
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
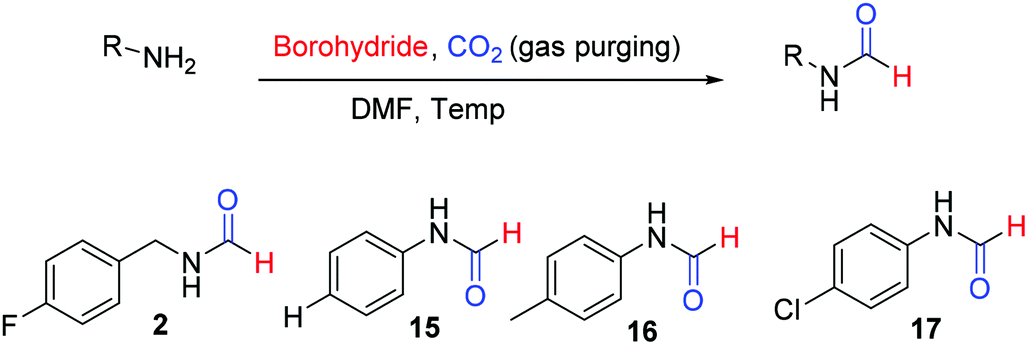
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceCatalyst free N-formylation of aromatic and aliphatic amines exploiting reductive formylation of CO2 using NaBH4†
Arun Kumar,
Pankaj Sharma,
Nidhi Sharma,
Yashwant Kumar and
Dinesh Mahajan *
*
Medicinal Chemistry and Pharmacology Lab, Translational Health Science and Technology Institute (THSTI), NCR Biotech Science Cluster, 3rd Milestone, Faridabad-Gurgaon Expressway, Faridabad-121001, India. E-mail: dinesh.mahajan@thsti.res.in; chemidinesh@gmail.com
First published on 27th July 2021
Abstract
Herein, we report a sustainable approach for N-formylation of aromatic as well as aliphatic amines using sodium borohydride and carbon dioxide gas. The developed approach is catalyst free, and does not need pressure or a specialized reaction assembly. The reductive formylation of CO2 with sodium borohydride generates formoxy borohydride species in situ, as confirmed by 1H and 11B NMR spectroscopy. The in situ formation of formoxy borohydride species is prominent in formamide based solvents and is critical for the success of the N-formylation reactions. The formoxy borohydride is also found to promote transamidation reactions as a competitive pathway along with reductive functionalization of CO2 with amine leading to N-formylation of amines.
Introduction
Formamides or N-formyls represent an important class of compounds in synthetic organic chemistry. Formamides are key intermediates for the synthesis of functional or biologically active molecules such as approved drugs1–3 and insecticides4 (Fig. 1). Formamides hold promise as solvents and organocatalysts5,6 and are the valuable raw material for various heterocycles,7–9 formamidines,10,11 as well as isocyanides.12,13 Formamides can also be exploited as surrogates of isocyanates for the synthesis of ureas and carbamates.14 The wide usage in synthetic organic chemistry as well as commercial application of N-formyls led to significant development for their synthesis. There is considerable interest in the synthesis of N-formyls directly from feedstock chemicals with high atom and economic efficiency. Methods have been developed where methanol,15–17 CO![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 18 and CO219–28 have been exploited as a formyl surrogate employing various catalysts including transition metals. Carbon dioxide (CO2) is a notorious greenhouse gas but an inexpensive, non-toxic, and abundant source of carbon in synthetic organic chemistry. Energy-efficient, green transformation or utilization of CO2 into value-added chemicals is a technical challenge of modern times and a solution to this will have a significant impact. CO2 gas can be a cost-efficient environment friendly raw material of the future if we develop energy and cost-efficient synthetic transformations to use it as a building block.
18 and CO219–28 have been exploited as a formyl surrogate employing various catalysts including transition metals. Carbon dioxide (CO2) is a notorious greenhouse gas but an inexpensive, non-toxic, and abundant source of carbon in synthetic organic chemistry. Energy-efficient, green transformation or utilization of CO2 into value-added chemicals is a technical challenge of modern times and a solution to this will have a significant impact. CO2 gas can be a cost-efficient environment friendly raw material of the future if we develop energy and cost-efficient synthetic transformations to use it as a building block.
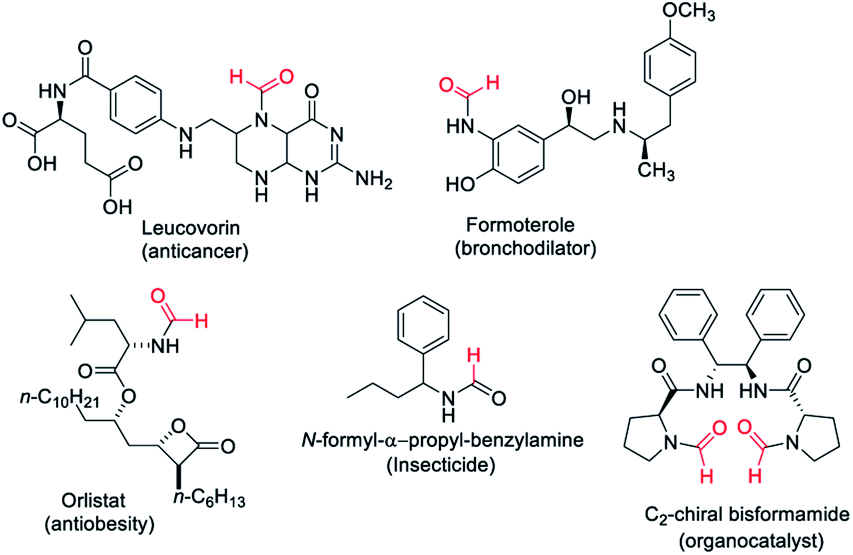 | ||
| Fig. 1 Examples of N-formyl containing molecules with their applications. | ||
There are many synthetic transformations reported in literature where CO2 has been exploited as a facile C1 source in the lab as well as at industrial scale.29–32 Recently, CO2 has been used as a formyl surrogate for the synthesis of N-formyls in the presence of a reductant.19–28 However, most of these reported methodologies require expensive or complex metal based catalysts20–22,24,26–28 as well as ligands along with hydrogen gas or hydrosilanes19,25 as a source of hydride under high temperature and pressure. Although these reported methods use substoichiometric amounts of catalysts, still these transformations are far from ideal due to limited product scope as well as poor energy and economic efficiency. This is majorly due to the high cost associated with metal based catalysts and specialized ligands along with there being no established method of their recycling. Additionally, a prerequisite of high pressure, high temperature and involvement of hazardous gases such as H2 as a hydride donor make these reported methodologies less appealing for general application despite their higher turnover number. The carbon atom in CO2 is electrophilic. Hydride ion is a unique nucleophile known in organic chemistry. Borohydrides such as sodium borohydride are stable and easily available sources of hydride in organic synthesis. Sodium borohydride (NaBH4) is one of the safest, inexpensive, most studied and exploited hydride sources.33 The commercial supply chain of sodium borohydride (NaBH4) is already established. Importantly, a very efficient regeneration process of NaBH4 using magnesium and CO2 has been reported recently. Zhu et al. have reported a facile and cost-efficient, green method for regeneration of NaBH4 from its aqueous hydrolytic product NaBO2 by reacting it with CO2 and Mg under ambient conditions without the use of H2 gas.34 This made, N-formylation of amines with CO2, using NaBH4 an attractive strategy that can provide both cost as well as energy efficiency. Hao et al.35 and Zou et al.36 have independently reported N-formylation of secondary amines using CO2 and NaBH4 (Fig. 2). Unfortunately, these two reported methods are not only limited to the use of secondary amines but also need high pressure of CO2 along with heating. Herein, we want to present a practically simple and facile method for the synthesis of N-formyls of primary as well as secondary amines using a sub-stoichiometric amount of NaBH4 in a regular glass line assembly under ambient pressure.
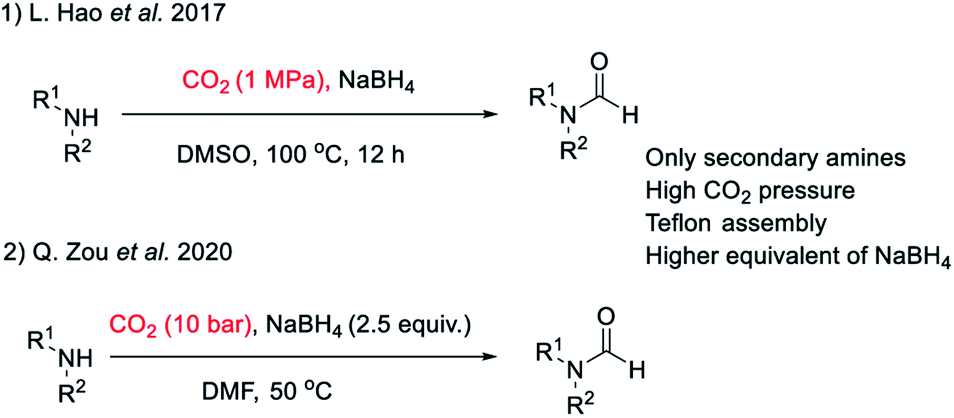 | ||
| Fig. 2 Literature reports on N-formylation of amines exploiting reductive functionalization of CO2 using NaBH4. | ||
Results and discussion
We attempted for N-formylation of a model substrate 4-fluorobenzylamine (1), under ambient conditions in a regular glass assembly using CO2 gas and NaBH4. Initial optimization efforts for the N-formylation reaction are summarized in Table 1. In our first attempt, NaBH4 was charged in acetonitrile and a stream of CO2 gas was bubbled through this solution at low temperature using ice bath for 10 minutes and reaction was sealed. Amine 1 was added to the reaction mixture as a solution in acetonitrile and the reaction was stirred for 24 h at room temperature. This typical procedure led to the formation of corresponding N-formyl product (2) in traces (Table 1, entry 1). A similar reaction using DMF as a solvent of choice provided a 23% conversion of the desired product (Table 1, entry 2). The outcome of the screening effort using other solvents such as tetrahydrofuran (THF), dimethylsulfoxide (DMSO), methanol (MeOH), toluene, 1,2-dichloroethane (DCE) and dimethylacetamide (DMA) was not encouraging. To improve conversion of 2 under ambient conditions, we attempted screening of various additives with DMF as a solvent of choice. While additives such as zinc acetate (entry 12) methanesulfonic acid (entry 13), triethylamine (entry 14), TBAF (entry 15) and water (entry 16) did have some positive outcomes but it was not significant and sufficient enough. There was no favorable effect of maintaining CO2 atmosphere using a CO2 balloon on overall yield of the reaction (entry 10). To our pleasant surprise, heating of the reaction mixture at 60 °C in DMF was found to have a significant effect on overall conversion resulting in 88% isolated yield of 2 (entry 17). The increase in conversion of 2 was also observed when the reaction mixture was heated at 60 °C using ACN or THF as solvent of choice (entry 18 and 20). But the positive effect of heating was more profound in DMF compared to THF or ACN. Interestingly, a two-fold increase in the formation of 2 was observed when a mixture of DMF and THF was used as a solvent compared to THF alone (entry 23 vs. 20). This indicated the role of DMF as a reaction promoter.
|
||||
|---|---|---|---|---|
| Entry | Solvent (30 volume) | Additive | Temp. | Isolated yieldb (%) |
| a Reaction conditions: NaBH4 (2.39 mmol, 1 equiv. to amine) dissolved in solvent 3 mL, CO2 gas sparging at 25 °C followed by addition of amine 1 (2.39 mmol, 300 mg); NA = no additive; NR = no reaction.b Reaction time of 24 h.c CO2 atmosphere maintained using balloon.d N2 atmosphere maintained using balloon.e THF:H2O (3:1).f THF:DMF (3:1). |
||||
| 1 | ACN | NA | RT | Traces |
| 2 | DMF | NA | RT | 23 |
| 3 | DMA | NA | RT | 5 |
| 4 | DMSO | NA | RT | 5 |
| 5 | THF | NA | RT | Traces |
| 6 | MeOH | NA | RT | NR |
| 7 | Toluene | NA | RT | NR |
| 8 | 1,2-DCE | NA | RT | NR |
| 9 | H2O | NA | RT | NR |
| 10 | DMFc | NA | RT | 20 |
| 11 | DMFd | NA | RT | 27 |
| 12 | DMF | Zinc acetate (0.01 equiv.) | RT | 41 |
| 13 | DMF | CH3SO2OH (0.01 equiv.) | RT | 47 |
| 14 | DMF | Et3N (1 equiv.) | RT | 40 |
| 15 | DMF | TBAF (0.01 equiv.) | RT | 35 |
| 16 | DMF | H2O (0.5 mL) | RT | 50 |
| 17 | DMF | NA | 60 °C | 88 |
| 18 | ACN | NA | 60 °C | 41 |
| 19 | DMSO | NA | 60 °C | 4 |
| 20 | THF | NA | 60 °C | 38 |
| 21 | MeOH | NA | 60 °C | NR |
| 22 | THFe | NA | 60 °C | 18 |
| 23 | THFf | NA | 60 °C | 80 |
The substrate scope of this reaction was studied by evaluation of different amines as starting materials. The summary of this study is documented in Scheme 1. All of the evaluated aliphatic amines provided high conversion to corresponding N-formyl products at 60 °C using the method of Table 1 (Scheme 1, 2–14) in DMF as a solvent. The various aromatic amines (Scheme 1, 15–21) provided fair to good yields of corresponding N-formyl products at elevated temperature i.e. 90–100 °C compare to aliphatic amines owing to their poor nucleophilicity. The formation of benzimidazole (22) in reasonably good yield from corresponding o-phenylenediamine indicated the potential of this approach for heterocycle synthesis. It was pleasant to see functional group tolerance for N-Boc (9) and double/triple bond (13 & 14) under these reaction conditions. The presence of hydroxyl group (12) or phenolic-OH (20) on the substrate did not interfere with the overall yield of N-formyl products. The mono N-formylation involving aliphatic amine in presence of aromatic –NH2 group (11) suggested a potential scope for chemo-selective N-formylation under these conditions.
