
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceCesium salt of 2-molybdo-10-tungstophosphoric acid as an efficient and reusable catalyst for the synthesis of uracil derivatives via a green route†
Kiran R. Khillare a,
Dipak S. Ahera,
Laxmikant D. Chavanb and
Sunil G. Shankarwar*a
a,
Dipak S. Ahera,
Laxmikant D. Chavanb and
Sunil G. Shankarwar*a
aDepartment of Chemistry, Dr Babasaheb Ambedkar Marathwada University, Aurangabad, 431 004, Maharashtra, India. E-mail: shankarwar_chem@yahoo.com
bJawaharlal Nehru Engineering College, Aurangabad, 431003, Maharashtra, India
First published on 20th October 2021
Abstract
A solid catalyst, cesium salt of 2-molybdo-10-tungstophosphoric acid (Cs2.3H0.7PW10Mo2O40) named as Cs-3, was synthesized by a simple, cheap, clean, and eco-friendly method. The physicochemical properties of the synthesized catalyst were studied via FTIR spectroscopy, XRD, EDX, ICP-AES, SEM-TEM, and BET techniques. The precursor 2-molybdo-10-tungstophosphoric acid (H3PW10Mo2O40) was easily soluble in water and other polar solvents. Moreover, their cesium salts CsxH3−xPW10Mo2 with Cs content in the range x = 2.0–2.5 were insoluble in water and other polar solvents. The surface area of the precursor (5.483 m2 g−1) increased after partial proton exchange by Cs+ ions (111.732 m2 g−1), and all samples with x > 1 were resistant to leaching of active components and can be recycled without obvious loss of activity. This catalyst used for the synthesis of uracil derivatives via a green route under solvent free conditions at 70 °C gives higher yield within a shorter reaction time. The catalyst was found to be more active and reusable over nine runs with a negligible loss of activity.
1. Introduction
Heterogeneous catalysis has been considered as one of the supreme pillars in the field of green chemistry. The development in heterogeneous processes was more conscious, with a suspicious image of the future, and it was supported since the establishment of green chemistry principles.1 Considering the past two decades, heteropoly acids have established a substantial dedication in heterogeneous catalysis due to their unique physicochemical attributes.1–5 They have very strong Brønsted acidity in the region of a superacid, and also, they are efficient oxidizers under mild conditions. Their structural adaptability is also emphasized since they are complex molecules that can be modified by changing the constituting elements to model their size, charge density, redox potentials, acidity, and solubility.6However, heteropoly acids show solubility in polar solvents, low surface area and efficiency, and these drawbacks are unappealing in organic transformations.7 But to outstrip such weaknesses, the partial exchange of protons by various cations (K+, Rb+, Cs+) has been presented, resulting in the formation of insoluble salts having high surface area and catalytic efficiency.8 These interesting compounds generally exhibit higher catalytic activities than their parent heteropoly acids and can be used in water and other polar organic solvents.9 The use of such insoluble salts as a catalyst could be an effective approach for organic transformations. They have been proven as efficient catalysts in many multi-component reactions with high capabilities in terms of reactivity and improved yields of products.10–17 As a final point, they can be used in less than the stoichiometric amount of a reagent due to high surface area and recycled without significant loss of catalytic activity, all of which postulate them as versatile, economical, and eco-friendly catalysts.18
Uracil derivatives have been reported to accommodate a diversity of pharmacological attributes, including antitumor,19 antimicrobial,20 antihypertensive,21 analgesic,22 and antibacterial.23 They can also act as calcium channel antagonist,24 AbI kinase inhibitor,25 adenosine kinase inhibitor,26 and tyrosine kinase inhibitor.27 Several approaches have been reported in the literature for the synthesis of uracil derivatives from 6-amino-1,3-dimethyl uracil, aldehydes, and 1,3-dicarbonyl compound condensation, such as microwave irradiation,28 and reflux conditions,29 Fe3O4@SiO2–SO3,30 thiourea dioxide,31 AcOH,32 H14[NaP5W30O110]/SiO2,33 SBA–Pr–SO3H,34 and BAIL@UiO-66.35 However, most of these reported methods have some demerits i.e., use of hazardous organic solvents, expensive reagents in stoichiometric amounts, extended reaction times, and high temperature with tedious work-up procedures.
Thus, there is a demand for an alternative greener and high-yielding approach for synthesizing pyrimido[4,5-b]quinoline derivatives, such as the use of a solid catalyst in a solvent-free condition. Solid catalysts are receiving a significant role in organic synthesis and transformation due to their various benefits, including chemical stability, high activity, non-toxicity, non-corrosiveness, high selectivity, easy separation, reusability without losing activity and carrying out reactions in an economical and green route.17
In this regard, we introduce a new and greener strategy to synthesize uracil derivatives from 6-amino-1,3-dimethyl uracil, aldehyde, and dimedone condensation under solvent-free conditions in a closed system at 70 °C with a high yield within a short period with easy workup. Here, we use multicomponent reactions (MCRs) as a tool, which is authorized as advanced tools for sustainable organic synthesis.36 The effectiveness of catalytic methodologies implemented in MCRs has simplified access to new libraries of organic compounds with a high potential for biological applications.37–41 We aim here to preserve green chemistry principles while performing this synthesis by simplifying the product recovery process, preventing waste generation, reducing reaction temperature, time, and the use of hazardous solvents. Already, the multicomponent reactions (MCRs) have approved all these provisions to an immense level.42,43
For this, here, we developed a modified heteropoly acid (2-molybdo-10-tungsto-phosphoricacid) and a series of insoluble salts using a partial exchange of protons by cesium. In our previous work, we reported some modified heteropolyacids by combining metals like Mo, V, and W with different ratios, which were used in some organic transformations showing good results.44–50 So here we studied a different ratio of Mo and W (2 Mo![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :10 W) and prepared our new catalyst. The whole synthesis for this catalyst i.e., Cs2.3H0.7PW10Mo2O40, was too simple and cheap than several others, which were used for the same organic transformation. This catalyst was found to be more active and reusable over nine runs with a slight loss of activity.
:10 W) and prepared our new catalyst. The whole synthesis for this catalyst i.e., Cs2.3H0.7PW10Mo2O40, was too simple and cheap than several others, which were used for the same organic transformation. This catalyst was found to be more active and reusable over nine runs with a slight loss of activity.
2. Experimental section
2.1. Materials and methods
Disodium phosphate (Na2HPO4), sodium molybdate (Na2MoO4·2H2O), sodium tungstate (Na2WO4·2H2O), and cesium carbonate (Cs2CO3) were purchased from Molychem in India and used without further purification. The other additional chemicals and solvents used in the organic synthesis were purchased from Alfa Aesar, Sigma Aldrich, and Merck.The functional integrity and primary Keggin structure of the modified heteropoly anions and after a partial exchange of the protons by cesium cations were investigated by FT-IR (Bruker ALPHA Eco-ATR). The elemental composition of catalysts for the derived molecular formula was investigated by EDX (FEI Nova Nano SEM 450 combined Bruker X Flash 6I30) element image mapping, and Inductively Coupled Plasma-Atomic Emission Spectrometry (ICP-AES, PerkinElmer Inc.). The specific surface area and pore volume were determined by nitrogen gas adsorption–desorption at 77.35 K using the BET method (NOVA Quanta chrome instruments version 11.05). XRD technique was used to define the crystallinity and secondary Keggin structure. The SEM-TEM analysis was used to study the microtopography.
In organic synthesis, the reaction progress was monitored by using thin-layer chromatography (Merck's silica plates), and imaging was accomplished by ultraviolet light or iodine. Melting points of all synthesized analogs were set in the open capillary tube. However, 1H and 13C NMR spectra were recorded to confirm the obtained product structure (Bruker Avance 400 Spectrometer).
2.2. Preparation of 2-molybdo-10-tungstophosphoric acid (H3[PW10Mo2O40])
The precursor for catalyst preparation was synthesized by the simple procedure as follows. Firstly, the desired amount of disodium phosphate (20.66 mmol) and sodium tungstate (206.64 mmol) were dissolved in deionized distilled water (25 mL). The obtained solution was stirred for 30 min at 90 °C. Then, the necessitated amount of aqueous sodium molybdate (41.33 mmol) was added to the above-heated solution. Subsequently, the addition of aqueous sulphuric acid (537.26 mmol) was done until the pH of the solution reached roughly in the range of 1.5 to 2. The resulting mixture was kept in the autoclave at 90 °C for 6 h. Finally, the reaction mixture was cooled and extracted with diethyl ether to get the etherate solution. The 2-molybdo-10-tungstophosphoric acid crystals were obtained by the recrystallization of the concentrated etherate solution in deionized distilled water.2.3. Preparation of cesium salts of 2-molybdo-10-tungstophosphoric acid as a catalyst
The synthesis of the catalyst was done by a simple titration method. Characteristically, a stoichiometric amount of Cs2CO3 was added to the aqueous solution of 2-molybdo-10-tungstophosphoric acid (3.7 mmol) at a constant rate. The Cs content, x in CsxH3−xPW10Mo2O40, was adjusted by the amount 1.85, 3.70, and 4.50 mmol of Cs2CO3 for Cs-1, Cs-2, and Cs-3, respectively, in solution. After complete addition, the procedure was done with continuous stirring and heating the resultant solution for 120 min at 60 °C. Then, the water was evaporated by heating, and the solid was dried in an oven at 100 °C, giving the cesium salts of heteropoly acid, denoted as Cs-1, Cs-2, and Cs-3.2.4. General procedure for the synthesis of pyrimido[4,5-b]quinolines(4a–4j)
A mixture of compounds consisting of aldehyde (1 mmol), dimedone (0.140 g, 1 mmol), 6-amino-1,3-dimethyluracil (0.155 g, 1 mmol), and Cs2.3H0.7PW10Mo2O40 (0.003 g) was stirred vigorously at 70 °C, and the progress of the reaction was monitored by TLC. After the completion of the reaction, the mixture was cooled to room temperature, EtOAc (20 mL) was added, and boiled for 5 min, and then centrifuged and decanted to separate the insoluble catalyst. The solvent of the solution obtained from the decanting was evaporated, and then the crude product was recrystallized from ethanol to give the pure product.2.5. Recyclability procedure of the catalyst
For studying the reusability of the catalyst, after completion of the reaction, the catalyst was recovered from the reaction mixture by simple filtration method. The recovered catalyst was washed with ethanol/water system (1:1) and dried in an oven for 60 min at 110 °C and then reused in the next run.
