
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceEfficient kinetic resolution in the asymmetric transfer hydrogenation of 3-aryl-indanones: applications to a short synthesis of (+)-indatraline and a formal synthesis of (R)-tolterodine†
Songsoon Parkab and
Hyeon-Kyu Lee *ab
*ab
aKorea Chemical Bank, Korea Research Institute of Chemical Technology, PO Box 107, Yuseong, Daejeon 305-600, Korea
bDepartment of Medicinal Chemistry and Pharmacology, University of Science and Technology, 113 Gwahango, Yuseong, Daejeon 305-333, Korea. E-mail: leehk@krict.re.kr
First published on 30th June 2021
Abstract
Efficient kinetic resolution (KR) occurs in asymmetric transfer hydrogenation (ATH) reactions of racemic 3-aryl-1-indanones using commercial (R,R)- or (S,S)-Ts-DENEB as a catalyst, a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :5 mixture of HCO2H and Et3N as a hydrogen source and MeOH as solvent. This process at room temperature produces near equal yields of cis-3-arylindanols with high dr and ee, and unreacted 3-arylindanones with excellent ee. Stereoselective transformations of 3-arylindanols and 3-arylindanones, generated by using the ATH-KR protocol, were carried out to form (+)-indatraline and synthetically valuable (R)-6-methyl-4-phenylcoumarine, which is a key intermediate in the preparation of (R)-tolterodine, (S)-4-aryl-3,4-dihydroquinoline-2(1H)-one and (S)-4-aryl-3,4-dihydroisoquinoline-1(2H)-one.
:5 mixture of HCO2H and Et3N as a hydrogen source and MeOH as solvent. This process at room temperature produces near equal yields of cis-3-arylindanols with high dr and ee, and unreacted 3-arylindanones with excellent ee. Stereoselective transformations of 3-arylindanols and 3-arylindanones, generated by using the ATH-KR protocol, were carried out to form (+)-indatraline and synthetically valuable (R)-6-methyl-4-phenylcoumarine, which is a key intermediate in the preparation of (R)-tolterodine, (S)-4-aryl-3,4-dihydroquinoline-2(1H)-one and (S)-4-aryl-3,4-dihydroisoquinoline-1(2H)-one.
Introduction
Indane frameworks are found in natural products that possess diverse biological activities and that serve as drug candidates.1–3 Among members of this family, 3-arylindanols and 3-arylindanones are privileged structural components of many pharmaceutical agents and key intermediates in the synthesis of a variety of bioactive compounds (Fig. 1).4–7 Typical examples of this type are (+)-indatraline used for the treatment of depression and cocaine addiction,8–10 the antihypertensive agent (+) irindalone,11 the neuroprotective agent (+)-quadrangularin A,12 (+)-isopaucifloral F used for the treatment of osteoporosis12 and α-diisoeugenol that has cytotoxic and antioxidant activities.13 An example of a bioactive indane bearing a 3-alkenyl group is (+)-multisianthol, which has antitumor activity.14,15 In addition, 3-arylindanoles and 3-arylindanones are valuable intermediates in routes for the synthesis medicinal agents.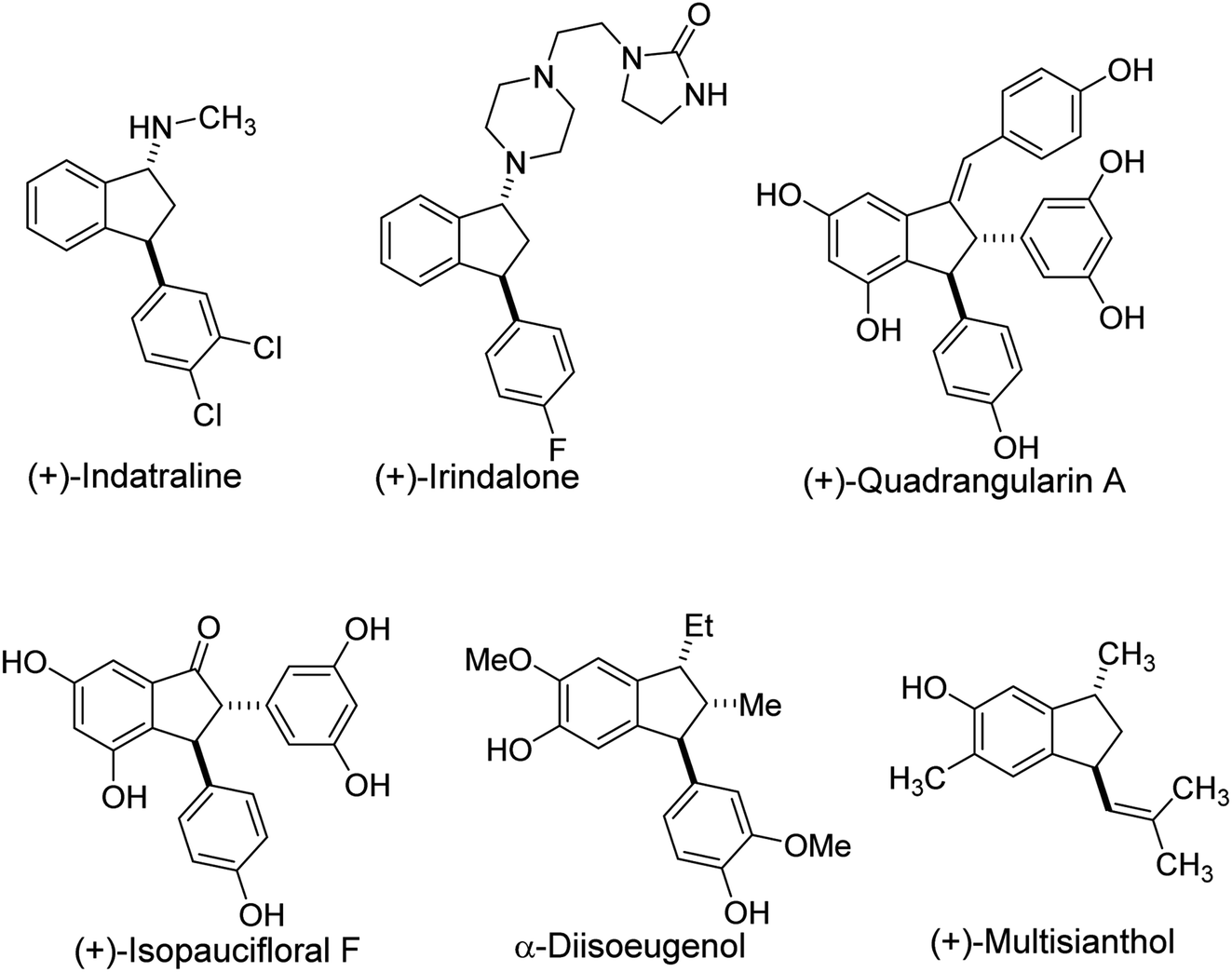 | ||
| Fig. 1 Examples of biologically active 3-(aryl)-substituted indanes. | ||
Consequently, the development of methods for convenient and stereoselective syntheses of 3-arylindanol and 3-arylindanone is an important goal in organic synthesis. Typically, enantioenriched 3-aryl-1-indanols are prepared by reduction7 (NaBH4 or K-selectride for 1,3-syn indanols) of enantioenriched 3-aryl-1-indanones. Also, Corey's oxazaborolidine-catalyzed reduction of racemic 3-aryl-1-indanones is known to produce mixtures containing almost equal amount of cis- and trans-3-aryl-1-indanols.16,17 Furthermore, resolution of racemic 3-aryl-1-indanol has been accomplished using a commercially available lipase (Novozyme 435®).18 Previous efforts have shown that enantioenriched 3-aryl-1-indanones can be prepared by intramolecular Friedel–Crafts acylation of enantioenriched 3,3-diaryl propanoic acids under strongly acidic conditions,19–22 or by Ir-catalysed asymmetric hydrogenation of 3-arylinden-1-ones (58–90% ee).23 Bakers' yeast-promoted conjugate reduction of 3-arylinden-1-ones to form enantioenriched 3-aryl-1-indanones has also been described.24,25 Recent approaches devised to generate enantioenriched 3-aryl-1-indanones rely on metal (Pd or Ni)-catalyzed asymmetric intramolecular reductive Heck reaction of 2′-halochalcones,7,26–28 and Rh-catalyzed asymmetric intramolecular 1,4-addition of aryl boronates to enones.29
Asymmetric transfer hydrogenation (ATH) reactions, using hydrogen sources other than molecular hydrogen, have proven to be among the most powerful processes for asymmetric reduction of ketones to produce enantioenriched alcohols. These processes have advantages associated with operational simplicity, ready availability of various hydrogen sources, and use of readily accessible and less sensitive catalysts.30–35 Indeed, stereoselective ATH of 1-indanones or 2-substituted-1-indanones to produce corresponding 1-indanols or 2-substituted-1-indanols, which utilize chiral transition metal (Ru, Rh) catalysts and a HCO2H/Et3N mixture as a hydrogen source, have already been described.36–38 However, no examples have been reported thus far of ATH promoted transformations of 3-aryl-1-indanones to 3-aryl-indanols having stereogenic centers at C-3. This deficiency encouraged us to explore the stereochemical outcome of ATH reactions of 3-aryl-indanones using enantioenriched chiral transition metal catalysts and a HCO2H/Et3N mixture as the hydrogen source.
Results and discussion
Previously, it was reported that ATH reaction of 3-methoxycarbonyl-1-indanone (3) with Mohar's Ru-catalyst (C4, Scheme 1d), containing benzosultam (syn-ULTAM) ligand and HCO2H/Et3N (5:2) as hydrogen source (40 °C, 6 h), produces cis-(1R,3S)-3-methoxycarbonyl-1-indanol with high levels of diastereoselectivity and enantioselectivity, owing to dynamic kinetic resolution (DKR) resulting from rapid racemization of the dually activated C-3 hydrogen (Scheme 1a).39,40 Because Mohar's catalyst C4 is not commercially available, we assessed whether the ATH reaction of 3-methoxycarbonyl-1-indanone (3) would take place efficiently employing commercial (R,R)-Ts-DENEB (C3) instead of C4 as catalyst under the same reaction conditions. The oxo-tethered Ru-catalysts (R,R)- and (S,S)-Ts-DENEB (C3) which were developed by T. Touge and T. Ikariya et al. generally showed, among the Noyori-type chiral Ru-catalysts, enhanced catalytic performance with excellent levels of stereoselectivity in the asymmetric transfer hydrogenation reactions of ketonic substrates.36,41
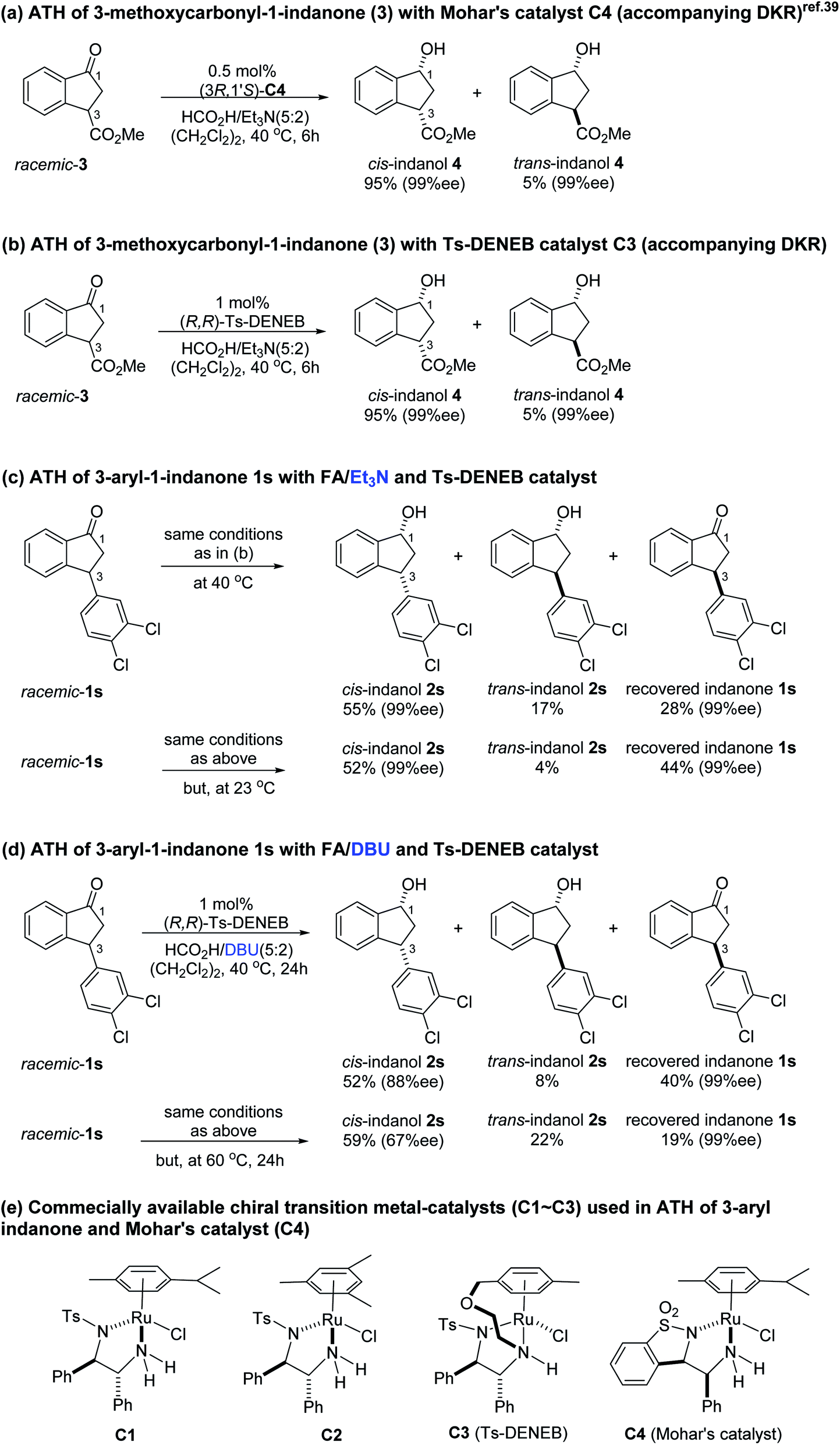 | ||
| Scheme 1 ATH reactions of 3-methoxycarbonyl-1-indanone (3) and 3-aryl-1-indanone 1s. | ||
The results show that C3 also promotes ATH reaction of 3 that forms the cis-(1R,3S)-indanol 4 with excellent levels of stereoselectivity (95%, 99% ee) accompanying DKR (Scheme 1b). This finding led us to speculate that ATH reaction of the 3-arylindanone 1s using C3 as catalyst under similar reaction conditions would also produce the corresponding 3-arylindanol stereoselectively, hopefully accompanying DKR.
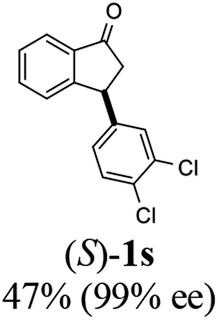
Contrary to expectation, subjection of 3-(3,4-dichlorophenyl)-1-indanone (1s) to the same conditions used for reduction of 3-methoxycarbonyl-1-indanone (3) (C3 as catalyst, FA/TEA = 5:2, 40 °C) leads to incomplete reaction (72%) and formation of a 76:24 mixture of cis (99% ee) and trans indanol 2s, and 28% of enantioenriched indanone 1s (99% ee) (Scheme 1c). In an attempt to find ATH reaction conditions which induce DKR, we employed stronger base of DBU (pKa = 24.3)42 instead of Et3N (pKa = 18.8) in the ATH reaction of 1s. However, ATH of 1s with FA/DBU (5:2) for 24 h, otherwise under the same reaction conditions, is still incomplete (60% conversion) affording 87:13 mixture of cis (87% ee) and trans indanol 2s, and 40% of unreacted indanone 1s (99% ee) (Scheme 1d). When the reaction temperature of ATH reaction of 1s with FA/DBU (5:2) was increased to 60 °C for 24 h, the conversion of the ATH reaction was increased to 81% but, dr (cis-2s: trans 2s = 73:27) and ee of cis-2s (67% ee) was rapidly decrease. Therefore, since ATH reaction of 1s in the presence of (R,R)-Ts-DENEB (C3) and FA/Et3N (5:2) as hydrogen source provided cis-indanol 2s (99% ee) and of enantioenriched indanone 1s (99% ee) in a single step we changed our attention to ATH accompanying kinetic resolution using FA/Et3N rather than attempted ATH-DKR employing FA/DBU (Scheme 1c).