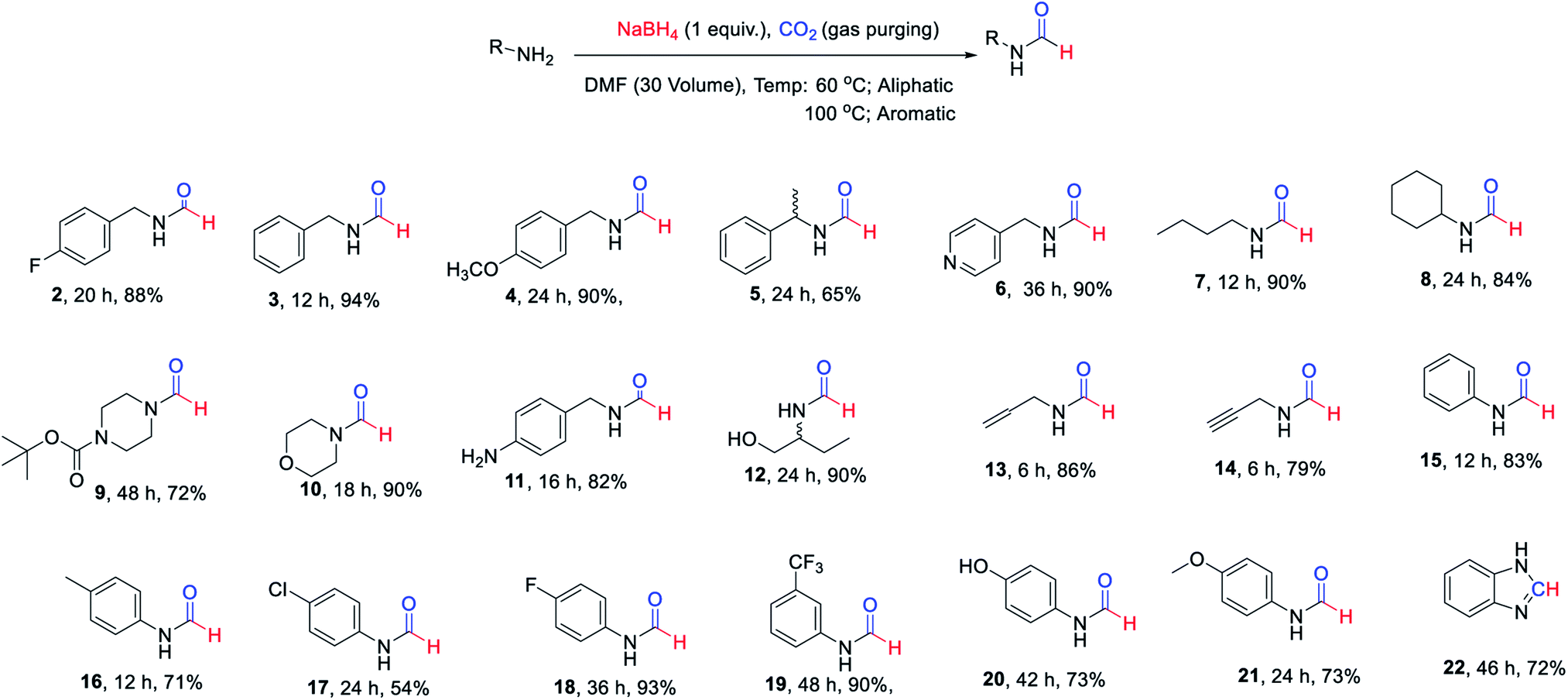 | ||
| Scheme 1 Isolated yields of different N-formyl amines evaluated to study substrate scope of the developed approach. | ||
An attempt was made to understand the mechanism by elucidating the role of NaBH4 and CO2 using proton (1H) and boron (11B) NMR spectroscopy. To a stirred solution of NaBH4 in DMF, a stream of commercial-grade CO2 gas was bubbled through for 10 minutes and an aliquot was extracted, dissolved in DMSO-d6 and NMR spectroscopy was performed (Fig. 3). The presence of a quartet at −6.43 to −9.43 and a doublet at 2.57 to 3.91 in 11B NMR spectrum (Fig. 3B) indicated the formation of bis-formoxy borohydride and tris-formoxy borohydride respectively as reported before.36,37 The presence of corresponding formyl protons was confirmed by three distinct singlets between 8.25–8.33 corresponding to bis-formoxy and tris-formoxy borohydrides in the 1H NMR spectrum (Fig. 3A). We also observed prominent signals corresponding to formoxy borohydride species in 1H and 11B NMR when DMF was replaced with DMA or ACN as a solvent of choice in a similar setup. However, other explored solvents such as THF, MeOH, toluene or DCE provided either no or very weak signals in 1H or 11B NMR spectrum corresponding to formoxy borohydride species (Fig. 3C). The poor to insignificant yield of N-formylated product in solvents such as THF, MeOH, DCE and toluene compared to DMF, DMA or ACN highlighted the critical role of in situ formed formoxy borohydride species for the success of the N-formylation (Table 1, entry 4–8).
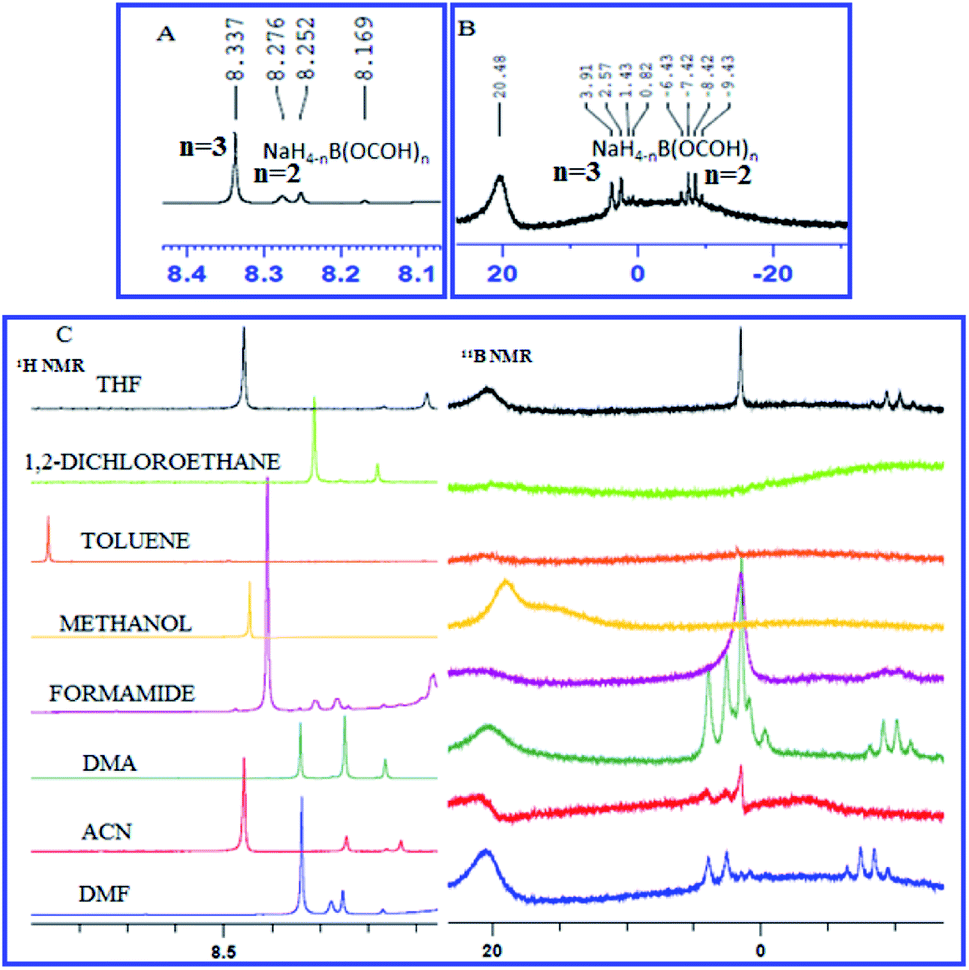 | ||
| Fig. 3 (A) 1H NMR spectrum and (B) 11B NMR spectrum of NaBH4 and CO2 dissolved in DMF (C) 1H NMR spectrum and 11B NMR spectrum of NaBH4 and CO2 dissolved in different solvents. See ESI† for complete NMR spectrum (S1 and S2†). | ||
We investigated the effect of the amount of DMF as well as NaBH4 on the yield of 2 as summarised in Table 2. The outcome of this investigation suggested that 10 volumes of DMF is optimum for conversion without the addition of any other solvent using 1 equivalent of NaBH4 (Table 2; entry 1–3). However higher volume of DMF was found to be beneficial for the overall yield of the reaction (Table 2, entry 1). The enhanced reaction yield of N-formylation in DMF is not unprecedented. DMF is reported to have a positive effect on yield in BH3·NH3 promoted N-formylation of amines using CO2 as a C1 source.23 The effect of the amount of borohydride provided intriguing insight. A quantitative conversion to N-formyl product 2 was not anticipated using less than 0.3 equivalent of NaBH4 (entry 5–8) as the reaction was understood to progress through formoxy borohydride species (vide supra). This made us speculate on the possibility of transamidation as one of the possible reaction mechanisms involving DMF as a source of formyl for N-formylation (vide infra). Evaluation of other borohydride reagents available on the shelf suggested that higher conversion of 1 to 2 is specific to NaBH4 (entry 9–11). The detrimental effect on reaction yields of 2 with a lower volume of DMF was profound with the substoichiometric amount of NaBH4 (entry 5 vs. 13–14) which was found to be consistent for the conversion of toluidine (entry 15 vs. 16) as well 4-chloroaniline (entry 18 vs. 19). Also, compared to 1, toluidine provided poor yield with a substoichiometric amount of NaBH4 (entry 16 vs. 17). Importantly, the absence of NaBH4 or CO2 did not provide any N-formyl product in either of the three tested anilines (entry 20–24), which highlighted the critical role of NaBH4 as well as CO2 and thus the involvement of formoxy borohydride species formed as a product of reductive formylation of CO2.