3. Result and discussion
3.1. FT-IR spectroscopy
The structural integrity of the modified Keggin-type heteropoly anions (i.e. HPWMo and CsPWMo) and their primary structures after the partial exchange of the protons by cesium cations was investigated by FT-IR.The literature describes that the FT-IR spectra provide information on the primary structure of heteropoly acids. The infrared spectra of both H3PW10Mo2 and their Cs salts (Fig. 1) exhibited four characteristic frequencies in the range of 1100–550 cm−1, which is called the fingerprint region of the Keggin structure.51 From Fig. 1, the spectra of the series showed (M–Oe–M) = 754 cm−1 for the asymmetric stretching of metal with edge oxygen, (M–Oc–M) = 879 cm−1 for the asymmetric stretching of corner oxygen, (M![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O) = 970 cm−1 for the asymmetric stretching of the terminal oxygen, (P–O) = 1080 cm−1 for the asymmetric stretching of oxygen with a central phosphorous atom, and (O–P–O) = 594 cm−1 for the bending of the central oxygen atom.52 From Fig. 1, the values for (M–Oe–M) = 754 cm−1 and (MO) = 970 cm−1 observed in spectra (a) were shifted towards higher values in the spectra of (b), (c), and (d) due to interaction with Cs+ ions, while no significant shift was observed in the main absorption bands of (P–O) = 1080 cm−1 and (O–P–O) = 594 cm−1. Since the absorption band of the P–O bond (1080 cm−1) was noticed at the same wavenumber in the spectra of parent acids and their salts, it is possible to conclude that no lacunar species were formed and that the Keggin anions preserved their structures intact after the partial exchange of protons by cesium.13 The observed broad bands at wavenumbers 2900–3600 cm−1 were assigned to ν(H5O2+).53 The peak between 1698–1618 cm−1 corresponds to the bending frequency of –OH, i.e., the water of crystallization.54 From Fig. 1, on increasing Cs loading in the series, the intensity of absorption band for –OH decreased, which specifies the decrease in the water of crystallization. There was no change observed in the functional integrity and their primary Keggin structure in the recovered Cs-3 catalyst after nine cycles (Fig. 1 (dr)).
O) = 970 cm−1 for the asymmetric stretching of the terminal oxygen, (P–O) = 1080 cm−1 for the asymmetric stretching of oxygen with a central phosphorous atom, and (O–P–O) = 594 cm−1 for the bending of the central oxygen atom.52 From Fig. 1, the values for (M–Oe–M) = 754 cm−1 and (MO) = 970 cm−1 observed in spectra (a) were shifted towards higher values in the spectra of (b), (c), and (d) due to interaction with Cs+ ions, while no significant shift was observed in the main absorption bands of (P–O) = 1080 cm−1 and (O–P–O) = 594 cm−1. Since the absorption band of the P–O bond (1080 cm−1) was noticed at the same wavenumber in the spectra of parent acids and their salts, it is possible to conclude that no lacunar species were formed and that the Keggin anions preserved their structures intact after the partial exchange of protons by cesium.13 The observed broad bands at wavenumbers 2900–3600 cm−1 were assigned to ν(H5O2+).53 The peak between 1698–1618 cm−1 corresponds to the bending frequency of –OH, i.e., the water of crystallization.54 From Fig. 1, on increasing Cs loading in the series, the intensity of absorption band for –OH decreased, which specifies the decrease in the water of crystallization. There was no change observed in the functional integrity and their primary Keggin structure in the recovered Cs-3 catalyst after nine cycles (Fig. 1 (dr)).
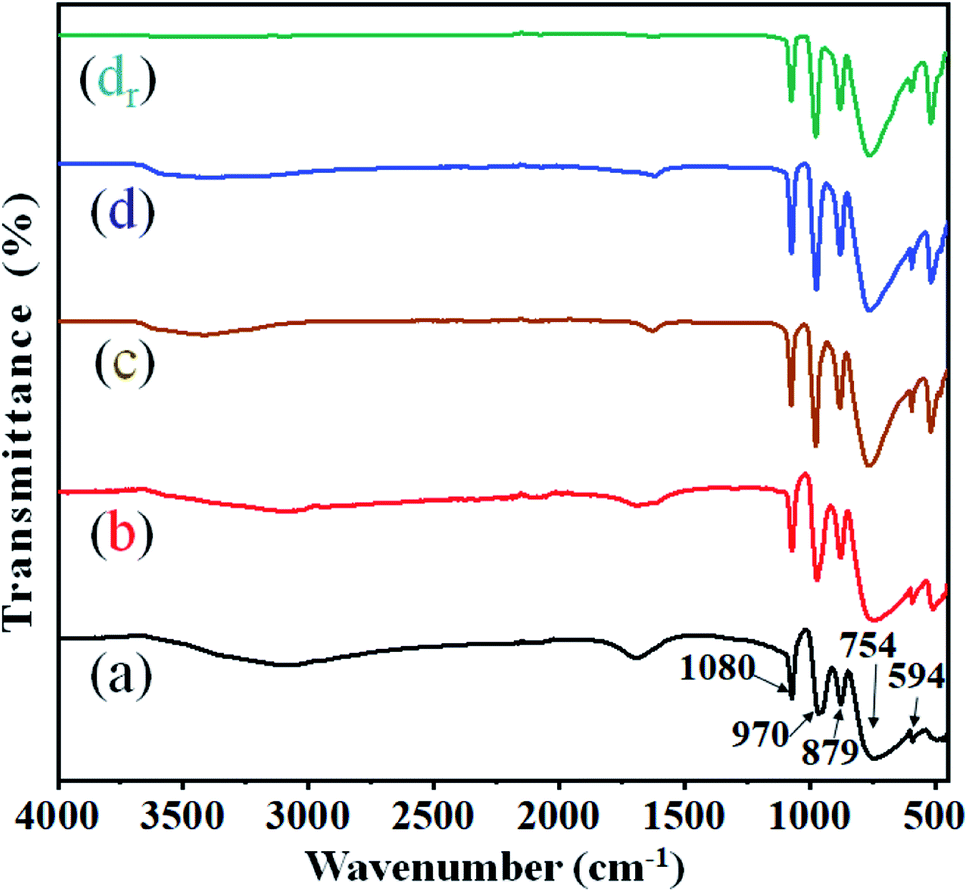 | ||
| Fig. 1 FT-IR spectra of (a) H3PW10Mo2, (b) Cs-1, (c) Cs-2, (d) Cs-3, and (dr) recovered Cs-3 after nine cycles. | ||
3.2. XRD analysis