To uncover conditions that would make this process more selective, ATH reaction of 1s was conducted under the same reaction conditions but at 23 °C rather than 40 °C for 24 h. Interestingly, the process was found to generate a 92:8 mixture of cis (99% ee) and trans indanol 2s in 56% yield along with 44% of enantioenriched 3-arylindanone 1s (99% ee). These results show that ATH reaction of 1s is not accompanied by DKR and that it proceeds with kinetic resolution (KR) to generate near equal amounts of enantioenriched 3-arylindanol 2s and 3-arylindanone 1s with excellent stereoselectivities for both of the cis-3-arylindanol 2s (99% ee) and recovered 3-arylindanone 1s (99% ee). Thus, the nature of 3-substituent governed lability of the proton at C-3 center controls whether or not the ATH process is attended by DKR. This is further demonstrated by the observation that treatment of enantioenriched (S)-3-methoxycarbonyl-1-indanone ((S)-3, 99% ee) with a 5:2 FA/TEA mixture in the absence of C3, as expected, does not promote formation of the reduction product 4 but instead leads to quantitative recovery of almost completely racemized indanone 3 (1.3% ee) (Scheme 2a). In contrast, reaction of enantioenriched (S)-3-arylindanone ((S)-1s) (99% ee) under the same conditions generates (S)-1s quantitatively without a noticeable decrease in enantiomeric purity (99% ee) (Scheme 2b).
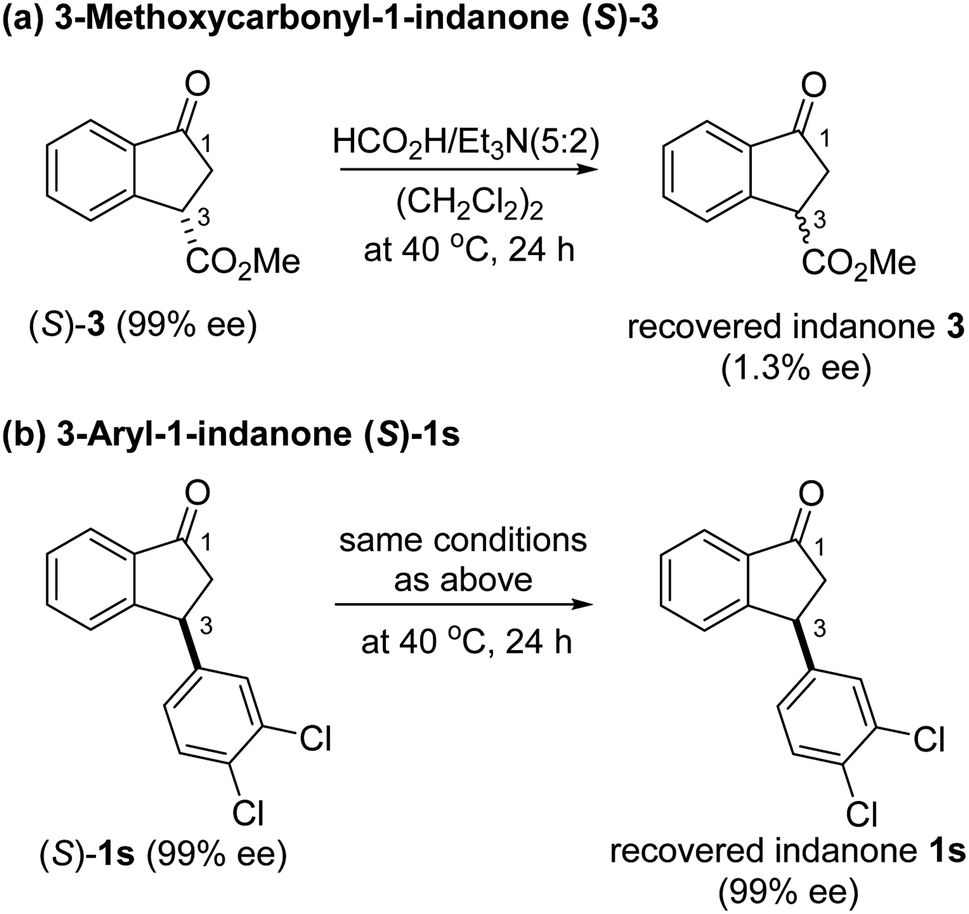 | ||
| Scheme 2 Racemization experiments of optically active 3-substituted-1-indanones. | ||
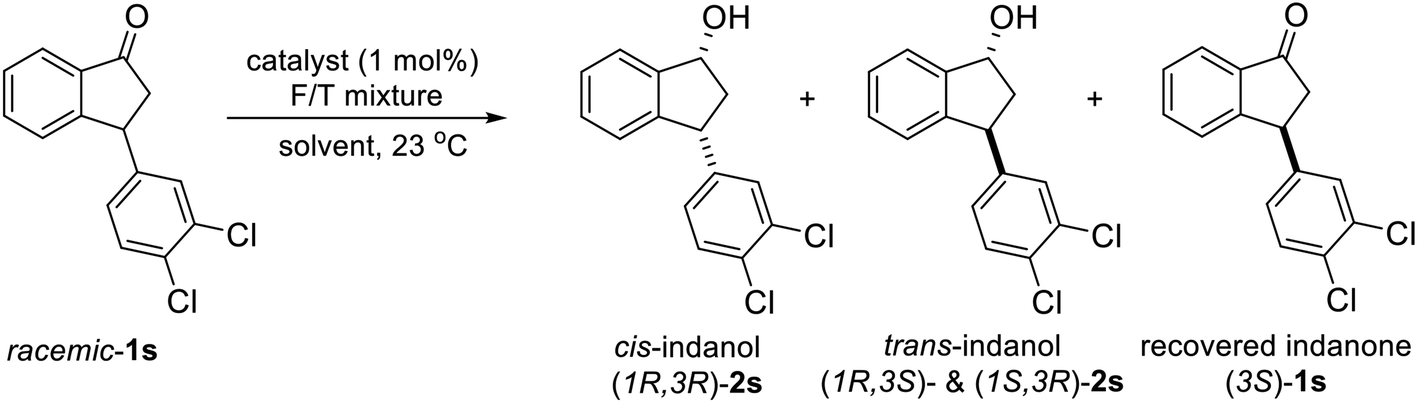
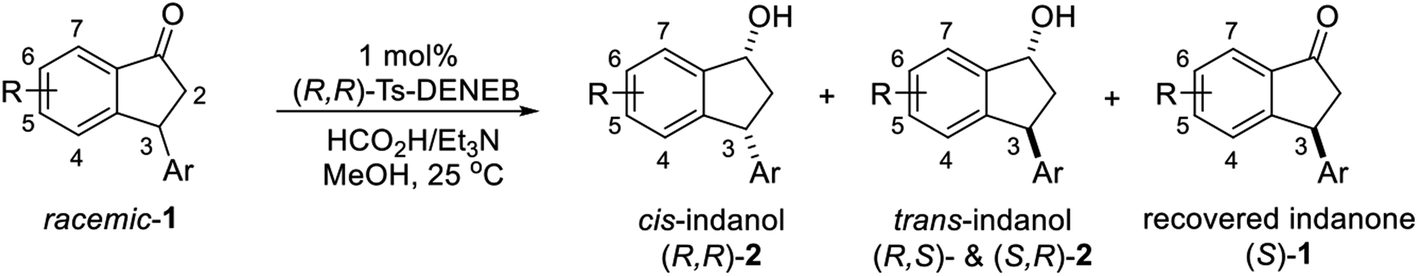
Because enantioenriched forms of variously substituted 3-arylindanols 2 and 3-arylindanones 1 are core motifs in many bioactive natural product and important intermediates in stereoselective syntheses of pharmaceuticals and biologically active compounds, we extended our study to uncover optimal conditions for ATH reactions of racemic 3-arylindanones 1 to produce 3-arylindanols and 3-arylindanones with high levels of stereoselectivity. In the first phase of this investigation, we explored the use of different commercially available chiral Ru-catalysts to promote ATH reaction of racemic 3-arylindanone 1s. ATH reaction of 1s (FA:TEA = 5:2) using Noyori catalyst (R,R)-RuCl[TsDPEN](cymene) (C1) or (R,R)-RuCl[TsDPEN](mesitylene) (C2) was found to occur for 6 h to form 3-arylindanol 2s with 53–56% conversions (entries 1 and 2, Table 1), and slightly higher conversions take place when the reaction time is extended to 24 h (65–77% conversion, entries 4 and 5). These processes produce slightly lower cis/trans ratios of 2s compared with those catalyzed by (R,R)-Ts-DENEB (C3) but the conversions of 1s to 2s in ATH reactions using C3 are nearly time independent (6 h, 56% and 24 h, 57%) (entries 3 and 6). Because longer time (>24 h) ATH reactions of 1s under acidic conditions provided by 5:2 FA:TEA are accompanied by formation of small quantities of undesired side-products (e.g., indenes resulting from dehydration of indanol 2s), the process was carried out under non-acidic conditions using 1:1 or 1:5 FA:TEA mixtures and C3 as catalyst. ATH reaction using 1:5 FA:TEA occurs in a 1s to 2s conversion of 53% after 6 h, which remains almost the same after 24 h (55%, entries 9 and 10). Moreover, no indene side-products are detected in the crude product mixture using 1H-NMR analysis. An investigation of the influence of solvents on the ATH reaction of 1s (entries 11–17) shows that reactions in the CH3CN, CH2Cl2, THF and EtOAc produce cis-indanol 2s in high ee (99% ee), but that the conversion of 1s to 2s is less than 50% after 6 h and the % ee of the recovered indanone 1s is not high (36–83% ee). However, reaction in MeOH for 6 h using 1:5 FA/TEA and C3 as the catalyst (entry 17) leads to reduction of only (3R)-1s to form (1R,3R)-2s (50%, 99% ee) and recovery of unreacted (3S)-1s (50%, 99% ee). In addition, when the time for reaction in MeOH is extended to 20 h, the yields and % ee's of (1R,3R)-2s (50%, 99% ee) and (3S)-1s (50%, 99% ee) remain the same (entry 18).
|
|||||||||
|---|---|---|---|---|---|---|---|---|---|
| Entry | Cat. | F/T ratio | Solvent | Rxn time (h) | Conv.b (%) | Indanol (2s) | Indanone (1s) | ||
| cis:transb |
ee (%) of cis-2sc | ee (%) of trans-2sc | ee (%) of recovered 1sc | ||||||
| a Reaction conditions: substrate (1 eq., 0.25 mmol), Cat. (1 mol%); FA:TEA = 10 eq.:4 eq. (5:2), 4 eq.:4 eq. (1:1), or 3 eq.:15 eq. (1:5); solvent (0.2 M); rt, under N2 atmosphere.b Determined by using 1H-NMR.c Determined by using chiral HPLC. |
|||||||||
| 1 | C1 | 5:2 |
DCE | 6 | 53 | 93:7 |
99 | 47 | 97 |
| 2 | C2 | 5:2 |
DCE | 6 | 56 | 92:8 |
99 | 15 | 99 |
| 3 | C3 | 5:2 |
DCE | 6 | 56 | 92:8 |
>99 | 40 | >99 |
| 4 | C1 | 5:2 |
DCE | 24 | 65 | 80:20 |
>99 | 58 | 96 |
| 5 | C2 | 5:2 |
DCE | 24 | 77 | 76:24 |
98 | 29 | 91 |
| 6 | C3 | 5:2 |
DCE | 24 | 57 | 88:12 |
>99 | 42 | >99 |
| 7 | C3 | 1:1 |
DCE | 6 | 53 | 83:17 |
>99 | 51 | 57 |
| 8 | C3 | 1:1 |
DCE | 24 | 58 | 87:13 |
>99 | 50 | 93 |
| 9 | C3 | 1:5 |
DCE | 6 | 53 | 89:11 |
>99 | 53 | 83 |
| 10 | C3 | 1:5 |
DCE | 24 | 55 | 91:9 |
>99 | 58 | 95 |
| 11 | C3 | 1:5 |
CH3CN | 6 | 32 | 100:0 |
99 | — | 48 |
| 12 | C3 | 1:5 |
CH2Cl2 | 6 | 28 | 99:1 |
>99 | — | 36 |
| 13 | C3 | 1:5 |
THF | 6 | 31 | 100:0 |
99 | — | 46 |
| 14 | C3 | 1:5 |
EtOAc | 6 | 44 | 99:1 |
>99 | — | 80 |
| 15 | C3 | 1:5 |
DMF | 6 | 46 | 100:0 |
99 | — | 83 |
| 16 | C3 | 1:5 |
Neat | 6 | 50 | 95:5 |
99 | — | 96 |
| 17 | C3 | 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/b_char_2009.gif) :5 :5 |
MeOH | 6 | 50 | 100:0 |
99 | — | 99 |
| 18 | C3 | 1:5 |
MeOH | 20 | 50 | 100:0 |
99 | — | 99 |
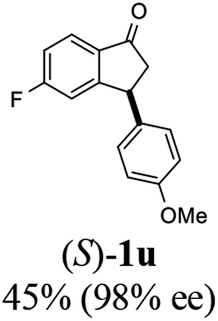
Having identified optimal conditions (C3 catalyst, 1:5 mixture of FA/TEA, in MeOH at rt for 6 h), the scope and limitations of the ATH-KR reaction were explored using variously substituted 3-aryl-indanones. The requisite racemic 3-aryl-indanones used for this purpose were prepared by triflic acid-catalyzed condensation reactions between the requisite of cinnamic acids and arenes (Scheme 3, Method A),43–45 or Pd-catalyzed intramolecular reductive Heck cyclization of the corresponding 2′-bromochalcones (Method B).46,47
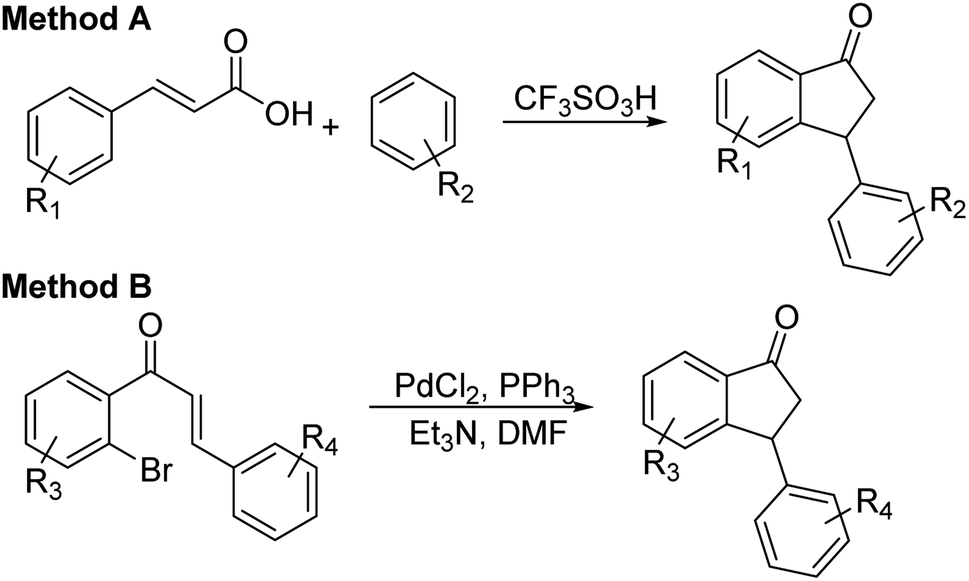 | ||
| Scheme 3 Synthesis of substituted 3-aryl-indanones 1. | ||
The results show that 3-arylindanones 1, containing an assortment of electron-donating and -withdrawing substituents, undergo ATH-KR reactions under the optimized conditions within 10 h to generate in most cases the corresponding cis-3-aryl-1-indanols (R,R)-2 and unreacted 3-aryl-1-indanones (S)-1 with excellent stereoselectivities (Table 2).