|
|||||||
|---|---|---|---|---|---|---|---|
| Entry | Product ID | DMFb (volume) | Borohydride reagent | Reagent (equiv.) | Temp. (°C) | Isolated yield (%) | Time (h) |
| a Reaction conditions: borohydride (specified equiv. to amine) dissolved in solvent (specified volume in reference to amine), CO2 gas sparging at 25 °C followed by addition of amine (300 mg) and then heating.b Volume of solvent in reference to amine substrate.c Corresponding acetylated product (32%) was also isolated.d Reaction performed without CO2 gas. | |||||||
| 1 | 2 | 50 | NaBH4 | 1 | 60 | 94 | 7 |
| 2 | 2 | 30 | NaBH4 | 1 | 60 | 88 | 24 |
| 3 | 2 | 10 | NaBH4 | 1 | 60 | 88 | 24 |
| 4 | 2 | 10 | NaBH4 | 1 | 25 | 23 | 24 |
| 5 | 2 | 10 | NaBH4 | 0.3 | 60 | 84 | 24 |
| 6 | 2 | 10 | NaBH4 | 0.25 | 60 | 84 | 24 |
| 7 | 2 | 10 | NaBH4 | 0.20 | 60 | 83 | 24 |
| 8 | 2 | 10 | NaBH4 | 0.1 | 60 | 80 | 24 |
| 9 | 2 | 10 | LiBH4 | 0.3 | 60 | 50 | 24 |
| 10 | 2 | 10 | NaBH3CN | 0.3 | 60 | 62 | 24 |
| 11 | 2 | 10 | Na(CH3COO)3BH | 0.3 | 60 | 58c | 24 |
| 12 | 2 | 10 | NaBH4 | 0.3 | 40 | 70 | 24 |
| 13 | 2 | 5 | NaBH4 | 0.3 | 60 | 72 | 24 |
| 14 | 2 | 3 | NaBH4 | 0.3 | 60 | 58 | 24 |
| 15 | 16 | 50 | NaBH4 | 1 | 90 | 71 | 12 |
| 16 | 16 | 10 | NaBH4 | 1 | 90 | 75 | 24 |
| 17 | 16 | 10 | NaBH4 | 0.3 | 90 | 47 | 24 |
| 18 | 17 | 50 | NaBH4 | 1 | 90 | 42 | 24 |
| 19 | 17 | 10 | NaBH4 | 0.3 | 90 | 13 | 24 |
| 20 | 16 | 10 | NaBH4 | 0 | 90 | 0 | 48 |
| 21 | 16d | 10 | NaBH4 | 1 | 90 | Traces | 48 |
| 22 | 16 | 10 | NaBH4 | 0 | 120 | 0 | 48 |
| 23 | 17 | 10 | NaBH4 | 0 | 120 | 0 | 24 |
| 24 | 15 | 10 | NaBH4 | 0 | 120 | 0 | 24 |
To investigate the involvement of transamidation reaction, isotope labelling experiments were performed (Fig. 4). In one reaction NaBH4 was replaced with NaBD4 (Fig. 4a) and in other, CO2 was replaced with 13CO2 (Fig. 4b). The relative quantification of deuterium-labelled product (16D; 53%) and 13C labelled N-formyl product (*16; 53%) in the isolated reaction products was done by LCMS analysis (Fig. S3–S11†). The significant but incomplete amounts of deuterium-labelled (16D; 53%) as well as 13C labelled N-formyl product (*16; 53%) of these two independent experiments 5a and 5b respectively, suggested the participation of transamidation from DMF as a contributing pathway along with reductive formylation. This was confirmed by another control reaction as shown in Fig. 4c. Using DMF-d7 as a solvent in place of DMF provided 37% of deuterated N-formyl product (Fig. 4c). This established transamidation reaction as a competitive contributing reaction pathway to the overall yield of N-formyl product. This was not expected due to the poor nucleophilicity of aniline for transamidation with DMF. Also, DMF has been reported as a challenging substrate for transamidation reaction at the operating temperature without the use of any catalyst. Generally, high heating and a catalyst are required to overcome this barrier. Gong et al.38 have reported transamidation of p-anisidine with DMF without any catalyst at 150 °C with 26% isolated yield. The significant amount of transamidation reaction in our protocols involving mild heating indicated the role of formoxy borohydride species as a reaction promoter. To confirm this hypothesis, another control reaction was performed by replacing NaBH4/CO2 with sodium triacetoxyborohydride as a homologous analogue of formoxy borohydride species in DMF (Fig. 4d). As anticipated, this control reaction employing sodium triacetoxyborohydride without CO2 gas in DMF provided considerable conversion to corresponding N-formyl product 16 through the transamidation route. This supported the role of formoxy borohydride species as a promoter of the transamidation process. Based on these control experiments and information gathered from NMR studies as well as data summarized in Table 2, we proposed a plausible mechanism as depicted in Fig. 5. The experimental evidence suggested transamidation (5A) as a complementary pathway along with reductive formylation (5B) for the yield of N-formyl product under these conditions.
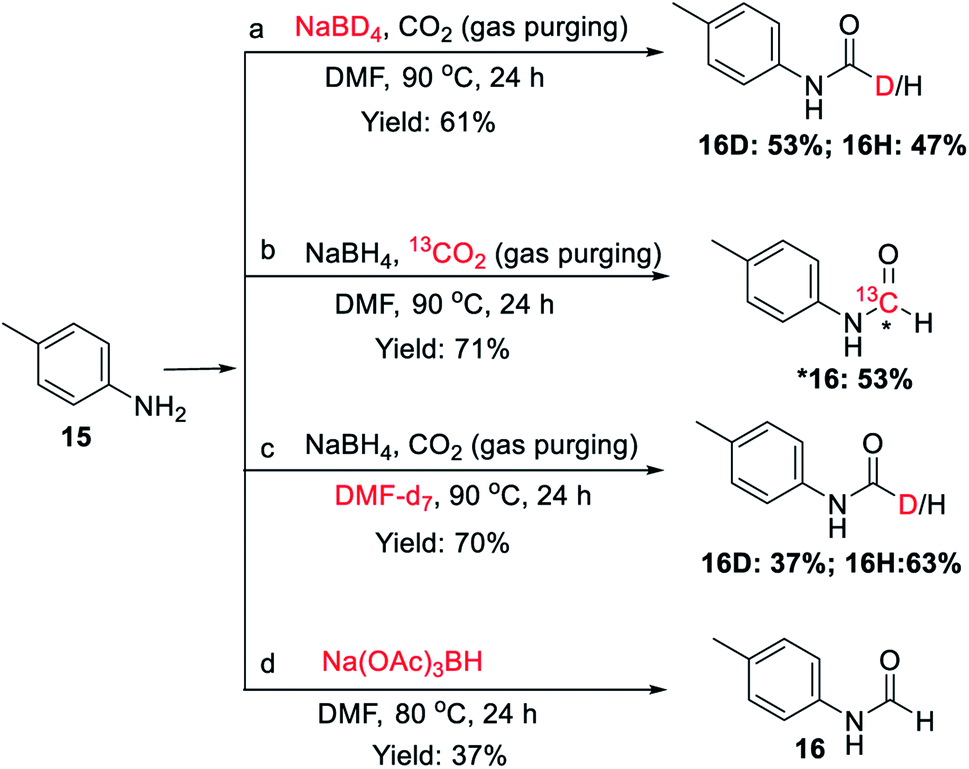 | ||
| Fig. 4 Control experiments to understand the involvement of transamidation reaction. | ||
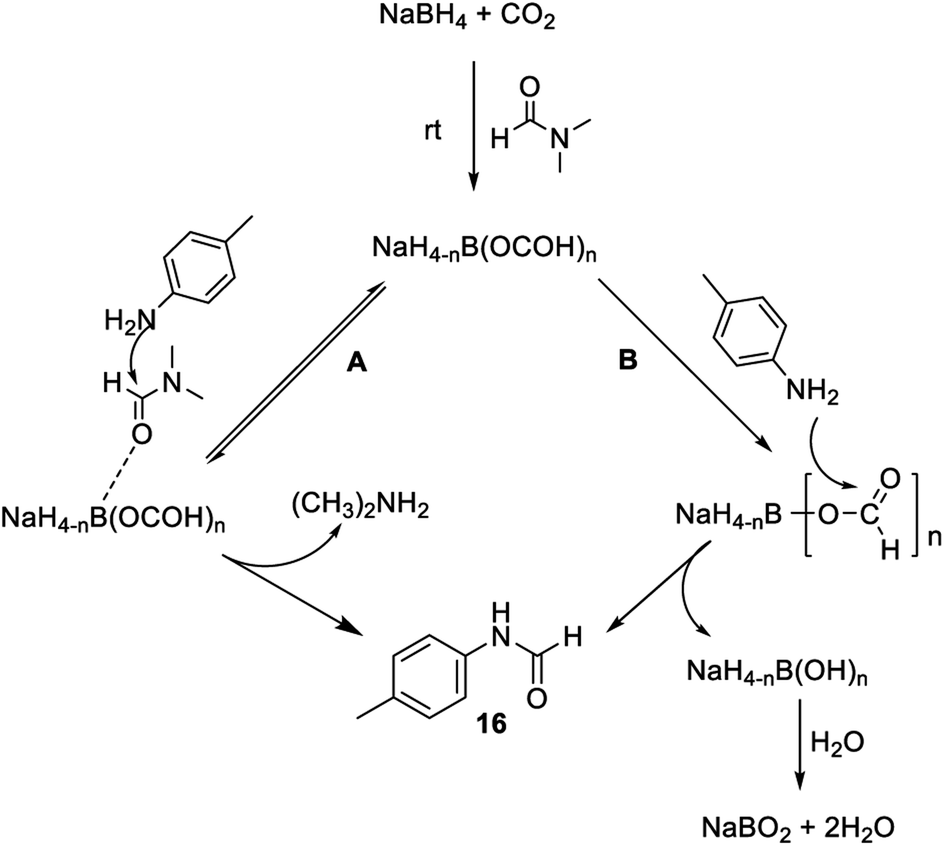 | ||
| Fig. 5 Plausible mechanism for N-formylation involving transamidation (A) and reductive formylation pathways (B). | ||
Different boron based reagents have been reported to catalyse the transamidation reactions in the recent past (Fig. S12†). However, among all tested reagents only boronic acid was reported to provide reasonable transamidation of DMF with aniline after 150 °C for 24 h as reported by Nguyen et al.39 Sheppard et al. reported the use of fluorinated borate ester in high stoichiometry for transamidation of DMF limited to aliphatic amines.40,41 Recently, Blanchet et al.42 reported borinic acid/AcOH based catalyst for transamidation of DMF with aliphatic amines under mild heating.
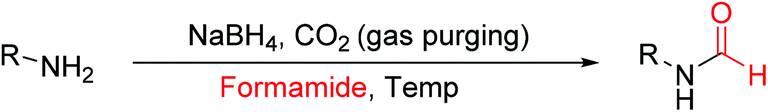
Our newly reported protocol involving NaBH4 and CO2 as a promoter of transamidation is unprecedented. Riding on the acquired mechanistic understanding, we attempted to study the effect of other solvents such as DMA and formamide. We envisaged that the N-formylation reaction should be more facile in a solvent such as formamide (NH2CHO) compared to DMF due to higher contribution through the transamidation process. A higher contribution of the transamidation reaction in formamide solvent was anticipated owing to favorable reaction kinetics due to the volatile nature of NH3 as a reaction by-product. An opposite was expected using N,N-dimethyl acetamide (DMA) as a solvent, as there is no scope for N-formylation through the transamidation pathway. The result of this study is documented in Tables 3 & 4. As anticipated, N-formylation reactions using formamide as a solvent in place of DMF were very facile (Table 3). All tested aromatic and aliphatic amines provided a higher yield of corresponding N-formamides in a shorter reaction time compared to DMF. For example, toluidine and 4-chloroaniline provided higher yields of corresponding N-formyl product in formamide (Table 3, entry 1 and 7) compared to the same reaction performed in DMF (Table 2, entry 16 and 18). Even substoichiometric use of NaBH4 (Table 3, entries 1–2) or reduced volume of formamide (Table 3, entry 5 and 6) as solvent was not found to be detrimental for N-formylation reactions. The relatively lower amount (only 10%) of deuterium incorporation using formamide as solvent employing NaBD4 confirmed the higher contribution of the transamidation process towards the overall yield of N-formyl product. In contrast, DMA as a solvent of choice provided fair to high yields of N-formamide products with different tested amines (Table 4) without any detectable amount of transamidation product. This indicates that DMA could be a solvent of choice for N-formylation reactions if there is a need to restrict transamidation in this newly developed approach.
