
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA novel Au/electroactive poly(amic acid) composite as an effective catalyst for p-nitrophenol reduction†
Guan-Hui Lai a,
Tsao-Cheng Huangb,
Bi-Sheng Huanga and
Yi-Chen Chou*a
a,
Tsao-Cheng Huangb,
Bi-Sheng Huanga and
Yi-Chen Chou*a
aDepartment of Cosmetic Science, Providence University, 43301 Taichung, Taiwan. E-mail: jacs1yichen@pu.edu.tw
bTechnical Department Plastics Division, Formosa Plastics Corporation, 814538 Kaohsiung, Taiwan
First published on 20th October 2021
Abstract
A Au/electroactive poly(amic acid) (Au/EPAA) composite was synthesized and characterized, and its catalytic ability was evaluated. EPAA was synthesized via oxidative coupling polymerization and Au nanoparticles were anchored to the amino and carboxyl groups. The Au/EPAA composite was characterized via X-ray diffraction analysis, X-ray photoelectron spectroscopy, and scanning electron microscopy, which confirmed that the Au nanoparticles were well dispersed on the EPAA surface. p-Nitrophenol was reduced to p-aminophenol within 5 min at room temperature, with a rate constant of 0.84 min−1. Cycling measurements showed that the Au/EPAA composite achieved higher than 92% conversion. The Au/EPAA composite showed excellent performance and stability as a catalyst for the reduction of p-nitrophenol to p-aminophenol.
1 Introduction
Increasing amounts of organic compounds are used in industry and produced daily; hence, water, as a commonly used solvent containing untreated organic agents or byproducts, is also generated continuously. Thus, proper treatment of wastewater has become an important issue. Heterogeneous catalysts based on metal nanoparticles (NPs) are promising tools for wastewater treatment. Typically, metal NPs are loaded onto a support such as carbon nanotube,1 graphene,2 boron nitride,3 clay,4 metal oxides,5 or polymers6 to prevent aggregation. Thus, improving the dispersion stability of metal NPs on a support is critical for the design and synthesis of high-performance heterogeneous catalysts.Polymers are among the most popular catalyst support materials as they can stabilize metal NPs to prevent aggregation.7,8 For example, Zhang et al. used an in situ method to prepare sulfhydryl-functionalized poly(glycidyl methacrylate) microspheres with surface sulfhydryl groups to adsorb Ag NPs. The sulfhydryl functional groups improved the monodispersity and recyclability of the Ag NPs in the catalyst system.9 Liu et al. anchored Au NPs on thin carbon sheets to prepare highly stable and efficient heterogeneous catalysts. The Au/carbon sheets were synthesized using sol–gel, calcination, and selective etching methods and featured many amino functional groups anchoring Au NPs stably on the surface.10 Zhang et al. fabricated porous Au/polyethyleneimine/graphene oxide composites via an in situ deposition and freeze-drying method. Polyethyleneimine functioned not only as a reducing and protective agent for the Au NPs, but also as a reducing agent and surface modifier for graphene oxide. The resulting porous functional composite exhibited excellent catalytic activity for the reduction of p-nitrophenol (PNP).11 In addition, Li has reported that dopamine-terminated poly(N-isopropylacrylamide)-modified carbon dots can easily anchor Au NPs on the material surface.12 Functional groups with a lone pair, such as –NH, –SH, –COOH, and –OH groups, can effectively adsorb metal NPs on a carrier uniformly without agglomeration, but material synthesis requires several complex steps, making it expensive, time-consuming, and highly energy-consuming.6,13,14
Polyaniline (PANI) is used as a polymer support for Au NPs because it is relatively cheap, easy to synthesize, and effective for immobilization of Au NPs and has special doping/dedoping properties and conductivity.15 However, it is difficult to dissolve in organic solvents, which limits its application. Thus, aniline oligomers and their derivative polymers have attracted attention because of their good solubility, mechanical strength, and biodegradability. Kaner et al. have demonstrated that tetraaniline is the smallest oligomer unit that can fully represent the structure of PANI, and single crystals of doped tetraaniline with different shapes can be produced through a simple self-assembly process.16 Chao and Berda synthesized a series of electroactive polymers and showed that their electrochromic behaviors