
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSolvent-free synthesis of symmetric methylene diesters via direct reaction of aromatic carboxylates with 1,n-dihaloalkanes†
Lin Bai *a,
Shenglong Dingb and
Xiaofang Maa
*a,
Shenglong Dingb and
Xiaofang Maa
aInstitute of Green Chemistry Experiment and Teaching, Lanzhou City University, Lanzhou, Gansu 730070, China. E-mail: bailin@lzcu.edu.cn
bCollege of Chemistry and Chemical Engineering, Lanzhou University, Lanzhou, Gansu 730000, China
First published on 25th August 2021
Abstract
An efficient methodology for the synthesis of symmetrical methylene diesters was developed through direct reaction of various aromatic carboxylates with 1,n-dihaloalkanes under solvent-free conditions. This strategy offers a high product yield, facile work-up and purification, and an environmentally friendly approach to obtain long-chain methylene carboxylate scaffolds with increased diversity.
Introduction
1,n-Methylene diesters are widely used as organic synthesis intermediates, and also in the pharmaceutical, combinatorial chemistry and materials industries due to their unique structural characteristics. Many natural and multifunctional compounds can be synthesized by the reaction of long-chain methylene diesters of carboxylic acids with other reagents.1–3 In polymer materials science, symmetric methylene diesters are frequently used to prepare high molecular-weight linear aliphatic polyesters, poly(methylene terephthalate),4 and crystalline polymer nanorods.5 In particular, various methylene-bridged diester derivatives have been found to possess versatile physiological activities such as anti-inflammatory and analgesic properties, and have potential for use as antitumor agents and other functions.6–9A variety of synthetic methods have been developed for the preparation of methylene diesters. The traditional route for the synthesis of methylene diesters is generally the direct esterification of alkanediols with carboxylic acid,10 the diesterification of diols through cyclic ketene acetal intermediates,11 or the acylation of alkanediols with acyl chloride12 or carboxylic anhydrides.13
The nucleophilic substitution of carboxylic acids with dihaloalkanes is a novel method for the synthesis of methylene diesters.14 The formation of aromatic methylene diesters by heating a mixture of a carboxylic acid, dihaloalkane and triethylamine has been previously reported.15 Improvements have also been reported, by replacement of the triethylamine by the quaternary alkyl ammonium ion (n-Bu4N+, anion exchange resin) in dipolar aprotic media.16 The reaction rate and yields increase quite dramatically for dibromoalkanes in NMP, HMPA, KF or acetonitrile.17 Dichloro-methane is a relatively inexpensive and inert compound that has a long reaction time,18 the microwave-assisted reaction of heterocyclic carboxylic acids with dichloromethane is recommended as a convenient method for the preparation of a variety of methylene diesters.19 Although these methods provide reliable routes for the preparation of methylene diester compounds, most of them require harsh conditions, long reaction times and the use of environmentally harmful solvents. Today, environmentally friendly synthesis and methodologies have become very important and widespread, owing to the drive towards the use of green chemistry methods. In particular, room-temperature ionic liquid has been successfully utilized for the formation of methylene diarylcarboxylates, in which the ionic liquid acts as a prime material, solvent and catalyst.20 The microwave-promoted diesterification of aromatic carboxylate with 1,4-dibromobutane offers an atom-efficient and environmentally friendly approach to synthesize butamethylene-diesters with excellent yields.21 The synthetic methods of dihaloalkanes used as methylene synthons are summarized in Scheme 1.