|
|||||||
|---|---|---|---|---|---|---|---|
| Entry | Substrate | NaBH4 | Formamide (volume) | Temp. (°C) | Rxn time (h) | Isolated yield (%) | Product ID |
| a Reaction conditions: NaBH4 (specified equiv. to amine) dissolved in solvent (specified volume in reference to amine), CO2 gas sparging at 25 °C followed by addition of amine (300 mg) and then heating. | |||||||
| 1 | p-Toluidine | 1 | 3 mL (10 volume) | 90 | 7 | 90 | 16 |
| 2 | p-Toluidine | 0.3 | 3 mL (10 volume) | 90 | 6 | 93 | 16 |
| 3 | p-Toluidine | 0.3 | 3 mL (10 volume) | 60 | 18 | 80 | 16 |
| 4 | p-Toluidine | 0.3 | 3 mL (10 volume) | 60 | 41 | 90 | 16 |
| 5 | 4-Methoxybenzylamine | 0.3 | 3 mL (10 volume) | 40 | 108 | 73 | 4 |
| 6 | 4-Methoxybenzylamine | 0.3 | 1.5 mL (5 volume) | 50 | 18 | 92 | 4 |
| 7 | 4-Chloroaniline | 1.0 | 3 mL (10 volume) | 90 | 24 | 90 | 17 |
| 8 | N-4-Dimethylaniline | 0.3 | 3 mL (10 volume) | 90 | 25 | 90 | 25 |
| 9 | 4-Fluoro-N-methylaniline | 1.0 | 3 mL (10 volume) | 90 | 24 | 74 | 23 |
| 10 | 4-Methoxy-N-methylaniline | 1.0 | 3 mL (10 volume) | 90 | 24 | 90 | 24 |
| 11 | 1-Boc-piperazine | 1.0 | 3 mL (10 volume) | 90 | 24 | 95 | 9 |
| 12 | (±)-α-Methylbenzylamine | 1.0 | 3 mL (10 volume) | 90 | 24 | 83 | 5 |
|
|||||||
|---|---|---|---|---|---|---|---|
| Entry | Substrate | NaBH4 (equiv.) | Acetamide (volume) | Temp. (°C) | Rxn time (h) | Isolated yield (%) | Product ID |
| a Reaction conditions: NaBH4 (1 equiv. to amine) dissolved in solvent (10 volume in reference to amine), CO2 gas sparging at 25 °C followed by addition of amine (300 mg) and then heating. | |||||||
| 1 | 4-Methoxybenzylamine | 1.0 | 3 mL (10 volume) | 90 | 24 | 99 | 4 |
| 2 | p-Toluidine | 1.0 | 3 mL (10 volume) | 100 | 48 | 75 | 16 |
| 3 | p-Toluidine | 1.0 | 3 mL (10 volume) | 90 | 6 | 87 | 16 |
| 4 | 4-Chloroaniline | 1.0 | 3 mL (10 volume) | 100 | 49 | 61 | 17 |
| 5 | Aniline | 1.0 | 3 mL (10 volume) | 100 | 49 | 75 | 15 |
| 6 | (±)-α-Methylbenzylamine | 1.0 | 3 mL (10 volume) | 60 | 24 | 32 | 5 |
| 7 | (±)-α-Methylbenzylamine | 1.0 | 3 mL (10 volume) | 90 | 24 | 78 | 5 |
| 8 | 1-Boc-piperazine | 1.0 | 3 mL (10 volume) | 90 | 6 | 86 | 9 |
| 9 | 4-Fluorobenzylamine | 1.0 | 3 mL (10 volume) | 90 | 24 | 74 | 2 |
| 10 | 4-Fluorobenzylamine | 1.0 | 3 mL (10 volume) | 25 | 20 | 4.0 | 2 |
| 11 | 4-Fluoro-N-methylaniline | 1.0 | 3 mL (10 volume) | 90 | 18 | 56 | 23 |
| 12 | N-4-Dimethylaniline | 1.0 | 3 mL (10 volume) | 90 | 18 | 43 | 25 |
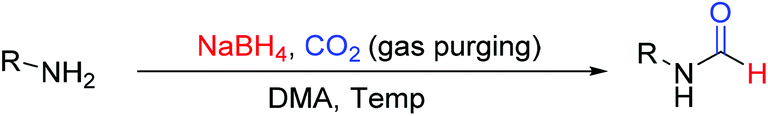
Conclusions
We have reported a catalyst free, facile and sustainable N-formylation approach of aliphatic and aromatic amines under mild heating and ambient pressure exploiting reductive formylation of CO2 with sodium borohydride. It is observed that the reaction of CO2 with NaBH4 led to the formation of formoxy borohydride species in situ as evident by proton and boron NMR spectroscopy. The formation of formoxy borohydride species is critical for the success of the desired transformation. It is also observed that this formoxy borohydride species can promote transamidation reaction of aromatic amines under mild conditions using formamide and DMF as a formyl source.Experimental
General procedure used for screening of solvents to optimize yield of 2
Sodium borohydride (2.39 mmol and 90.69 mg) was dissolved in a solvent (3 mL; 30 volumes to amine) inside a round bottom flask (rbf) at 25 °C and a stream of CO2 was bubbled through this stirred solution for 10 min. After CO2 sparging, 4-fluorobenzylamine (2.39 mmol, 300 mg, 1 equivalent to borohydride) was added and the reaction mixture was stirred at 25–60 °C. The progress of the reaction was monitored through TLC. After maximum conversion (as monitored by TLC), the excess of the solvent was evaporated from reaction mixture under reduced pressure and crude product thus obtained is subjected to column chromatography using increasing volume of ethyl acetate in hexane to isolate pure product.General procedure for N-formylation of different amines (Scheme 1) using DMF as solvent
Sodium borohydride (1 equivalent to amine) was dissolved in DMF (3 mL, 30 volumes to amine) inside a round bottom flask (rbf) at 25 °C and a stream of CO2 was bubbled through this stirred solution for 10 min. After CO2 sparging, an amine (100 mg) was added and the reaction mixture was stirred at 60–100 °C. The progress of the reaction was monitored through TLC. After maximum conversion (as monitored by TLC), the excess of the solvent was evaporated from the reaction mixture and crude product is subjected to column chromatography using increasing volume of ethyl acetate in hexane to isolate pure product.Specific procedure for characterization of in situ formed formoxy borohydride species using NMR spectroscopy
Sodium borohydride (2.39 mmol and 90.69 mg) was dissolved in a solvent (3 mL) inside a round bottom flask (rbf) at 25 °C and a stream of CO2 was bubbled through this stirred solution for 10 min. After 10 min of CO2 bubbling, white precipitates were formed in the reaction mixture. A homogeneous aliquot was taken and dissolved in DMSO-d6 to prepare NMR sample. For detailed NMR spectrum see Fig. S1 and S2 in ESI.†General procedure for N-formylation of different amines using formamide or DMA as solvent
Sodium borohydride (1 equivalent to amine) was dissolved in formamide or DMA (10 volumes to amine) inside a round bottom flask (rbf) at 25 °C and a stream of CO2 was bubbled through this stirred solution for 10 min. After CO2 sparging, amine (300 mg, 1 equiv. to NaBH4) was added and the reaction mixture was stirred at 60–100 °C. The progress of the reaction was monitored through TLC. After maximum conversion (as monitored by TLC), the reaction mixture was evaporated and subjected to column chromatography using increasing volume of ethyl acetate in hexane to isolate pure product. The isolated pure products were characterized as a mixture of two rotamers as observed by NMR spectroscopy.43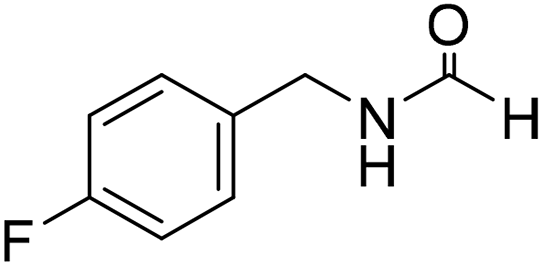 The title compound was synthesized from 4-fluorobenzylamine (91.32 μL, 0.799 mmol) and sodium borohydride (30.22 mg, 0.799 mmol) according to general procedure. Brown crystalline solid (108 mg, 88% yield); m.p. 65 °C; 1H-NMR (300 MHz, CDCl3) δ (ppm) major rotamer: 8.10 (s, 1H), 7.09–7.14 (m, 2H), 6.85–6.94 (m, 2H), 5.98 (br, 1H), 4.30 (d, J = 6.0 Hz, 2H), minor rotamer: 8.02 (d, J = 11.9 Hz, 1H), 7.09–7.14 (m, 2H), 6.85–6.94 (m, 2H), 4.24 (d, J = 6.4 Hz, 2H); 13C{1H} NMR (75 MHz, CDCl3) δ mixture of rotamers: 164.5, 163.8, 161.03, 160.6, 133.4, 133.3, 129.5, 129.4, 128.7, 128.6, 115.7, 115.5, 45.0, 41.4. The analytical data was found to be consistent with literature.16
The title compound was synthesized from 4-fluorobenzylamine (91.32 μL, 0.799 mmol) and sodium borohydride (30.22 mg, 0.799 mmol) according to general procedure. Brown crystalline solid (108 mg, 88% yield); m.p. 65 °C; 1H-NMR (300 MHz, CDCl3) δ (ppm) major rotamer: 8.10 (s, 1H), 7.09–7.14 (m, 2H), 6.85–6.94 (m, 2H), 5.98 (br, 1H), 4.30 (d, J = 6.0 Hz, 2H), minor rotamer: 8.02 (d, J = 11.9 Hz, 1H), 7.09–7.14 (m, 2H), 6.85–6.94 (m, 2H), 4.24 (d, J = 6.4 Hz, 2H); 13C{1H} NMR (75 MHz, CDCl3) δ mixture of rotamers: 164.5, 163.8, 161.03, 160.6, 133.4, 133.3, 129.5, 129.4, 128.7, 128.6, 115.7, 115.5, 45.0, 41.4. The analytical data was found to be consistent with literature.16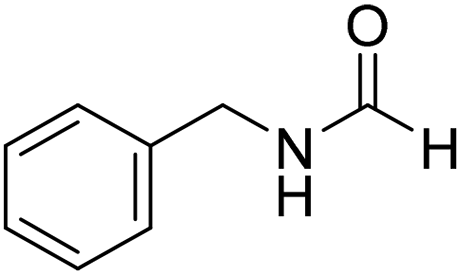 The title compound was synthesized from benzylamine (101.93 μL, 0.939 mmol) and sodium borohydride (35.52 mg, 0.939 mmol) according to general procedure. Off white solid (118 mg, 94% yield); m.p. 52 °C; 1H-NMR (300 MHz, CDCl3) δ (ppm) major rotamer: 8.07 (s, 1H), 7.04–7.20 (m, 5H), 5.64–5.65 (br, 1H), 4.29 (d, J = 5.9 Hz, 2H), minor rotamer: 8.0 (d, J = 11.9 Hz, 1H), 7.04–7.20 (m, 5H), 4.22 (d, J = 6.5 Hz, 2H); 13C{1H} NMR (75 MHz, CDCl3) δ mixture of rotamers: 164.6, 161.0, 137.5, 128.9, 128.8, 127.8, 127.7, 126.9, 45.6, 42.2. The analytical data was found to be consistent with literature.16
The title compound was synthesized from benzylamine (101.93 μL, 0.939 mmol) and sodium borohydride (35.52 mg, 0.939 mmol) according to general procedure. Off white solid (118 mg, 94% yield); m.p. 52 °C; 1H-NMR (300 MHz, CDCl3) δ (ppm) major rotamer: 8.07 (s, 1H), 7.04–7.20 (m, 5H), 5.64–5.65 (br, 1H), 4.29 (d, J = 5.9 Hz, 2H), minor rotamer: 8.0 (d, J = 11.9 Hz, 1H), 7.04–7.20 (m, 5H), 4.22 (d, J = 6.5 Hz, 2H); 13C{1H} NMR (75 MHz, CDCl3) δ mixture of rotamers: 164.6, 161.0, 137.5, 128.9, 128.8, 127.8, 127.7, 126.9, 45.6, 42.2. The analytical data was found to be consistent with literature.16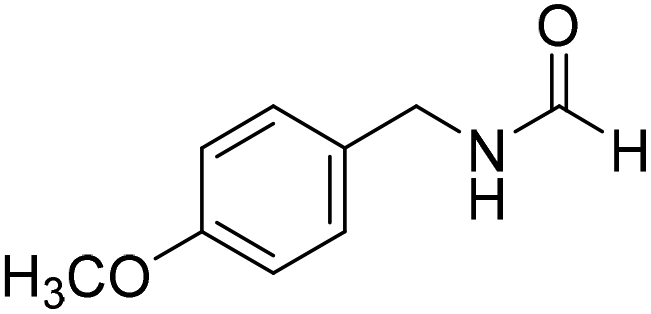 The title compound was synthesized from 4-methoxy benzylamine (95.23 μL, 0.728 mmol) and sodium borohydride (27.54 mg, 0.728 mmol) according to general procedure. Brown crystalline solid (108 mg, 90% yield); m.p. 67–70 °C; 1H-NMR (300 MHz, CDCl3) δ (ppm) major rotamer: 8.21 (s, 1H), 7.16–7.28 (m, 2H), 6.85–6.91 (m, 2H), 6.05 (br, 1H), 4.40 (d, J = 5.61 Hz, 2H), 3.80 (s, 3H), minor rotamer: 8.15 (d, J = 12.0 Hz, 1H), 7.16–7.28 (m, 2H), 6.85–6.91 (m, 2H), 6.05 (br, 1H), 4.34 (d, J = 6.3 Hz, 2H), 3.80 (s, 3H); 13C{1H} NMR (75 MHz, DMSO-d6) δ mixture of rotamers: 165.2, 161.3, 158.7, 132.0, 131.3, 129.1, 128.8, 114.1, 55.5, 44.4. The analytical data was found to be consistent with literature.16
The title compound was synthesized from 4-methoxy benzylamine (95.23 μL, 0.728 mmol) and sodium borohydride (27.54 mg, 0.728 mmol) according to general procedure. Brown crystalline solid (108 mg, 90% yield); m.p. 67–70 °C; 1H-NMR (300 MHz, CDCl3) δ (ppm) major rotamer: 8.21 (s, 1H), 7.16–7.28 (m, 2H), 6.85–6.91 (m, 2H), 6.05 (br, 1H), 4.40 (d, J = 5.61 Hz, 2H), 3.80 (s, 3H), minor rotamer: 8.15 (d, J = 12.0 Hz, 1H), 7.16–7.28 (m, 2H), 6.85–6.91 (m, 2H), 6.05 (br, 1H), 4.34 (d, J = 6.3 Hz, 2H), 3.80 (s, 3H); 13C{1H} NMR (75 MHz, DMSO-d6) δ mixture of rotamers: 165.2, 161.3, 158.7, 132.0, 131.3, 129.1, 128.8, 114.1, 55.5, 44.4. The analytical data was found to be consistent with literature.16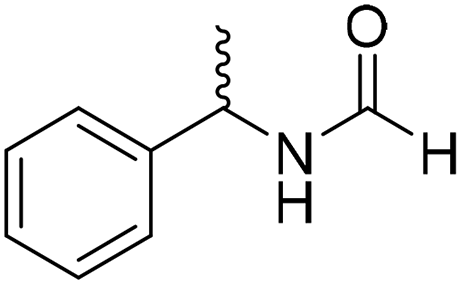 The title compound was synthesized from (±)-α-methylbenzylamine (105.04 μL, 0.825 mmol) and sodium borohydride (31.21 mg, 0.825 mmol) according to general procedure. Brown oil (80 mg, 65% yield); 1H-NMR (300 MHz, DMSO-d6) δ (ppm) major rotamer: 8.54–8.57 (br, 1H), 8.03 (s, 1H), 7.23–7.35 (m, 5H), 4.94–5.04 (m, 1H), 1.35 (d, J = 6.99 Hz, 3H), minor rotamer: 8.30–8.33 (br, 1H), 8.08 (d, J = 11.5 Hz, 1H), 7.23–7.35 (m, 5H), 4.67–4.72 (m, 1H), 1.41 (d, J = 6.90 Hz, 3H); 13C{1H} NMR (75 MHz, DMSO-d6) δ mixture of rotamers: 164.4, 160.5, 145.2, 144.6, 128.9, 128.7, 127.3, 127.2, 126.4, 126.3, 51.2, 47.0, 23.9, 22.9. The analytical data was found to be consistent with literature.44
The title compound was synthesized from (±)-α-methylbenzylamine (105.04 μL, 0.825 mmol) and sodium borohydride (31.21 mg, 0.825 mmol) according to general procedure. Brown oil (80 mg, 65% yield); 1H-NMR (300 MHz, DMSO-d6) δ (ppm) major rotamer: 8.54–8.57 (br, 1H), 8.03 (s, 1H), 7.23–7.35 (m, 5H), 4.94–5.04 (m, 1H), 1.35 (d, J = 6.99 Hz, 3H), minor rotamer: 8.30–8.33 (br, 1H), 8.08 (d, J = 11.5 Hz, 1H), 7.23–7.35 (m, 5H), 4.67–4.72 (m, 1H), 1.41 (d, J = 6.90 Hz, 3H); 13C{1H} NMR (75 MHz, DMSO-d6) δ mixture of rotamers: 164.4, 160.5, 145.2, 144.6, 128.9, 128.7, 127.3, 127.2, 126.4, 126.3, 51.2, 47.0, 23.9, 22.9. The analytical data was found to be consistent with literature.44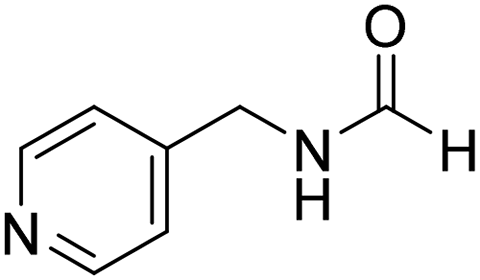 The title compound was synthesized from 4-aminomethyl pyridine (93.89 μL, 0.924 mmol) and sodium borohydride (34.95 mg, 0.924 mmol) according to general procedure. Colourless oil (114 mg, 90% yield); 1H-NMR (300 MHz, DMSO-d6) δ (ppm) mixture of rotamers: 8.61 (br, 1H), 8.49–8.51 (m, 2H), 8.19 (d, J = 1.41 Hz, 1H), 7.25–7.27 (m, 2H), 4.33 (d, J = 6.2 Hz, 2H); 13C{1H} NMR (75 MHz, DMSO-d6) δ mixture of rotamers: 165.6, 161.9, 150.6, 150.1, 149.0, 148.4, 122.5, 121.8, 43.9; HRMS (ESI-TOF) (m/z): [M + H]+ calcd for C7H9N2O 137.0715; found 137.07112.