can be controlled by adjusting the pH and the applied potential.17 Wang and co-workers prepared electroactive polymers containing aniline oligomers in the main and side chains, which exhibited electrochemical, dielectric, and photoresponsive properties.18,19 Huang and co-workers investigated the anti-corrosion properties of electroactive polymers and attributed them to the passive metal oxide layers on the metal electrode surface induced by the redox catalytic capabilities of the aniline oligomer units in the electroactive polymer.20–22 In addition, electroactive polymers have been utilized as sensing materials because of their unique properties, such as their acid–base properties, doping-dedoping chemistry, and redox behavior.23–25
In our previous studies, aniline oligomers and electroactive polymers incorporating graphene oxide, SiO2, Au NPs, or carbon nanotubes were used as catalysts,26 anti-corrosion coating materials,27 and chemical sensors.28,29 Poly(amic acid) (PAA), containing amide and carboxyl groups, is a typical precursor for polyimides. In this study, we synthesized electroactive PAA (EPAA) via oxidative coupling polymerization and anchored Au NPs directly on it via the amino and carboxyl groups in the polymer chain. This Au/EPAA composite was used to reduce PNP, one of the water pollutants prioritized by the US Environmental Protection Agency.30 PNP is toxic to humans and animals, whereas the reduction product, p-aminophenol (PAP), is an important intermediate of paracetamol.31
2 Experimental
2.1 Materials
4,4′-Oxydiphthalic anhydride (ODPA, 98%, TCI), N-phenyl-p-phenylenediamine (98%, Aldrich), 1,4-phenylenediamine (99%, Aldrich), ammonium persulfate (APS, Aldrich), N-methyl-2-pyrrolidone (NMP, Tedia), dichloromethane (Tedia), hydrochloric acid (HCl, Showa), ammonium hydroxide (Showa), hydrazine (Acros), tetrachloroauric(III) acid trihydrate (Acros), sodium citrate (Aldrich), PNP (Aldrich), and sodium borohydride (NaBH4, Aldrich) were obtained from various sources.2.2 Synthesis of EPAA
 | ||
| Fig. 1 Synthesis of EPAA. | ||
2.3 Synthesis of Au/EPAA composites
The Au NPs were prepared using the thermal reflux method. A solution of 0.0394 g (1.0 mM) of tetrachloroauric(III) acid trihydrate in 100 mL of DI water was heated, and 0.1141 g (38.8 mM) of sodium citrate was dissolved in 10 mL of DI water. After the solution had boiled, the sodium citrate solution was added and the resulting mixture was stirred for 10 min to obtain a solution of Au NPs. The Au/EPAA composite was prepared by dispersing 10 mg of EPAA in 50 mL of the Au NP solution and stirring overnight. The Au/EPAA composites were recovered via centrifugation at 6000 rpm for 10 min and washed three times with DI water.2.4 Catalytic activity of the Au/EPAA composite
The catalytic properties of the Au/EPAA composite were monitored through ultraviolet-visible (UV-vis) spectroscopy via the conversion of PNP to PAP using NaBH4 as a reductant. In a quartz cell, 3.0 mL of a PNP solution (1.0 mM) was mixed with 0.30 mL of a NaBH4 solution (1.0 M), and the color of the solution changed from light yellow to dark yellow. The solution became colorless with the addition of 1.0 mg of the Au/EPAA composite.2.5 Characterization
Liquid chromatography-mass spectra were obtained using a triple quadrupole mass spectrometer (TSQ Quantum) with an electrospray ionization source. The chemical structures of the oligomer and EPAA were determined via 1H nuclear magnetic resonance (NMR) spectroscopy using a Bruker 400 spectrometer with deuterated dimethyl sulfoxide (DMSO-d6) as the solvent. The weight average molecular weight (Mw) and number average molecular weight (Mn) were determined through gel permeation chromatography (GPC) using a Waters-150CV pump and a differential refractometer. Fourier-transform infrared (FTIR) spectroscopy was performed using a Jasco FT/IR-4600 spectrometer at room temperature with a resolution of 4 cm−1 and 64 scans. UV-vis spectroscopy was performed on a Jasco V-750 UV-visible spectrophotometer. Electrochemical experiments were performed on a CHI 6273E electrochemical analyzer using a Ag/AgCl reference electrode and Pt counter electrode at a scan rate of 50 mV s−1. Thermogravimetric analysis (TGA) was conducted on a TA Instruments Q500 from 50 to 800 °C in air at a heating rate of 20 °C min−1. X-ray diffraction (XRD) measurements were performed with a PANalytical X'Pert 3 powder diffractometer to investigate the crystalline structure of the Au NPs. X-ray photoelectron spectroscopy (XPS) was performed using a PHI 1600 ESCA spectrometer (Physical Electronics, Inc.). A JEOL JSM-7100F scanning electron microscope and JEM-1400 transmission electron microscope were employed to investigate the morphologies and structures of the typical products.3 Results and discussion