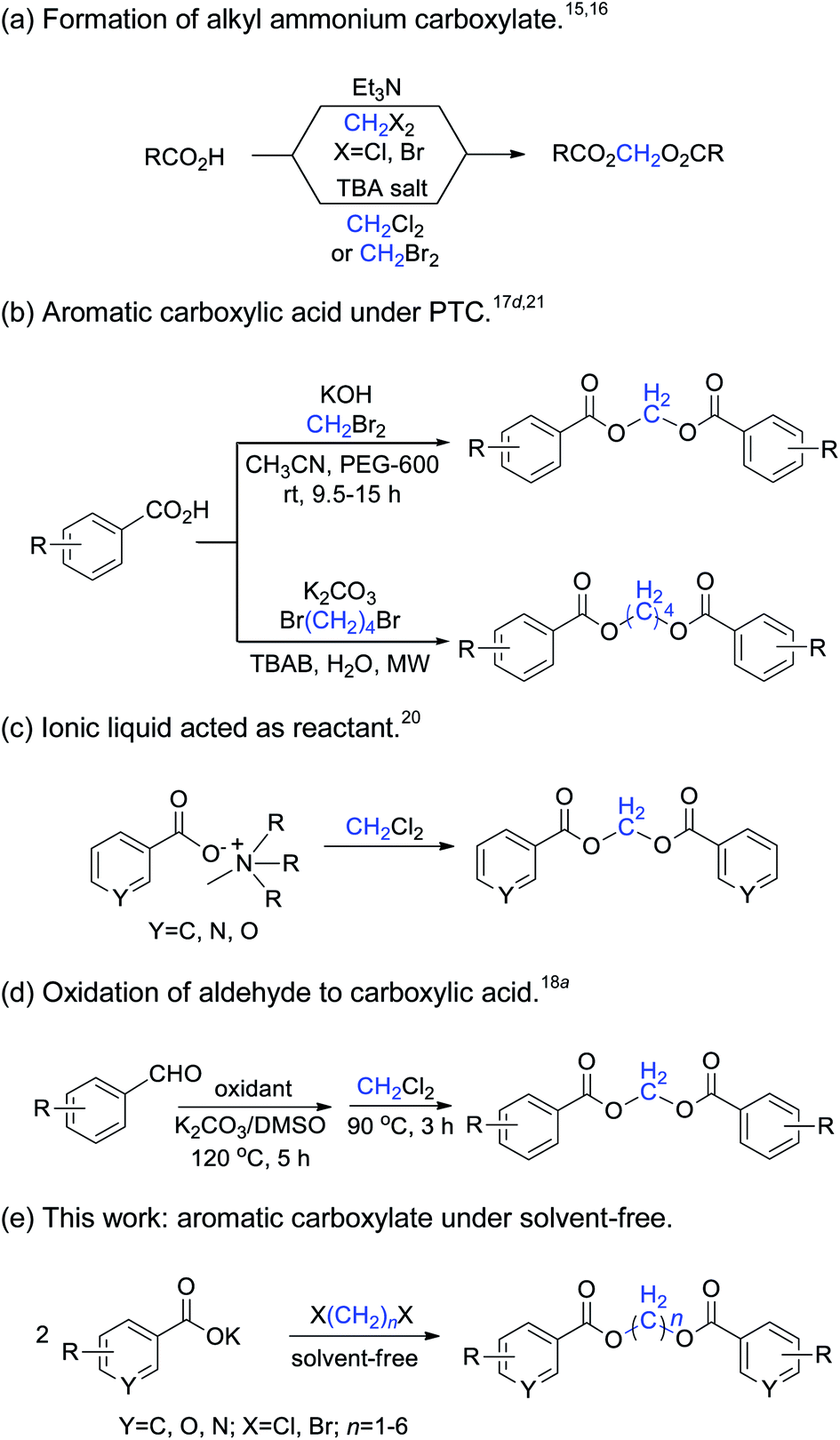 | ||
| Scheme 1 Methods used for the synthesis of symmetric methylene diesters. | ||
This ongoing interest in dihaloalkanes inspired us to investigate their novel application in organic synthetic chemistry.21,22 Herein, we wish to explore and develop a convenient, efficient and environmentally friendly method to synthesize symmetric methylene diesters from various carboxylates and 1,n-dihaloalkanes under solvent-free conditions.