The title compound was synthesized from 4-aminomethyl pyridine (93.89 μL, 0.924 mmol) and sodium borohydride (34.95 mg, 0.924 mmol) according to general procedure. Colourless oil (114 mg, 90% yield); 1H-NMR (300 MHz, DMSO-d6) δ (ppm) mixture of rotamers: 8.61 (br, 1H), 8.49–8.51 (m, 2H), 8.19 (d, J = 1.41 Hz, 1H), 7.25–7.27 (m, 2H), 4.33 (d, J = 6.2 Hz, 2H); 13C{1H} NMR (75 MHz, DMSO-d6) δ mixture of rotamers: 165.6, 161.9, 150.6, 150.1, 149.0, 148.4, 122.5, 121.8, 43.9; HRMS (ESI-TOF) (m/z): [M + H]+ calcd for C7H9N2O 137.0715; found 137.07112.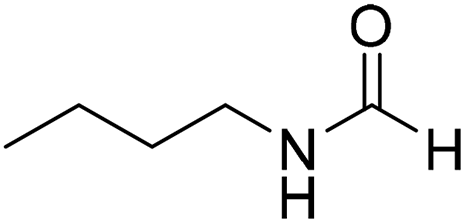 The title compound was synthesized from N-butyl amine (135.13 μL, 1.367 mmol) and sodium borohydride (51.71 mg, 1.367 mmol) according to general procedure. Colourless oil (125 mg, 90% yield); 1H-NMR (300 MHz, CDCl3) δ (ppm) major rotamer: 8.15 (s, 1H), 5.58 (br, 1H), 3.20–3.33 (m, 2H), 1.47–1.53 (m, 2H), 1.24–1.39 (m, 2H), 0.92 (t, J = 7.2 Hz, 3H), minor rotamer: 8.03 (d, J = 12.03 Hz, 1H) 5.58 (br, 1H), 3.20–3.33 (m, 2H), 1.47–1.53 (m, 2H), 1.24–1.39 (m, 2H), 0.92 (t, J = 7.2 Hz, 3H); 13C{1H} NMR (75 MHz, CDCl3) δ mixture of rotamers: 164.6, 161.2, 41.4, 37.9, 33.2, 31.5, 20.0, 19.5, 13.7, 13.5. The analytical data was found to be consistent with literature.17,45
The title compound was synthesized from N-butyl amine (135.13 μL, 1.367 mmol) and sodium borohydride (51.71 mg, 1.367 mmol) according to general procedure. Colourless oil (125 mg, 90% yield); 1H-NMR (300 MHz, CDCl3) δ (ppm) major rotamer: 8.15 (s, 1H), 5.58 (br, 1H), 3.20–3.33 (m, 2H), 1.47–1.53 (m, 2H), 1.24–1.39 (m, 2H), 0.92 (t, J = 7.2 Hz, 3H), minor rotamer: 8.03 (d, J = 12.03 Hz, 1H) 5.58 (br, 1H), 3.20–3.33 (m, 2H), 1.47–1.53 (m, 2H), 1.24–1.39 (m, 2H), 0.92 (t, J = 7.2 Hz, 3H); 13C{1H} NMR (75 MHz, CDCl3) δ mixture of rotamers: 164.6, 161.2, 41.4, 37.9, 33.2, 31.5, 20.0, 19.5, 13.7, 13.5. The analytical data was found to be consistent with literature.17,45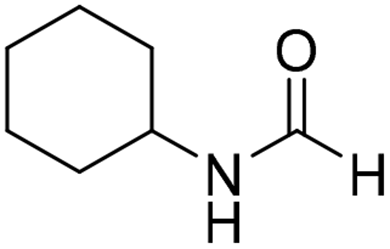 The title compound was synthesized from cyclohexyl amine (115.60 μL, 1.008 mmol) and sodium borohydride (38.13 mg, 1.008 mmol) according to general procedure. Colourless oil (108 mg, 84% yield); 1H-NMR (300 MHz, DMSO-d6) δ (ppm) major rotamer: 7.91 (s, 1H + NH), 3.57–3.61 (m, 1H), 1.11–1.72 (m, 10H), minor rotamer: 7.96–8.0 (m, 2H), 3.05–3.07 (m, 1H), 1.11–1.72 (m, 10H); 13C{1H} NMR (75 MHz, CDCl3) δ mixture of rotamers: 163.6, 160.3, 50.9, 47.0, 34.6, 33.0, 25.4, 25.0, 24.7. The analytical data was found to be consistent with literature.16
The title compound was synthesized from cyclohexyl amine (115.60 μL, 1.008 mmol) and sodium borohydride (38.13 mg, 1.008 mmol) according to general procedure. Colourless oil (108 mg, 84% yield); 1H-NMR (300 MHz, DMSO-d6) δ (ppm) major rotamer: 7.91 (s, 1H + NH), 3.57–3.61 (m, 1H), 1.11–1.72 (m, 10H), minor rotamer: 7.96–8.0 (m, 2H), 3.05–3.07 (m, 1H), 1.11–1.72 (m, 10H); 13C{1H} NMR (75 MHz, CDCl3) δ mixture of rotamers: 163.6, 160.3, 50.9, 47.0, 34.6, 33.0, 25.4, 25.0, 24.7. The analytical data was found to be consistent with literature.16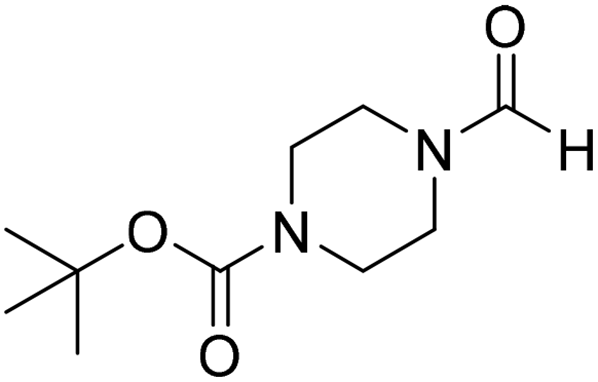 The title compound was synthesized from 1-boc-piperazine (100 mg, 0.536 mmol) and sodium borohydride (20.27 mg, 0.536 mmol) according to general procedure. White solid (83 mg, 72% yield); m.p. 108–110 °C; 1H-NMR (300 MHz, CDCl3) δ (ppm) mixture of rotamers: 8.07 (s, 1H), 3.33–3.53 (m, 8H), 1.46 (s, 9H); 13C{1H} NMR (75 MHz, CDCl3) δ mixture of rotamers: 160.9, 154.4, 80.5, 45.4, 39.9, 28.3; HRMS (ESI-TOF) (m/z): [M + Na]+ calcd for C10H18N2O3Na 237.1215; found 237.12095.
The title compound was synthesized from 1-boc-piperazine (100 mg, 0.536 mmol) and sodium borohydride (20.27 mg, 0.536 mmol) according to general procedure. White solid (83 mg, 72% yield); m.p. 108–110 °C; 1H-NMR (300 MHz, CDCl3) δ (ppm) mixture of rotamers: 8.07 (s, 1H), 3.33–3.53 (m, 8H), 1.46 (s, 9H); 13C{1H} NMR (75 MHz, CDCl3) δ mixture of rotamers: 160.9, 154.4, 80.5, 45.4, 39.9, 28.3; HRMS (ESI-TOF) (m/z): [M + Na]+ calcd for C10H18N2O3Na 237.1215; found 237.12095.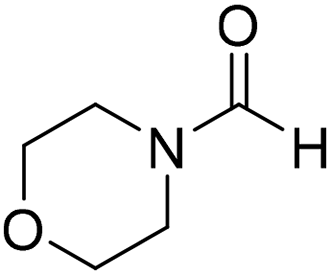 The title compound was synthesized from morpholine (99.00 μL, 1.147 mmol) and sodium borohydride (43.39 mg, 1.147 mmol) according to general procedure. Colourless oil (119 mg, 90% yield); 1H-NMR (300 MHz, CDCl3) δ (ppm) 8.04 (s, 1H), 3.63–3.70 (m, 4H), 3.54–3.57 (m, 2H), 3.38 (t, J = 4.8 Hz, 2H); 13C{1H} NMR (75 MHz, CDCl3) δ 159.2, 65.6, 64.8, 44.2, 39.0, 28.0. The analytical data was found to be consistent with literature.46
The title compound was synthesized from morpholine (99.00 μL, 1.147 mmol) and sodium borohydride (43.39 mg, 1.147 mmol) according to general procedure. Colourless oil (119 mg, 90% yield); 1H-NMR (300 MHz, CDCl3) δ (ppm) 8.04 (s, 1H), 3.63–3.70 (m, 4H), 3.54–3.57 (m, 2H), 3.38 (t, J = 4.8 Hz, 2H); 13C{1H} NMR (75 MHz, CDCl3) δ 159.2, 65.6, 64.8, 44.2, 39.0, 28.0. The analytical data was found to be consistent with literature.46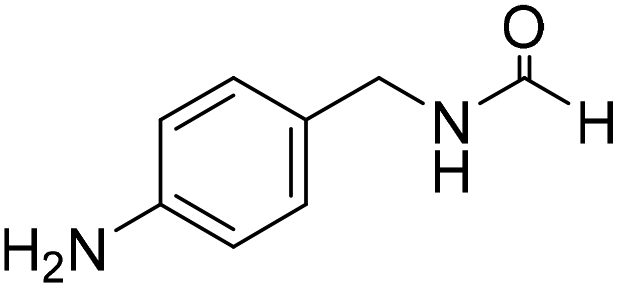 The title compound was synthesized from 4-aminomethyl aniline (92.76 μL, 0.818 mmol) and sodium borohydride (30.94 mg, 0.818 mmol) according to general procedure. Brown oil (101 mg, 82% yield); 1H-NMR (300 MHz, CDCl3) δ (ppm) major rotamer: 8.21 (s, 1H), 7.07 (d, J = 8.4 Hz, 2H), 6.64 (d, J = 8.4 Hz, 2H), 5.67–5.70 (br, 1H), 4.35 (d, J = 5.7 Hz, 2H), 3.68 (br, 2H), minor rotamer: 8.17 (d, J = 12.1 Hz, 1H), 7.02 (d, J = 8.4 Hz, 2H), 6.64 (d, J = 8.4 Hz, 2H), 4.29 (d, J = 6.3 Hz, 2H); 13C{1H} NMR (75 MHz, CDCl3) δ mixture of rotamers: 164.5, 160.9, 146.0, 129.2, 128.3, 127.3, 115.3, 115.2, 45.3, 41.8; HRMS (ESI-TOF) (m/z): [M + H]+ calcd for C8H11N2O 151.0871; found 151.12318.
