercomputing facilities.
References
- N. J. Hestand and F. C. Spano, Chem. Rev., 2018, 118, 7069–7163 CrossRef CAS PubMed
 .
. - R. Ramakrishnan, M. Niyas, M. Lijina and M. Hariharan, Acc. Chem. Res., 2019, 52, 3075–3086 CrossRef CAS PubMed .
- S. Birudula, D. D. Prabhu, T. Ghosh, A. B, S. Das and R. K. Vijayaraghavan, Chem.–Eur. J., 2020, 26, 11135–11140 CrossRef CAS PubMed .
- G. Gryn'ova, K.-H. Lin and C. Corminboeuf, J. Am. Chem. Soc., 2018, 140, 16370–16386 CrossRef PubMed .
- C. Wang, H. Dong, L. Jiang and W. Hu, Chem. Soc. Rev., 2018, 47, 422–500 RSC .
- X. Zhang, H. Dong and W. Hu, Adv. Mater., 2018, 30, 1801048 CrossRef PubMed .
- S. K. Park, J. H. Kim and S. Y. Park, Adv. Mater., 2018, 30, 1704759 CrossRef PubMed .
- S. Varghese and S. Das, J. Phys. Chem. Lett., 2011, 2, 863–873 CrossRef CAS PubMed .
- M. J. Aliaga-Gosalvez, N. Demitri, M. Dohr, J. C. Roldao, S. K. Park, S. Oh, S. Varghese, S. Y. Park, Y. Olivier, B. Milián-Medina, R. Resel and J. Gierschner, Adv. Opt. Mater., 2019, 7, 1900749 CrossRef CAS .
- R. V. Ambili, D. Sasikumar, P. Hridya and M. Hariharan, Chem.–Eur. J., 2019, 25, 1992–2002 CrossRef CAS PubMed .
- M. C. R. Delgado, E.-G. Kim, D. A. d. S. Filho and J.-L. Bredas, J. Am. Chem. Soc., 2010, 132, 3375–3387 CrossRef PubMed .
- S. Varghese, S. K. Park, S. Casado, R. Resel, R. Wannemacher, L. Lüer, S. Y. Park and J. Gierschner, Adv. Funct. Mater., 2016, 26, 2349–2356 CrossRef CAS .
- T. Ghosh, S. Birudula, K. J. Kalita and R. K. Vijayaraghavan, Chem.–Eur. J., 2020, 26, 10501–10509 CrossRef CAS PubMed .
- J. T. E. Quinn, J. Zhu, X. Li, J. Wang and Y. Li, J. Mater. Chem. C, 2017, 5, 8654–8681 RSC .
- T. Okamoto, S. Kumagai, E. Fukuzaki, H. Ishii, G. Watanabe, N. Niitsu, T. Annaka, M. Yamagishi, Y. Tani, H. Sugiura, T. Watanabe, S. Watanabe and J. Takeya, Sci. Adv., 2020, 6, eaaz0632 CrossRef CAS PubMed .
- B. A. Jones, M. J. Ahrens, M.-H. Yoon, A. Facchetti, T. J. Marks and M. R. Wasielewski, Angew. Chem., Int. Ed., 2004, 43, 6363–6366 CrossRef CAS PubMed .
- J. Dhar, U. Salzner and S. Patil, J. Mater. Chem. C, 2017, 5, 7404–7430 RSC .
- G. R. Desiraju and A. Gavezzotti, Acta Crystallogr., Sect. B: Struct. Sci., 1989, 45, 473–482 CrossRef .
- D. Zhang, S. Yokomori, R. Kameyama, C. Zhao, A. Ueda, L. Zhang, R. Kumai, Y. Murakami, H. Meng and H. Mori, ACS Appl. Mater. Interfaces, 2021, 13, 989–998 CrossRef CAS PubMed .
- F. Zhang, Y. Hu, T. Schuettfort, C.-a. Di, X. Gao, C. R. McNeill, L. Thomsen, S. C. B. Mannsfeld, W. Yuan, H. Sirringhaus and D. Zhu, J. Am. Chem. Soc., 2013, 135, 2338–2349 CrossRef CAS PubMed .
- Y. Hu, D. X. Cao, A. T. Lill, L. Jiang, C.-A. Di, X. Gao, H. Sirringhaus and T.-Q. Nguyen, Adv. Electron. Mater., 2018, 4, 1800175 CrossRef .
- T.-T. Do, Y. Takeda, S. Manzhos, J. Bell, S. Tokito and P. Sonar, J. Mater. Chem. C, 2018, 6, 3774–3786 RSC .