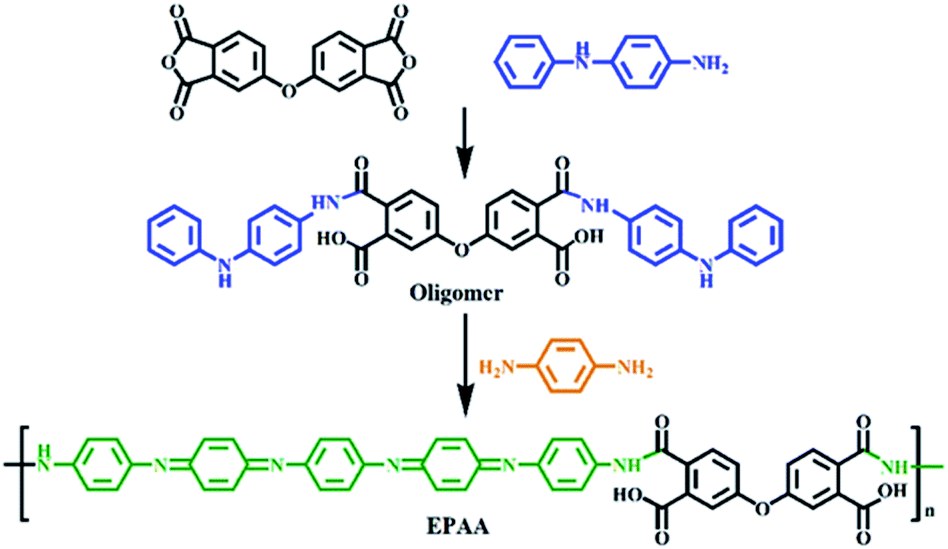
Fig. 2(a) shows the FTIR spectra of the oligomer and EPAA between 4000 and 600 cm−1; the characteristic N–H and O–H peaks were observed at 3500–3200 cm−1. The peaks at 1711, 1311, and 744 cm−1 correspond to C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O symmetric stretching, C–N bending, and C–H bending, respectively.32 The peaks at 1589 and 1509 cm−1 correspond to NQN and NBN bonding, where Q represents the quinoid ring and B represents the benzene ring structure.33,34 The chemical oxidation of EPAA is shown in Fig. 2(b). The obtained EPAA (0.4 g) was dispersed in a solution of 4 mL of hydrazine hydrate in 40 mL of 1.0 M ammonium hydroxide and the resulting mixture was stirred for 10 h. The reaction mixture was then filtered, and the residue was washed with distilled water several times and dried under dynamic vacuum at 40 °C for 24 h. Finally, EPAA was reduced to the leucoemeraldine oxidation state (0.34 g, 85%). The leucoemeraldine base (LB) form of EPAA was dissolved in NMP. Subsequently, trace amounts of the oxidant, (NH4)2S2O8, were gradually introduced into the EPAA solution for in situ monitoring of the sequential oxidation of the conjugated aniline pentamer components through UV-vis absorption spectroscopy. The LB form gave only one absorption peak at 310 nm, which is associated with the π–π* transition of the conjugated ring system.35 During oxidation, this absorption peak of EPAA was blue-shifted to 297 nm. A new absorption peak was observed at 591 nm, which was assigned to the benzenoid-to-quinoid excitonic transition.36 Cyclic voltammetry (CV) studies have been used to examine the redox properties of electroactive polymers. In this study, EPAA was examined using CV with a typical three-electrode electrochemical cell. As shown in Fig. 2(c), the CV curve shows redox peaks for EPAA at 0.37, 0.51, and 0.70 V.37 Fig. 2(d) shows that EPAA starts in the LB state containing a benzene ring, and is oxidized to emeraldine base I (EB I), then to emeraldine base II (EB II), and finally to the pernigraniline base (PNB), with the quinoid ring peak (591 nm) growing until equilibrium is reached.
O symmetric stretching, C–N bending, and C–H bending, respectively.32 The peaks at 1589 and 1509 cm−1 correspond to NQN and NBN bonding, where Q represents the quinoid ring and B represents the benzene ring structure.33,34 The chemical oxidation of EPAA is shown in Fig. 2(b). The obtained EPAA (0.4 g) was dispersed in a solution of 4 mL of hydrazine hydrate in 40 mL of 1.0 M ammonium hydroxide and the resulting mixture was stirred for 10 h. The reaction mixture was then filtered, and the residue was washed with distilled water several times and dried under dynamic vacuum at 40 °C for 24 h. Finally, EPAA was reduced to the leucoemeraldine oxidation state (0.34 g, 85%). The leucoemeraldine base (LB) form of EPAA was dissolved in NMP. Subsequently, trace amounts of the oxidant, (NH4)2S2O8, were gradually introduced into the EPAA solution for in situ monitoring of the sequential oxidation of the conjugated aniline pentamer components through UV-vis absorption spectroscopy. The LB form gave only one absorption peak at 310 nm, which is associated with the π–π* transition of the conjugated ring system.35 During oxidation, this absorption peak of EPAA was blue-shifted to 297 nm. A new absorption peak was observed at 591 nm, which was assigned to the benzenoid-to-quinoid excitonic transition.36 Cyclic voltammetry (CV) studies have been used to examine the redox properties of electroactive polymers. In this study, EPAA was examined using CV with a typical three-electrode electrochemical cell. As shown in Fig. 2(c), the CV curve shows redox peaks for EPAA at 0.37, 0.51, and 0.70 V.37 Fig. 2(d) shows that EPAA starts in the LB state containing a benzene ring, and is oxidized to emeraldine base I (EB I), then to emeraldine base II (EB II), and finally to the pernigraniline base (PNB), with the quinoid ring peak (591 nm) growing until equilibrium is reached.