|
|||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Entry | Substrate (1) | Time (h) | Convb (%) | Indanol (2)c | Recovered indanonec | Entry | Substrate (1) | Time (h) | Convb (%) | Indanol (2)c | Recovered indanonec |
| a Reaction conditions: substrate 1 (1 eq., 0.5 mmol), (R,R)-Ts-DENEB catalyst (1 mol%), FA:TEA (3 eq.:15 eq.), MeOH (0.2 M, 2.5 mL), rt (23 °C) under N2 atmosphere.b Determined by using 1H NMR.c Yields correspond to isolated yields, % ee's were determined by chiral HPLC. Absolute stereochemistry was determined by comparison with optical rotation of known compounds.d 2 mol% of (R,R)-Ts-DENEB catalyst was used.e (S,S)-Ts-DENEB catalyst (1 mol%) was used. |
|||||||||||
| 1 | 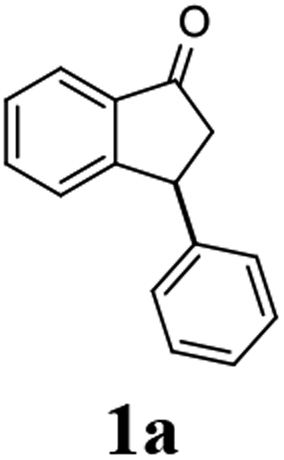 |
9 | 50 | 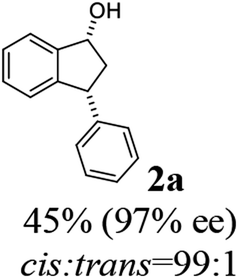 |
 |
15 | 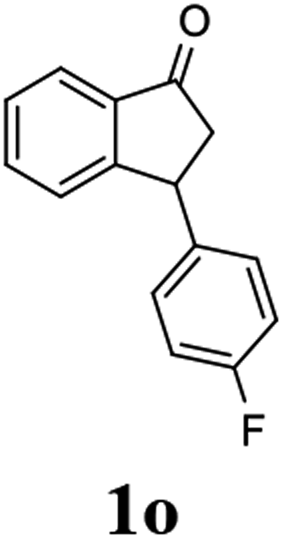 |
6 | 50 | 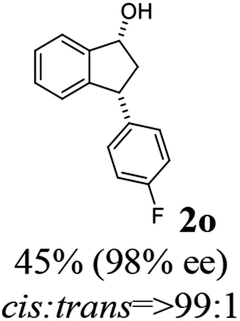 |
 |
| 2 | 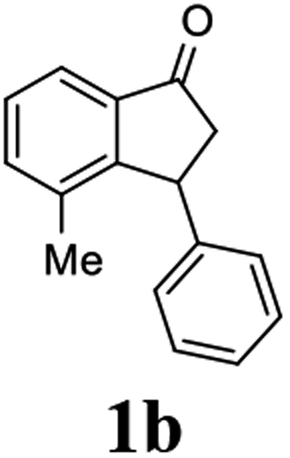 |
6d | 50 | 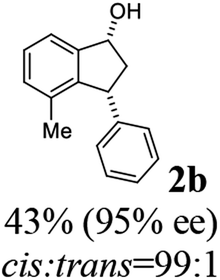 |
 |
16 | 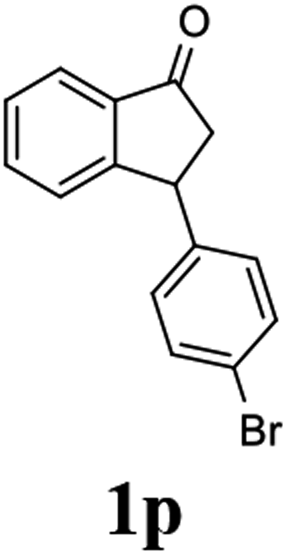 |
8 | 51 | 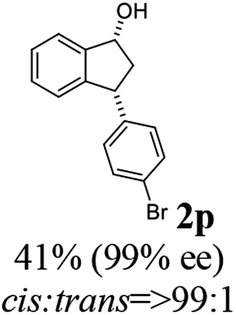 |
 |
| 3e | 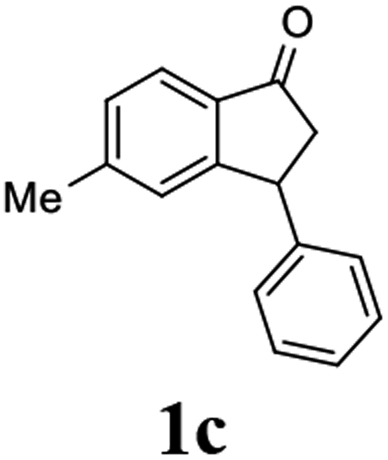 |
10 | 50 | 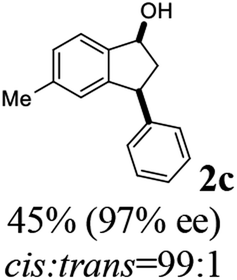 |
 |
17 | 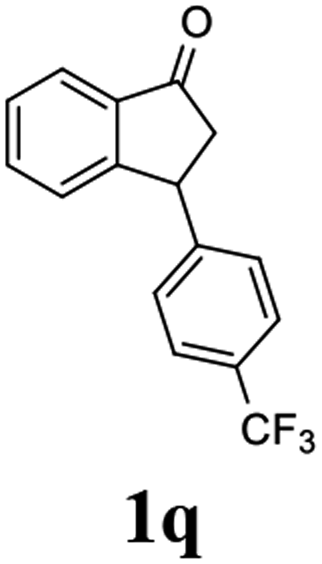 |
8 | 52 | 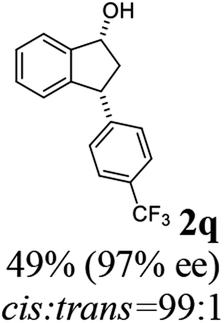 |
 |
| 4 | 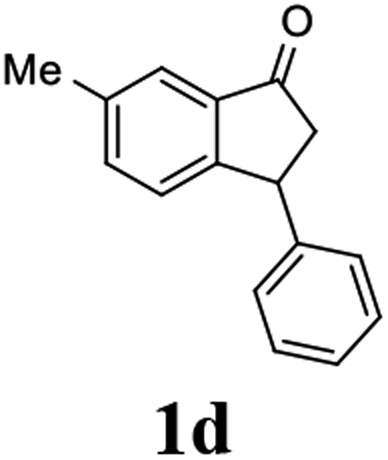 |
10 | 50 | 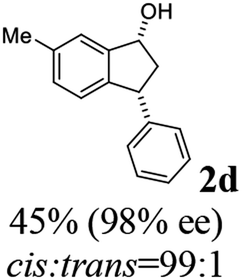 |
 |
18 | 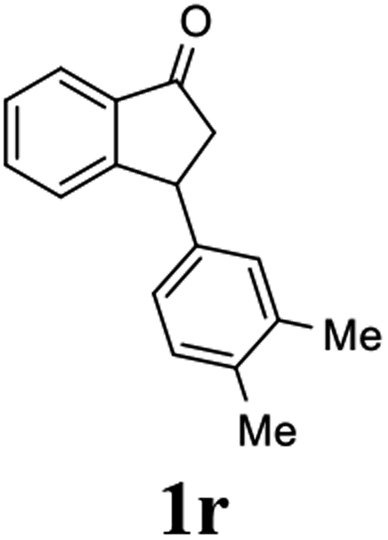 |
9 | 50 | 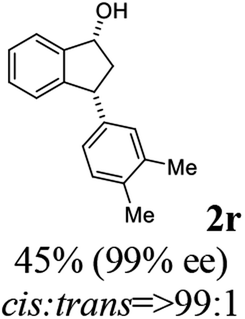 |
 |
| 5 | 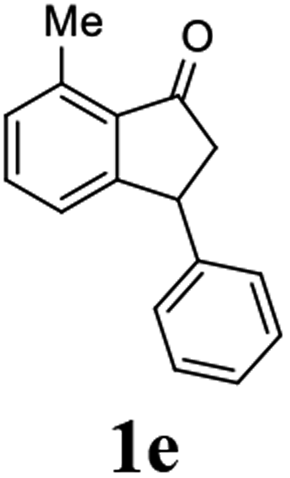 |
6d | 50 | 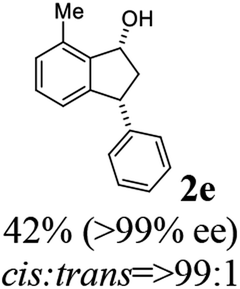 |
 |
19 | 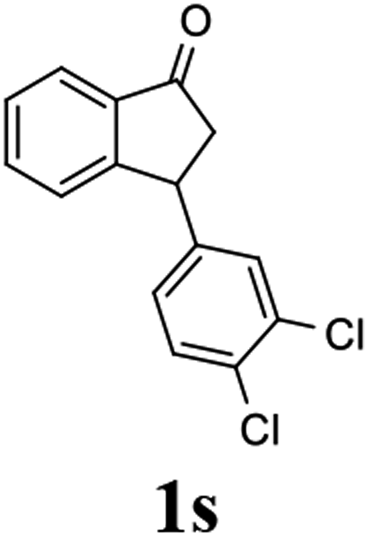 |
6 | 50 | 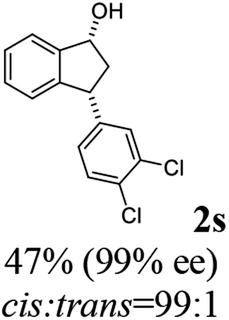 |
 |
| 6 |  |
10 | 50 | 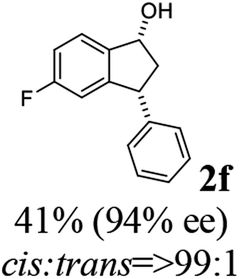 |
 |
20 | 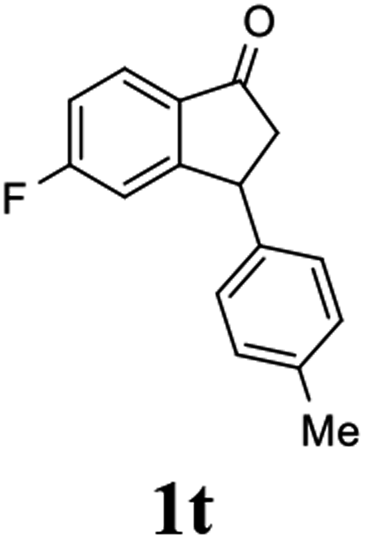 |
11 | 50 | 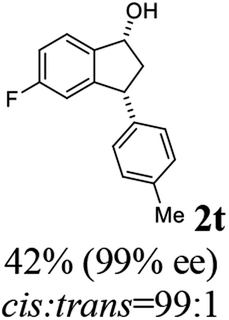 |
 |
| 7 | 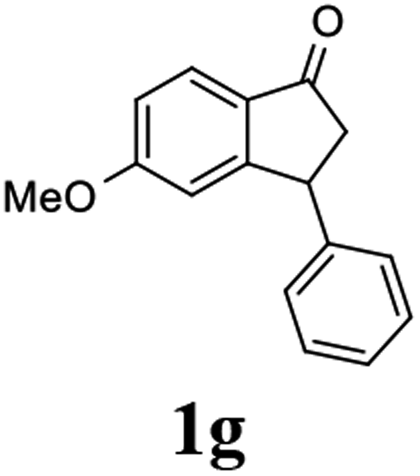 |
17d | 50 | 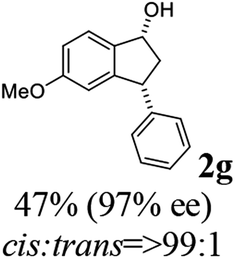 |
 |
21 | 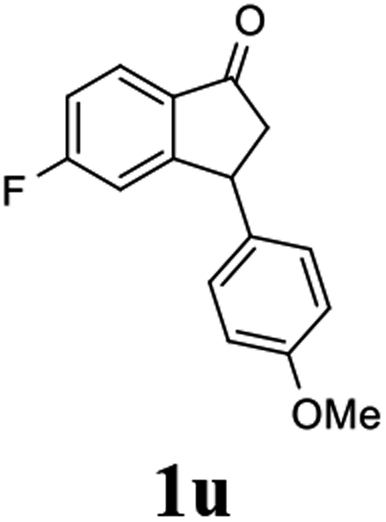 |
11 | 52 | 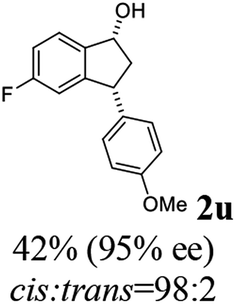 |
 |
| 8 | 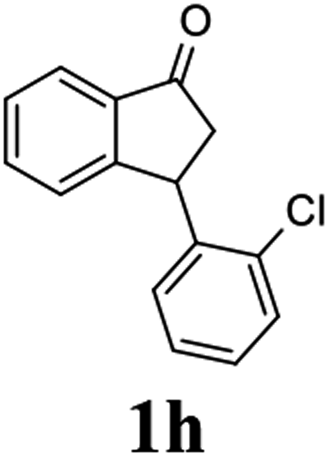 |
7 | 50 | 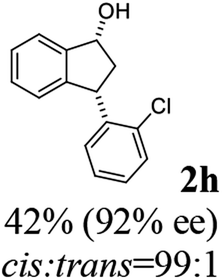 |
 |
22 | 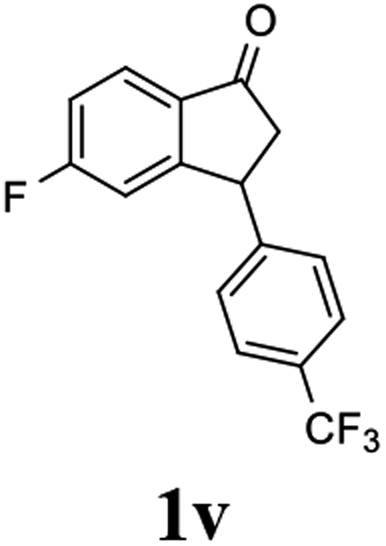 |
10 | 52 | 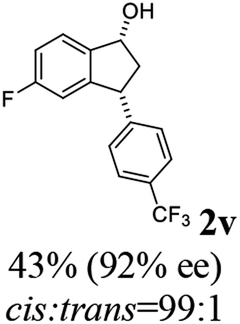 |
 |
| 9 |  |
7 | 50 | 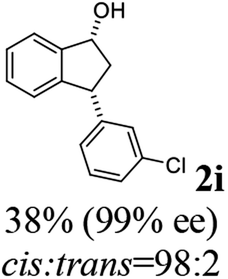 |
 |
23 | 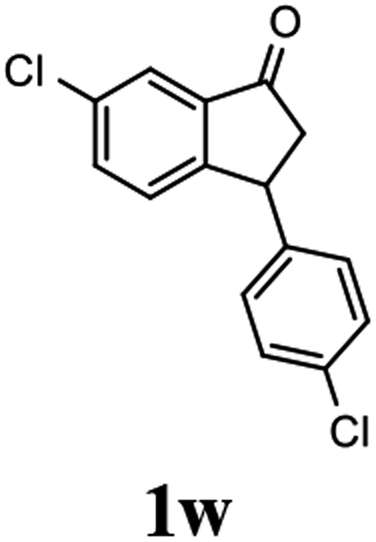 |
5 | 51 | 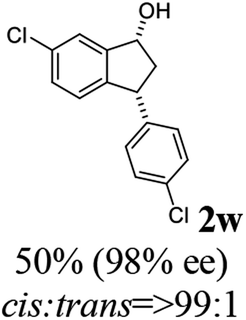 |
 |
| 10 | 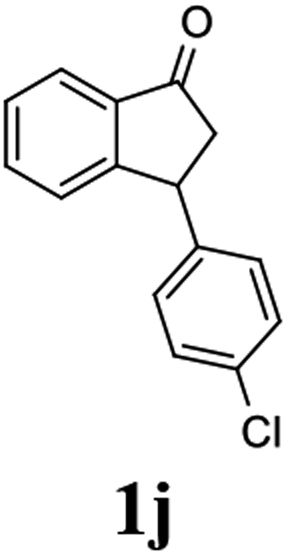 |
7 | 50 | 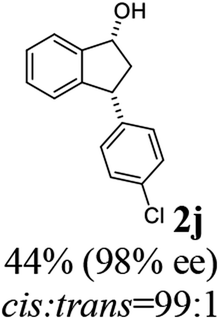 |
 |
24 | 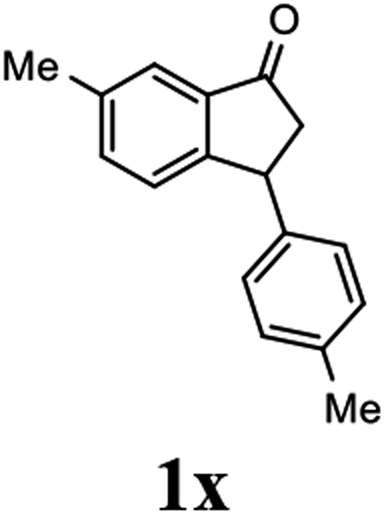 |
10 | 51 | 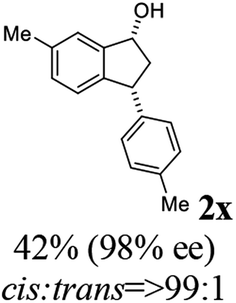 |
 |
| 11 | 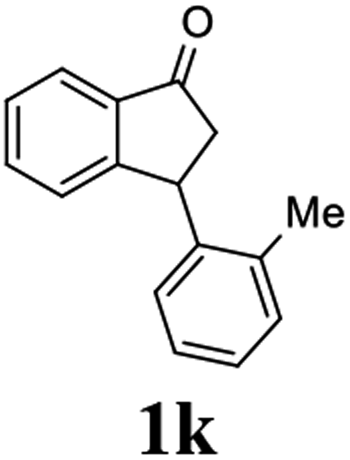 |
8 | 50 | 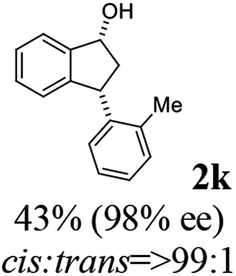 |
 |
25 | 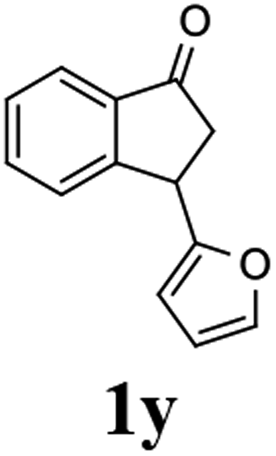 |
7 | 51 | 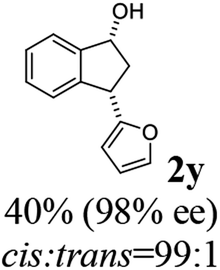 |
 |
| 12 |  |
7 | 50 | 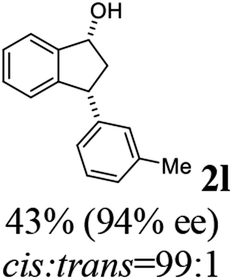 |
 |
26 |  |
8 | 50 | 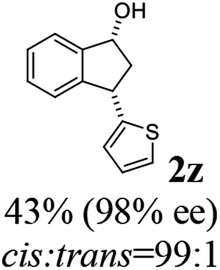 |
 |
| 13 | 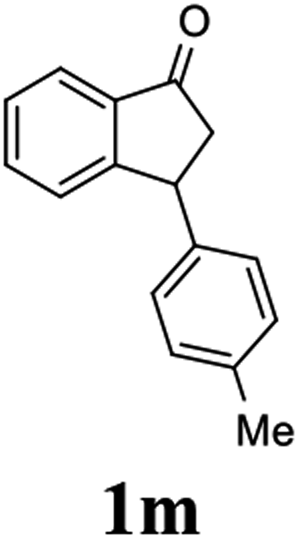 |
7 | 49 | 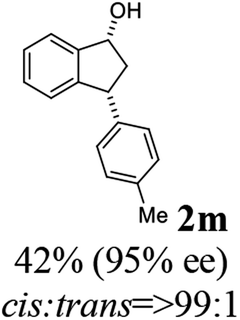 |
 |
27e | 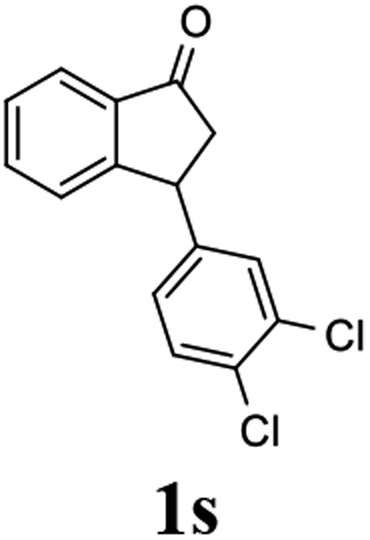 |
6 | 50 | 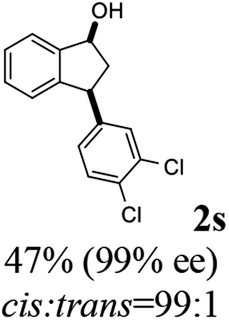 |
 |
| 14 | 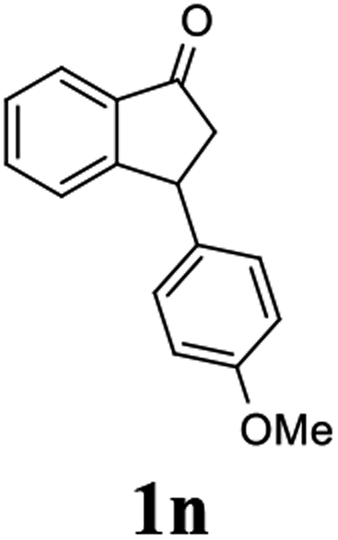 |
7 | 51 | 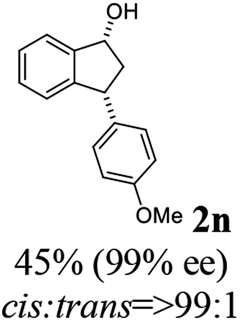 |
 |
||||||
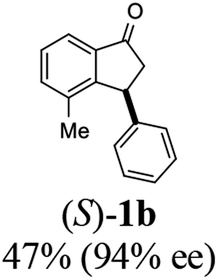
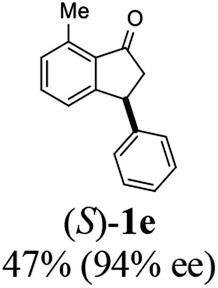
Most reactions reach to 50% conversion within 10 h at room temperature and produce almost equal amounts of the corresponding indanols and indanones. Moreover, extending the reaction times to more than 10 h does not affect the conversion ratios and stereoselectivities (Table 1, entries 17 and 18). However, in contrast to that of the unsubstituted analog, reactions of 3-arylindanones, containing electron-donating substituents such as Me or OMe on the indanone aromatic ring (Table 2, entries 2–5 and 7), require slightly longer reaction times to attain 50% conversions but the stereoselectivities for both 3-aryl-1-indanol and unreacted 3-aryl-1-indanone formation are excellent. Moreover, ATH-KR reactions of 4-Me-3-phenylindanone (1b, Table 2, entry 2) and 7-Me-3-phenylindanone (1e, Table 2, entry 5) which have Me substituents near to carbonyl moiety or 3-phenyl substituent are not complete (<50% conversion) even after 20 h. However, by carrying out these reactions using 2 mol% of C3 as catalyst, 50% conversions are attained after 6 h for 1b and 1e (entries 2 and 5). These observations suggest that not only electronic nature but also steric factor of substituents on the indanone ring have an influence on the ATH-KR process, perhaps by affecting formation of the catalyst–substrate complex.