Results and discussion
The diesterification of two equivalent carboxylates with 1,n-dihaloalkanes was performed in a sealed vessel (microwave synthesis system accessory) under conventional heating conditions, in which the volatilization of substrates was avoided and the reaction temperature was easily controlled resulting in a better reproducibility. To elucidate the reaction, potassium benzoate and CH2Br2 were chosen for the model reaction to optimize the diesterification conditions. Table 1 presents some selected results, which illustrate the effects of the phase transfer catalysts (PTCs), temperature, and reaction time on the transformation outcome.
|
||||
|---|---|---|---|---|
| Entry | PTC | Time (h) | Temp. (°C) | Yieldb (%) |
| a Reaction conditions: potassium benzoate (10 mmol), CH2Br2 (5 mmol), PTC (1 mmol).b Average isolated yield from two reactions. | ||||
| 1 | TBAB | 3 | 100 | 76 |
| 2 | TBAB | 3 | 110 | 87 |
| 3 | TBAB | 3 | 120 | 93 |
| 4 | TBAB | 3 | 130 | 89 |
| 5 | TBAB | 3 | 140 | 85 |
| 6 | TBAB | 2 | 120 | 71 |
| 7 | TBAB | 4 | 120 | 89 |
| 8 | TBAB | 5 | 120 | 87 |
| 9 | TBAC | 3 | 120 | 80 |
| 10 | TMBAC | 3 | 120 | 83 |
| 11 | HDTMAB | 3 | 120 | 71 |
| 12 | PEG-400 | 3 | 120 | 25 |
| 13 | PEG-1000 | 3 | 120 | 38 |
| 14 | 0 | 3 | 120 | Trace |
As it stands, the solubility of organic substrates in the heterogeneous phase can be enhanced with the aid of a phase transfer catalyst (PTC). Quaternary-ammonium salts such as tetrabutylammonium chloride (TBAC), tetrabutylammonium bromide (TBAB), trimethylbenzylammonium chloride (TMBAC), hexadecyltrimethylammonium bromide (HDTMAB) and polyethylene glycol (PEG-400 and PEG-1000) were applied herein as PTCs in the model reaction. Table 1 indicates that using quaternary ammonium salts as catalysts gave significantly better results than those obtained for the polyethylene glycols (PEGs). The reaction proceeded very slowly or not at all in the absence of PTC. Among the quaternary ammonium salts, TBAB gave an excellent yield and enhanced reaction efficiency at synthetically useful levels.
With the optimal reaction conditions in hand, a series of 1,n-dihaloalkanes (X(CH2)nX, X = Br, Cl; n = 1–6) and various aromatic carboxylates were employed to explore the scope and limitations of this diesterification reaction. As summarized in Table 2, the reactivity was observed with both electron-donating and electron-withdrawing groups on the aromatic carboxylate rings, and the reaction can tolerate many functional groups, including methyl, methoxy, halo and nitro groups. In general, the reaction between the dihaloalkanes and aromatic carboxylate derivatives with electron-donating groups such as CH3 and OCH3 gave the desired products in higher yields. Halogen substituents, such as Br in the para position were well tolerated, leading to a series of halo-substituted products under the optimized reaction conditions. Higher yields were obtained when an electron-withdrawing NO2 group was present in benzoate, which reacted with the dihaloalkanes and gave the desired products. Gratifyingly, the steric hinderance of substituents in the ortho-position had no appreciable impact on the reaction efficiency, as reflected by the yields of 20, 21 and 22. It is worth highlighting that for the reaction of cinnamate with dihaloalkanes all of the products (23–26) were trans-isomers, which were identified using 1H NMR and infrared spectroscopy (IR).