 | ||
| Fig. 2 (a) FTIR spectra of oligomer and EPAA. (b) UV-vis spectra monitoring chemical oxidation of EPAA. (c) CV measurements for EPAA. (d) Redox states of EPAA. | ||
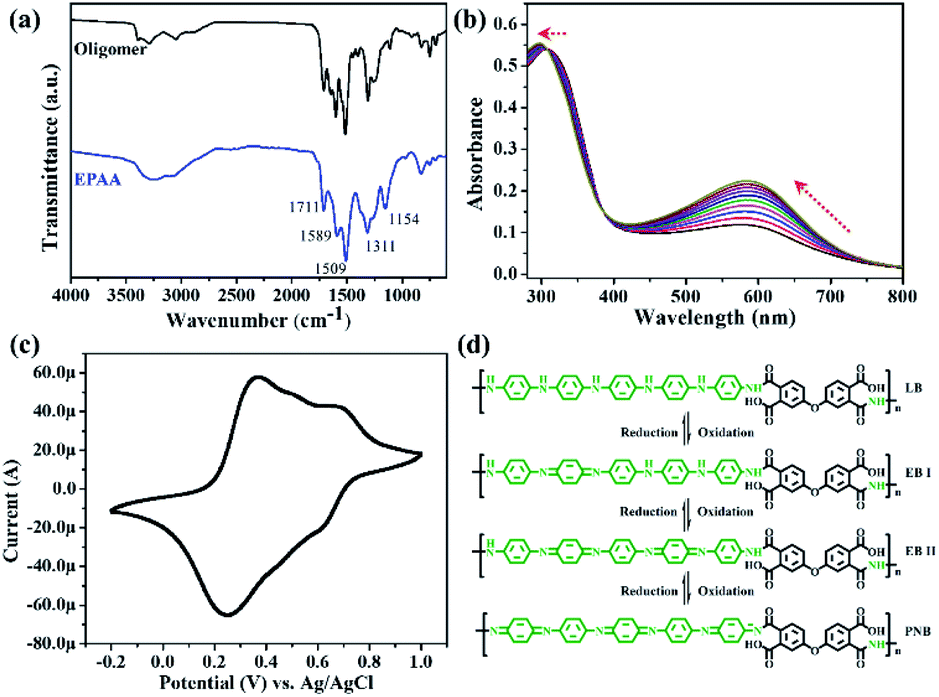
When gold is nanometer-sized, there is a special plasma resonance effect.38 The peak at 523 nm in the UV-vis spectrum corresponded to Au NPs approximately 15 nm in size,39 as shown in Fig. 3(a). The morphologies of the Au NPs were examined through their transmission electron microscopy (TEM) images, and their diameter was 15.8 ± 1.7 nm (Fig. S4†). After the Au NPs were adsorbed on the EPAA surface to afford the Au/EPAA composite, the peak remained at 524 nm. The crystallinities of EPAA and the Au/EPAA composite were investigated using XRD, and the results are shown in Fig. 3(b). The broad peak at 20.5° indicates the amorphous phase in the EPAA polymer. For the Au/EPAA composite, the diffraction peaks at 38.2°, 44.4°, 64.8°, 77.7°, and 81.8° can be ascribed to the (111), (200), (220), (311), and (222) crystal planes of Au, respectively.40–42 The XPS survey spectra of EPAA and the Au/EPAA composite are shown in Fig. 3(c). EPAA gave significantly intense C 1s, N 1s, and O 1s peaks at 284.30, 399.81, and 531.43 eV, respectively.43 After the adsorption of Au NPs, the Au/EPAA composite gave an additional Au 4f peak, which was attributed to the anchoring of Au NPs on EPAA, confirming successful adsorption. The peaks at binding energies of 86.79 and 82.99 eV in Fig. 3(d) can be attributed to Au 4f7/2 and Au 4f5/2, respectively, confirming the loading of Au NPs on the surface of EPAA.44
 | ||
| Fig. 3 (a) UV-vis spectra of Au and Au/EPAA composite. (b) XRD patterns of EPAA and Au/EPAA composite. (c) XPS spectra of EPAA and the Au/EPAA composite; (d) high-resolution Au 4f XPS spectrum of the Au/EPAA composite. | ||
The morphologies of EPAA and the Au/EPAA composites are shown in Fig. 4(a–c). Au NPs can be clearly observed in the image of the Au/EPAA composite, but not in that of EPAA, which showed that the Au NPs were well dispersed. The TEM image of the Au/EPAA composite shows that the Au NPs are well dispersed on the EPAA surface with an average size of 16.8 ± 1.3 nm (Fig. 4(c)). Fig. 4(d) displays the TGA results for the Au/EPAA composite. To the best of our knowledge, the polymer decomposes into CO and CO2 when heated to 800 °C in air. With the anchored Au NPs, the combustion residue of the Au/EPAA composite had 16% of the initial weight owing to the Au NP content.