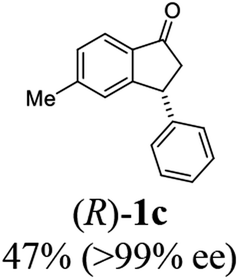
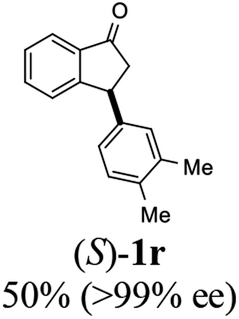
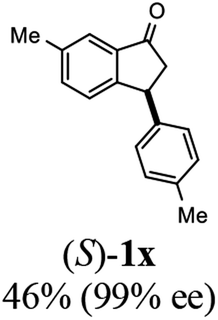
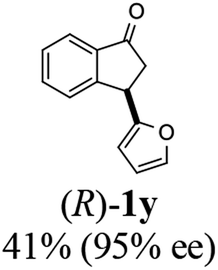
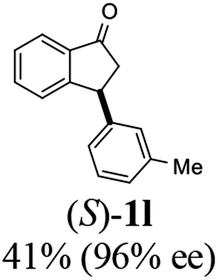
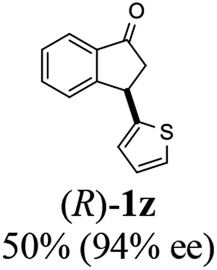
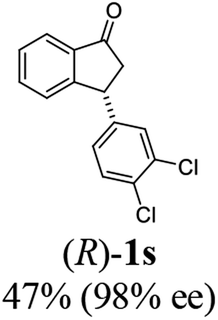
Unlike substituents on the indanone ring, the electronic nature and position of substituents on the 3-aryl ring do not noticeably affect the times required to reach 50% conversion, and stereoselectivities of the 3-arylindanols 2 and recovered 3-arylindanones 1 products remain high. For example, ATH-KR reactions of 3-(2-Cl-phenyl), 3-(3-Cl-phenyl)-, or 3-(4-Cl-phenyl)-1-indanones (1h–1j) and 3-(2-Me-phenyl), 3-(3-Me-phenyl)-, or 3-(4-Me-phenyl)-1-indanones (1k–1m) reach 50% conversion after 7–8 h and produce the corresponding 3-arylindanols and 3-arylindanones with excellent stereoselectivities (entries 8–13). ATH-KR reactions of 3-arylindanones containing diverse electron-rich or electron-deficient 3-aryl groups (1n–1q) proceed in a similar manner to form the corresponding 3-aryl-indanols and unreacted 3-aryl-indanones after 6–8 h with good stereoselectivities (entries 14–17). 3-Arylindanones possessing various substituents on both the indanone and 3-phenyl rings also are suitable substrates for the ATH-KR reaction (entries 20–24). The results show that ATH-KR reactions of 3-arylindanones containing 3-furan (1y) and 3-thiophene (1z) substituents also produce the corresponding 3-arylindanols and unreacted 3-arylindanones with similar efficiencies and stereoselectivities (entries 25 and 26). In addition, ATH-KR reaction of 1c under the same conditions, except employing (S,S)-Ts-DENEB instead of (R,R)-Ts-DENEB as catalyst, yields the antipodal (S,S)-3-arylindanol 2c (45%, 98% ee) and unreacted (R)-3-arylindanone 1c (47%, 99% ee) with excellent levels of stereoselectivity (entry 3). Similarly, ATH-KR reaction of 1s under the same conditions using (S,S)-Ts-DENEB as catalyst forms (S,S)-3-arylindanol 2s (47%, >99% ee) and unreacted (R)-3-arylindanone 1s (47%, 98% ee) (entries 19 and 27).
The absolute configurations of resulting 2a as (1R,3R) and recovered 1a as (S) were determined by comparison of optical rotations and NMR data with those of the known compounds.7,23,27 The stereochemical outcomes of the ATH reaction can be rationalized by the well-known attractive C–H/π interaction48,49 in the transition state between η6 of (R,R)-Ts-DENEB catalyst (C3) and the aromatic ring moiety of indanone 1a as shown in Fig. 2. The cis product of 2a might be favored as a consequence of 3-phenyl substituent of 1a keeping away from the reaction site in the transition state.41
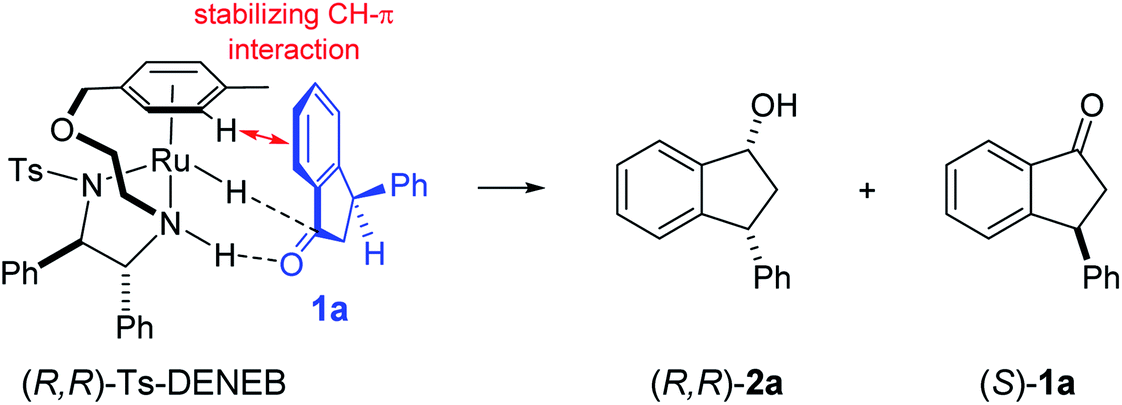 | ||
| Fig. 2 Proposed asymmetric induction model41 in the ATH-KR of racemic-1a to (R,R)-2a and (S)-1a. | ||
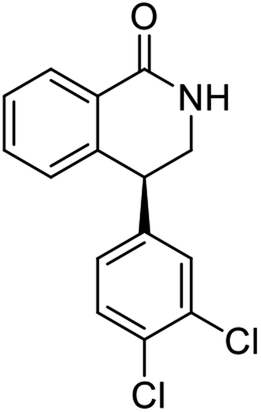
Highly enantiomerically enriched 3-aryl-1-indanols 2 and 3-aryl-1-indanones 1 produced in the ATH-KR reactions are valuable intermediates for the synthesis of medicinally important compounds such as (+)-indatraline8 or (R)-tolterodine.50 To demonstrate this assertion, (S,S)-3-(3,4-dichlorophenyl)-1-indanol (2s), formed by ATH-KR reaction of 1s, was converted to (+)-indatraline via mesylation and subsequent reaction of the formed mesylate with methylamine in the same flask (Scheme 4a).7,18 In a route for the synthesis of (R)-tolterodine, a potent and competitive muscarinic antagonist that is currently used for the treatment of urinary urge incontinence,50 (R)-5-methyl-3-phenyl-1-indanone (1c) obtained from ATH-KR reaction of 1c was transformed to (R)-6-methyl-4-phenylcoumarine (5) via Baeyer–Villiger oxidation without deterioration of optical purity (Scheme 4b). Because the conversion of (R)-5 to (R)-tolterodine via DIBAL-H reduction to a lactol and subsequent reductive amination with diisopropylamine has been reported,7,51–53 this route constitutes a formal synthesis of (R)-tolterodine (Scheme 4b). Finally, to demonstrate applications to the synthesis of quinoline derivatives, treatment of (S)-3-(3,4-dichlorophenyl)-1-indanone oxime O-tosylate (6), obtained from (S)-1s, using 1.5 equiv. of AlCl3 at room temperature,54 produces the readily separable mixture of (S)-4-aryl-3,4-dihydroquinoline-2(1H)-one ((S)-7) and (S)-4-aryl-3,4-dihydroisoquinoline-1(2H)-one ((S)-8).
 | ||
| Scheme 4 Synthetic applications. | ||
Conclusions
In summary, this effort demonstrates that efficient kinetic resolution (KR) attends asymmetric transfer hydrogenation (ATH) reactions of diverse racemic 3-aryl-1-indanones when commercial (R,R)- or (S,S)-Ts-DENEB is employed as catalyst, a 1:5 mixture of HCO2H and Et3N is used as a hydrogen source and MeOH is utilized as solvent. These processes, carried out at room temperature, produce near equal amounts of the corresponding cis-3-arylindanols and unreacted 3-arylindanones with excellent levels of diastereo- and enantio-selectivity. The key merit of the process is that it forms both highly enantiomerically enriched cis-3-arylindanols and 3-arylindanones in a single step. In addition, selected stereoselective transformations of 3-arylindanol and 3-arylindanones generated by the ATH-KR process, demonstrate the usefulness of this method in producing key intermediates for the preparation of (+)-indatraline, (R)-tolterodine, (S)-4-aryl-3,4-dihydroquinoline-2(1H)-one and (S)-4-aryl-3,4-dihydroisoquinoline-1(2H)-one.
Experimental section
General
Synthetic procedure and characterization data of starting 3-aryl-1-indanones 1a–1z are included in the ESI.† All reactions were conducted under an inert atmosphere of nitrogen using anhydrous solvents. Mixtures of HCO2H/Et3N (5:2 and 1:1) are commercially available and 1:5 mixture of HCO2H/Et3N was prepared by adding 1 equiv. of Et3N to 5 equiv. of HCO2H at 0 °C under a nitrogen atmosphere and used as such. Chiral transition metal catalysts C1–C3 were purchased from commercial vendors. The progress of reactions was monitored using thin layer chromatography (TLC) and visualized using UV light and by staining with ethanolic phosphomolybdic acid (PMA) solution or ninhydrin solution followed by heating. Flash column chromatography was carried out on silica gel (38–75 μm). Analytical thin layer chromatography (TLC) was performed on Merck silica gel 60 F254 plates. Preparative thin layer chromatography (PLC) was performed on Merck silica gel 60 F254 2 mm plates. Syntheses under microwave system were conducted by using CEM Discover SP. Nuclear magnetic resonance (NMR) spectra were recorded using Bruker 500 MHz NMR instrument (1H NMR at 500 MHz and 13C NMR at 125 MHz) or Bruker 400 MHz NMR instrument (1H NMR at 400 MHz and 13C NMR at 101 MHz). 1H NMR data are reported as follows: chemical shift (δ, ppm), multiplicity (s = singlet, d = doublet, t = triplet, q = quartet, m = multiplet, br = broad), integration, coupling constants (Hz). Data for 13C NMR are reported in terms of chemical shift (δ, ppm). High performance liquid chromatography (HPLC) was carried out on a Young Lin HPLC system (7725i Injector, SDV 30 Plus Solvent Degassor & Valve Module (Helium Sparging), SP930D Solvent Delivery Pump, UV 730D Absorbance Detector) equipped with a Chiralpak IA, IB, IC, ID or Chiralpak AD-H, Chiralcel OD-H column. Specific rotations were measured on a Rudolph Autopol IV (Automatic polarimeter). High-resolution mass spectra and elemental analysis were obtained from the Korea Research Institute of Chemical Technology. HR-MS were measured with electron impact (EI) via double focusing mass analyzer (magnetic and electric fields) or electrospray ionization (ESI) via time of flight (TOF) analyzer.
Representative procedure for the ATH of 3-phenyl-1-indanone (1a) accompanying kinetic resolution
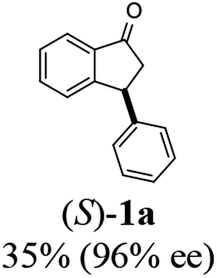
To a solution of 3-phenyl-1-indanone (1a, 104 mg, 0.5 mmol) and triethylamine (1.06 mL, 7.5 mmol) dissolved in methanol (1.5 mL) was added formic acid (63.4 μL, 1.5 mmol) followed by (R,R)-Ts-DENEB catalyst (3.2 mg, 0.005 mmol dissolved in 1.0 mL of methanol). The reaction mixture was stirred at 25 °C under of N2 atmosphere. After the reaction time specified in the Table 2 (6–14 h), the reaction mixture was diluted with chloroform (30 mL) and washed with water and brine (20 mL) successively. The organic layer was dried with MgSO4, filtered and concentrated by rotary evaporation. The resulting mixture of 3-phenyl-1-indanol (2a) and unreacted remaining 1a were easily separated by flash column chromatography (ethyl acetate:n-hexane 1:7). dr and ee's of the resulting indanol 2a and unreacted remaining indanone 1a were determined by chiral HPLC. (Racemic cis- and trans-3-phenyl-1 indanols (2a) were obtained by NaBH4 reduction of 1a in MeOH.) Absolute configurations were determined by comparison of optical rotations and NMR data with those of the known compounds.