|
|
|---|---|
| a Conditions: potassium carboxylate (10 mmol), dihaloalkane (5 mmol), TBAB (1 mmol), 120 °C for 3 h.b Average isolated yield from two reactions of the dibromoalkanes.c Average isolated yield from two reactions of the dichloroalkanes.d 105 °C for 3 h.e 110 °C for 3 h. | |
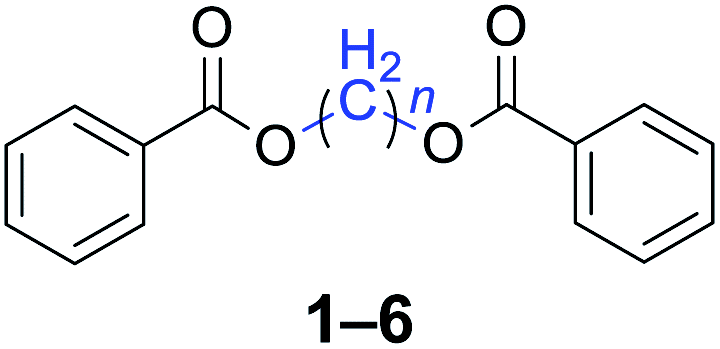 |
n = 1, 1, 93%b (91%)c |
| n = 2, 2, 88%b (85%)c | |
| n = 3, 3, 89%b (87%)c | |
| n = 4, 4, 85%b (84%)c | |
| n = 5, 5, 90%b (91%)c | |
| n = 6, 6, 92%b (90%)c | |
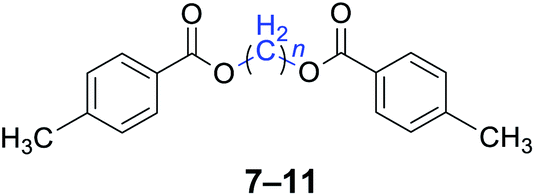 |
n = 1, 7, 96%b (93%)c |
| n = 3, 8, 92%b (87%)c | |
| n = 4, 9, 95%b (91%)c | |
| n = 5, 10, 90%b (86%)c | |
| n = 6, 11, 92%b (87%)c | |
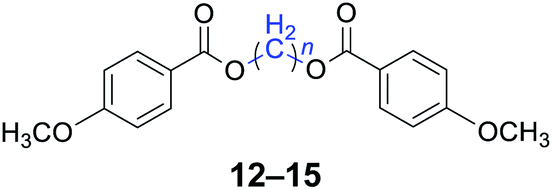 |
n = 1, 12, 96%b (93%)c |
| n = 4, 13, 98%b (96%)c | |
| n = 5, 14, 97%b (94%)c | |
| n = 6, 15, 96%b (92%)c | |
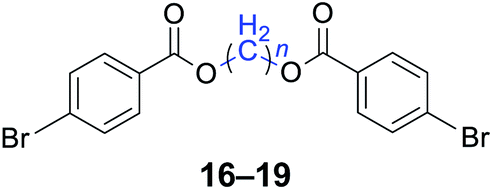 |
n = 1, 16, 95%b (93%)c |
| n = 3, 17, 91%b (87%)c | |
| n = 4, 18, 88%b (85%)c | |
| n = 5, 19, 85%b (81%)c | |
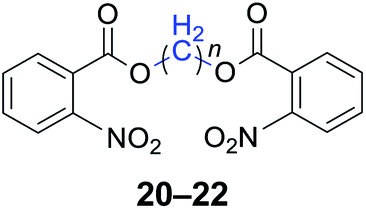 |
n = 1, 20, 91%b (88%)c |
| n = 2, 21, 93%b (90%)c | |
| n = 4, 22, 94%b (91%)c | |
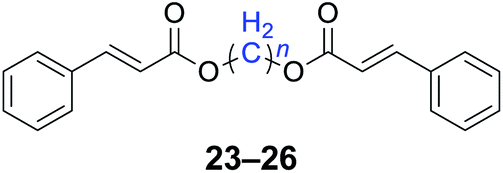 |
n = 1, 23, 92%b (87%)c |
| n = 2, 24, 89%b (85%)c | |
| n = 3, 25, 87%b (81%)c | |
| n = 4, 26, 90%b (84%)c | |
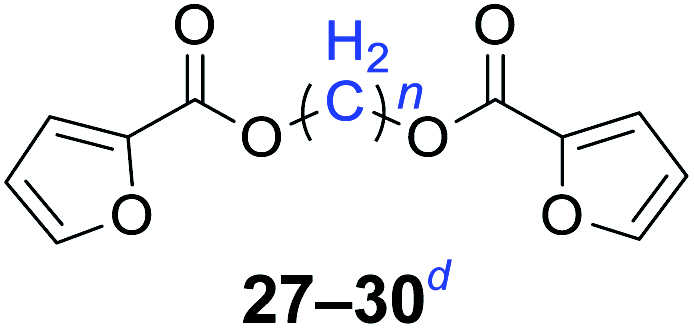 |
n = 1, 27, 87%b (81%)c |
| n = 2, 28, 84%b (77%)c | |
| n = 4, 29, 81%b (74%)c | |
| n = 6, 30, 78%b (76%)c | |
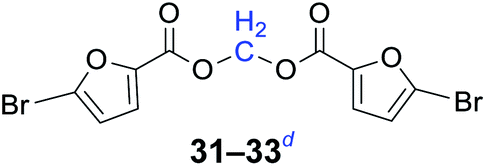 |
n = 1, 31, 93%b (89%)c |
| n = 3, 32, 95%b (90%)c | |
| n = 5, 33, 96%b (88%)c | |
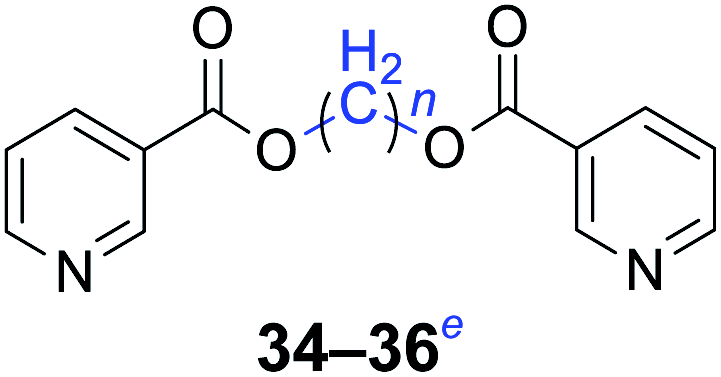 |
n = 1, 34, 68%b (72%)c |
| n = 2, 35, 62%b (74%)c | |
| n = 4, 36, 60%b (70%)c | |