A comparison of diffractograms of cesium salts with their parent heteropoly acids (Fig. 2) can help to determine the effect of proton exchange on the level of crystallinity.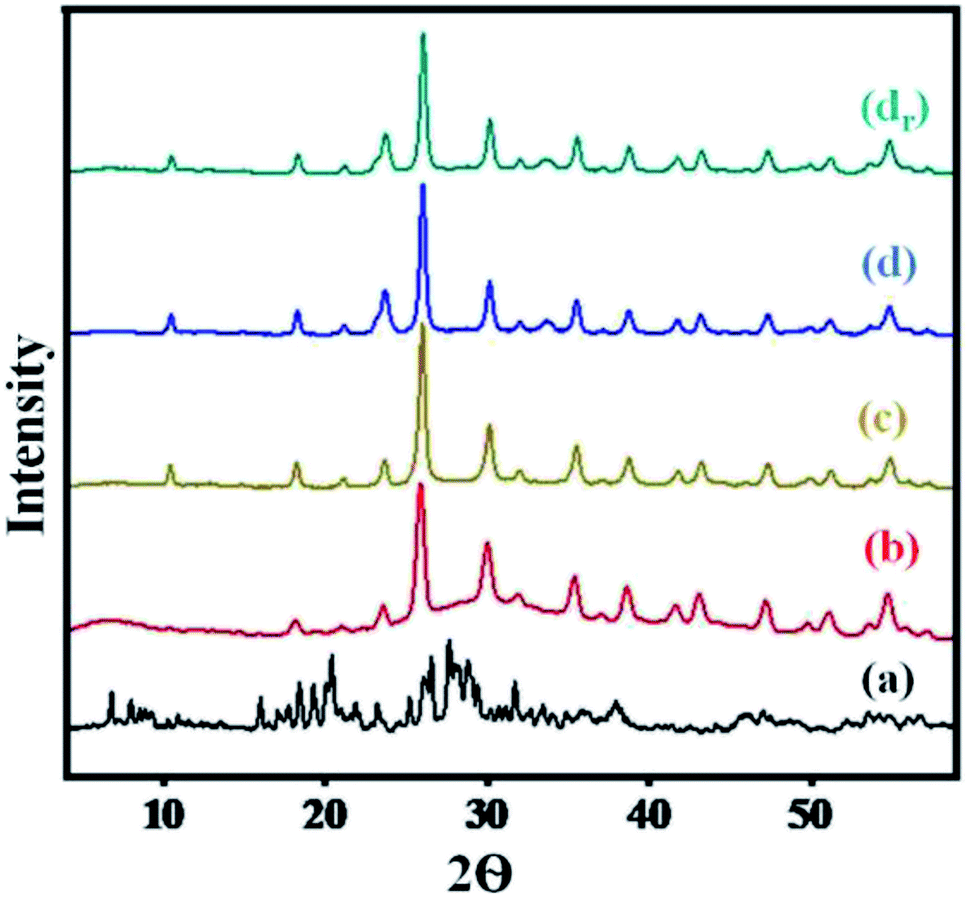 | ||
| Fig. 2 XRD pattern of (a) H3PW10Mo2, (b) Cs-1, (c) Cs-2, (d) Cs-3, and (dr) recovered Cs-3 after nine cycles. | ||
The diffractograms of Cs heteropoly salts and their precursor indicated that the synthesized Cs salts were crystalline. While the diffractograms of the precursor showed the main XRD 2θ peaks in the region of 5° to 45° (Fig. 2), which can be attributed to the characteristic peaks of the secondary structure of the Keggin anion.55 Moreover, the Cs salts presented well-defined diffraction peaks along the series (Fig. 2) that showed increased crystallinity with the increase in Cs content. All 2θ angle range values i.e., 10.58°, 18.42°, 23.71°, 25.90°, 30.26°, 35.6°, and 38.72°, indicated intact secondary keggin structure after proton exchange. From Fig. 2, the peak was found to be broader in graph (a), while comparing the peak intensity in the region of 5° to 10° with graph (b). Later on, all peaks were sharper as compared to graph (a) with increasing Cs loading in the series. There was no change observed in the level of crystallinity and secondary Keggin structure in the recovered Cs-3 catalyst after nine cycles (Fig. 2).
3.3. EDS analysis
EDS spectra were recorded to confirm the molecular formula of parent HPAs, i.e., H3[PW10Mo2O40]. The results of EDX elemental image mapping revealed that the atomic percentage ratio (P:W:Mo) is preserved to be 7.7:76.9:15.4, which corresponds to the formula H3[PW10Mo2O40]. The cesium load is incorporated into the parent HPAs, and this effect can be evidenced by the slight increase in the intensity of Cs characteristic peaks between 4 and 5 keV, indicating the stoichiometry of the reaction was successfully controlled.56 From Fig. 3, there is no peak between 4 and 5 keV for cesium in graph (a). While comparing the graphs (b), (c), and (d), the peak intensity between 4 and 5 keV increased as cesium load increases in the series of Cs salts. The contents of tungsten, molybdenum, phosphorus, and cesium were also confirmed by ICP-AES investigations, and all the results are found to be in good agreement with accepted results (Table S1 ESI†).
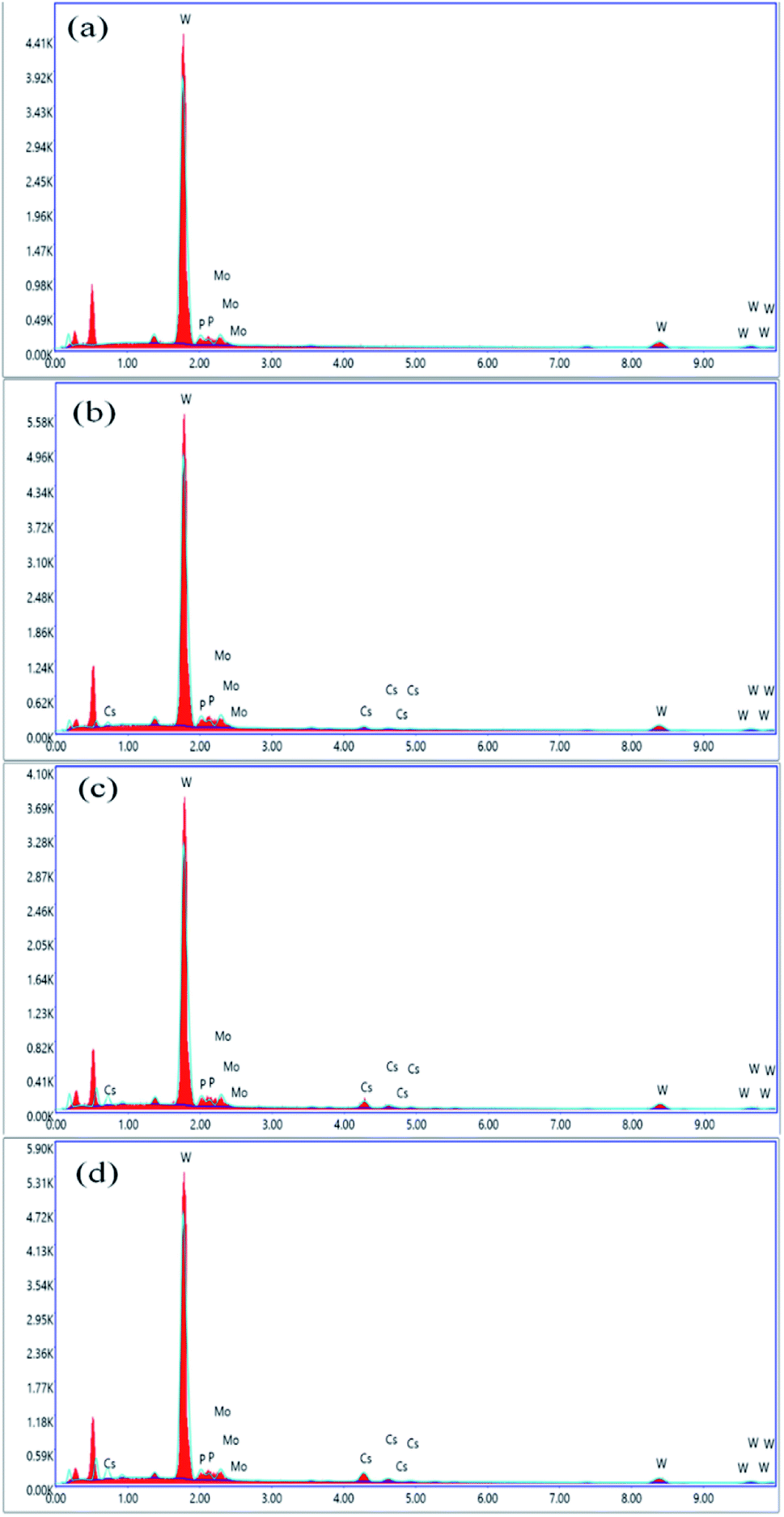 | ||
| Fig. 3 EDX spectra of (a) H3PW10Mo2, (b) Cs-1, (c) Cs-2, and (d) Cs-3. | ||
3.4. BET analysis
Low-temperature nitrogen adsorption analysis was performed for studying the textural properties of the catalyst. The given data shows the specific surface area, SBET, of Cs-salts varying between 51.456 and 111.732 m2 g−1 and pore volume between 0.020 and 0.083 cm3 g−1, which were higher than those of parent HPA (5.483 m2 g−1 and 0.010 cm3 g−1) (Table 1). The specific surface area improved in the series H3PW10Mo2 < Cs-1 < Cs-2 < Cs-3 with the increase in Cs loading.57 The resulting salts are expected to have higher activities and good recyclability, but the degree of the exchange should be moderate because the partially exchanged Cs salts are more active than the fully exchanged analogues (Table 1).58 The investigation on insoluble cesium salts of CsxH3−xPW10Mo2 shows that those with Cs content in the range x = 2.0–2.5 have surface areas of ∼100 m2 g−1 and Brønsted acid strengths similar to those of the parent HPA.57 All samples with x > 1 are resistant to leaching of active components and can be recycled without obvious loss of activity.2| Catalyst | H3PW10Mo2 | Cs-1 | Cs-2 | Cs-3 |
|---|---|---|---|---|
| Pore volume | 0.010 cm3 g−1 | 0.020 cm3 g−1 | 0.075 cm3 g−1 | 0.083 cm3 g−1 |
| Surface area | 5.483 m2 g−1 | 51.456 m2 g−1 | 90.052 m2 g−1 | 111.732 m2 g−1 |