(1R,3R)-3-Phenyl-2,3-dihydro-1H-inden-1-ol (2a)
Yield 45% (46.7 mg as white solid); mp 95.2–95.9 °C; 97% ee (Chiralpak IB, 0 to 6% IPA for 7 min in n-hexane, 1 mL min−1, 270 nm, tR(major) = 23.2 min, tR(minor) = +16.9 min); [α]24D = −15.6 (c 1.13, CH2Cl2). Literature values: [α]23D = −11 (c 1, CHCl3 for 95% ee).27 [α]23D = +16.1 (c 0.1, CH2Cl2 for 86% ee)23 for (1S,3S)-2a; 1H NMR (CDCl3, 500 MHz): δ 7.48 (d, 1H, J = 7.5 Hz), 7.36–7.27 (m, 3H), 7.27–7.18 (m, 4H), 6.95 (d, 1H, J = 7.5 Hz), 5.34–5.23 (m, 1H), 4.19 (t, 1H, J = 8.4 Hz), 3.03 (dt, 1H, J = 12.9, 7.2 Hz), 2.07–1.90 (m, 2H); 13C{1H} NMR (101 MHz, CDCl3) δ 145.6, 145.2, 144.2, 128.6, 128.4, 128.3, 127.2, 126.6, 125.1, 123.7, 75.1, 48.3, 47.2; HRMS (EI, double focusing) m/z: [M]+ calcd for C15H14O 210.1045; found 210.1054.
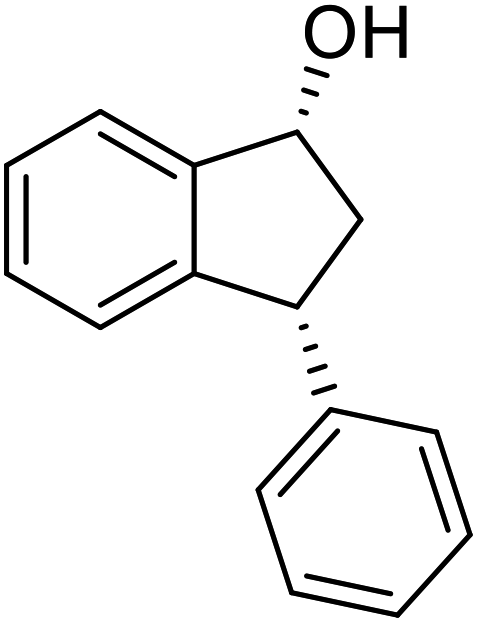
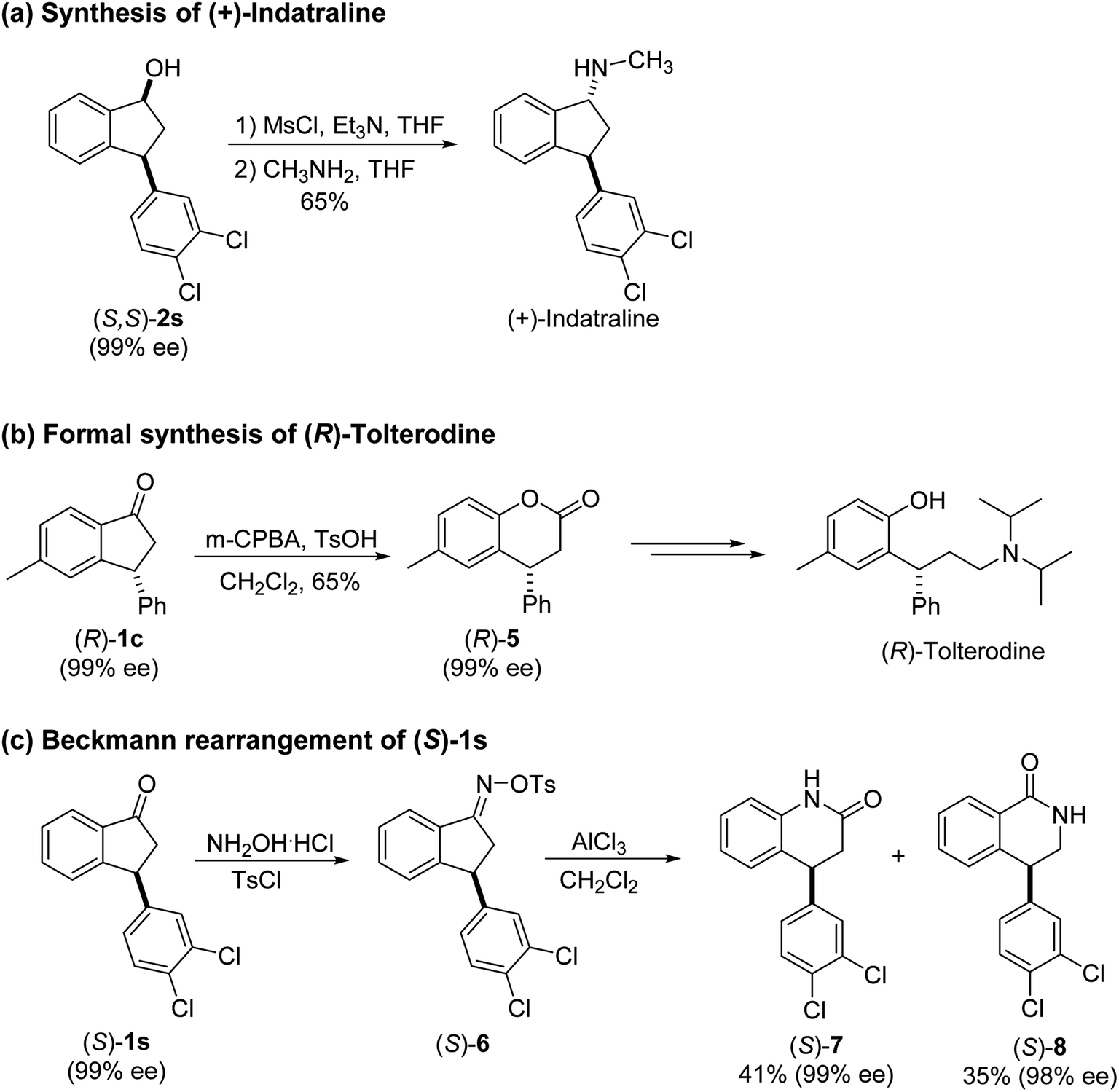
(S)-3-Phenyl-2,3-dihydro-1H-inden-1-one, (S)-1a
Yield 35% (36.4 mg as white solid); 98% ee (Chiralpak IB, 0 to 6% IPA for 7 min in n-hexane, 1 mL min−1, 270 nm, tR(major) = 15.0 min, tR(minor) = 16.2 min); [α]20D = +72.8 (c 1.5, CH2Cl2). Literature values: [α]25D = +64.9 (c 0.4, CH2Cl2 for 86% ee).23 [α]23D = −49 (c 1.0, CHCl3) for 91% ee of (R)-1a;7 1H NMR (CDCl3, 500 MHz): δ 7.82 (d, 1H, J = 7.7 Hz), 7.57 (t, 1H, J = 7.4 Hz), 7.42 (t, 1H, J = 7.4 Hz), 7.32 (t, 2H, J = 7.4 Hz), 7.29–7.24 (m, 2H), 7.2–7.09 (m, 2H), 4.58 (dd, 1H, J = 8.1, 3.9 Hz), 3.24 (dd, 1H, J = 19.2, 8.1 Hz), 2.70 (dd, 1H, J = 19.2, 3.9 Hz); 13C{1H} NMR (CDCl3,101 MHz) δ 206.0, 158.0, 143.7, 136.7, 135.1, 128.9, 127.9, 127.6, 127.0, 126.9, 123.4, 46.8, 44.5; HRMS (EI, double focusing) m/z: [M]+ calcd for C15H12O 208.0888; found 208.0883.
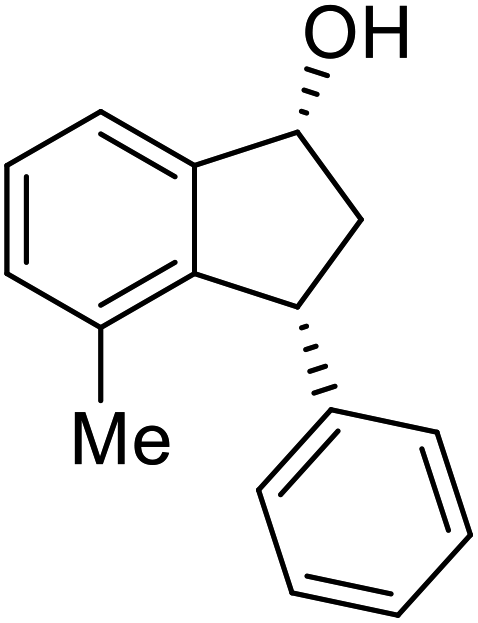
(1R,3R)-4-Methyl-3-phenyl-2,3-dihydro-1H-inden-1-ol (2b)
Yield 43% (48.2 mg as white solid); mp 122.6–123.3 °C; 95% ee (Chiralpak IB, 0 to 6% IPA for 7 min in n-hexane, 1 mL min−1, 270 nm, tR(major) = 16.6 min, tR(minor) = 25.4 min); [α]29D = −5.6 (c 2.4, CH2Cl2); 1H NMR (CDCl3, 500 MHz): δ 7.35 (d, 1H, J = 7.5 Hz), 7.30–7.23 (m, 3H), 7.18 (t, 1H, J = 7.3 Hz), 7.13 (d, 2H, J = 7.1 Hz), 7.09 (d, 1H, J = 7.3 Hz), 5.21 (s, 1H), 4.29 (dd, 1H, J = 8.6, 5.3 Hz), 3.00 (dt, 1H, J = 13.8, 8.6, 7.3 Hz), 1.97 (dt, 1H, J = 13.8, 4.8 Hz), 1.89 (s, 3H), 1.81 (s, 1H); 13C{1H} NMR (101 MHz, CDCl3) δ 145.8, 145.4, 143.4, 135.5, 130.4, 128.6, 127.9, 127.8, 126.2, 121.9, 75.6, 48.4, 46.3, 19.1; HRMS (EI, double focusing) m/z: [M]+ calcd for C16H16O 224.1201; found 224.1191.
(S)-4-Methyl-3-phenyl-2,3-dihydro-1H-inden-1-one, (S)-1b
Yield 47% (52.1 mg as white solid); 94% ee (Chiralpak IB, 0 to 6% IPA for 7 min in n-hexane, 1 mL min−1, 270 nm, tR(major) = 14.6 min, tR(minor) = 16.1 min); [α]29D = +36.3 (c 2.77, CH2Cl2); 1H NMR (CDCl3, 500 MHz): δ 7.69 (d, 1H, J = 6.6 Hz), 7.41–7.34 (m, 2H), 7.29–7.25 (m, 2H), 7.21 (t, 1H, J = 7.3 Hz), 7.02 (d, 2H, J = 7.1 Hz), 4.58 (dd, 1H, J = 8.3, 2.6 Hz), 3.24 (dd, 1H, J = 19.2, 8.3 Hz), 2.60 (dd, 1H, J = 19.2, 2.6 Hz), 2.02 (s, 3H); 13C{1H} NMR (101 MHz, CDCl3) δ 206.6, 155.6, 143.7, 137.2, 136.8, 136.4, 128.9, 128.4, 127.4, 126.7, 121.0, 47.6, 43.9, 18.4; HRMS (EI, double focusing) m/z: [M]+ calcd for C16H14O 222.1045; found 222.1037.
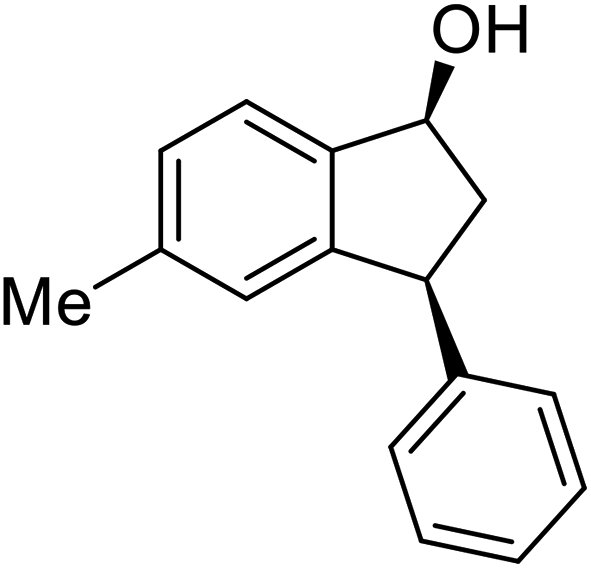
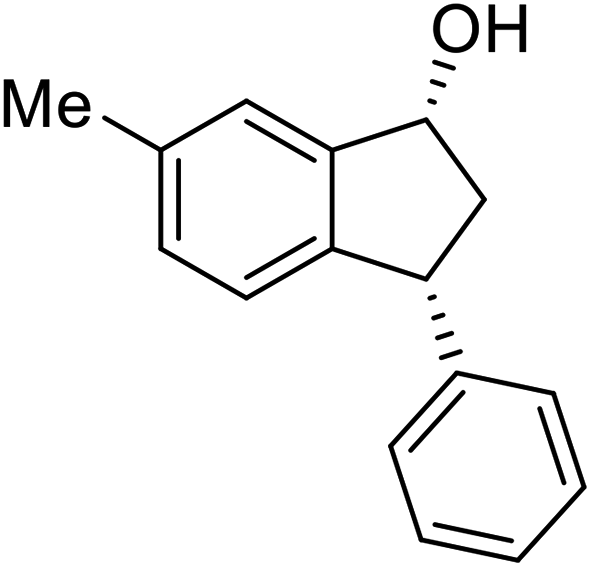
(1S,3S)-5-Methyl-3-phenyl-2,3-dihydro-1H-inden-1-ol (2c)
Yield 44% (49 mg as white solid); mp 118.3–118.7 °C; 97% ee (Chiralpak IB, 0 to 6% IPA for 7 min in n-hexane, 1 mL min−1, 270 nm, tR(major) = 16.6 min, tR(minor) = 23.9 min); [α]25D = +7.3 (c 1.2, CH2Cl2); 1H NMR (CDCl3, 500 MHz): δ 7.38–7.30 (m, 3H), 7.28–7.21 (m, 3H), 7.11 (d, 1H, J = 7.7 Hz), 6.75 (s, 1H), 5.28–5.22 (m, 1H), 4.15 (t, 1H, J = 8.3 Hz), 3.01 (dt, 1H, J = 12.9, 7.3 Hz), 2.28 (s, 3H), 1.99–1.90 (m, 2H); 13C{1H} NMR (101 MHz, CDCl3) δ 145.9, 144.4, 142.4, 138.3, 128.6, 128.3, 128.1, 126.5, 125.6, 123.5, 74.9, 48.3, 47.2, 21.4; HRMS (EI, double focusing) m/z: [M]+ calcd for C16H16O 224.1201; found 224.1207.
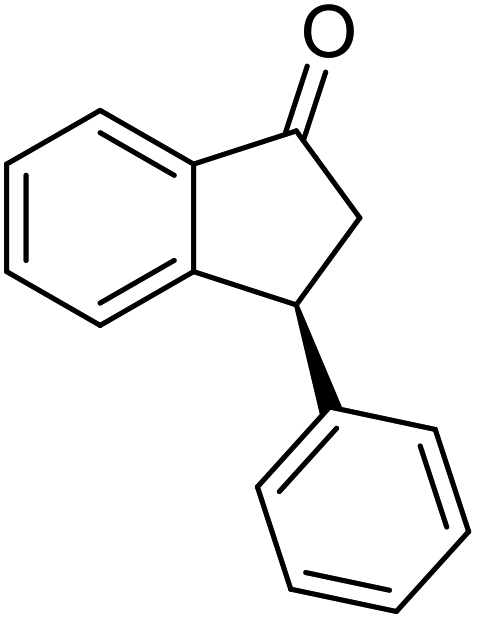
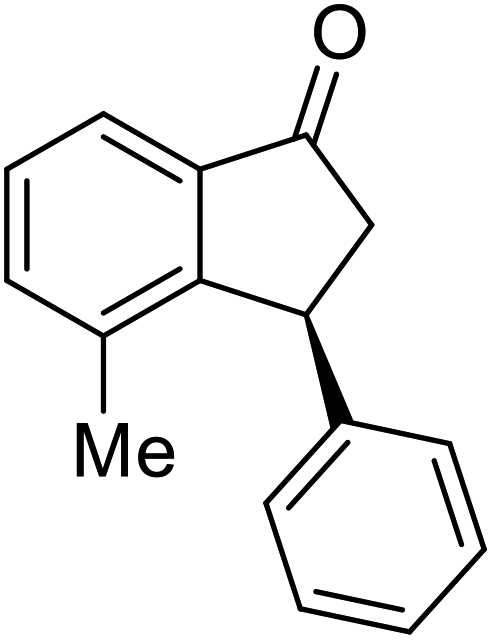
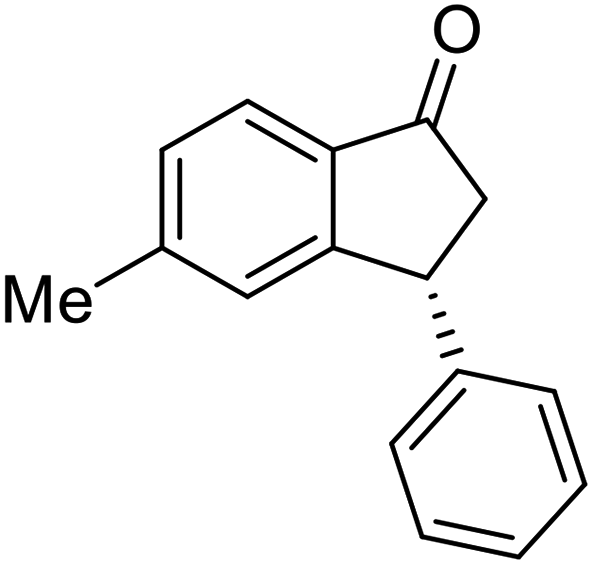
(R)-5-Methyl-3-phenyl-2,3-dihydro-1H-inden-1-one, (R)-1c
Yield 47% (52.2 mg as white solid); >99% ee (Chiralpak IB, 0 to 6% IPA for 7 min in n-hexane, 1 mL min−1, 270 nm, tR(major) = 16.0 min, tR(minor) = 15.1 min); [α]21D = −29.4 (c 1.7, CH2Cl2). Literature values for (S)-1c: [α]23D = +28.9° (c 1.0, CHCl3 for 97% ee).7 [α]25D = +20.3 (c 0.1, CH2Cl2 for 86% ee);23 1H NMR (CDCl3, 500 MHz): δ 7.70 (d, 1H, J = 7.9 Hz), 7.34–7.28 (m, 2H), 7.28–7.24 (m, 1H), 7.22 (d, 1H, J = 7.9 Hz), 7.12 (d, 2H, J = 7.1 Hz), 7.05 (s, 1H), 4.51 (dd, 1H, J = 8.0, 3.8 Hz), 3.21 (dd, 1H, J = 19.1, 8.0 Hz), 2.67 (dd, 1H, J = 19.1, 3.8 Hz), 2.37 (s, 3H); 13C{1H} NMR (101 MHz, CDCl3) δ 205.6, 158.5, 146.4, 143.9, 134.5, 129.2, 128.9, 127.7, 127.1, 126.9, 123.2, 47.0, 44.3, 22.1; HRMS (EI, double focusing) m/z: [M]+ calcd for C16H14O 222.1045; found 222.1038.