Furthermore, heteroaromatic carboxylates with furyl and pyridyl moieties were investigated, they also formed the corresponding products with satisfactory yields. The diesterification of potassium 3-pyridinecarboxylate with 1,n-dihaloalkanes resulted in lower yields compared with other products, due in part to the decreased electron density of nicotinoyl carboxylate induced by the effect of the heteroatom (34–36). When the temperature was raised to 120 °C, the yields of the reaction of 2-furoyl carboxylate, 3-pyridoyl carboxylate and dibromoalkanes decreased. Therefore, reducing the reaction temperature could give a better result. The transformation yields with using benzoate derivatives were generally better than those obtained by employing heteroaryl carboxylates.
Compared with dibromoalkane, dichloroalkane is a simpler and cheaper halogenated molecule that is widely used as a solvent in the laboratory. Dichloroalkane is a relatively inert compound, its electrophilicity is enhanced under solvent-free and phase transfer catalysis conditions. It can be seen from Table 2 that a wide array of 1,n-dihaloalkanes can be efficiently transformed into the corresponding products in high yields. In all cases examined, the reactions proceeded very cleanly, essentially forming pure symmetrical methylene diesters in a short reaction time. The analytically pure product was obtained by filtration and recrystallization. The procedure can readily be scaled up to a quantity of 10 g, and there was no decrease in the yield.
In addition, aliphatic carboxylates, such as potassium acetate, potassium chloroacetate, potassium stearate, potassium phenylacetate and potassium pyruvate did not react with 1,n-dibromoalkanes under the model conditions. A reaction employing dichloromethane as an alternative substrate for dibromoalkane was also performed, but the target product of methylene dibenzoate was not formed.
On the basis of the above described results and the previously published literature,19,23 a plausible mechanism of phase transfer catalysis for this diesterification is illustrated in Scheme 2.