The title compound was synthesized from 4-aminomethyl aniline (92.76 μL, 0.818 mmol) and sodium borohydride (30.94 mg, 0.818 mmol) according to general procedure. Brown oil (101 mg, 82% yield); 1H-NMR (300 MHz, CDCl3) δ (ppm) major rotamer: 8.21 (s, 1H), 7.07 (d, J = 8.4 Hz, 2H), 6.64 (d, J = 8.4 Hz, 2H), 5.67–5.70 (br, 1H), 4.35 (d, J = 5.7 Hz, 2H), 3.68 (br, 2H), minor rotamer: 8.17 (d, J = 12.1 Hz, 1H), 7.02 (d, J = 8.4 Hz, 2H), 6.64 (d, J = 8.4 Hz, 2H), 4.29 (d, J = 6.3 Hz, 2H); 13C{1H} NMR (75 MHz, CDCl3) δ mixture of rotamers: 164.5, 160.9, 146.0, 129.2, 128.3, 127.3, 115.3, 115.2, 45.3, 41.8; HRMS (ESI-TOF) (m/z): [M + H]+ calcd for C8H11N2O 151.0871; found 151.12318. The title compound was synthesized from (±)-2-aminobutan-1-ol (106.04 μL, 1.121 mmol) and sodium borohydride (42.40 mg, 1.121 mmol) according to general procedure. Colourless oil (118 mg, 90% yield); 1H-NMR (300 MHz, CDCl3) δ (ppm) major rotamer: 8.18 (d, J = 1.2 Hz, 1H), 6.07 (br, 1H), 3.91–3.92 (m, 1H), 3.47–3.71 (m, 2H), 1.45–1.61 (m, 2H), 0.94 (t, J = 7.4 Hz, 3H), minor rotamer: 8.02 (d, J = 11.9 Hz, 1H), 6.36 (br, 1H), 3.47–3.71 (m, 2H), 3.27–3.29 (m, 1H), 1.45–1.61 (m, 1H), 0.94 (t, J = 7.4 Hz, 3H); 13C{1H} NMR (75 MHz, CDCl3) δ mixture of rotamers: 165.6, 162.3, 64.7, 64.2, 56.8, 24.5, 24.0, 10.46; HRMS (ESI-TOF) (m/z): [M + Na]+ calcd for C5H11NO2Na 140.0687; found 140.06829.
The title compound was synthesized from (±)-2-aminobutan-1-ol (106.04 μL, 1.121 mmol) and sodium borohydride (42.40 mg, 1.121 mmol) according to general procedure. Colourless oil (118 mg, 90% yield); 1H-NMR (300 MHz, CDCl3) δ (ppm) major rotamer: 8.18 (d, J = 1.2 Hz, 1H), 6.07 (br, 1H), 3.91–3.92 (m, 1H), 3.47–3.71 (m, 2H), 1.45–1.61 (m, 2H), 0.94 (t, J = 7.4 Hz, 3H), minor rotamer: 8.02 (d, J = 11.9 Hz, 1H), 6.36 (br, 1H), 3.47–3.71 (m, 2H), 3.27–3.29 (m, 1H), 1.45–1.61 (m, 1H), 0.94 (t, J = 7.4 Hz, 3H); 13C{1H} NMR (75 MHz, CDCl3) δ mixture of rotamers: 165.6, 162.3, 64.7, 64.2, 56.8, 24.5, 24.0, 10.46; HRMS (ESI-TOF) (m/z): [M + Na]+ calcd for C5H11NO2Na 140.0687; found 140.06829. The title compound was synthesized from allyl amine (131.06 μL, 1.751 mmol) and sodium borohydride (66.24 mg, 1.751 mmol) according to general procedure. Colourless oil (128 mg, 86% yield); 1H-NMR (300 MHz, CDCl3) δ (ppm) major rotamer: 8.19 (s, 1H), 5.77–5.86 (m, 2H), 5.12–5.22 (m, 2H), 3.90 (t, J = 5.6 Hz, 2H), minor rotamer: 8.02 (d, J = 12.0 Hz, 1H), 5.77–5.86 (m, 2H), 5.12–5.22 (m, 2H), 3.83 (t, J = 3.2 Hz, 2H); 13C{1H} NMR (75 MHz, CDCl3) δ mixture of rotamers: 159.5, 132.0, 115.4, 39.0, 28.2. The analytical data was found to be consistent with literature.47
The title compound was synthesized from allyl amine (131.06 μL, 1.751 mmol) and sodium borohydride (66.24 mg, 1.751 mmol) according to general procedure. Colourless oil (128 mg, 86% yield); 1H-NMR (300 MHz, CDCl3) δ (ppm) major rotamer: 8.19 (s, 1H), 5.77–5.86 (m, 2H), 5.12–5.22 (m, 2H), 3.90 (t, J = 5.6 Hz, 2H), minor rotamer: 8.02 (d, J = 12.0 Hz, 1H), 5.77–5.86 (m, 2H), 5.12–5.22 (m, 2H), 3.83 (t, J = 3.2 Hz, 2H); 13C{1H} NMR (75 MHz, CDCl3) δ mixture of rotamers: 159.5, 132.0, 115.4, 39.0, 28.2. The analytical data was found to be consistent with literature.47 The title compound was synthesized from propargyl amine (116.27 μL, 1.815 mmol) and sodium borohydride (68.66 mg, 1.815 mmol) according to general procedure. Colourless oil (119 mg, 79% yield); 1H-NMR (300 MHz, CDCl3) δ (ppm) major rotamer: 8.18 (s, 1H), 5.97 (br, 1H), 4.08–4.11 (m, 2H), 2.25 (t, J = 2.5 Hz, 1H), minor rotamer: 8.13 (d, J = 11.9 Hz, 1H) 4.0–4.03 (m, 2H), 2.35 (t, J = 2.5 Hz, 1H); 13C{1H} NMR (75 MHz, CDCl3) δ mixture of rotamers: 164.4, 160.8, 78.8, 73.0, 71.8, 31.4, 27.8. The analytical data was found to be consistent with literature.46
The title compound was synthesized from propargyl amine (116.27 μL, 1.815 mmol) and sodium borohydride (68.66 mg, 1.815 mmol) according to general procedure. Colourless oil (119 mg, 79% yield); 1H-NMR (300 MHz, CDCl3) δ (ppm) major rotamer: 8.18 (s, 1H), 5.97 (br, 1H), 4.08–4.11 (m, 2H), 2.25 (t, J = 2.5 Hz, 1H), minor rotamer: 8.13 (d, J = 11.9 Hz, 1H) 4.0–4.03 (m, 2H), 2.35 (t, J = 2.5 Hz, 1H); 13C{1H} NMR (75 MHz, CDCl3) δ mixture of rotamers: 164.4, 160.8, 78.8, 73.0, 71.8, 31.4, 27.8. The analytical data was found to be consistent with literature.46 The title compound was synthesized from aniline (98.03 μL, 1.073 mmol) and sodium borohydride (40.59 mg, 1.073 mmol) according to general procedure. Colourless oil (108 mg, 83% yield); 1H-NMR (300 MHz, DMSO-d6) δ (ppm) major rotamer: 10.17 (br, 1H), 8.26 (d, J = 1.8 Hz, 1H), 7.58 (d, J = 7.5 Hz, 2H), 7.31 (t, J = 7.8 Hz, 3H), minor rotamer: 8.78 (d, J = 10.9 Hz, 1H), 7.19 (d, J = 7.5 Hz, 2H), 7.04–7.09 (m, 3H); 13C{1H} NMR (75 MHz, CDCl3) δ mixture of rotamers: 162.8, 159.1, 136.8, 136.7, 129.7, 129.1, 125.3, 124.8, 120.0, 118.8. The analytical data was found to be consistent with literature.48
The title compound was synthesized from aniline (98.03 μL, 1.073 mmol) and sodium borohydride (40.59 mg, 1.073 mmol) according to general procedure. Colourless oil (108 mg, 83% yield); 1H-NMR (300 MHz, DMSO-d6) δ (ppm) major rotamer: 10.17 (br, 1H), 8.26 (d, J = 1.8 Hz, 1H), 7.58 (d, J = 7.5 Hz, 2H), 7.31 (t, J = 7.8 Hz, 3H), minor rotamer: 8.78 (d, J = 10.9 Hz, 1H), 7.19 (d, J = 7.5 Hz, 2H), 7.04–7.09 (m, 3H); 13C{1H} NMR (75 MHz, CDCl3) δ mixture of rotamers: 162.8, 159.1, 136.8, 136.7, 129.7, 129.1, 125.3, 124.8, 120.0, 118.8. The analytical data was found to be consistent with literature.48 The title compound was synthesized from p-toluidine (100 mg, 0.933 mmol) and sodium borohydride (35.29 mg, 0.933 mmol) according to general procedure. Brown crystalline solid (90 mg, 71% yield); m.p. 45 °C; 1H-NMR (300 MHz, CDCl3) δ (ppm) mixture of rotamers: 8.55 (d, J = 11.4 Hz, 1H), 8.27 (d, J = 1.6 Hz, 1H), 8.07 (br, 1H), 7.35 (d, J = 8.4 Hz, 2H), 7.07 (t, J = 7.8 Hz, 4H), 6.91 (d, J = 8.3 Hz, 2H), 2.26/2.24 (each s, 6H); 13C{1H} NMR (75 MHz, CDCl3) δ mixture of rotamers: 162.9, 159.1, 135.1, 134.4, 134.3, 134.1, 130.2, 129.5, 120.0, 119.1, 20.9, 20.8. The analytical data was found to be consistent with literature.46
The title compound was synthesized from p-toluidine (100 mg, 0.933 mmol) and sodium borohydride (35.29 mg, 0.933 mmol) according to general procedure. Brown crystalline solid (90 mg, 71% yield); m.p. 45 °C; 1H-NMR (300 MHz, CDCl3) δ (ppm) mixture of rotamers: 8.55 (d, J = 11.4 Hz, 1H), 8.27 (d, J = 1.6 Hz, 1H), 8.07 (br, 1H), 7.35 (d, J = 8.4 Hz, 2H), 7.07 (t, J = 7.8 Hz, 4H), 6.91 (d, J = 8.3 Hz, 2H), 2.26/2.24 (each s, 6H); 13C{1H} NMR (75 MHz, CDCl3) δ mixture of rotamers: 162.9, 159.1, 135.1, 134.4, 134.3, 134.1, 130.2, 129.5, 120.0, 119.1, 20.9, 20.8. The analytical data was found to be consistent with literature.46 The title compound was synthesized from 4-chloroaniline (100 mg, 0.783 mmol) and sodium borohydride (29.62, 0.783 mmol) according to general procedure. Brown crystalline solid (66 mg, 54% yield); m.p. 84 °C; 1H-NMR (300 MHz, DMSO-d6) δ (ppm) major rotamer: 10.33 (br, 1H), 8.27 (d, J = 1.8 Hz, 1H), 7.58–7.62 (m, 2H), 7.34–7.39 (m, 2H), minor rotamer: 10.20–10.24 (br, 1H), 8.77 (d, J = 10.8 Hz, 1H), 7.34–7.39 (m, 2H), 7.21 (d, J = 8.8 Hz, 2H); 13C{1H} NMR (75 MHz, DMSO-d6) δ mixture of rotamers: 163.0, 160.2, 137.6, 129.7, 129.2, 127.6, 121.1, 119.4. The analytical data was found to be consistent with literature.48
The title compound was synthesized from 4-chloroaniline (100 mg, 0.783 mmol) and sodium borohydride (29.62, 0.783 mmol) according to general procedure. Brown crystalline solid (66 mg, 54% yield); m.p. 84 °C; 1H-NMR (300 MHz, DMSO-d6) δ (ppm) major rotamer: 10.33 (br, 1H), 8.27 (d, J = 1.8 Hz, 1H), 7.58–7.62 (m, 2H), 7.34–7.39 (m, 2H), minor rotamer: 10.20–10.24 (br, 1H), 8.77 (d, J = 10.8 Hz, 1H), 7.34–7.39 (m, 2H), 7.21 (d, J = 8.8 Hz, 2H); 13C{1H} NMR (75 MHz, DMSO-d6) δ mixture of rotamers: 163.0, 160.2, 137.6, 129.7, 129.2, 127.6, 121.1, 119.4. The analytical data was found to be consistent with literature.48 The title compound was synthesized from 4-fluoroaniline (85.25 μL, 0.90 mmol) and sodium borohydride (34.04 mg, 0.90 mmol) according to general procedure. Brown crystalline solid (116 mg, 93% yield); m.p. 52–55 °C; 1H-NMR (300 MHz, DMSO-d6) δ (ppm) major rotamer: 10.23 (br, 1H), 8.25 (d, J = 1.9 Hz, 1H), 7.57–7.62 (m, 2H), 7.12–7.20 (m, 2H), minor rotamer: 10.11–10.15 (br, 1H), 8.69 (d, J = 10.9 Hz, 1H), 7.12–7.20 (m, 4H); 13C{1H} NMR (75 MHz, DMSO-d6) δ mixture of rotamers: 163.1, 160.1, 159.9, 156.9, 135.1, 121.3, 121.2, 119.9, 119.8, 116.6, 116.3, 116.0, 115.7. The analytical data was found to be consistent with literature.48