(1R,3R)-6-Methyl-3-phenyl-2,3-dihydro-1H-inden-1-ol (2d)
Yield 45% (49.9 mg as white solid); mp 130.1–130.7 °C; 98% ee (Chiralpak IB, 0 to 6% IPA for 7 min in n-hexane, 1 mL min−1, 270 nm, tR(major) = 24.5 min, tR(minor) = 16.3 min); [α]25D = −32.7 (c 1.3, CH2Cl2); 1H NMR (CDCl3, 500 MHz): δ 7.35–7.27 (m, 3H), 7.27–7.20 (m, 3H), 7.05 (d, 1H, J = 7.7 Hz), 6.84 (d, 1H, J = 7.7 Hz), 5.29–5.21 (m, 1H), 4.15 (t, 1H, J = 8.3 Hz), 3.01 (dt, 1H, J = 12.9, 7.2 Hz), 2.38 (s, 3H), 2.01–1.89 (m, 2H); 13C{1H} NMR (101 MHz, CDCl3) δ 145.4, 144.5, 142.7, 137.0, 129.3, 128.6, 128.2, 126.5, 124.8, 124.2, 75.1, 48.0, 47.4, 21.3; HRMS (EI, double focusing) m/z: [M]+ calcd for C16H16O 224.1201; found 224.1192.
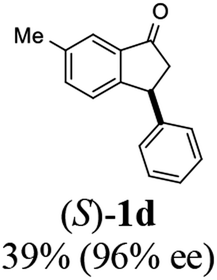
(S)-6-Methyl-3-phenyl-2,3-dihydro-1H-inden-1-one, (S)-1d
Yield 39.4% (43.7 mg, white solid); mp 92.8–92.9 °C; 96% ee (Chiralpak IB, 0 to 6% IPA for 7 min in n-hexane, 1 mL min−1, 270 nm, tR(major) = 14.4 min, tR(minor) = 15.4 min); [α]23D = +60.1 (c 1.7, CH2Cl2); 1H NMR (CDCl3, 500 MHz): δ 7.61 (s, 1H), 7.39 (d, 1H, J = 7.8 Hz), 7.30 (t, 2H, J = 7.4 Hz), 7.24 (t, 1H, J = 7.4 Hz), 7.16 (d, 1H, J = 7.8 Hz), 7.11 (d, 2H, J = 7.1 Hz), 4.53 (dd, 1H, J = 7.9, 3.7 Hz), 3.22 (dd, 1H, J = 19.2, 8.0 Hz), 2.68 (dd, 1H, J = 19.2, 3.8 Hz), 2.42 (s, 3H); 13C{1H} NMR (101 MHz, CDCl3) δ 206.1, 155.4, 143.9, 137.9, 137.0, 136.4, 128.9, 127.6, 126.9, 126.5, 123.3, 47.2, 44.1, 21.1; HRMS (EI, double focusing) m/z: [M]+ calcd for C16H14O 222.1045; found 222.1038.
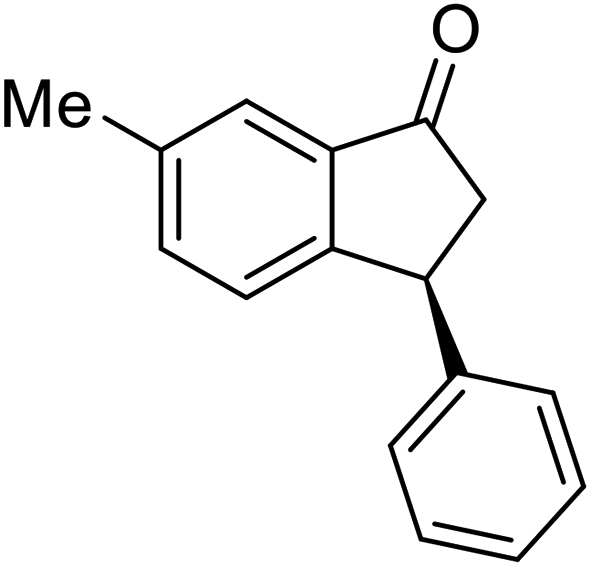
(1R,3R)-7-Methyl-3-phenyl-2,3-dihydro-1H-inden-1-ol (2e)
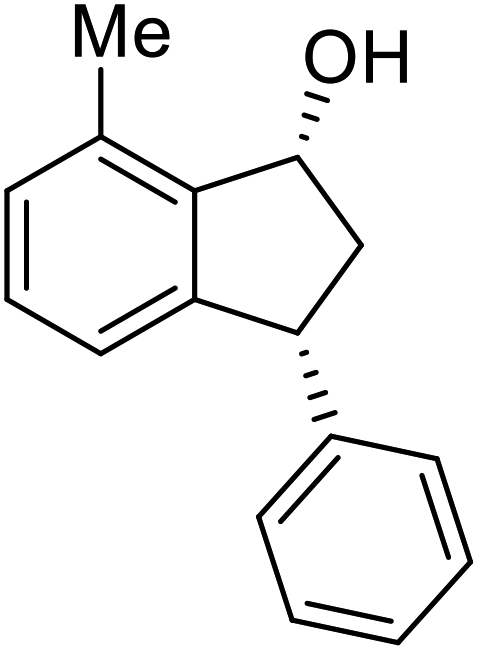
Yield 42% (46 mg as white solid); mp 87.5–88.9 °C; >99% ee (Chiralpak IB, 0 to 6% IPA for 7 min in n-hexane, 1 mL min−1, 270 nm, tR(major) = 21.3 min, tR(minor) = 15.9 min); [α]26D = −88.7 (c 0.73, CH2Cl2); 1H NMR (CDCl3, 500 MHz): δ 7.34–7.26 (m, 2H), 7.24–7.19 (m, 3H), 7.16 (t, 1H, J = 7.5 Hz), 7.07 (d, 1H, J = 7.4 Hz), 6.85 (d, 1H, J = 7.5 Hz), 5.37 (s, 1H), 4.26–4.17 (m, 1H), 3.01 (dt, 1H, J = 13.7, 8.6, 7.2 Hz), 2.49 (s, 3H), 2.02 (ddd, 1H, J = 13.7, 6.4, 4.8 Hz), 1.78 (s, 1H); 13C{1H} NMR (101 MHz, CDCl3) δ 146.0, 145.5, 142.7, 135.4, 128.9, 128.8, 128.6, 128.0, 126.4, 122.9, 75.1, 48.7, 45.7, 18.4; HRMS (EI, double focusing) m/z: [M]+ calcd for C16H16O 224.1201; found 224.1209.
(S)-7-Methyl-3-phenyl-2,3-dihydro-1H-inden-1-one, (S)-1e
Yield: 47% (52.6 mg, white solid); mp 90.0–90.2 °C; 94% ee (Chiralpak IB, 0 to 6% IPA for 7 min in n-hexane, 1 mL min−1, 270 nm, tR(major) = 13.7 min, tR(minor) = 14.1 min); [α]24D = +127.4 (c 2.0, CH2Cl2); 1H NMR (CDCl3, 500 MHz): δ 7.40 (t, 1H, J = 7.5 Hz), 7.30 (t, 2H, J = 7.4 Hz), 7.24 (d, 1H, J = 7.4 Hz), 7.13 (t, 3H, J = 7.7 Hz), 7.06 (d, 1H, J = 7.7 Hz), 4.50 (dd, 1H, J = 8.2, 4.0 Hz), 3.19 (dd, 1H, J = 19.0, 8.2 Hz), 2.74–2.63 (m, 4H); 13C{1H} NMR (101 MHz, CDCl3) δ 206.9, 158.8, 144.1, 138.5, 134.3, 134.1, 129.6, 128.8, 127.7, 126.8, 124.2, 47.3, 43.9, 18.4; HRMS (EI, double focusing) m/z: [M]+ calcd for C16H14O 222.1045; found 222.1038.
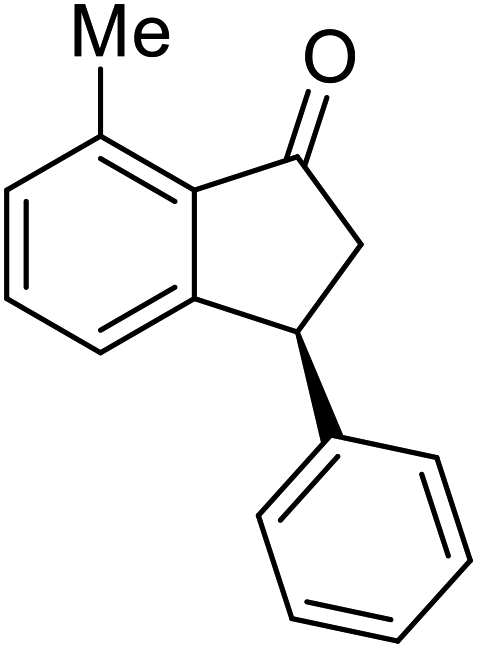
(1R,3R)-5-Fluoro-3-phenyl-2,3-dihydro-1H-inden-1-ol (2f)
Yield 41% (47 mg as white solid); mp 88.5–89.0 °C; 94% ee (Chiralpak IB, 0 to 6% IPA for 7 min in n-hexane, 1 mL min−1, 270 nm, tR(major) = 24.9 min, tR(minor) = 17.3 min); [α]27D = −27.4 (c 1.9, CH2Cl2); 1H NMR (CDCl3, 400 MHz): δ 7.41 (dd, 1H, J = 8.3, 5.2 Hz), 7.38–7.30 (m, 2H), 7.30–7.19 (m, 3H), 7.08–6.93 (m, 1H), 6.62 (d, 1H, J = 8.9 Hz), 5.26 (t, 1H, J = 7.1 Hz), 4.16 (t, 1H, J = 8.4 Hz), 3.04 (dt, 1H, J = 13.0, 7.3 Hz), 2.08–1.82 (m, 2H); 13C{1H} NMR (101 MHz, CDCl3) δ 163.4 (d, JC–F = 245.5 Hz), 148.0 (d, JC–F = 8.0 Hz), 143.5, 140.8 (d, JC–F = 2.4 Hz), 128.7, 128.2, 126.9, 125.1 (d, JC–F = 9.0 Hz), 114.5 (d, JC–F = 23.0 Hz), 111.9 (d, JC–F = 22.4 Hz), 74.4, 48.2, 47.4; HRMS (EI, double focusing) m/z: [M]+ calcd for C15H13FO 228.0950; found 228.0948.
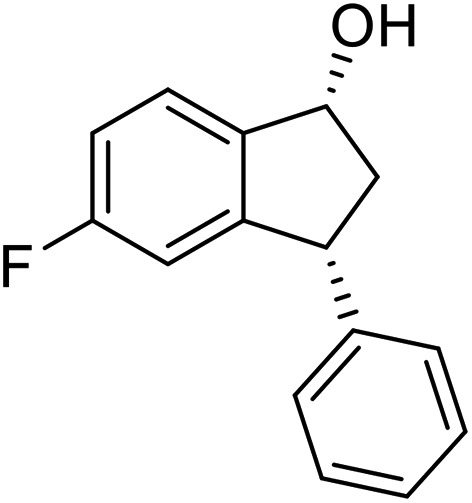
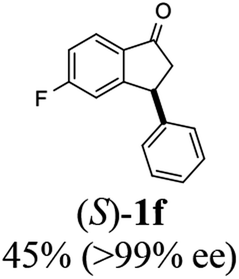
(S)-5-Fluoro-3-phenyl-2,3-dihydro-1H-inden-1-one, (S)-1f
Yield 45% (50.8 mg, yellow solid); mp 107.5–108.3 °C; >99% ee (Chiralpak IB, 0 to 6% IPA for 7 min in n-hexane, 1 mL min−1, 270 nm, tR(major) = 15.3 min, tR(minor) = 17.2 min); [α]24D = +54.9 (c 2.1, CH2Cl2); 1H NMR (CDCl3, 400 MHz): δ 7.82 (dd, 1H, J = 8.5, 5.3 Hz), 7.37–7.30 (m, 2H), 7.30–7.26 (m, 1H), 7.16–7.07 (m, 3H), 6.92 (d, 1H, J = 8.5 Hz), 4.54 (dd, 1H, J = 8.1, 3.9 Hz), 3.25 (dd, 1H, J = 19.2, 8.1 Hz), 2.73 (dd, 1H, J = 19.2, 3.9 Hz); 13C{1H} NMR (101 MHz, CDCl3) δ 204.0, 168.7–166.1 (d, JC–F = 256.5 Hz), 160.9–160.8 (d, JC–F = 9.5 Hz), 142.9, 133.2 (d, JC–F = 1.6 Hz), 129.1, 127.6, 127.3, 125.8–125.7 (d, JC–F = 10.3 Hz), 116.5–116.2 (d, JC–F = 23.9 Hz), 113.6–113.3(d, JC–F = 22.7 Hz), 46.9, 44.3; HRMS (EI, double focusing) m/z: [M]+ calcd for C15H11FO 226.0794; found 226.0796.
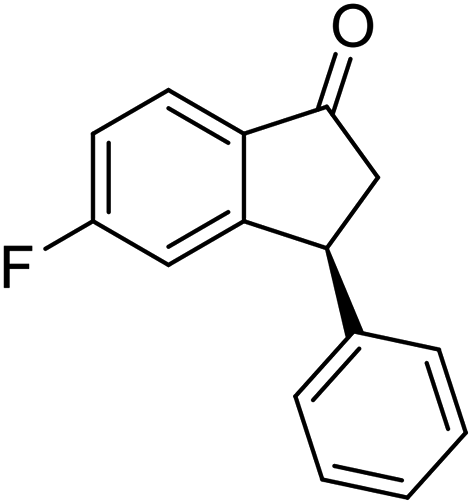
(1R,3R)-5-Methoxy-3-phenyl-2,3-dihydro-1H-inden-1-ol (2g)
Yield 48% (57 mg as white solid); mp 127.9–128.3 °C; 97% ee (Chiralpak IB, 0 to 6% IPA for 9 min in n-hexane, 0.9 mL min−1, 270 nm, tR(major) = 25.9 min, tR(minor) = 22.4 min); [α]27D = +17.4 (c 2.9, CH2Cl2); 1H NMR (CDCl3, 400 MHz): δ 7.37 (d, 1H, J = 8.3 Hz), 7.35–7.29 (m, 2H), 7.28–7.20 (m, 3H), 6.85 (dd, 1H, J = 8.3, 2.3 Hz), 6.47 (s, 1H), 5.24 (s, 1H), 4.16 (t, 1H, J = 8.2 Hz), 3.71 (s, 3H), 3.02 (dt, 1H, J = 13.0, 7.3 Hz), 2.00–1.83 (m, 2H); 13C{1H} NMR (101 MHz, CDCl3) δ 160.3, 147.4, 144.2, 137.6, 128.6, 128.2, 126.6, 124.6, 113.8, 109.9, 74.7, 55.4, 48.5, 47.4; HRMS (EI, double focusing) m/z: [M]+ calcd for C16H16O2 240.1150; found 240.1155.