The nitrogen adsorption–desorption isotherms of all samples were studied, which shows the quick initial increase corresponding to the formation of the first layer; therefore, an increase in pressure forms the second layer of the adsorbed molecules, followed by another layer (Fig. 4). The irreversibility of nitrogen adsorption–desorption isotherms (i.e., presence of a hysteresis cycle) was observed for all samples. From Fig. 4, graphs (a), (b), (c), and (d) of all these isotherms can be classified as Type-IV physisorption isotherms and associated hysteresis loops H3, which is usually observed for mesoporous materials.13
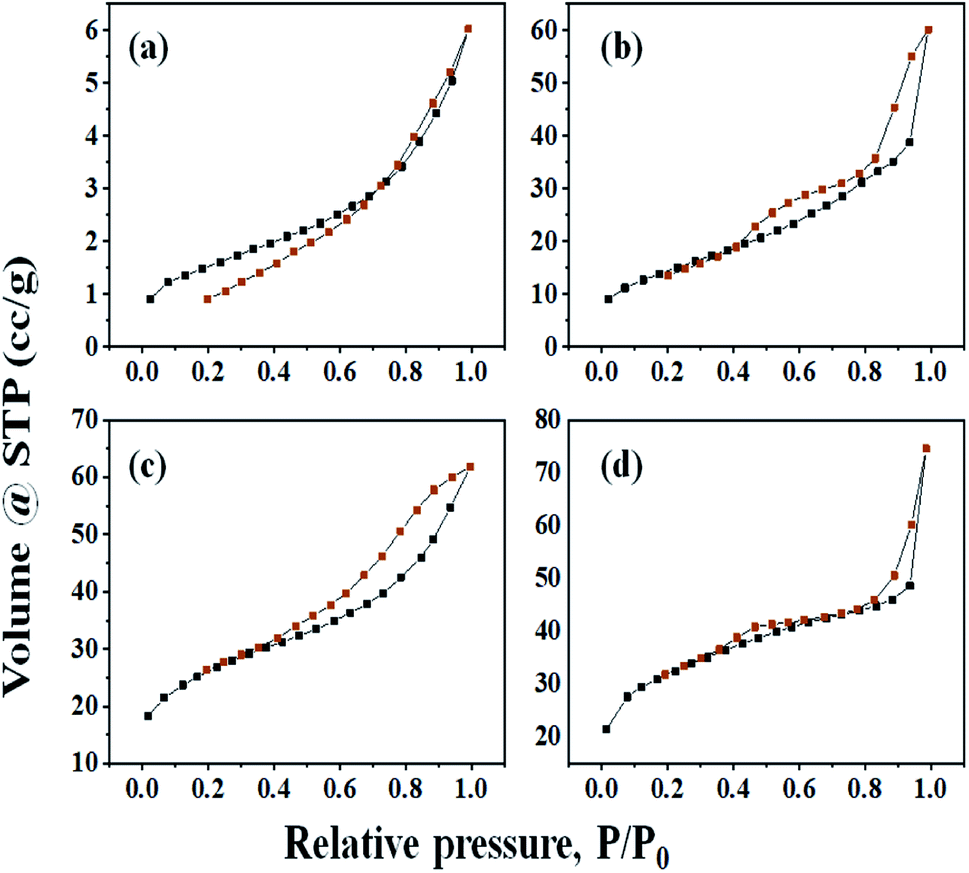 | ||
| Fig. 4 Nitrogen adsorption–desorption isotherms of (a) H3PW10Mo2, (b) Cs-1, (c) Cs-2, and (d) Cs-3. | ||
3.5. SEM-TEM analysis
The topography of HPAs and their Cs salts was examined with SEM-TEM image analysis. The image of HPAs did not specify particles of a particular shape, while the Cs salt particles were spherical (Fig. 5), having a diameter in the range of 50 to 1000 nm. | ||
| Fig. 5 SEM-TEM images of (a) H3PW10Mo2, (b) Cs-1, (c) Cs-2, and (d) Cs-3. | ||
The images were selected to show the influence of cesium load on the morphological properties of materials. The observed result reveals that the crystallite size was gradually reduced in the series of Cs salts while the cesium loading was increased (Fig. 5).57 Fig. 5 shows the crystallites size of about 500–1000 nm (image b), 200–400 nm (image c), and the crystallites size reduced than 200 nm in (image d) of Cs-3. These results support the results obtained from XRD and BET investigations because crystallinity and surface area increase as the Cs content increases (Table 1).
4. Catalyst testing for the synthesis of pyrimido[4,5-b]quinolines
The efficiency of synthesized catalysts (0.01 g) were evaluated by multicomponent reaction for the synthesis of pyrimido[4,5-b]quinolines (Scheme 1) from 6-amino-1,3-dimethyl uracil, benzaldehyde, and dimedone as the model reaction and the results are presented in Table 2. It is clear from Table 2 that Cs-3 is found to be superior to its precursor HPA and other series members possibly due to higher surface area.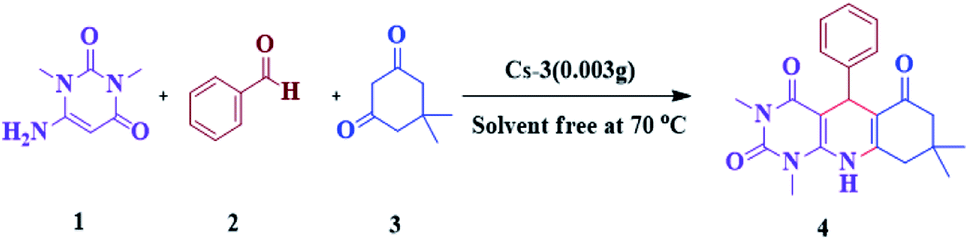 | ||
| Scheme 1 Model reaction for the synthesis of pyrimido[4,5-b]quinolines 4a. | ||
After that, the model reaction was conducted using varying amounts of Cs-3 to confirm the optimum amount i.e., 0.003 g (Fig. 6). When the amount was increased more than 0.003 g, there was no change in the yield percentage, while reducing the catalyst amount resulted in a reduction in the product yield. Therefore, the optimum amount of Cs-3 as 0.003 g in the model reaction was confirmed (Fig. 6).
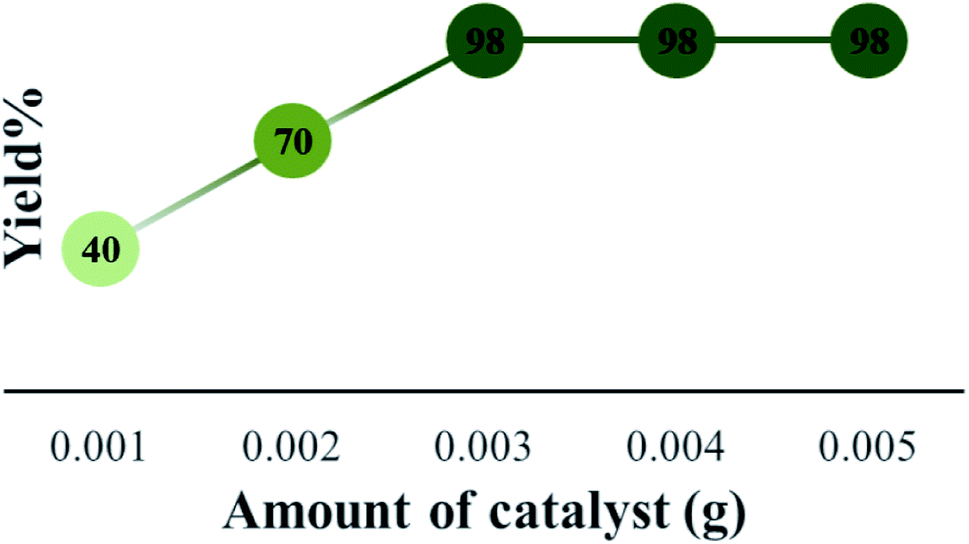 | ||
| Fig. 6 Optimization of the amount of Cs-3 and comparison of yield (%) for the synthesis of 4a under solvent free condition. | ||
Next, we conducted the model reaction in the presence of Cs-3 using different solvents and solvent-free conditions at varying temperatures ranging from 25 °C (room temperature) to 90 °C (Table 3). As shown in Table 3, under solvent-free conditions at 70 °C, the best outcomes were achieved (Table 3, entry 8). In solvent-free conditions, the higher availability of the reactant molecules results in effortless access to active sites through the pores of the catalyst, and this porous structure gives the best results than in a solvent. In a solvent, the availability of reactant molecules decreases due to the presence of solvent molecules, which decreases the yield of the product. Table 3 shows that the poor results were obtained at low temperatures, and no change was found in yield percentage at temperatures higher than 70 °C. However, 0.003 g of the catalyst was sufficient at 70 °C within 10 min in a solvent-free condition, and these reaction conditions were optimal.