 | ||
| Fig. 4 Scanning electron microscopy (SEM) images of (a) EPAA and (b) Au/EPAA; the insert shows the energy dispersive X-ray spectroscopy (EDS) spectrum of the Au/EPAA composite. (c) TEM image of Au/EPAA; the insert shows the particle size distribution. (d) TGA results for the Au/EPAA composite. | ||
The catalytic activity of the Au/EPAA composite for the reduction of PNP to PAP with NaBH4 was evaluated. When NaBH4 was added, the peak for PNP shifted from 317 to 400 nm, indicating the formation of p-nitrophenolate ions, as shown in Fig. 5(a). This reaction did not occur in the absence of the catalyst. Fig. 5(b) displays the typical UV-spectra at 300 nm,45 which show the formation of PAP from the reduction of the p-nitrophenolate ion.46 After the Au/EPAA composite was added, the solution changed from dark yellow to colorless, and the reduction reaction was completed within 270 s without ultrasound or stirring. This behavior was expected because the Au NPs in the Au/EPAA composites adsorb hydrogen from NaBH4 and efficiently release it via reduction. Hence, Au NPs act as hydrogen carriers between NaBH4 and PNP. Since the concentration of NaBH4 exceeded that of PNP, this reduction reaction can be assumed to be a pseudo-first-order reaction that can be described as follows:47
| ln(Ct/C0) = −kt |
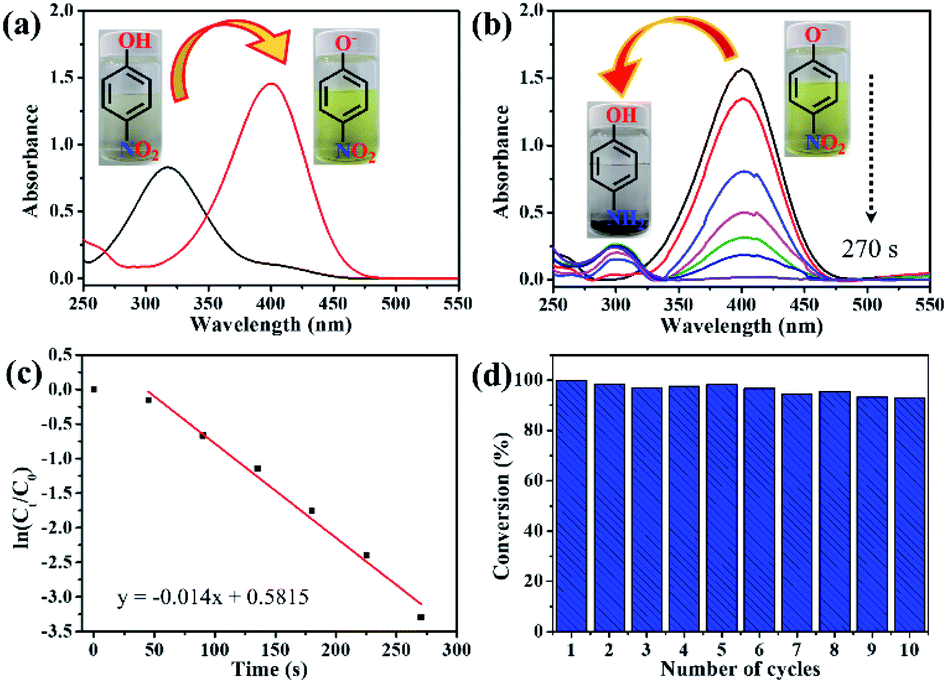 | ||
| Fig. 5 UV-vis spectra of (a) PNP before and after adding NaBH4 and (b) reduction of PNP with NaBH4 using Au/EPAA composite. (c) Linear relationship between ln(Ct/C0) and reaction time with Au/EPAA composite as a catalyst. (d) Cycling performance of Au/EPAA composite for PNP reduction. | ||
| Catalyst | Weight (mg) | Reduction time (s) | Rate constant (min−1) | 4-NP![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :NaBH4 :NaBH4 |
Conversion | Ref. |
|---|---|---|---|---|---|---|
| a S-g-C3N4: sulfur doped graphitic carbon nitride.b HMSC: hollow microsphere supported.c RGO: reduced graphene oxide.d NC: nanoporous carbon. | ||||||
| Fe3O4–Cu | 0.2 | 15 | NA | 1:40 |
90%, 6 times | 50 |
| Fe3O4–Au | NA | 120 | 1.82 | 1:40 |
>90% | 51 |
| Au/EPAA | 1.0 | 270 | 0.84 | 1:100 |
>92%, 10 times | This work |
| Au@S-g-C3N4a | 2.5 | 300 | 0.75 | 1:100 |
95%, 5 times | 52 |
| PdO/ZnO | 1 | 360 | 0.37 | 1:100 |
>93%, 5 times | 49 |
| HMSCb–NiCoP | 100 | 585 | 2.24 | 1:100 |
97%, 6 times | 53 |
| PtNi/RGOc | 0.05 | 600 | 6.48 | 1:2380 |
86%, 5 times | 54 |
| Ag@NCd | 1 | 840 | 0.25 | 1:1250 |
>90%, 5 times | 55 |
The mechanism for the reduction of PNP using the Au/EPAA composite can be described using the Langmuir–Hinshelwood model.47–49 The PNP and BH4− ions are first adsorbed on the surface of the Au/EPAA composite, where BH4− acts as a hydrogen source. Then, the p-nitrophenolate ion is converted into a p-aminophenolate ion via the transfer of hydrogen and electrons. Finally, PAP is desorbed from the surface of the Au/EPAA composite, as shown in Fig. 6. At the same time, the Au/EPAA composite returns to its initial state to maintain stability.