The title compound was synthesized from 4-fluoroaniline (85.25 μL, 0.90 mmol) and sodium borohydride (34.04 mg, 0.90 mmol) according to general procedure. Brown crystalline solid (116 mg, 93% yield); m.p. 52–55 °C; 1H-NMR (300 MHz, DMSO-d6) δ (ppm) major rotamer: 10.23 (br, 1H), 8.25 (d, J = 1.9 Hz, 1H), 7.57–7.62 (m, 2H), 7.12–7.20 (m, 2H), minor rotamer: 10.11–10.15 (br, 1H), 8.69 (d, J = 10.9 Hz, 1H), 7.12–7.20 (m, 4H); 13C{1H} NMR (75 MHz, DMSO-d6) δ mixture of rotamers: 163.1, 160.1, 159.9, 156.9, 135.1, 121.3, 121.2, 119.9, 119.8, 116.6, 116.3, 116.0, 115.7. The analytical data was found to be consistent with literature.48 The title compound was synthesized from 3-trifluoromethylaniline (77.51 μL, 0.620 mmol) and sodium borohydride (23.45 mg, 0.620 mmol) according to general procedure. Brown crystalline solid (106 mg, 90% yield); m.p. 40 °C; 1H-NMR (300 MHz, DMSO-d6) δ (ppm) major rotamer: 10.54 (br, 1H), 8.35 (d, J = 1.7 Hz, 1H), 8.08 (s, 1H), 7.76 (d, J = 8.2 Hz, 1H), 7.51–7.60 (m, 1H), 7.43 (d, J = 7.0 Hz, 1H), minor rotamer: 10.35–10.39 (br, 1H), 8.92 (d, J = 10.8 Hz, 1H), 7.51–7.60 (m, 3H), 7.43 (d, J = 7.0 Hz, 1H); 13C{1H} NMR (75 MHz, DMSO-d6) δ mixture of rotamers: 163.2, 160.6, 139.8, 139.4, 131.0, 130.6, 130.2, 129.8, 126.2, 123.1, 122.6, 121.3, 120.4, 120.3, 115.7, 115.6, 114.0, 113.9. The analytical data was found to be consistent with literature.48
The title compound was synthesized from 3-trifluoromethylaniline (77.51 μL, 0.620 mmol) and sodium borohydride (23.45 mg, 0.620 mmol) according to general procedure. Brown crystalline solid (106 mg, 90% yield); m.p. 40 °C; 1H-NMR (300 MHz, DMSO-d6) δ (ppm) major rotamer: 10.54 (br, 1H), 8.35 (d, J = 1.7 Hz, 1H), 8.08 (s, 1H), 7.76 (d, J = 8.2 Hz, 1H), 7.51–7.60 (m, 1H), 7.43 (d, J = 7.0 Hz, 1H), minor rotamer: 10.35–10.39 (br, 1H), 8.92 (d, J = 10.8 Hz, 1H), 7.51–7.60 (m, 3H), 7.43 (d, J = 7.0 Hz, 1H); 13C{1H} NMR (75 MHz, DMSO-d6) δ mixture of rotamers: 163.2, 160.6, 139.8, 139.4, 131.0, 130.6, 130.2, 129.8, 126.2, 123.1, 122.6, 121.3, 120.4, 120.3, 115.7, 115.6, 114.0, 113.9. The analytical data was found to be consistent with literature.48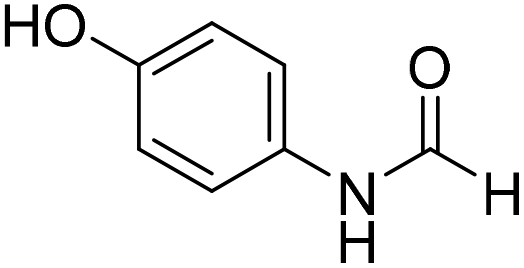 The title compound was synthesized from 4-aminophenol (100 mg, 0.916 mmol) and sodium borohydride (34.65 mg, 0.916 mmol) according to general procedure. Brown solid (92 mg, 73% yield); m.p. 123 °C; 1H-NMR (300 MHz, DMSO-d6) δ (ppm) major rotamer: 9.89 (br, 1H), 9.23 (s, 1H), 8.15 (d, J = 1.98 Hz, 1H), 7.36 (d, J = 8.85 Hz, 2H), 6.69 (d, J = 8.88 Hz, 2H), minor rotamer: 9.82–9.86 (br, 1H), 9.26 (s, 1H), 8.49 (d, J = 11.19 Hz, 1H), 6.97 (d, J = 8.73 Hz, 2H), 6.69 (d, J = 8.88 Hz, 2H); 13C{1H} NMR (75 MHz, DMSO-d6) δ mixture of rotamers: 163.0, 159.2, 154.6, 153.9, 130.4, 130.1, 121.2, 120.6, 116.2, 115.6. The analytical data was found to be consistent with literature.49
The title compound was synthesized from 4-aminophenol (100 mg, 0.916 mmol) and sodium borohydride (34.65 mg, 0.916 mmol) according to general procedure. Brown solid (92 mg, 73% yield); m.p. 123 °C; 1H-NMR (300 MHz, DMSO-d6) δ (ppm) major rotamer: 9.89 (br, 1H), 9.23 (s, 1H), 8.15 (d, J = 1.98 Hz, 1H), 7.36 (d, J = 8.85 Hz, 2H), 6.69 (d, J = 8.88 Hz, 2H), minor rotamer: 9.82–9.86 (br, 1H), 9.26 (s, 1H), 8.49 (d, J = 11.19 Hz, 1H), 6.97 (d, J = 8.73 Hz, 2H), 6.69 (d, J = 8.88 Hz, 2H); 13C{1H} NMR (75 MHz, DMSO-d6) δ mixture of rotamers: 163.0, 159.2, 154.6, 153.9, 130.4, 130.1, 121.2, 120.6, 116.2, 115.6. The analytical data was found to be consistent with literature.49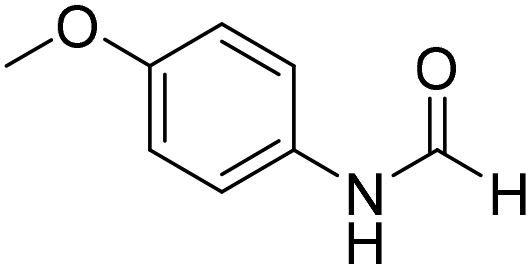 The title compound was synthesized from p-anisidine (100 mg, 0.811 mmol) and sodium borohydride (30.71 mg, 0.811 mmol) according to general procedure. Brown solid (90 mg, 73% yield); m.p. 48–50 °C; 1H-NMR (300 MHz, DMSO-d6) δ (ppm) major rotamer: 10.04 (br, 1H), 8.19 (d, J = 1.95 Hz, 1H), 7.50 (d, J = 9.03 Hz, 2H), 6.88 (d, J = 9.06 Hz, 2H), 3.71 (s, 3H), minor rotamer: 9.90 (br, 1H), 8.59 (d, J = 11.13 Hz, 1H), 7.11 (d, J = 8.94 Hz, 2H), 6.88 (d, J = 9.06 Hz, 2H), 3.71 (s, 3H); 13C{1H} NMR (75 MHz, DMSO-d6) δ mixture of rotamers: 163.0, 159.5, 156.4, 155.8, 131.9, 121.0, 120.1, 115.0, 114.4, 55.7, 55.6. The analytical data was found to be consistent with literature.50
The title compound was synthesized from p-anisidine (100 mg, 0.811 mmol) and sodium borohydride (30.71 mg, 0.811 mmol) according to general procedure. Brown solid (90 mg, 73% yield); m.p. 48–50 °C; 1H-NMR (300 MHz, DMSO-d6) δ (ppm) major rotamer: 10.04 (br, 1H), 8.19 (d, J = 1.95 Hz, 1H), 7.50 (d, J = 9.03 Hz, 2H), 6.88 (d, J = 9.06 Hz, 2H), 3.71 (s, 3H), minor rotamer: 9.90 (br, 1H), 8.59 (d, J = 11.13 Hz, 1H), 7.11 (d, J = 8.94 Hz, 2H), 6.88 (d, J = 9.06 Hz, 2H), 3.71 (s, 3H); 13C{1H} NMR (75 MHz, DMSO-d6) δ mixture of rotamers: 163.0, 159.5, 156.4, 155.8, 131.9, 121.0, 120.1, 115.0, 114.4, 55.7, 55.6. The analytical data was found to be consistent with literature.50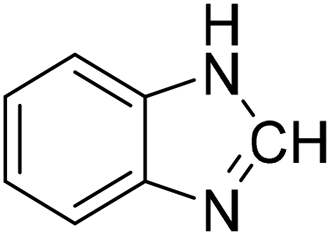 The title compound was synthesized from o-phenylene diamine (100 mg, 0.924 mmol) and sodium borohydride (34.95 mg, 0.924 mmol) according to general procedure. Brown solid (78 mg, 72% yield); m.p. 128–130 °C; 1H-NMR (300 MHz, CDCl3) δ (ppm) 8.10 (s, 1H), 7.65–7.68 (m, 2H), 7.28–7.31 (m, 2H); 13C{1H} NMR (75 MHz, CDCl3) δ 140.5, 137.6, 122.9, 115.5. The analytical data was found to be consistent with literature.51
The title compound was synthesized from o-phenylene diamine (100 mg, 0.924 mmol) and sodium borohydride (34.95 mg, 0.924 mmol) according to general procedure. Brown solid (78 mg, 72% yield); m.p. 128–130 °C; 1H-NMR (300 MHz, CDCl3) δ (ppm) 8.10 (s, 1H), 7.65–7.68 (m, 2H), 7.28–7.31 (m, 2H); 13C{1H} NMR (75 MHz, CDCl3) δ 140.5, 137.6, 122.9, 115.5. The analytical data was found to be consistent with literature.51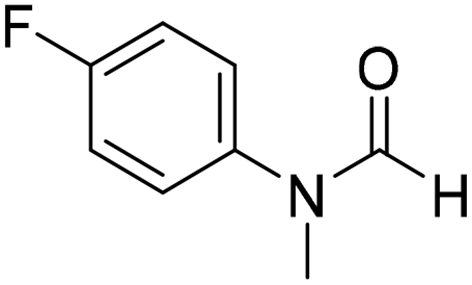 The title compound was synthesized from 4-fluoro-N-methylaniline (288.46 μL, 2.39 mmol) and sodium borohydride (90.41 mg, 2.39 mmol) according to general procedure. Brown oil (272 mg, 74% yield); 1H-NMR (300 MHz, DMSO-d6) δ (ppm) major rotamer: 8.42 (s, 1H), 7.35–7.39 (m, 2H), 7.22–7.27 (m, 2H), 3.18 (s, 3H), minor rotamer: 8.31 (s, 1H), 7.47–7.51 (m, 2H), 7.22–7.27 (m, 2H), 3.28 (s, 3H); 13C{1H} NMR (75 MHz, CDCl3) δ mixture of rotamers: 162.6, 162.2, 159.4, 138.3, 138.2, 125.6, 125.5, 124.6, 124.5, 116.6, 116.3, 116.0, 115.7, 37.0, 32.5, 29.7. The analytical data was found to be consistent with literature.52
The title compound was synthesized from 4-fluoro-N-methylaniline (288.46 μL, 2.39 mmol) and sodium borohydride (90.41 mg, 2.39 mmol) according to general procedure. Brown oil (272 mg, 74% yield); 1H-NMR (300 MHz, DMSO-d6) δ (ppm) major rotamer: 8.42 (s, 1H), 7.35–7.39 (m, 2H), 7.22–7.27 (m, 2H), 3.18 (s, 3H), minor rotamer: 8.31 (s, 1H), 7.47–7.51 (m, 2H), 7.22–7.27 (m, 2H), 3.28 (s, 3H); 13C{1H} NMR (75 MHz, CDCl3) δ mixture of rotamers: 162.6, 162.2, 159.4, 138.3, 138.2, 125.6, 125.5, 124.6, 124.5, 116.6, 116.3, 116.0, 115.7, 37.0, 32.5, 29.7. The analytical data was found to be consistent with literature.52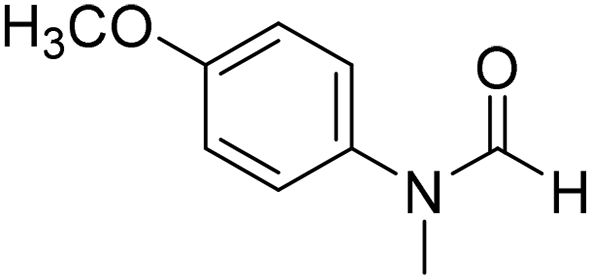 The title compound was synthesized from 4-methoxy-N-methylaniline (300 mg, 2.19 mmol) and sodium borohydride (82.84 mg, 2.19 mmol) according to general procedure. Brown oil (325 mg, 90% yield); 1H-NMR (300 MHz, CDCl3) δ (ppm) mixture of rotamers: 8.34 (s, 1H), 7.10 (d, J = 8.4 Hz, 2H), 6.93 (d, J = 8.5 Hz, 2H), 3.82 (s, 3H), 3.27 (s, 3H); 13C{1H} NMR (75 MHz, DMSO-d6) δ mixture of rotamers: 162.7, 162.4, 157.8, 157.3, 135.5, 133.8, 125.3, 124.3, 115.0, 114.3, 55.7, 55.6, 36.9, 32.1. The analytical data was found to be consistent with literature.21