| Entry | Solvent | Reaction condition | Time (min) | Yieldb (%) |
|---|---|---|---|---|
| a Reaction substrate: 6-amino-1,3-dimethyl uracil (1 mmol), benzaldehyde (1 mmol), and dimedone (1 mmol).b Isolated yields. | ||||
| 1 | EtOH | Reflux | 60 | 55 |
| 2 | CH3CN | Reflux | 80 | 45 |
| 3 | Toluene | Reflux | 90 | 40 |
| 4 | Water | Reflux | 60 | 45 |
| 5 | Solvent-free | 25 °C | 40 | 50 |
| 6 | Solvent-free | 50 °C | 25 | 75 |
| 7 | Solvent-free | 60 °C | 10 | 85 |
| 8 | Solvent-free | 70 °C | 10 | 98 |
| 9 | Solvent-free | 80 °C | 10 | 98 |
| 10 | Solvent-free | 90 °C | 10 | 98 |
Further, the catalytic system was explored for the synthesis of various pyrimido[4,5-b]quinoline derivatives using the optimized reaction conditions, and good to excellent results were obtained, which are summarized in Table 4. The results depicted the optimal conditions, which worked well for aromatic aldehydes bearing both electron withdrawing and electron donating groups.
|
|||||
|---|---|---|---|---|---|
| Entry | R | Time (min) | Yieldb (%) | Melting point (°C) observed | Meting point (°C) literature |
| a Reaction substrates: 6-amino-1,3-dimethyl uracil (1 mmol), benzaldehyde (1 mmol), and dimedone (1 mmol).b Isolated yields. | |||||
| 1 | H | 10 | 98 | >300 | >300 (ref. 32) |
| 2 | 4-Cl | 10 | 95 | >300 | >300 (ref. 32) |
| 3 | 4-Br | 11 | 96 | >300 | >300 (ref. 24) |
| 4 | 4-N(Me)2 | 11 | 96 | >300 | >300 |
| 5 | 3-NO2 | 10 | 97 | >300 | >300 (ref. 32) |
| 6 | 4-OH | 11 | 97 | >300 | >300 (ref. 24) |
| 7 | 2-OH-4-OMe | 12 | 96 | >300 | >300 |
| 8 | 4-OMe | 12 | 93 | >300 | >300 (ref. 32) |
| 9 | 4-Me | 14 | 92 | >300 | >300 (ref. 32) |
| 10 | 3,4-(OMe)2 | 13 | 94 | >300 | >300 |
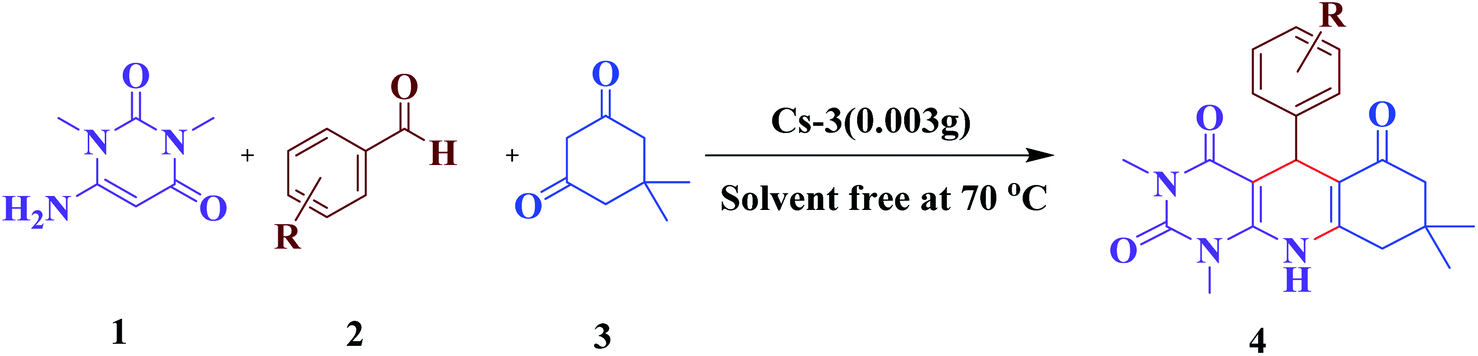
Finally, to demonstrate the effectiveness of our developed strategy for the synthesis of pyrimido[4,5-b]quinolines, a comparison of catalytic activity of Cs-3 with some of the reported catalysts was made, and the results are illustrated in Table 5. From the observed outcomes, we concluded that Cs-3 is indeed much efficient for this MCR under optimized conditions in terms of shorter reaction times in solvent-free conditions and at a lower temperature than most of the reported catalysts.
| Entry | Catalyst | Conditions | Time | Yield (%) |
|---|---|---|---|---|
| 1 | KF/Al2O3 | EtOH/80 °C | 8 h | 70–95 (ref. 32) |
| 2 | — | i-PrOH/reflux | 8 h | 75–88 (ref. 29) |
| 3 | Thiourea dioxide | H2O/50 °C | 8 h | 90–95 (ref. 31) |
| 4 | SBA–Pr–SO3H | CH3CN/reflux | 45 min | 80–93 (ref. 34) |
| 5 | Nano Fe3O4@SiO2–SO3H | H2O/stir./70 °C | 25 min | 81–94 (ref. 30) |
| 6 | H14[NaP5W30O110]/SiO2 | H2O/reflux | 20 min | 80–94 (ref. 33) |
| 7 | Wang-merrifield resins | CH3COOH/MW-irradiation/rt | 15 min | 82–92 (ref. 28) |
| 8 | BAIL@UiO-66 | Solvent-free/100 °C | 15 min | 87–98 (ref. 35) |
| 9 | Cs2.3H0.7PW10Mo2O40 | Solvent-free/70 °C | 10 min | 92–98 (this work) |
Then, to depict the advantages of the present work, a correlation of catalytic efficiency of Cs-3 with other catalysts was performed in the model study. We observed (Table 5), Cs-3 showed excellent catalytic activity due to the porous structure of Cs-3, which serves the effortless access of reactants to Cs+ ions in the secondary keggin structure as active cites. The presence of Cs+ ions might be assisting to enhance the electrophilic character of carbonyl carbons of reactants than other reported catalysts. The corresponding product 4 generated an excellent yield (98%) within a short reaction time (10 min) under solvent-free conditions at a temperature of 70 °C and with a lesser amount of the catalyst. The whole synthesis for Cs-3 was straightforward and economical than several others.
5. Recyclability of the catalyst
At the end of the reaction, the catalyst was recovered from the reaction mixture by a simple filtration method. The recovered catalyst was washed with an ethanol/water system (1:1) and then reused in the next run. To confirm that the catalyst was insoluble during reaction workup, the filtered catalyst was weighed before reuse. The results indicated that the catalysts were not soluble in the solvent, and the yields of reactions using these catalysts over 9 runs indicated only a slight loss of activity (Fig. 7).