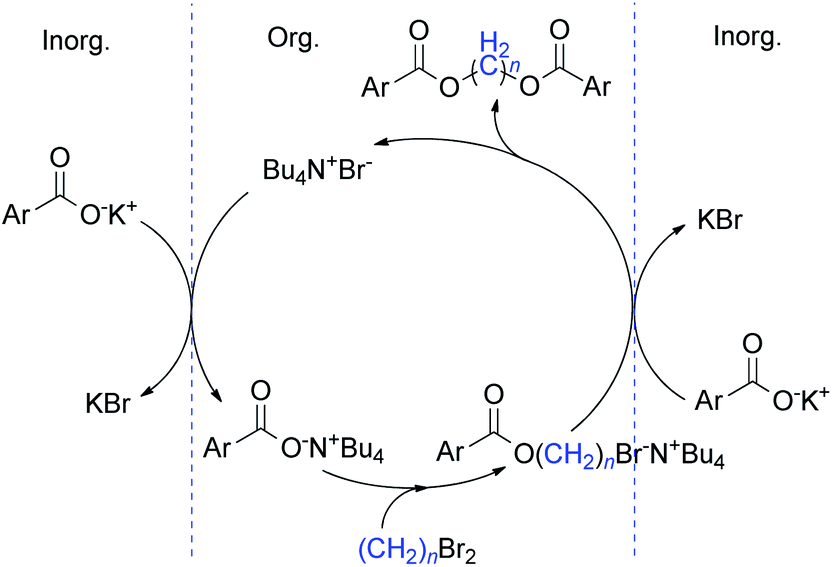 | ||
| Scheme 2 Mechanism proposed for the diesterification. | ||
Initially, the carboxylate salt was converted into ArCOO−N+Bu4 by anion exchange with the TBAB and transferred to the organic phase. Subsequently, the negatively charged oxygen belonging to the carboxylate anion underwent a nucleophilic attack on the considerably electrophilic carbon of the methylene bromide, the methylene diester was generated with the liberation of TBAB which established the catalytic cycle of the PTC. This direct diesterification most probably proceeded via successive SN2 processes.
Conclusions
In this work, we have developed a practical methodology for the synthesis of methylene-bridged biaryl carboxylates by the direct reaction of the aromatic carboxylate with 1,n-dihaloalkanes under solvent-free conditions. This method has the advantages of a short reaction time and higher yields, as well as being environmentally friendly. It provides a novel route for increasing the synthetic diversification of the methylene diester products with simple commercial reagents. From a synthetic perspective, it also provides a feasible method for the synthesis of long-chain symmetrical methylene diesters and is expected to be used in the total synthesis of natural products and multifunctional compounds.Experimental section
Materials and methods
All carboxylic acids, phase transfer catalysts and bases were obtained from Sinopharm Chemical Reagent Co., Ltd. and were analytically pure. Dihaloalkanes were obtained from Aladdin with a purity of more than 98% and were used as received. The potassium salt of carboxylic acid was prepared by dissolving carboxylic acid (2 mol) and potassium carbonate (1 mol) in water, then evaporating the solvent and drying the salt in an oven at 100 °C to obtain a constant weigh. Melting points were determined on a WRS-1A digital instrument and the temperature was uncorrected. Fourier transform infrared spectroscopy (FT-IR) spectra were recorded on a Nicolet 5700 spectrometer. Samples were prepared by grinding the products with KBr and compressing the mixture to form discs. 1H, and 13C spectra were recorded on an Agilent-DD2 400 MHz spectrometer in acetone-d6 using trimethylsilane (TMS) as an internal standard at room temperature (1H NMR: acetone-d6 at 2.05 ppm; and 13C NMR: acetone-d6 at 29.0 ppm), unless otherwise indicated. High resolution mass spectra (HRMS) were determined on a Thermo Orbitrap Elite instrument (or Thermoscientific Q Exactive) using the electrospray ionization mode.General synthetic procedures
In an 80 mL Pyrex vessel were placed aromatic carboxylate (10 mmol), dihaloalkane (5 mmol), TBAB (1 mmol), and a magnetic stir bar. The vessel was sealed with a locking cover and placed into the oil bath. Once the temperature reached 120 °C, the reaction mixture was held at this temperature for 3 h. After cooling the mixture to room temperature, the reaction vessel was opened and the contents poured into a beaker with 50 mL of cold water. After stirring, the product was chilled and precipitation occurred, the product was then washed with water and filtered under vacuum. The product was recrystallized from ethanol, giving the analytically pure products.Selected spectral data
Methylene dibenzoate (1), white crystal, (1.192 g, yield-93%), mp 97–98 °C. 1H NMR (400 MHz, acetone-d6) δ (ppm): 8.05–8.06 (m, 4H), 7.68 (m, 2H), 7.52–7.67 (m, 4H), 6.26 (s, 2H). 13C NMR (100 MHz, acetone-d6) δ (ppm): 205.97, 164.79, 133.81, 129.66, 129.08, 128.72, 80.54. HRMS calcd for C15H12O4Na [M + Na]+, 279.0634, found 279.0626.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was financially supported by the Doctor Research Initiation Fund Project of Lanzhou City University (Grant Number LZCU-BS2019-04).Notes and references
- P. Wessig and A. Matthes, J. Am. Chem. Soc., 2011, 133, 2642–2650 CrossRef CAS PubMed.