- J. Min, Y. N. Luponosov, N. Gasparini, M. Richter, A. V. Bakirov, M. A. Shcherbina, S. N. Chvalun, L. Grodd, S. Grigorian, T. Ameri, S. A. Ponomarenko and C. J. Brabec, Adv. Energy Mater., 2015, 5, 1500386 CrossRef .
- C. Röger and F. Würthner, J. Org. Chem., 2007, 72, 8070–8075 CrossRef PubMed .
- L. Loots and L. J. Barbour, CrystEngComm, 2012, 14, 300–304 RSC .
- A. Benny, D. Sasikumar and M. Hariharan, J. Phys. Chem. C, 2019, 123, 26758–26768 CrossRef CAS .
- B. Baumeier, J. Kirkpatrick and D. Andrienko, Phys. Chem. Chem. Phys., 2010, 12, 11103–11113 RSC .
- E. A. Komissarova, D. I. Dominskiy, V. E. Zhulanov, G. G. Abashev, A. Siddiqui, S. P. Singh, A. Y. Sosorev and D. Y. Paraschuk, Phys. Chem. Chem. Phys., 2020, 22, 1665–1673 RSC .
- T. Kakinuma, H. Kojima, M. Ashizawa, H. Matsumoto and T. Mori, J. Mater. Chem. C, 2013, 1, 5395–5401 RSC .
- E. F. Valeev, V. Coropceanu, D. A. da Silva Filho, S. Salman and J.-L. Brédas, J. Am. Chem. Soc., 2006, 128, 9882–9886 CrossRef CAS PubMed .
- C. Sutton, M. S. Marshall, C. D. Sherrill, C. Risko and J.-L. Brédas, J. Am. Chem. Soc., 2015, 137, 8775–8782 CrossRef CAS PubMed .
- K. J. Thorley and C. Risko, J. Mater. Chem. C, 2016, 4, 4040–4048 RSC .
- C. D. Sherrill, Acc. Chem. Res., 2013, 46, 1020–1028 CrossRef CAS PubMed .
- M. A. Niyas, R. Ramakrishnan, V. Vijay and M. Hariharan, Chem.–Eur. J., 2018, 24, 12318–12329 CrossRef CAS PubMed .
- A. Nicolaï, H. Liu, R. Petraglia and C. Corminboeuf, J. Phys. Chem. Lett., 2015, 6, 4422–4428 CrossRef PubMed .
- R. Devarapalli, S. B. Kadambi, C.-T. Chen, G. R. Krishna, B. R. Kammari, M. J. Buehler, U. Ramamurty and C. M. Reddy, Chem. Mater., 2019, 31, 1391–1402 CrossRef CAS .
- M. J. Turner, S. P. Thomas, M. W. Shi, D. Jayatilaka and M. A. Spackman, Chem. Commun., 2015, 51, 3735–3738 RSC .
- C. Sutton, C. Risko and J.-L. Brédas, Chem. Mater., 2016, 28, 3–16 CrossRef CAS .
- P. de Silva and C. Corminboeuf, J. Chem. Theory Comput., 2014, 10, 3745–3756 CrossRef CAS PubMed .
- J. Contreras-García, E. R. Johnson, S. Keinan, R. Chaudret, J.-P. Piquemal, D. N. Beratan and W. Yang, J. Chem. Theory Comput., 2011, 7, 625–632 CrossRef PubMed .
- W.-Q. Deng, L. Sun, J.-D. Huang, S. Chai, S.-H. Wen and K.-L. Han, Nat. Protoc., 2015, 10, 632 CrossRef CAS PubMed .
- P.-P. Lin, S.-F. Zhang, N.-X. Zhang, J.-X. Fan, L.-F. Ji, J.-F. Guo and A.-M. Ren, Phys. Chem. Chem. Phys., 2019, 21, 3044–3058 RSC .
- H. Ma, N. Liu and J.-D. Huang, Sci. Rep., 2017, 7, 331 CrossRef PubMed .
- X.-Y. Zhang, J.-D. Huang, J.-J. Yu, P. Li, W.-P. Zhang and T. Frauenheim, Phys. Chem. Chem. Phys., 2015, 17, 25463–25470 RSC .
- G.-Y. Qin, L.-F. Ji, J.-X. Fan, N.-X. Zhang, P.-P. Lin, S.-F. Zhang, L.-Y. Zou and A.-M. Ren, J. Phys. Chem. A, 2019, 123, 3300–3314 CrossRef CAS PubMed .
- https://Github.Com/JoshuaSBrown/QC_Tools.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1ra05274h |
|
| This journal is © The Royal Society of Chemistry 2021 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *
*