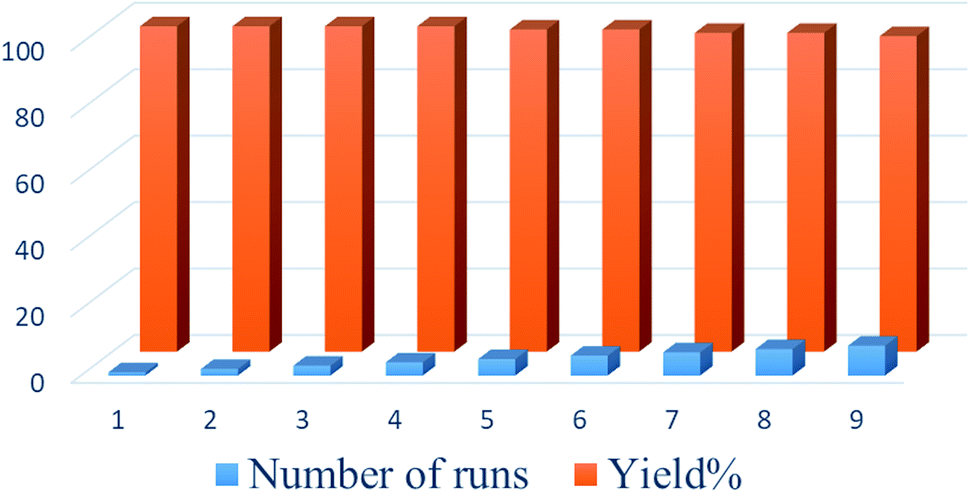 | ||
| Fig. 7 Reusability of Cs-3 catalyst. | ||
6. Proposed plausible mechanism
The synthesis of pyrimido[4,5-b]quinolines using Cs-3 as a catalyst is shown by a plausible mechanism in Scheme 2. The Cs-3 assists as a Brønsted acid, increasing the carbonyl group's electrophilicity of the aldehyde. The first stage is the Knoevenagel condensation between the aldehyde (2) and enol form of dimedone (3) to yield adduct A with the removal of a water molecule. Then, the second stage is the Michael addition between the 6-amino-1,3-dimethyl uracil (1) and adduct A for producing an intermediate B. Intramolecular cyclization occurs, followed by dehydration and aromatization of the intermediate resulting in the final product (4).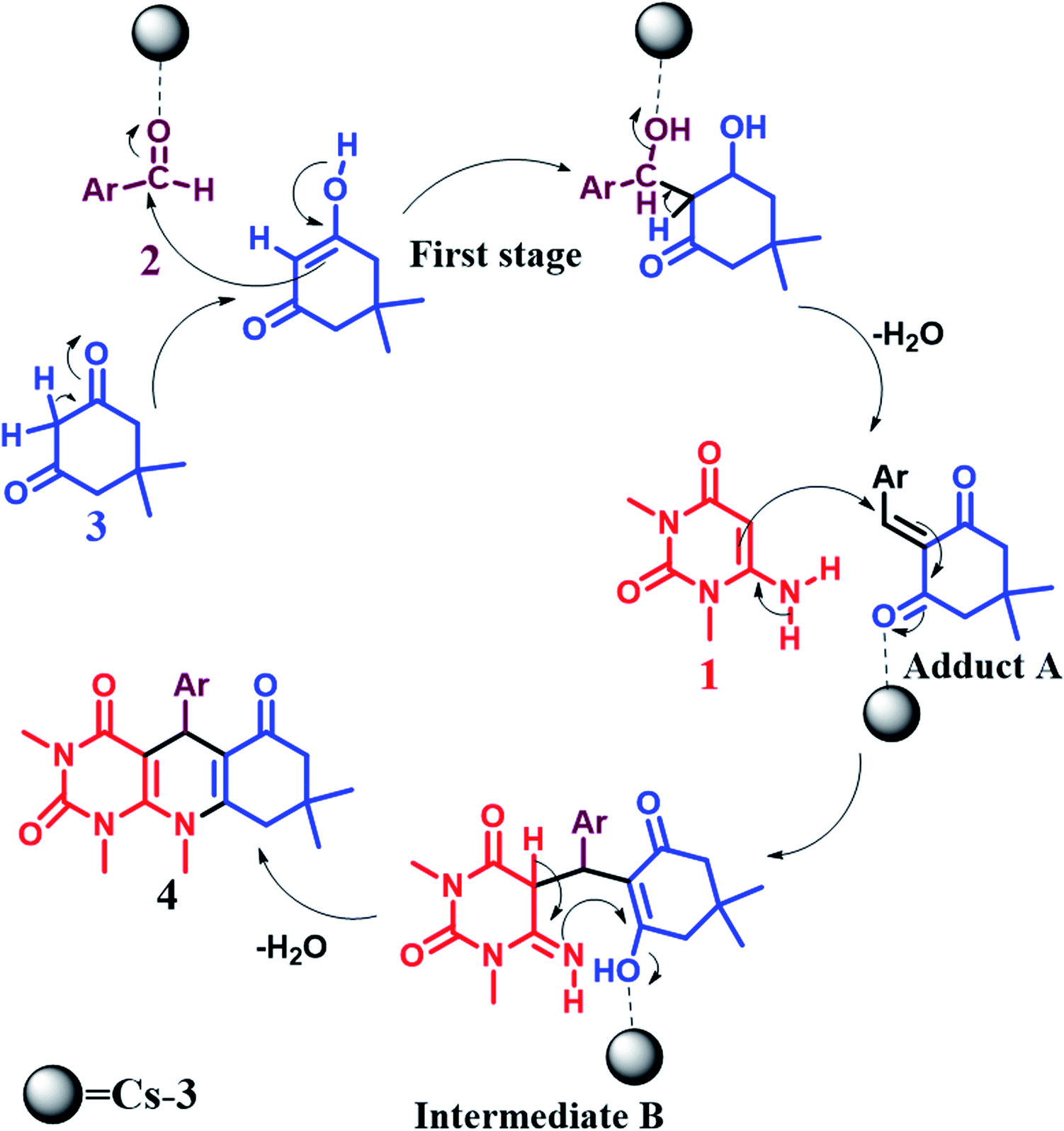 | ||
| Scheme 2 Plausible mechanism for the synthesis of pyrimido[4,5-b]quinolones. | ||
7. Conclusions
The properties of the synthesized catalysts were studied by FTIR, XRD, EDX, ICP-AES, SEM-TEM, and BET techniques. The detailed surface and bulk characterization suggest these materials are composed of a CsHPWMo core. The excellent achievement of Cs-3 is accredited to the partial exchange of protons by Cs+ ions with high surface area leading to more active sites with insolubility. The Cs-3 (Cs2.3H0.7PW10Mo2O40) have been investigated for uracil derivative reactions showing high efficiency, high product yields (98%) with high purity in short reaction time (10 min) under solvent-free conditions at 70 °C by employing less than the stoichiometric amount of the catalyst. The whole synthesis for the Cs-3 catalyst was too simple, cheap, clean, and eco-friendly. Moreover, Cs-3 was resistant to leaching, can be recycled and reused in nine runs of reactions without significant loss of activity, all of which postulate them as versatile, economical, and eco-friendly catalysts. It is expected that this strategy would be extensively serviceable via green route in many other organic transformations for drug synthesis.8. Spectral data
8.1. 1,3,8,8-Tetramethyl-5-phenyl-5,8,9,10-tetrahydropyrimido[4,5-b]quinoline-2,4,6(1H,3H,7H)-trione (4a)
MS 365 (M+1), IR (KBr) 3015, 2962, 1712, 1652, 1558, 1541, 1508, 1482 cm−1. 1H NMR (400 MHz, DMSO-d6): δ (ppm) 0.89 (s, 3H), 1.05 (s, 3H), 2.05 (d, J = 16.0 Hz, 1H), 2.23 (d, J = 16.1 Hz, 1H), 2.52–2.65 (distorted AB system, 2H), 3.10 (s, 3H), 3.47 (s, 3H), 4.89 (s, 1H), 7.09 (t, J = 7.1 Hz, 1H), 7.17–7.25 (m, 4H), 9.02 (s, 1H). 13C NMR (100 MHz, DMSO-d6) δ = 204.4, 160.3, 159.6, 151.2, 150.4, 149.5, 139.7, 132.4, 130.2, 127.4, 120.9, 105.5, 31.7, 29.9, 28.3, 23.5.8.2. 5-(4-Chlorophenyl)-1,3,8,8-tetramethyl-5,8,9,10-tetrahydropyrimido[4,5-b]quinoline-2,4,6(1H,3H,7H)-trione (4b)
MS 399 (M+1), IR (KBr) 3005, 2948, 1709, 1660, 1588, 1548, 1512, 1480 cm−1. 1H NMR (400 MHz, DMSO-d6): δ (ppm) 0.85 (s, 3H), 1.00 (s, 3H), 2.01 (d, J = 16.1 Hz, 1H), 2.19 (d, J = 16.1 Hz, 1H), 2.47–2.55 (distorted AB system, 2H), 3.06 (s, 3H), 3.42 (s, 3H), 4.83 (s, 1H), 7.14–7.28 (m, 5H). 13C NMR (100 MHz, DMSO-d6) δ = 204.5, 160.5, 159.7, 151.2, 151.4, 150.2, 148.2, 143.4, 129.7, 122.2, 121.5, 105.5, 31.6, 29.9, 28.3, 23.5.8.3. 5-(4-Bromophenyl)-1,3,8,8-tetramethyl-5,8,9,10-tetrahydropyrimido[4,5-b]quinoline-2,4,6(1H,3H,7H)-trione (4c)
MS 443 (M+1), IR (KBr) 3025, 2965, 1712, 1675, 1558, 1536, 1513, 1470 cm−1. 1H NMR (400 MHz, DMSO-d6): δ (ppm) 0.90 (s, 3H), 1.03 (s, 3H), 2.03–2.10 (distorted AB system, 2H), 2.20–2.28 (distorted AB system, 2H), 3.09 (s, 3H), 3.45 (s, 3H), 4.85 (s, 1H), 7.18 (d, J = 8.4 Hz, 2H), 7.37 (d, J = 8.4 Hz, 2H), 9.04 (s, 1H). 13C NMR (100 MHz, DMSO-d6) δ = 204.4, 160.3, 159.6, 151.2, 150.4, 149.5, 139.7, 132.4, 130.2, 127.4, 120.9, 105.5, 31.7, 29.9, 28.3, 23.5.8.4. (5-(4-(Dimethylamino)phenyl)-1,3,8,8-tetramethyl-7,8,9,10-tetrahydropyrimido[4,5-b]quinoline-2,4,6(1H,3H,5H)-trione) (4d)
MS 408 (M+1), IR (KBr) 3020, 2960, 1717, 1670, 1555, 1532, 1512, 1472 cm−1. 1H NMR (400 MHz, DMSO-d6): δ (ppm) 7.00 (d, J = 8.7 Hz, 7H), 6.52 (d, J = 8.8 Hz, 7H), 4.72 (s, 3H), 3.41 (s, 10H), 3.07 (s, 10H), 2.77 (s, 18H), 2.20 (d, J = 16.2 Hz, 4H), 2.01 (d, J = 16.3 Hz, 5H), 1.02 (s, 9H), 0.89 (s, 9H). 13C NMR (100 MHz, DMSO-d6) δ = 195.69, 161.53, 151.07, 149.34, 135.03, 128.74, 112.57, 91.41, 50.61, 40.72, 32.93, 32.48, 30.52, 29.51, 28.12, 26.89.8.5. 1,3,8,8-Tetramethyl-5-(3-nitrophenyl)-5,8,9,10-tetrahydropyrimido [4,5-b]quinoline-2,4,6(1H,3H,7H)-trione (4e)