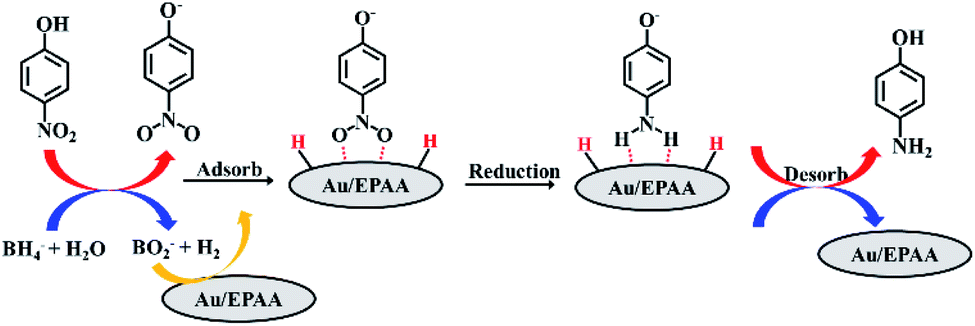 | ||
| Fig. 6 Mechanism for reduction of PNP. | ||
4 Conclusions
In conclusion, a new Au/EPAA composite was synthesized to catalyze the reduction of PNP to PAP. Au NPs were anchored and well dispersed on the EPAA surface via the carboxyl and amino groups of EPAA. This Au/EPAA composite exhibited excellent catalytic properties for the reduction of PNP, with a rate constant of 0.84 min−1. After ten cycles, the conversion with Au/EPAA was still higher than 92%. The Au/EPAA composite exhibited high catalytic activity; hence, it has potential for new applications.Author contributions
Guan-Hui Lai: conceptualization, methodology, investigation, data curation, writing – original draft. Tsao-Cheng Huang: methodology, supervision, writing – review & editing. Bi-Sheng Huang: investigation, data curation. Yi-Chen Chou: writing – review & editing, supervision, project administration, funding acquisition.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors would like to thank the Ministry of Science and Technology, Taiwan under grant number MOST 109-2637-E-241-004, and MOST 110-2637-E-241-003 for their financial support. Special thanks are given to the National Chung Hsing University (MOST 108-2731-M-005-001) for the LC-Mass, SEM, TEM, and XPS support.References
- X. Gu, W. Qi, X. Xu, Z. Sun, L. Zhang, W. Liu, X. Pan and D. Su, Nanoscale, 2014, 6, 6609–6616 RSC.
- X.-Y. Zhu, Z.-S. Lv, J.-J. Feng, P.-X. Yuan, L. Zhang, J.-R. Chen and A.-J. Wang, J. Colloid Interface Sci., 2018, 516, 355–363 CrossRef CAS PubMed.
- B. Yu, B. Han, X. Jiang, C. Zhou, K. Xia, Q. Gao and J. Wu, J. Phys. Chem. C, 2019, 123, 10389–10397 CrossRef CAS.
- G.-H. Lai, T.-C. Huang, Y.-H. Pai, B.-S. Huang, M.-H. Tsai, T.-I. Yang and Y.-H. Chung, J. Taiwan Inst. Chem. Eng., 2019, 95, 525–531 CrossRef CAS.
- Y. Xu, X. Shi, R. Hua, R. Zhang, Y. Yao, B. Zhao, T. Liu, J. Zheng and G. Lu, Appl. Catal., B, 2020, 260, 118142 CrossRef CAS.
- B. Hao, G. Lu, S. Zhang, Y. Li, A. Ding and X. Huang, Polym. Chem., 2020, 11, 4094–4104 RSC.
- Q. Geng and J. Du, RSC Adv., 2014, 4, 16425–16428 RSC.
- R. Rarima and G. Unnikrishnan, Mater. Chem. Phys., 2020, 241, 122389 CrossRef.
- W. Zhang, Y. Sun and L. Zhang, Ind. Eng. Chem. Res., 2015, 54, 6480–6488 CrossRef CAS.