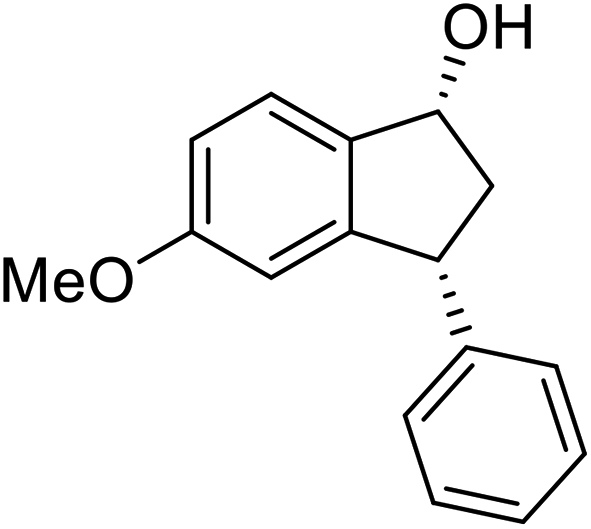
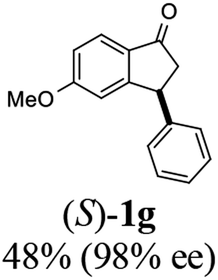
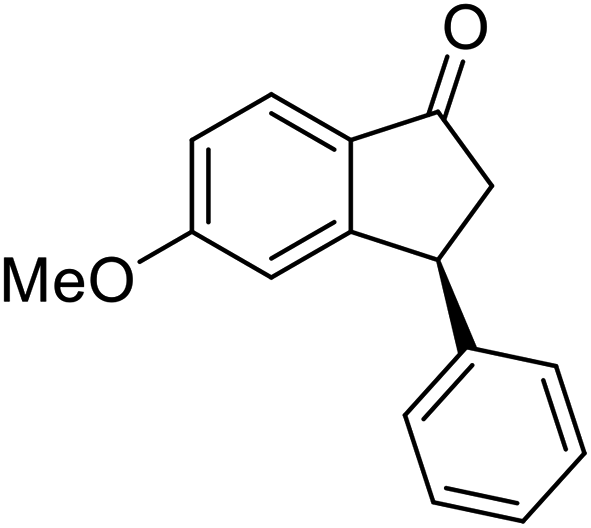
(S)-5-Methoxy-3-phenyl-2,3-dihydro-1H-inden-1-one, (S)-1g
Yield 47.9% (54.1 mg, white solid); mp 129.8–130.0 °C; 98% ee; (Chiralpak IB, 0 to 6% IPA for 9 min in n-hexane, 0.9 mL min−1, 270 nm, tR(major) = 20.8 min, tR(minor) = 20.3 min); [α]25D = −11.8 (c 3.1, CH2Cl2). Literature values: [α]25D = −10.0 (c 0.2, CH2Cl2 for 84% ee).23 [α]23D = +17 (c 1.0, CHCl3 for 96% ee) for (R)-1g;27 1H NMR (CDCl3, 400 MHz): δ 7.74 (d, 1H, J = 8.5 Hz), 7.31 (t, 2H, J = 7.3 Hz), 7.29–7.21 (m, 1H), 7.13 (d, 2H, J = 7.0 Hz), 6.94 (dd, 1H, J = 8.5, 2.2 Hz), 6.65 (s, 1H), 4.50 (dd, 1H, J = 8.0, 3.8 Hz), 3.78 (s, 3H), 3.20 (dd, 1H, J = 19.0, 8.1 Hz), 2.66 (dd, 1H, J = 19.0, 3.8 Hz); 13C{1H} NMR (101 MHz, CDCl3) δ 204.1, 165.6, 160.9, 143.7, 130.2, 128.9, 127.6, 127.0, 125.1, 116.0, 109.8, 55.7, 47.1, 44.5; HRMS (EI, double focusing) m/z: [M]+ calcd for C16H14O2 238.0994; found 238.1006.
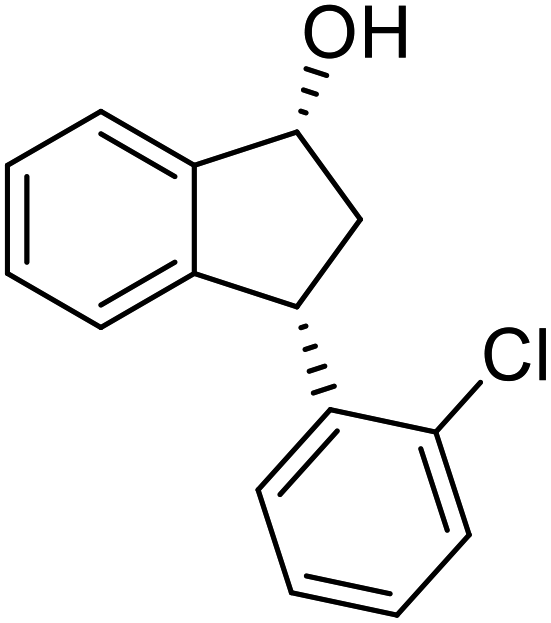
(1R,3S)-3-(2-Chlorophenyl)-2,3-dihydro-1H-inden-1-ol (2h)
Yield 42% (51 mg as white solid); mp 103.2–104.3 °C; 92% ee (Chiralpak IB, 0 to 3% IPA for 7 min in n-hexane, 1 mL min−1, 270 nm, tR(major) = 27.5 min, tR(minor) = 19.8 min); [α]27D = +42.8 (c 1.4, CH2Cl2); 1H NMR (CDCl3, 400 MHz): δ 7.50 (d, 1H, J = 7.4 Hz), 7.44–7.38 (m, 1H), 7.35–7.25 (m, 2H), 7.21–7.14 (m, 2H), 7.14–7.07 (m, 1H), 7.02 (d, 1H, J = 7.5 Hz), 5.31 (s, 1H), 4.77 (t, 1H, J = 8.1 Hz), 3.09 (dt, 1H, J = 13.1, 7.4 Hz), 2.01–1.79 (m, 2H); 13C{1H}NMR (101 MHz, CDCl3) δ 145.4, 144.4, 142.1, 134.1, 129.4, 129.1, 128.5, 127.7, 127.4, 127.2, 125.2, 124.1, 75.1, 45.3, 44.5; HRMS (EI, double focusing) m/z: [M]+ calcd for C15H13ClO 244.0655; found 244.0654.
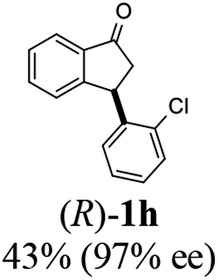
(R)-3-(2-Chlorophenyl)-2,3-dihydro-1H-inden-1-one, (R)-1h
Yield 42.6% (51.7 mg, pale yellow solid); mp 56.5–57.7 °C; 97% ee (Chiralpak IB, 0 to 3% IPA for 7 min in n-hexane, 1 mL min−1, 270 nm, tR(major) = 16.5 min, tR(minor) = 16.9 min); [α]26D = −56.6 (c 1.7, CH2Cl2). Literature value [α]25D = −36.0 (c = 0.4, CH2Cl2 for 70% ee);23 1H NMR (CDCl3, 400 MHz): δ 7.83 (d, 1H, J = 7.7 Hz), 7.61 (t, 1H, J = 7.5 Hz), 7.49–7.40 (m, 2H), 7.34 (d, 1H, J = 7.7 Hz), 7.24–7.10 (m, 2H), 6.88 (s, 1H), 5.12 (s, 1H), 3.30 (dd, 1H, J = 19.2, 8.2 Hz), 2.61 (d, 1H, J = 18.7 Hz); 13C{1H} NMR (101 MHz, CDCl3) δ 205.5, 156.6, 141.2, 137.3, 135.1, 134.0, 129.8, 128.4, 128.2, 128.1, 127.4, 126.9, 123.7, 45.4, 41.0; HRMS (EI, double focusing) m/z: [M]+ calcd for C15H11ClO 242.0498; found 242.0499.
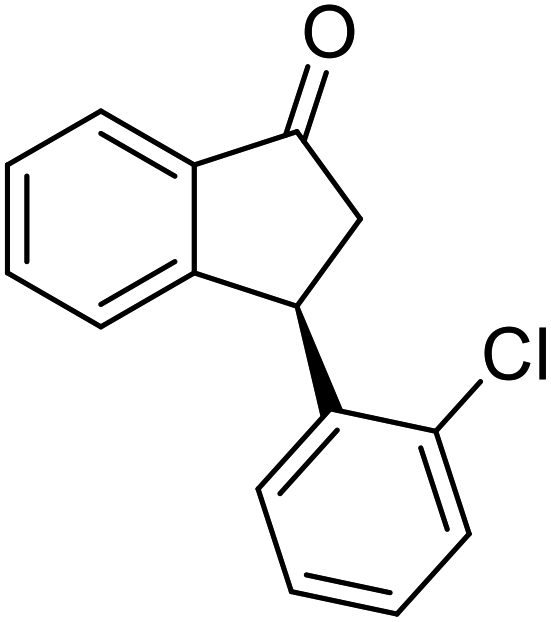
(1R,3R)-3-(3-Chlorophenyl)-2,3-dihydro-1H-inden-1-ol (2i)
Yield 38% (46 mg as white solid); mp 104.2–104.6 °C; 99% ee (Chiralpak IB, 0 to 3% IPA for 7 min in n-hexane, 1 mL min−1, 270 nm, tR(major) = 24.3 min, tR(minor) = 20.3 min); [α]28D = −15.1 (c 1.1, CH2Cl2); 1H NMR (CDCl3, 400 MHz): δ 7.48 (d, 1H, J = 7.5 Hz), 7.31 (t, 1H, J = 7.4 Hz), 7.27 (s, 1H), 7.24–7.19 (m, 3H), 7.14–7.08 (m, 1H), 6.95 (d, 1H, J = 7.5 Hz), 5.34–5.24 (m, 1H), 4.17 (t, 1H, J = 8.4 Hz), 3.02 (dt, 1H, J = 13.0, 7.1 Hz), 2.06–1.86 (m, 2H); 13C{1H}NMR (101 MHz, CDCl3) δ 146.4, 145.2, 144.8, 134.4, 129.9, 128.6, 128.4, 127.5, 126.8, 126.5, 125.0, 123.8, 75.0, 48.0, 46.9; HRMS (EI, double focusing) m/z: [M]+ calcd for C15H13ClO 244.0655; found 244.0659.
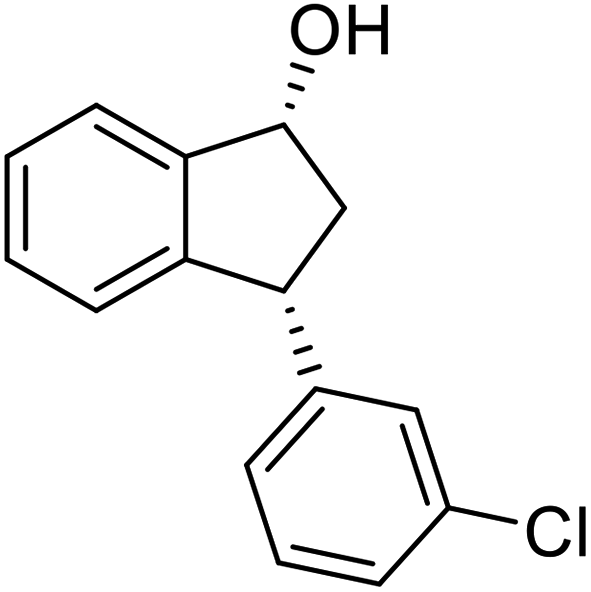
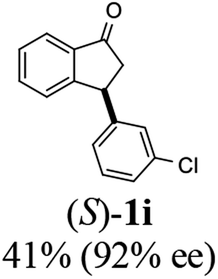
(S)-3-(3-Chlorophenyl)-2,3-dihydro-1H-inden-1-one, (S)-1i
Yield 41.4% (50.2 mg, white solid); mp 108.5–109.1 °C; 92% ee (Chiralpak IB, 0 to 3% IPA for 7 min in n-hexane, 1 mL min−1, 270 nm, tR(major) = 17.2 min, tR(minor) = 19.1 min); [α]26D = +66.7 (c 2.33, CH2Cl2). Literature value: [α]25D = +41.4 (c 0.6, CH2Cl2 for 84% ee);23 1H NMR (CDCl3, 400 MHz): δ 7.82 (d, 1H, J = 7.7 Hz), 7.60 (t, 1H, J = 7.5 Hz), 7.45 (t, 1H, J = 7.5 Hz), 7.30–7.26 (m, 1H), 7.25–7.21 (m, 2H), 7.12 (s, 1H), 7.04–6.97 (m, 1H), 4.56 (dd, 1H, J = 8.1, 3.9 Hz), 3.23 (dd, 1H, J = 19.2, 8.1 Hz), 2.66 (dd, 1H, J = 19.2, 3.9 Hz); 13C{1H} NMR (101 MHz, CDCl3) δ 205.3, 157.0, 145.7, 136.8, 135.3, 134.7, 130.2, 128.2, 127.8, 127.2, 126.8, 125.8, 123.6, 46.6, 44.1; HRMS (EI, double focusing) m/z: [M]+ calcd for C15H11ClO 242.0498; found 242.0505.
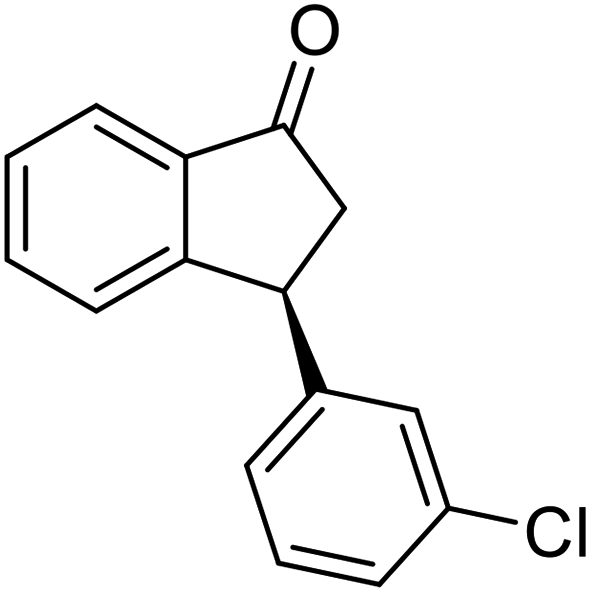
(1R,3R)-3-(4-Chlorophenyl)-2,3-dihydro-1H-inden-1-ol (2j)
Yield 44% (56 mg as white solid); mp 111.9–120.2 °C; 98% ee (Chiralpak IB, 0 to 3% IPA for 7 min in n-hexane, 1 mL min−1, 270 nm, tR(major) = 23.8 min, tR(minor) = 21.6 min); [α]28D = −30.9 (c 1.7, CH2Cl2); 1H NMR (CDCl3, 400 MHz): δ 7.48 (d, 1H, J = 7.5 Hz), 7.35–7.26 (m, 3H), 7.25–7.21 (m, 1H), 7.17 (d, 2H, J = 8.4 Hz), 6.93 (d, 1H, J = 7.5 Hz), 5.35–5.23 (m, 1H), 4.17 (t, 1H, J = 8.3 Hz), 3.02 (dt, 1H, J = 12.9, 7.3 Hz), 2.01–1.82 (m, 2H); 13C{1H}NMR (101 MHz, CDCl3) δ 145.2, 145.1, 142.8, 132.4, 129.6, 128.7, 128.5, 127.4, 125.0, 123.8, 75.0, 47.8, 47.0; HRMS (EI, double focusing) m/z: [M]+ calcd for C15H13ClO 244.0655; found 244.0651.
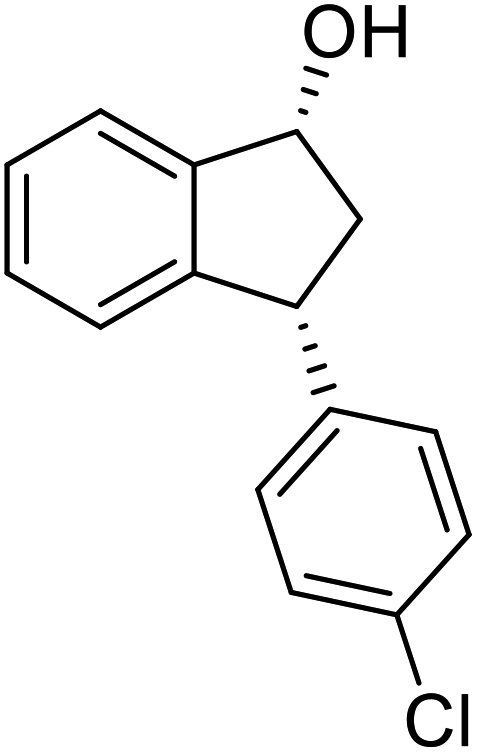
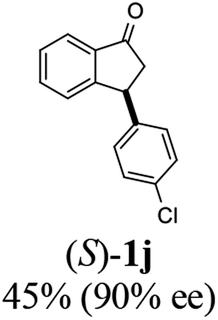
(S)-3-(4-Chlorophenyl)-2,3-dihydro-1H-inden-1-one, (S)-1j
Yield 44.6% (54.1 mg, white solid); mp 75.9–76.5 °C; 90% ee (Chiralpak IB, 0 to 3% IPA for 7 min in n-hexane, 1 mL min−1, 270 nm, tR(major) = 17.5 min, tR(minor) = 18.4 min); [α]27D = +37.9 (c 2.6, CH2Cl2). Literature values: [α]27D = +42.9 (c 0.6, CHCl3 for 77% ee).26 [α]25D = +48.5 (c 0.4, CH2Cl2 for 90% ee);23 1H NMR (CDCl3, 500 MHz): δ 7.82 (d, 1H, J = 7.7 Hz), 7.59 (t, 1H, J = 7.7, 1.1 Hz), 7.44 (t, 1H, J = 7.5 Hz), 7.31–7.22 (m, 3H), 7.06 (d, 2H, J = 8.3 Hz), 4.56 (dd, 1H, J = 8.1, 3.8 Hz), 3.23 (dd, 1H, J = 19.2, 8.1 Hz), 2.63 (dd, 1H, J = 19.2, 3.8 Hz); 13C{1H} NMR (101 MHz, CDCl3) δ 205.5, 157.3, 142.2, 136.8, 135.2, 132.8, 129.1, 129.0, 128.1, 126.8, 123.5, 46.7, 43.8; HRMS (EI, double focusing) m/z: [M]+ calcd for C15H11ClO 242.0498; found 242.0501.