The title compound was synthesized from 4-methoxy-N-methylaniline (300 mg, 2.19 mmol) and sodium borohydride (82.84 mg, 2.19 mmol) according to general procedure. Brown oil (325 mg, 90% yield); 1H-NMR (300 MHz, CDCl3) δ (ppm) mixture of rotamers: 8.34 (s, 1H), 7.10 (d, J = 8.4 Hz, 2H), 6.93 (d, J = 8.5 Hz, 2H), 3.82 (s, 3H), 3.27 (s, 3H); 13C{1H} NMR (75 MHz, DMSO-d6) δ mixture of rotamers: 162.7, 162.4, 157.8, 157.3, 135.5, 133.8, 125.3, 124.3, 115.0, 114.3, 55.7, 55.6, 36.9, 32.1. The analytical data was found to be consistent with literature.21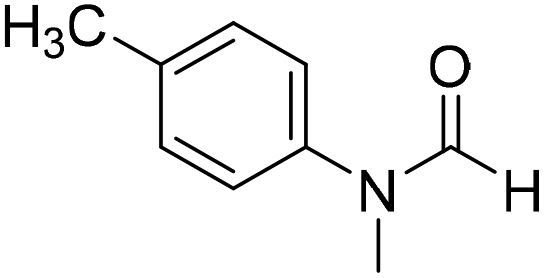 The title compound was synthesized from N,4-dimethylaniline (313.15 μL, 2.47 mmol) and sodium borohydride (93.65 mg, 2.47 mmol) according to general procedure. Brown oil (332 mg, 90% yield); 1H-NMR (300 MHz, CDCl3) δ (ppm) major rotamer: 8.41 (s, 1H), 7.20 (d, J = 8.04 Hz, 2H), 7.05 (d, J = 8.37 Hz, 2H), 3.28 (s, 3H), 2.35 (s, 3H), minor rotamer: 8.32 (s, 1H), 7.20 (d, J = 8.04 Hz, 2H), 7.05 (d, J = 8.37 Hz, 2H), 3.28 (s, 3H), 2.35 (s, 3H); 13C{1H} NMR (75 MHz, CDCl3) δ mixture of rotamers: 159.9, 137.2, 133.9, 127.7, 120.1, 29.7, 27.2, 18.4. The analytical data was found to be consistent with literature.21
The title compound was synthesized from N,4-dimethylaniline (313.15 μL, 2.47 mmol) and sodium borohydride (93.65 mg, 2.47 mmol) according to general procedure. Brown oil (332 mg, 90% yield); 1H-NMR (300 MHz, CDCl3) δ (ppm) major rotamer: 8.41 (s, 1H), 7.20 (d, J = 8.04 Hz, 2H), 7.05 (d, J = 8.37 Hz, 2H), 3.28 (s, 3H), 2.35 (s, 3H), minor rotamer: 8.32 (s, 1H), 7.20 (d, J = 8.04 Hz, 2H), 7.05 (d, J = 8.37 Hz, 2H), 3.28 (s, 3H), 2.35 (s, 3H); 13C{1H} NMR (75 MHz, CDCl3) δ mixture of rotamers: 159.9, 137.2, 133.9, 127.7, 120.1, 29.7, 27.2, 18.4. The analytical data was found to be consistent with literature.21Author contributions
DM conceived the idea and proposed and planned the study. AK did most of the experiments and compiled the data for analysis. PS and NS did some of the experiments. YK performed the LCMS experiments. DM analysed the compiled data. DM and AK drafted the manuscript. All authors read and approved the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
DM and PS acknowledge the financial support from DBT as a grant (BT/PR28766/BRB/10/1701/2018) and fellowship respectively. DM, AK and NS acknowledge the financial support from DBT under Translational Research Program (TRP) at THSTI.Notes and references
- R. A. Forsch and A. Rosowsky, J. Org. Chem., 1985, 50, 2582–2583 CrossRef CAS.
- R. Hett, Q. K. Fang, Y. Gao, S. A. Wald and C. H. Senanayake, Org. Process Res. Dev., 1998, 2, 96–99 CrossRef CAS.
- G. Ma, M. Zancanella, Y. Oyola, R. D. Richardson, J. W. Smith and D. Romo, Org. Lett., 2006, 8, 4497–4500 CrossRef CAS PubMed.
- Y. S. Sadanandam, K. R. Reddy and S. S. H. Qadri, J. Plant Dis. Prot., 1982, 89, 518–522 CAS.
- Y. Wen, Y. Xiong, L. Chang, J. Huang, X. Liu and X. Feng, J. Org. Chem., 2007, 72, 7715–7719 CrossRef CAS PubMed.
- S. B. Jagtap and S. B. Tsogoeva, Chem. Commun., 2006, 4747–4749 RSC.
- R. Sun, X. Yang, Q. Li, K. Xu, J. Tang, X. Zheng, M. Yuan, H. Fu, R. Li and H. Chen, Org. Lett., 2019, 21, 9425–9429 CrossRef CAS PubMed.
- B.-C. Chen, M. S. Bednarz, R. Zhao, J. E. Sundeen, P. Chen, Z. Shen, A. P. Skoumbourdis and J. C. Barrish, Tetrahedron Lett., 2000, 41, 5453–5456 CrossRef CAS.
- A. Jackson and O. Meth-Cohn, J. Chem. Soc., Chem. Commun., 1995, 1319 RSC.
- C.-H. Chang, H. J. Tsai, Y.-Y. Huang, H.-Y. Lin, L.-Y. Wang, T.-S. Wu and F. F. Wong, Tetrahedron, 2013, 69, 1378–1386 CrossRef CAS.
- Y. Han and L. Cai, Tetrahedron Lett., 1997, 38, 5423–5426 CrossRef CAS.
- P. Patil, M. Ahmadian-Moghaddam and A. Domling, Green Chem., 2020, 22, 6902–6911 RSC.
- N. Liu, F. Chao, M.-G. Liu, N.-Y. Huang, K. Zou and L. Wang, J. Org. Chem., 2019, 84, 2366–2371 CrossRef CAS PubMed.
- J. Bruffaerts, N. von Wolff, Y. Diskin-Posner, Y. Ben-David and D. Milstein, J. Am. Chem. Soc., 2019, 141, 16486–16493 CrossRef CAS PubMed.
- H. Yu, Z. Wu, Z. Wei, Y. Zhai, S. Ru, Q. Zhao, J. Wang, S. Han and Y. Wei, Commun. Chem., 2019, 2(15), 1–7 CAS.
- N. Ortega, C. Richter and F. Glorius, Org. Lett., 2013, 15, 1776–1779 CrossRef CAS PubMed.
- H. Tumma, N. Nagaraju and K. V. Reddy, J. Mol. Catal. A: Chem., 2009, 310, 121–129 CrossRef CAS.
- Y. Chen, J. Mao, R. Shen, D. Wang, Q. Peng, Z. Yu, H. Guo and W. He, Nano Res., 2017, 10, 890–896 CrossRef CAS.
- Z. Li, Z. Yu, X. Luo, C. Li, H. Wu, W. Zhao, H. Li and S. Yang, RSC Adv., 2020, 10, 33972–34005 RSC.
- Y. Shen, Q. Zheng, Z.-N. Chen, D. Wen, J. H. Clark, X. Xu and T. Tu, Angew. Chem., Int. Ed., 2021, 60, 4125–4132 CrossRef CAS PubMed.
- Q. Zhang, X.-T. Lin, N. Fukaya, T. Fujitani, K. Sato and J.-C. Choi, Green Chem., 2020, 22, 8414–8422 RSC.
- X. Jiang, Z. Huang, M. Makha, C.-X. Du, D. Zhao, F. Wang and Y. Li, Green Chem., 2020, 22, 5317–5324 RSC.
- T.-X. Zhao, G.-W. Zhai, J. Liang, P. Li, X.-B. Hu and Y.-T. Wu, Chem. Commun., 2017, 53, 8046–8049 RSC.
- P. Daw, S. Chakraborty, G. Leitus, Y. Diskin-Posner, Y. Ben-David and D. Milstein, ACS Catal., 2017, 7, 2500–2504 CrossRef CAS.
- F. D. Bobbink, S. Das and P. J. Dyson, Nat. Protoc., 2017, 12, 417–428 CrossRef CAS PubMed.
- L. Zhang, Z. Han, X. Zhao, Z. Wang and K. Ding, Angew. Chem., 2015, 127, 6284–6287 CrossRef.
- A. Tlili, E. Blondiaux, X. Frogneux and T. Cantat, Green Chem., 2015, 17, 157–168 RSC.
- C. Federsel, A. Boddien, R. Jackstell, R. Jennerjahn, P. J. Dyson, R. Scopelliti, G. Laurenczy and M. Beller, Angew. Chem., Int. Ed., 2010, 49, 9777–9780 CrossRef CAS PubMed.
- Q. Liu, L. Wu, R. Jackstell and M. Beller, Nat. Commun., 2015, 6(5933), 1–15 Search PubMed.
- A. Otto, T. Grube, S. Schiebahn and D. Stolten, Energy Environ. Sci., 2015, 8, 3283–3297 RSC.
- C. Maeda, Y. Miyazaki and T. Ema, Catal. Sci. Technol., 2014, 4, 1482–1497 RSC.
- M. Aresta and A. Dibenedetto, Dalton Trans., 2007, 2975–2992 RSC.
- L. Banfi, E. Narisano, R. Riva, N. Stiasni, M. Hiersemann, T. Yamada and T. Tsubo, Sodium Borohydride: Encyclopedia of Reagents for Organic Synthesis, 2014, https://onlinelibrary.wiley.com/doi/epdf/10.1002/047084289X.rs052.pub3 Search PubMed.
- Y. Zhu, L. Ouyang, H. Zhong, J. Liu, H. Wang, H. Shao, Z. Huang and M. Zhu, Angew. Chem., Int. Ed., 2020, 59, 8623–8629 CrossRef CAS PubMed.
- L. Hao, H. Zhang, X. Luo, C. Wu, Y. Zhao, X. Liu, X. Gao, Y. Chen and Z. Liu, J. CO2 Util., 2017, 22, 208–211 CrossRef CAS.
- Q. Zou, G. Long, T. Zhao and X. Hu, Green Chem., 2020, 22, 1134–1138 RSC.
- I. Knopf and C. C. Cummins, Organometallics, 2015, 34, 1601–1603 CrossRef CAS.
- J. Yin, J. Zhang, C. Cai, G.-J. Deng and H. Gong, Org. Lett., 2019, 21, 387–392 CrossRef CAS PubMed.
- T. B. Nguyen, J. Sorres, M. Q. Tran, L. Ermolenko and A. Al-Mourabit, Org. Lett., 2012, 14, 3202–3205 CrossRef CAS PubMed.
- R. M. Lanigan, P. Starkov and T. D. Sheppard, J. Org. Chem., 2013, 78, 4512–4523 CrossRef CAS PubMed.
- P. Starkov and T. D. Sheppard, Org. Biomol. Chem., 2011, 9, 1320–1323 RSC.
- T. M. E. Dine, D. Evans, J. Rouden and J. Blanchet, Chem. - Eur. J., 2016, 22, 5894–5898 CrossRef PubMed.
- R. Quintanilla-Licea, J. F. Colunga-Valladares, A. Caballero-Quintero, C. Rodriguez-Padilla, R. Tamez-Guerra, R. Gomez-Flores and N. Waksman, Molecules, 2002, 7, 662–673 CrossRef CAS.
- V. Pace, K. de la Vega-Hernandez, E. Urban and T. Langer, Org. Lett., 2016, 18, 2750–2753 CrossRef CAS PubMed.
- B.-X. Leong, Y.-C. Teo, C. Condamines, M.-C. Yang, M.-D. Su and C.-W. So, ACS Catal., 2020, 10, 14824–14833 CrossRef CAS.
- S. N. Rao, D. C. Mohan and S. Adimurthy, Org. Lett., 2013, 15, 1496–1499 CrossRef CAS PubMed.
- B. Thiedemann, C. M. L. Schmitz and A. Staubitz, J. Org. Chem., 2014, 79, 10284–10295 CrossRef CAS PubMed.
- D.-Z. Lin and J.-M. Huang, Org. Lett., 2018, 20, 2112–2115 CrossRef CAS PubMed.
- M. Hosseini-Sarvari and H. Sharghi, J. Org. Chem., 2006, 71, 6652–6654 CrossRef CAS PubMed.
- C. Zhang, Z. Xu, T. Shen, G. Wu, L. Zhang and N. Jiao, Org. Lett., 2012, 14, 2362–2365 CrossRef CAS PubMed.
- Z. G. Wang, X. H. Cao, Y. Yang and M. Lu, Synth. Commun., 2015, 45, 1476–1483 CrossRef CAS.
- X. Ma, S. Deng and Q. Song, Org. Chem. Front., 2018, 5, 3505–3509 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1ra04848a |
| This journal is © The Royal Society of Chemistry 2021 |