- F. D. Lewis, J. D. Oxman, L. L. Gibson, H. L. Hampsch and S. L. Quillen, J. Am. Chem. Soc., 1986, 108, 3005–3015 CrossRef CAS.
- P. Benitez, A. Delgado, J.-A. Farrera and J. M. Ribó, Synth. Commun., 1997, 27, 1697–1702 CrossRef CAS.
- (a) G. C. East and M. Morshed, Polymer, 1982, 23, 1555–1557 CrossRef CAS; (b) N. Lacoudre, A. Leborgne, M. Sepulchre, N. Spassky, J. Djonlagic and M. Jacovic, Mukromol. Chem., 1986, 187, 341–350 CrossRef CAS; (c) A. L. Cimecioglu, G. C. East, M. Morshed and S. H. Zeronian, J. Polym. Sci., Polym. Chem. Ed., 1988, 26, 2129–2139 CrossRef CAS; (d) A. L. Cimecioglu and G. C. East, Makromol. Chem., Rapid Commun., 1989, 10, 319–324 CrossRef CAS; (e) S. Paszkiewicz, A. Szymczyk, D. Pawlikowska, I. Irska, I. Taraghi, R. Pilawka, J. Gu, X. Li, Y. Tu and E. Piesowicz, RSC Adv., 2017, 7, 41745–41754 RSC.
- R. O. Al-Kaysi, R. J. Dillon, J. M. Kaiser, L. J. Mueller, G. Guirado and C. J. Bardeen, Macromolecules, 2007, 40, 9040–9044 CrossRef CAS.
- C. M. Svahn, F. Merenyi, L. Karlson, L. Widlund and M. Grallst, J. Med. Chem., 1986, 29, 448–453 CrossRef CAS PubMed.
- A. Nudelman, I. Levovich, S. M. Cutts, D. R. Phillips and A. Rephaeli, J. Med. Chem., 2005, 48, 1042–1054 CrossRef CAS PubMed.
- L. Lazzarato, M. Donnola, B. Rolando, K. Chegaev, E. Marini, C. Cena, A. Di Stilo, R. Fruttero, S. Biondi, E. Ongini and A. Gasco, J. Med. Chem., 2009, 52, 5058–5068 CrossRef CAS.
- A. Korolyov, S. Dorbes, J. Azéma, B. Guidetti, M. Danel, D. Lamoral-Theys, T. Gras, J. Dubois, R. Kiss, R. Martino and M. Malet-Martino, Bioorg. Med. Chem., 2010, 18, 8537–8548 CrossRef CAS.
- (a) J. Otera and J. Nishikido, Esterification. Methods, Reactions, and Applications, Wiley-VCH, Weinheim, Germany, 2nd edn, 2010 Search PubMed; (b) Z.-W. Guo and J. S. Charles, J. Am. Chem. Soc., 1988, 110, 1999–2001 CrossRef CAS; (c) N.-X. Wang, J. Baoji Coll. Arts Sci., Nat. Sci., 2000, 20, 36–38 CAS; (d) P. Ciuffreda, S. Casati and E. Santaniello, Tetrahedron Lett., 2003, 44, 3663–3665 CrossRef CAS.