MS 410 (M+1), IR (KBr) 3017, 2960, 1713, 1655, 1560, 1540, 1508, 1482 cm−1. 1H NMR (400 MHz, DMSO-d6): δ (ppm) 0.91 (s, 3H), 1.06 (s, 3H), 2.06 (d, J = 16.2 Hz, 1H), 2.26 (d, J = 16.4 Hz, 1H), 2.57–2.69 (distorted AB system, 2H), 3.09 (s, 3H), 3.47 (s, 3H), 4.98 (s, 1H), 7.52 (m, 1H), 7.72 (d, J = 7.7 Hz, 1H), 7.98 (dd, J = 8.2, 1.4 Hz, 1H), 8.05 (s, 1H), 9.18 (s, 1H). 13C NMR (100 MHz, DMSO-d6) δ = 199.5, 161.5, 158.5, 150.8, 142.0, 141.8, 137.1, 128.8, 128.4, 113.9, 91.5, 55.1, 37.4, 29.8, 28.7, 28.2, 19.8.8.6. 5-(4-Hydroxyphenyl)-1,3,8,8-tetramethyl-7,8,9,10-tetrahydropyrimido[4,5-b]quinoline-2,4,6(1H,3H,5H)-trione (4f)
MS 381 (M+1), IR (KBr) 3020, 2950, 1726, 1660, 1552, 1523, 1522, 1460 cm−1. 1H NMR (400 MHz, DMSO-d6): δ (ppm) 6.98 (d, J = 8.5 Hz, 2H), 6.54 (d, J = 8.5 Hz, 2H), 4.73 (s, 1H), 3.40 (s, 3H), 3.06 (s, 3H), 2.52 (s, 2H), 2.18 (d, J = 16.1 Hz, 1H), 2.00 (d, J = 16.4 Hz, 1H), 1.00 (s, 3H), 0.85 (s, 3H). 13C NMR (100 MHz, DMSO-d6) δ = 195.64, 161.31, 155.57, 151.05, 149.83, 143.97, 137.56, 128.96, 114.82, 112.46, 91.11, 70.15, 50.48, 40.19, 39.52, 33.16, 32.45, 30.52, 29.53, 28.12, 26.72.8.7. 5-(2-Hydroxy-4-methoxyphenyl)-1,3,8,8-tetramethyl-7,8,9,10-tetrahydropyrimido [4,5-b]quinoline-2,4,6(1H,3H,5H)-trione (4g)
MS 411 (M+1), IR (KBr) 3020, 2930, 1712, 1662, 1570, 1532, 1525, 1462 cm−1. 1H NMR (400 MHz, DMSO-d6): δ (ppm) 6.66 (t, J = 5.7 Hz, 2H), 6.63 (s, 1H), 6.56 (s, 1H), 4.95 (s, 1H), 3.67 (s, 3H), 3.41 (s, 3H), 3.08 (s, 3H), 2.56 (d, J = 14.2 Hz, 2H), 2.23 (d, J = 16.3 Hz, 1H), 2.03 (d, J = 16.2 Hz, 1H), 1.02 (s, 3H), 0.90 (s, 3H). 13C NMR (100 MHz, DMSO-d6) δ = 196.47, 162.59, 151.61, 150.70, 149.15, 143.17, 134.09, 120.93, 119.88, 111.43, 110.44, 90.26, 70.14, 55.86, 50.23, 40.18, 32.43, 30.72, 29.40, 28.73, 28.36, 26.88.8.8. 5-(4-Methoxyphenyl)-1,3,8,8-tetramethyl-7,8,9,10-tetrahydropyrimido[4,5-b]quinoline-2,4,6(1H,3H,5H)-trione (4h)
MS 395 (M+1), IR (KBr) 3020, 2950, 1726, 1660, 1540, 1550, 1502, 1456 cm−1. 1H NMR (400 MHz, DMSO-d6): δ (ppm) 7.10 (d, J = 8.7 Hz, 1H), 6.71 (d, J = 8.7 Hz, 1H), 4.78 (s, 1H), 3.64 (s, 1H), 3.41 (s, 1H), 3.06 (s, 1H), 2.53 (d, J = 5.5 Hz, 1H), 2.10 (d, J = 59.3 Hz, 1H), 1.01 (s, 2H), 0.86 (s, 1H). 13C NMR (100 MHz, DMSO-d6) δ = 195.43, 161.09, 157.64, 150.85, 128.80, 113.32, 112.05, 90.73, 55.13, 50.25, 33.07, 32.26, 30.34, 29.26, 27.91, 26.60.8.9. 1,3,8,8-Tetramethyl-5-(p-tolyl)-7,8,9,10-tetrahydropyrimido[4,5-b]quinoline-2,4,6(1H,3H,5H)-trione (4i)
MS 379 (M+1), IR (KBr) 3020, 2950, 1722, 1666, 1559, 1542, 1510, 1470 cm−1. 1H NMR (400 MHz, DMSO-d6): δ (ppm) 7.06 (d, 7H), 6.95 (d, 7H), 4.78 (s, 3H), 3.41 (s, 9H), 3.05 (s, 9H), 2.53 (s, 5H), 2.19 (d, 4H), 2.16 (s, 9H), 2.00 (d, 4H), 1.01 (s, 9H), 0.85 (s, 9H). 13C NMR (101 MHz, DMSO-d6) δ = 195.43, 161.09, 157.64, 150.85, 128.80, 113.32, 112.05, 90.73, 55.13, 50.25, 33.07, 32.26, 30.34, 29.26, 27.91, 26.60.8.10. 5-(3,4-Dimethoxyphenyl)-1,3,8,8-tetramethyl-7,8,9,10-tetrahydropyrimido[4,5-b]quinoline-2,4,6(1H,3H,5H)-trione (4j)
MS 425 (M+1), IR (KBr) 3022, 2970, 1715, 1640, 1556, 1525, 1520, 1460 cm−1. 1H NMR (400 MHz, DMSO-d6): δ (ppm) 6.81 (s, 1H), 6.70 (dd, J = 22.3, 8.2 Hz, 2H), 4.78 (s, 1H), 3.64 (d, J = 7.5 Hz, 7H), 3.40 (s, 3H), 3.07 (s, 3H), 2.53 (d, J = 9.0 Hz, 2H), 2.20 (d, J = 16.2 Hz, 1H), 2.01 (d, J = 16.2 Hz, 1H), 1.01 (s, 3H), 0.88 (s, 3H). 13C NMR (100 MHz, DMSO-d6) δ = 195.70, 161.37, 151.03, 150.16, 148.38, 147.50, 143.98, 139.41, 119.85, 112.24, 111.71, 90.97, 55.80, 50.44, 33.58, 32.43, 30.53, 29.60, 28.14, 26.67.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors Mr KRK and Prof. SGS gratefully acknowledge Dr Babasaheb Ambedkar Marathwada University, Aurangabad (MS), India (STAT/VI/RG/DEPT/2019-20/337-38) and UGC-DST SAP for financial assistance. We are also thankful to the Department of Chemistry, Dr Babasaheb Ambedkar Marathwada University, Aurangabad (MS), India for providing laboratory facilities.References
- A. M. Escobar, G. Blustein, R. Luque and G. P. Romanelli, Catalysts, 2021, 11, 291 CrossRef CAS.
- S. S. Wang and G. Y. Yang, Chem. Rev., 2015, 115, 4893–4962 CrossRef CAS PubMed.
- L. Mouheb, L. Dermeche, N. Essayem and C. Rabia, Catal. Lett., 2020, 150, 3327–3334 CrossRef CAS.
- M. M. Heravi and S. Sadjadi, J. Iran. Chem. Soc., 2009, 6, 1–54 CrossRef CAS.
- I. V. Kozhevnikov, Chem. Rev., 1998, 98, 171–198 CrossRef CAS PubMed.
- M. B. Colombo Migliorero, V. Palermo, E. Alexis Alarcon Durango, A. Luz Villa Holguin, P. Graciela Vazquez, A. Gabriel Sathicq and G. Pablo Romanelli, Lett. Org. Chem., 2018, 15, 826–832 CrossRef CAS.
- A. Popa, V. Sasca and I. Holclajtner-Antunović, Microporous Mesoporous Mater., 2012, 156, 127–137 CrossRef CAS.
- E. Rafiee and M. Kahrizi, S. Afr. J. Chem., 2013, 66, 145–149 CAS.
- S. Soled, S. Miseo, G. McVicker, W. E. Gates, A. Gutierrez and J. Paes, Chem. Eng. J. Biochem. Eng. J., 1996, 64, 247–254 CrossRef CAS.
- K. Y. Lee, S. Oishi, H. Igarashi and M. Misono, Catal. Today, 1997, 33, 183–189 CrossRef CAS.
- X. Li and Y. Zhang, ACS Catal., 2016, 6, 2785–2791 CrossRef CAS.