- X. Liu, X. Cui, Y. Liu and Y. Yin, Nanoscale, 2015, 7, 18320–18326 RSC.
- P. Deng, Z. Xu, Y. Feng and J. Li, Sens. Actuators, B, 2012, 168, 381–389 CrossRef CAS.
- L. Li, T. Zhang, J. Lü and C. Lü, Appl. Surf. Sci., 2018, 454, 181–191 CrossRef CAS.
- J. Zhou, C. Zhang and Y. Wang, Polym. Chem., 2019, 10, 1642–1649 RSC.
- G. Wu, X. Liu, P. Zhou, L. Wang, M. Hegazy, X. Huang and Y. Huang, Mater. Sci. Eng., C, 2019, 94, 524–533 CrossRef CAS PubMed.
- X. Feng, C. Mao, G. Yang, W. Hou and J.-J. Zhu, Langmuir, 2006, 22, 4384–4389 CrossRef CAS PubMed.
- Y. Wang, H. D. Tran, L. Liao, X. Duan and R. B. Kaner, J. Am. Chem. Soc., 2010, 132, 10365–10373 CrossRef CAS PubMed.
- Y. Wang, X. Jia, E. B. Berda, J. Zhao, X. Liu and D. Chao, Eur. Polym. J., 2020, 138, 109979 CrossRef CAS.
- Y. Yan, N. Sun, X. Jia, X. Liu, C. Wang and D. Chao, Polymer, 2018, 134, 1–7 CrossRef CAS.
- Y. Yan, N. Sun, F. Li, X. Jia, C. Wang and D. Chao, ACS Appl. Mater. Interfaces, 2017, 9, 6497–6503 CrossRef CAS PubMed.
- H.-Y. Huang, T.-C. Huang, J.-C. Lin, J.-H. Chang, Y.-T. Lee and J.-M. Yeh, Mater. Chem. Phys., 2013, 137, 772–780 CrossRef CAS.
- T.-C. Yeh, T.-C. Huang, H.-Y. Huang, Y.-P. Huang, Y.-T. Cai, S.-T. Lin, Y. Wei and J.-M. Yeh, Polym. Chem., 2012, 3, 2209–2216 RSC.
- T. C. Huang, J. M. Yeh and C. Y. Lai, in Advances in Polymer Nanocomposites, ed. F. Gao, Woodhead Publishing, 2012, pp. 605–638 Search PubMed.
- T. C. Huang, L. C. Yeh, G. H. Lai, F. Y. Lai, T. I. Yang, Y. J. Huang, A. Y. Lo and J. M. Yeh, eXPRESS Polym. Lett., 2016, 10, 450–461 CrossRef CAS.
- L.-C. Yeh, T.-C. Huang, Y.-P. Huang, H.-Y. Huang, H.-H. Chen, T.-I. Yang and J.-M. Yeh, Electrochim. Acta, 2013, 94, 300–306 CrossRef CAS.
- L.-C. Yeh, T.-C. Huang, F.-Y. Lai, G.-H. Lai, A.-Y. Lo, S.-C. Hsu, T.-I. Yang and J.-M. Yeh, Surf. Coat. Technol., 2016, 303, 154–161 CrossRef CAS.
- G.-H. Lai, Y.-C. Chou, B.-S. Huang, T.-I. Yang and M.-H. Tsai, RSC Adv., 2021, 11, 71–77 RSC.
- Y.-C. Chou, P.-C. Lee, T.-F. Hsu, W.-Y. Huang, L. Zi-Han, C.-Y. Chuang, T.-I. Yang and J.-M. Yeh, Polym. Compos., 2014, 35, 617–625 CrossRef CAS.
- T.-C. Huang, L.-C. Yeh, H.-Y. Huang, Z.-Y. Nian, Y.-C. Yeh, Y.-C. Chou, J.-M. Yeh and M.-H. Tsai, Polym. Chem., 2014, 5, 630–637 RSC.
- M. H. Tsai, S. H. Lu, Y. H. Lai, G. H. Lai, G. V. Dizon, T. I. Yang, Y. J. Lin and Y. C. Chou, eXPRESS Polym. Lett., 2018, 12, 71–81 CrossRef CAS.
- S. A. Hira, M. Nallal and K. H. Park, Sens. Actuators, B, 2019, 298, 126861 CrossRef CAS.
- M. T. Islam, N. Dominguez, M. A. Ahsan, H. Dominguez-Cisneros, P. Zuniga, P. J. J. Alvarez and J. C. Noveron, J. Environ. Chem. Eng., 2017, 5, 4185–4193 CrossRef CAS.
- T.-C. Huang, S.-T. Lin, L.-C. Yeh, C.-A. Chen, H.-Y. Huang, Z.-Y. Nian, H.-H. Chen and J.-M. Yeh, Polymer, 2012, 53, 4373–4379 CrossRef CAS.