- Z. Wu, R. R. Stanley and C. U. Pittman Jr, J. Org. Chem., 1999, 64, 8386–8395 CrossRef CAS PubMed.
- (a) Y. G. Gololobov and M. A. Galkina, Russ. Chem. Bull., 1995, 44, 760–761 CrossRef; (b) H. Ogawa, M. Amano and T. Chihara, Chem. Commun., 1998, 4, 495–496 RSC.
- Z. Maeno, S. Yamada, T. Mitsudome, T. Mizugaki and K. Jitsukawa, Green Chem., 2017, 19, 2612–2619 RSC.
- E. Haslam, Tetrahedron, 1980, 36, 2409–2433 CrossRef CAS.
- (a) R. H. Mills, M. W. Farrar and O. J. Weinkauff, Chem. Ind., 1962, 40, 2144 Search PubMed; (b) C. K. Lyon and V. H. Garrett, J. Am. Oil Chem. Soc., 1970, 47, 145–146 CrossRef CAS.
- (a) K. Holmberg and B. Hansen, Tetrahedron Lett., 1975, 16, 2303–2306 CrossRef; (b) K. Arimoto and D. F. Zinkel, J. Am. Oil Chem. Soc., 1982, 59, 166–168 CrossRef CAS; (c) D. G. Salunkhe, M. H. Jagdale, M. M. Salunkhe and P. P. Wadgaonkar, Bull. Soc. Chim. Belg., 1987, 96, 247–248 CrossRef CAS.
- (a) A. L. Cimecioglu and G. C. East, Makromol. Chem., Rapid Commun., 1987, 8, 141–146 CrossRef CAS; (b) J. E. Shaw and D. C. Kunerth, J. Org. Chem., 1974, 39, 1968–1970 CrossRef CAS; (c) J. H. Clark, J. Emsley and O. P. A. Hoyte, J. Chem. Soc., Perkin Trans. 1, 1977, 9, 1091–1094 RSC; (d) B. P. Kavitake, M. M. Salunkhe and P. P. Wadgaonkar, Synth. Commun., 1997, 27, 1703–1710 CrossRef CAS.
- (a) F. Lin, Q. Feng, X. Cui and Q. Song, RSC Adv., 2013, 3, 20246–20253 RSC; (b) J. Xiong, G. Zhong, L. Zou and Y. Liu, ChemistrySelect, 2018, 3, 8291–8293 CrossRef CAS; (c) S. Wang, Z. Fu, Z. Huang, Y. Jiang, S. Guo and H. Cai, Chin. Chem. Lett., 2019, 30, 1173–1177 CrossRef CAS.
- Y. Ma, G. Wu, N. Jiang, S. Ge, Q. Zhou and B. Sun, Chin. Chem. Lett., 2015, 26, 81–84 CrossRef CAS.
- (a) N. V. Likhanova, I. V. Lijanova, L. P. M. Alvarado, M. M. Garcia, S. Hernandez-Ortega and O. O. Xometl, Curr. Org. Chem., 2013, 17, 79–82 CrossRef CAS; (b) A. H. Jadhav, K. Lee, S. Koo and J. G. Seo, RSC Adv., 2015, 5, 26197–26208 RSC; (c) D. Gómora-Herrera, I. V. Lijanova, O. Olivares-Xometl, A. Toscano and N. V. Likhanova, Can. J. Chem., 2017, 95, 744–750 CrossRef.
- S.-L. Ding, L. Bai, Y. Cong and T. Cheng, Synth. Commun., 2018, 48, 2559–2565 CrossRef CAS.
- L. Bai, X.-Q. Lu and P.-P. Mao, Journal of Gansu Normal Colleges, 2019, 24, 15–18 Search PubMed.
- E. V. Dehmlow and S. S. Dehmlow, Phase Transfer Catalysis, Verlag Chemie, Weinheim, 2nd edn, 1983 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental installation, characterization data for all the synthesized compounds, and scanned copies of their respective 1H-NMR, 13C-NMR and HRMS spectra. See DOI: 10.1039/d1ra04814g |
| This journal is © The Royal Society of Chemistry 2021 |