- L. Zhou, L. Wang, Y. Diao, R. Yan and S. Zhang, Mol. Catal., 2017, 433, 153–161 CrossRef CAS.
- M. J. da Silva, N. P. G. Lopes, S. O. Ferreira, R. C. da Silva, R. Natalino, D. M. Chaves and M. G. Texeira, Chem. Pap., 2021, 75, 153–168 CrossRef CAS.
- N. V. Gromov, T. B. Medvedeva, Y. A. Rodikova, D. E. Babushkin, V. N. Panchenko, M. N. Timofeeva, E. G. Zhizhina, O. P. Taran and V. N. Parmon, RSC Adv., 2020, 10, 28856–28864 RSC.
- H. Firouzabadi and A. A. Jafari, J. Iran. Chem. Soc., 2005, 2, 85–114 CrossRef CAS.
- S. Liu, L. Chen, G. Wang, J. Liu, Y. Gao, C. Li and H. Shan, J. Energy Chem., 2016, 25, 85–92 CrossRef.
- P. Gupta and S. Paul, Catal. Today, 2014, 236, 153–170 CrossRef CAS.
- N. V. Gromov, T. B. Medvedeva, Y. A. Rodikova, D. E. Babushkin, V. N. Panchenko, M. N. Timofeeva, E. G. Zhizhina, O. P. Taran and V. N. Parmon, RSC Adv., 2020, 10, 28856–28864 RSC.
- E. M. Grivsky, S. Lee, C. W. Sigel, D. S. Duch and C. A. Nichol, J. Med. Chem., 1980, 23, 327–329 CrossRef CAS PubMed.
- I. O. Donkor, C. L. Klein, L. Liang, N. Zhu, E. Bradley and A. M. Clark, J. Pharm. Sci., 1995, 84, 661–664 CrossRef CAS PubMed.
- L. R. Bennett, C. J. Blankley, R. W. Fleming, R. D. Smith and D. K. Tessman, J. Med. Chem., 1981, 24, 382–389 CrossRef CAS PubMed.
- H. Hafez, H.-A. Abbas and A.-R. El-Gazzar, Acta Pharm., 2008, 58, 359–378 CAS.
- S. Ravi Kanth, G. Venkat Reddy, K. Hara Kishore, P. Shanthan Rao, B. Narsaiah and U. Surya Narayana Murthy, Eur. J. Med. Chem., 2006, 41, 1011–1016 CrossRef PubMed.
- X. Wang, Z. Zeng, D. Shi, X. Wei and Z. Zong, Synth. Commun., 2004, 34, 4331–4338 CrossRef CAS.
- D. R. Huron, M. E. Gorre, A. J. Kraker, C. L. Sawyers, N. Rosen and M. M. Moasser, Clin. Cancer Res., 2003, 9, 1267–1273 CAS.
- G. Z. Zheng, C. Lee, J. K. Pratt, R. J. Perner, M. Q. Jiang, A. Gomtsyan, M. A. Matulenko, Y. Mao, J. R. Koenig, K. H. Kim, S. Muchmore, H. Yu, K. Kohlhaas, K. M. Alexander, S. McGaraughty, K. L. Chu, C. T. Wismer, J. Mikusa, M. F. Jarvis, K. Marsh, E. A. Kowaluk, S. S. Bhagwat and A. O. Stewart, Bioorg. Med. Chem. Lett., 2001, 11, 2071–2074 CrossRef CAS PubMed.
- A. J. Kraker, B. G. Hartl, A. M. Amar, M. R. Barvian, H. D. H. Showalter and C. W. Moore, Biochem. Pharmacol., 2000, 60, 885–898 CrossRef CAS PubMed.
- A. Agarwal and P. M. S. Chauhan, Tetrahedron Lett., 2005, 46, 1345–1348 CrossRef CAS.
- M. Kajino and K. Meguro, Heterocycles, 1990, 31, 2153 CrossRef CAS.
- F. Nemati and R. Saeedirad, Chin. Chem. Lett., 2013, 24, 370–372 CrossRef CAS.
- S. Verma and S. L. Jain, Tetrahedron Lett., 2012, 53, 2595–2600 CrossRef CAS.
- A. Agarwal and P. M. S. Chauhan, Synth. Commun., 2004, 34, 4447–4461 CrossRef CAS.
- B. Alimadadi, M. Heravi, N. Nazari, H. Abdi Oskooie and F. Bamoharram, Sci. Iran., 2016, 23, 2717–2723 Search PubMed.
- A. B. G. Mohammadi Ziarani, N. Hosseini Nasab, M. Rahimifard and A. Abolhassani Soorki, Sci. Iran., 2015, 22, 2319–2325 Search PubMed.
- B. Mirhosseini-Eshkevari, M. Esnaashari and M. A. Ghasemzadeh, ACS Omega, 2019, 4, 10548–10557 CrossRef CAS PubMed.
- A. Dömling, W. Wang and K. Wang, Chem. Rev., 2012, 112, 3083–3135 CrossRef PubMed.
- M. M. Heravi, F. Derikvand and F. F. Bamoharram, J. Mol. Catal. A: Chem., 2007, 263, 112–114 CrossRef CAS.
- H. G. O. Alvim, G. A. Bataglion, L. M. Ramos, A. L. de Oliveira, H. C. B. de Oliveira, M. N. Eberlin, J. L. de Macedo, W. A. da Silva and B. A. D. Neto, Tetrahedron, 2014, 70, 3306–3313 CrossRef CAS.
- H. G. O. Alvim, J. R. Correa, J. A. F. Assumpção, W. A. da Silva, M. O. Rodrigues, J. L. de Macedo, M. Fioramonte, F. C. Gozzo, C. C. Gatto and B. A. D. Neto, J. Org. Chem., 2018, 83, 4044–4053 CrossRef CAS PubMed.
- G. Raveendra, A. Rajasekhar, M. Srinivas, P. S. Sai Prasad and N. Lingaiah, Appl. Catal., A, 2016, 520, 105–113 CrossRef CAS.
- M. M. Heravi and Z. Faghihi, J. Iran. Chem. Soc., 2014, 11, 209–224 CrossRef CAS.
- C. K. Jadhav, A. S. Nipate, A. V. Chate, V. S. Dofe, J. N. Sangshetti, V. M. Khedkar and C. H. Gill, ACS Omega, 2020, 5, 29055–29067 CrossRef CAS PubMed.
- C. K. Jadhav, A. S. Nipate, A. V. Chate, V. D. Songire, A. P. Patil and C. H. Gill, ACS Omega, 2019, 4, 22313–22324 CrossRef CAS PubMed.
- L. D. Chavan, B. B. Nagolkar, T. K. Chondhekar and S. G. Shankarwar, J. Catal., 2016, 6, 99–105 CAS.
- L. D. Chavan and S. G. Shankarwar, Chin. J. Catal., 2015, 36, 1054–1059 CrossRef CAS.
- D. S. Aher, K. R. Khillare, L. D. Chavan and S. G. Shankarwar, ChemistrySelect, 2020, 5, 7320–7331 CrossRef CAS.
- D. S. Aher, K. R. Khillare, L. D. Chavan and S. G. Shankarwar, RSC Adv., 2021, 11, 2783–2792 RSC.
- D. S. Aher, K. R. Khillare and S. G. Shankarwar, RSC Adv., 2021, 11, 11244–11254 RSC.
- L. D. Chavan, S. N. Deshmukh and S. G. Shankarwar, Orbital, 2019, 11, 314–320 CAS.
- L. D. Chavan, B. B. Nagolkar, T. K. Chondhekar and S. G. Shankarwar, Orbital, 2017, 9, 210–218 CAS.
- N. Essayem, A. Holmqvist, P. Y. Gayraud, J. C. Vedrine and Y. Ben Taarit, J. Catal., 2001, 197, 273–280 CrossRef CAS.
- N. Dimitratos and J. C. Védrine, Appl. Catal., A, 2003, 256, 251–263 CrossRef CAS.
- M. J. da Silva and C. M. de Oliveira, Curr. Catal., 2018, 7, 26–34 CrossRef.
- A. Srikanth, B. Viswanadham, V. P. Kumar, N. R. Anipindi and K. V. R. Chary, Appl. Petrochem. Res., 2016, 6, 145–153 CrossRef CAS.
- X. Li and Y. Zhang, ACS Catal., 2016, 6, 2785–2791 CrossRef CAS.
- A. Akbari, M. Chamack and M. Omidkhah, J. Mater. Sci., 2020, 55, 6513–6524 CrossRef CAS.
- K. Narasimharao, D. Brown, A. Lee, A. Newman, P. Siril, S. Tavener and K. Wilson, J. Catal., 2007, 248, 226–234 CrossRef CAS.
- B. Y. Giri, K. N. Rao, B. L. A. P. Devi, N. Lingaiah, I. Suryanarayana, R. B. N. Prasad and P. S. S. Prasad, Catal. Commun., 2005, 6, 788–792 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental section, FTIR, XRD, EDS, SEM-TEM, BET, ICP-AES, and NMR. See DOI: 10.1039/d1ra05190c |
| This journal is © The Royal Society of Chemistry 2021 |