- T.-C. Huang, L.-C. Yeh, G.-H. Lai, B.-S. Huang, T.-I. Yang, S.-C. Hsu, A.-Y. Lo and J.-M. Yeh, Int. J. Green Energy, 2017, 14, 113–120 CrossRef CAS.
- H.-Y. Huang, J.-W. Jian, Y.-T. Lee, Y.-T. Li, T.-C. Huang, J.-H. Chang, L.-C. Yeh and J.-M. Yeh, Polymer, 2012, 53, 4967–4976 CrossRef CAS.
- R. Yang, D. Chao, H. Liu, E. B. Berda, S. Wang, X. Jia and C. Wang, Electrochim. Acta, 2013, 93, 107–113 CrossRef CAS.
- T.-C. Huang, T.-C. Yeh, H.-Y. Huang, W.-F. Ji, T.-C. Lin, C.-A. Chen, T.-I. Yang and J.-M. Yeh, Electrochim. Acta, 2012, 63, 185–191 CrossRef CAS.
- T.-C. Huang, T.-C. Yeh, H.-Y. Huang, W.-F. Ji, Y.-C. Chou, W.-I. Hung, J.-M. Yeh and M.-H. Tsai, Electrochim. Acta, 2011, 56, 10151–10158 CrossRef CAS.
- T. Wu, Y. Kou, H. Zheng, J. Lu, N. R. Kadasala, S. Yang, C. Guo, Y. Liu and M. Gao, Nanomaterials, 2020, 10, 48 CrossRef CAS PubMed.
- N. G. Bastús, J. Comenge and V. Puntes, Langmuir, 2011, 27, 11098–11105 CrossRef PubMed.
- S. S. Kumar, C. S. Kumar, J. Mathiyarasu and K. L. Phani, Langmuir, 2007, 23, 3401–3408 CrossRef CAS PubMed.
- T. Wu, H. Zheng, Y. Kou, X. Su, N. R. Kadasala, M. Gao, L. Chen, D. Han, Y. Liu and J. Yang, Microsyst. Nanoeng., 2021, 7, 23 CrossRef CAS PubMed.
- J.-M. Yeh, K.-Y. Huang, S.-Y. Lin, Y.-Y. Wu, C.-C. Huang and S.-J. Liou, J. Nanotechnol., 2009, 2009, 217469 Search PubMed.
- H. Yao, T.-C. Huang and H.-J. Sue, RSC Adv., 2014, 4, 61823–61830 RSC.
- M.-Q. Yang, X. Pan, N. Zhang and Y.-J. Xu, CrystEngComm, 2013, 15, 6819–6828 RSC.
- H. Zheng, J. Huang, T. Zhou, Y. Jiang, Y. Jiang, M. Gao and Y. Liu, Catalysts, 2020, 10, 1437 CrossRef CAS.
- Q. Cheng, Q. Li, Z. Yuan, S. Li, J. H. Xin and D. Ye, ACS Appl. Mater. Interfaces, 2021, 13, 4410–4418 CrossRef CAS PubMed.
- S. K. Ghosh, M. Mandal, S. Kundu, S. Nath and T. Pal, Appl. Catal., A, 2004, 268, 61–66 CrossRef CAS.
- L. Qin, Z. Zeng, G. Zeng, C. Lai, A. Duan, R. Xiao, D. Huang, Y. Fu, H. Yi, B. Li, X. Liu, S. Liu, M. Zhang and D. Jiang, Appl. Catal., B, 2019, 259, 118035 CrossRef CAS.
- M. Liu, F. Cui, Q. Ma, L. Xu, J. Zhang, R. Zhang and T. Cui, New J. Chem., 2020, 44, 4042–4048 RSC.
- W. Zhao, S. Yang, C. Guo, J. Yang and Y. Liu, Mater. Chem. Phys., 2021, 260, 124144 CrossRef CAS.
- Y. Chen, T. Wu, G. Xing, Y. Kou, B. Li, X. Wang, M. Gao, L. Chen, Y. Wang, J. Yang, Y. Liu, Y. Zhang and D. Wang, Ind. Eng. Chem. Res., 2019, 58, 15151–15161 CrossRef CAS.
- V. Balakumar, H. Kim, J. W. Ryu, R. Manivannan and Y.-A. Son, J. Mater. Sci. Technol., 2020, 40, 176–184 CrossRef.
- G. Du, B. Liao, R. Liu, Z. An and J. Zhang, RSC Adv., 2020, 10, 35287–35294 RSC.
- P. Song, J.-J. Feng, S.-X. Zhong, S.-S. Huang, J.-R. Chen and A.-J. Wang, RSC Adv., 2015, 5, 35551–35557 RSC.
- V. Veeramani, N. Van Chi, Y.-L. Yang, N. T. Hong Huong, T. Van Tran, T. Ahamad, S. M. Alshehri and K. C. W. Wu, RSC Adv., 2021, 11, 6614–6619 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1ra05347g |
| This journal is © The Royal Society of Chemistry 2021 |