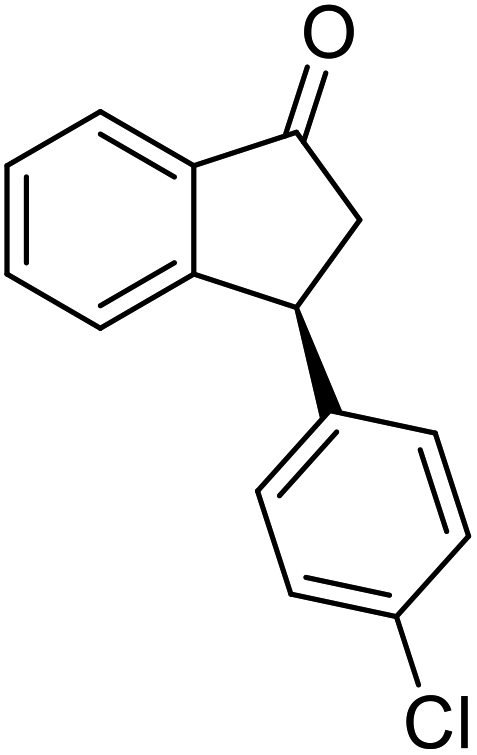
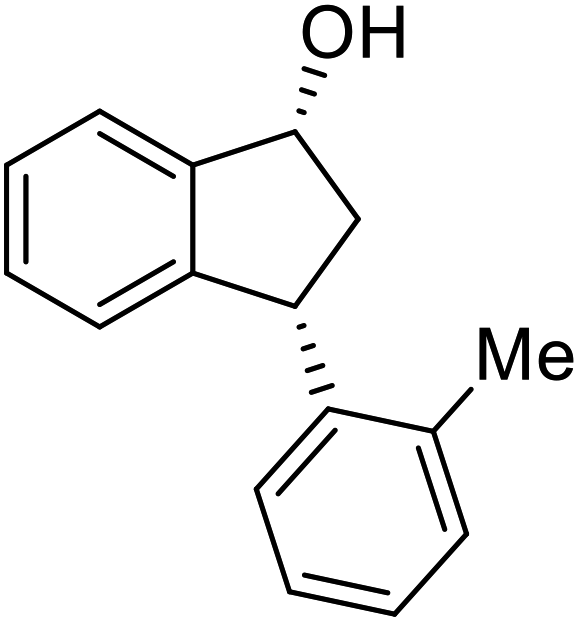
(1R,3R)-3-(o-Tolyl)-2,3-dihydro-1H-inden-1-ol (2k)
Yield 41% (45 mg as white solid); mp 122.6–123.9 °C; 98% ee (Chiralpak IB, 0 to 4% IPA for 7 min in n-hexane, 1 mL min−1, 270 nm, tR(major) = 26.1 min, tR(minor) = 18.0 min); [α]28D = +49.3 (c 2.5, CH2Cl2); 1H NMR (CDCl3, 400 MHz): δ 7.49 (d, 1H, J = 7.5 Hz), 7.31 (t, 1H, J = 7.3 Hz), 7.25 (t, 1H, J = 7.0 Hz), 7.23–7.17 (m, 1H), 7.17–7.08 (m, 2H), 7.04–6.94 (m, 2H), 5.29 (s, 1H), 4.45 (t, 1H, J = 8.3 Hz), 3.03 (dt, 1H, J = 12.9, 7.3 Hz), 2.42 (s, 3H), 2.00–1.81 (m, 2H); 13C{1H} NMR (101 MHz, CDCl3) δ 145.4, 145.3, 142.7, 136.1, 130.3, 128.4, 127.5, 127.1, 126.4, 126.4, 125.2, 123.9, 75.2, 45.9, 44.1, 19.8; HRMS (EI, double focusing) m/z: [M]+ calcd for C16H16O 224.1201; found 224.1203.
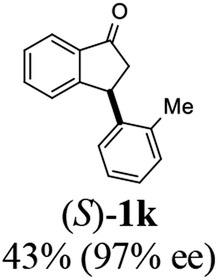
(S)-3-(o-Tolyl)-2,3-dihydro-1H-inden-1-one, (S)-1k
Yield 43.4% (48.2 mg, yellow solid); mp 55.7–56.5 °C; 97% ee (Chiralpak IB, 0 to 4% IPA for 7 min in n-hexane, 1 mL min−1, 270 nm, tR(major) = 15.8 min, tR(minor) = 16.9 min); [α]28D = −72.5 (c 2.4, CH2Cl2). Literature value for (R)-1k: [α]23D = +56 (c 1.0, CHCl3 for 98% ee);27 1H NMR (CDCl3, 400 MHz): δ 7.83 (d, 1H, J = 7.7 Hz), 7.60 (t, 1H, J = 7.5 Hz), 7.44 (t, 1H, J = 7.5 Hz), 7.30 (d, 1H, J = 7.7 Hz), 7.22 (d, 1H, J = 7.4 Hz), 7.15 (t, 1H, J = 7.4 Hz), 7.08 (t, 1H, J = 7.4 Hz), 6.77 (d, 1H, J = 7.0 Hz), 4.84 (dd, 1H, J = 8.1, 3.9 Hz), 3.25 (dd, 1H, J = 19.1, 8.1 Hz), 2.57 (dd, 1H, J = 19.1, 3.9 Hz), 2.43 (s, 3H); 13C{1H} NMR (101 MHz, CDCl3) δ 206.0, 157.8, 142.0, 137.3, 135.9, 135.0, 130.6, 127.8, 127.0, 126.8, 126.6, 123.5, 45.8, 29.7, 19.9; HRMS (EI, double focusing) m/z: [M]+ calcd for C16H14O 222.1045; found 222.1036.
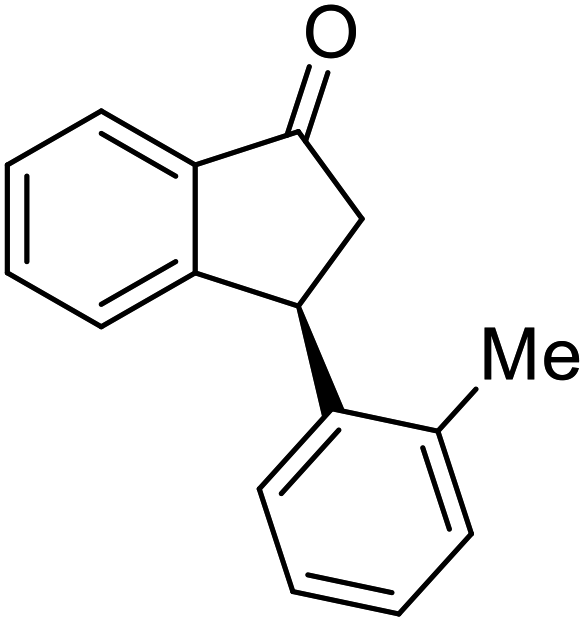
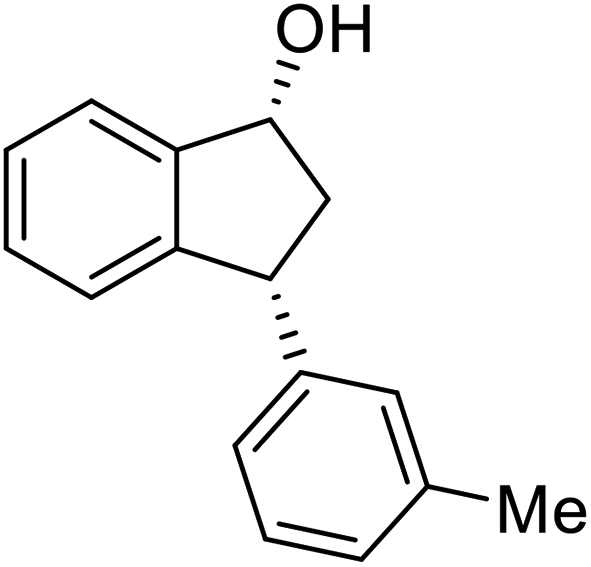
(1R,3R)-3-(m-Tolyl)-2,3-dihydro-1H-inden-1-ol (2l)
Yield 43% (47 mg as white solid); mp 77.0–77.3 °C; 94% ee (Chiralpak IB, 0 to 4% IPA for 7 min in n-hexane, 1 mL min−1, 270 nm, tR(major) = 30.0 min, tR(minor) = 19.4 min); [α]28D = −22.0 (c 0.7, CH2Cl2); 1H NMR (CDCl3, 400 MHz): δ 7.47 (d, 1H, J = 7.4 Hz), 7.29 (t, 1H, J = 7.4 Hz), 7.26–7.17 (m, 2H), 7.09–6.99 (m, 3H), 6.95 (d, 1H, J = 7.5 Hz), 5.27 (t, 1H, J = 7.2 Hz), 4.14 (t, 1H, J = 8.4 Hz), 3.01 (dt, 1H, J = 12.8, 7.2 Hz), 2.32 (s, 3H), 2.11–1.87 (m, 2H); 13C{1H} NMR (101 MHz, CDCl3) δ 145.7, 145.2, 144.2, 138.2, 129.0, 128.5, 128.3, 127.4, 127.1, 125.3, 125.1, 123.6, 75.1, 48.2, 47.2, 21.4; HRMS (EI, double focusing) m/z: [M]+ calcd for C16H16O 224.1201; found 224.1200.
(S)-3-(m-Tolyl)-2,3-dihydro-1H-inden-1-one, (S)-1l
Yield 41.2% (45.8 mg, pale yellow solid); mp 62.7–63.7 °C; 96% ee (Chiralpak IB, 0 to 4% IPA for 7 min in n-hexane, 1 mL min−1, 270 nm, tR(major) = 15.8 min, tR(minor) = 16.3 min); [α]28D = +74.2 (c 2.1, CH2Cl2). Literature value for (R)-1l: [α]23D = +33.2 (c 0.9, CHCl3 for 95% ee);7 1H NMR (CDCl3, 400 MHz): δ 7.81 (d, 1H, J = 7.7 Hz), 7.57 (t, 1H, J = 7.5 Hz), 7.42 (t, 1H, J = 7.5 Hz), 7.28 (d, 1H, J = 7.7 Hz), 7.20 (t, 1H, J = 7.9 Hz), 7.06 (d, 1H, J = 7.5 Hz), 6.96–6.88 (m, 2H), 4.54 (dd, 1H, J = 8.0, 3.9 Hz), 3.22 (dd, 1H, J = 19.2, 8.0 Hz), 2.69 (dd, 1H, J = 19.2, 3.9 Hz), 2.31 (s, 3H); 13C{1H} NMR (101 MHz, CDCl3) δ 206.2, 158.1, 143.6, 138.6, 136.7, 135.1, 128.8, 128.3, 127.8, 127.7, 126.9, 124.7, 123.4, 46.8, 44.4, 21.4; HRMS (EI, double focusing) m/z: [M]+ calcd for C16H14O 222.1045; found 222.1032.
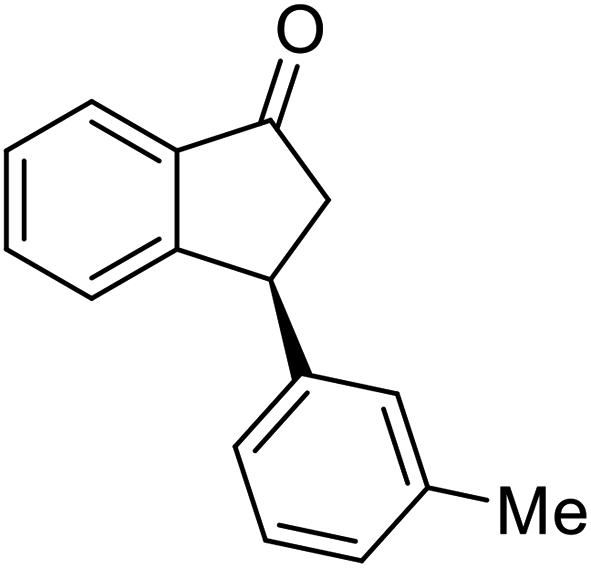
(1R,3R)-3-(p-Tolyl)-2,3-dihydro-1H-inden-1-ol (2m)
Yield 42% (47 mg as white solid); mp 97.7–98.0 °C; 95% ee (Chiralpak IB, 6% IPA in n-hexane, 1 mL min−1, 270 nm, tR(major) = 12.4 min, tR(minor) = 7.1 min); [α]28D = −21.7 (c 1.5, CH2Cl2); 1H NMR (CDCl3, 400 MHz): δ 7.47 (d, 1H, J = 7.4 Hz), 7.29 (t, 1H, J = 7.4 Hz), 7.22 (t, 1H, J = 7.3 Hz), 7.17–7.10 (m, 4H), 6.95 (d, 1H, J = 7.5 Hz), 5.28 (s, 1H), 4.16 (d, 1H, J = 8.2 Hz), 3.01 (dt, 1H, J = 12.8, 7.2 Hz), 2.34 (s, 3H), 2.00–1.85 (m, 2H); 13C{1H} NMR (101 MHz, CDCl3) δ 145.8, 145.2, 141.2, 136.2, 129.3, 128.3, 128.1, 127.1, 125.1, 123.6, 75.1, 47.9, 47.3, 21.0; HRMS (EI, double focusing) m/z: [M]+ calcd for C16H16O 224.1201; found 224.1190.
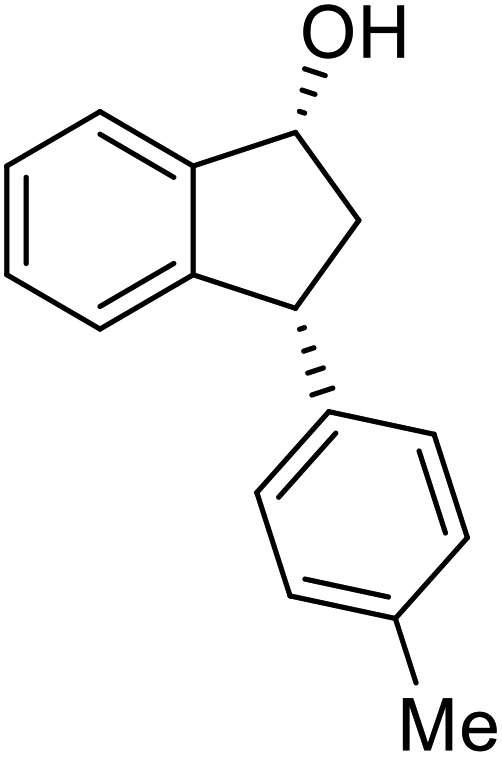
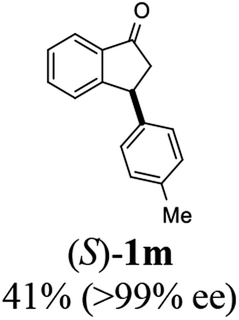
(S)-3-(p-Tolyl)-2,3-dihydro-1H-inden-1-one, (S)-1m
Yield 41.5% (46.1 mg, yellow solid); mp 78.3–79.1 °C; >99% ee (Chiralpak IB, 6% IPA in n-hexane, 1 mL min−1, 270 nm, tR(major) = 6.1 min, tR(minor) = 6.4 min); [α]28D = +40.8 (c 2.5, CH2Cl2). Literature values: [α]25D = +109.0 (c 0.2, CH2Cl2 for 84% ee).23 [α]23D = −62.9 (c 0.7, CHCl3 for 90% ee) for (R)-1m;7 1H NMR (CDCl3, 500 MHz): δ 7.80 (d, 1H, J = 7.7 Hz), 7.56 (t, 1H, J = 7.5 Hz), 7.41 (t, 1H, J = 7.4 Hz), 7.28 (d, 1H, J = 7.5 Hz), 7.12 (d, 2H, J = 7.8 Hz), 7.02 (d, 2H, J = 7.8 Hz), 4.54 (dd, 1H, J = 8.0, 3.8 Hz), 3.22 (dd, 1H, J = 19.2, 8.0 Hz), 2.67 (dd, 1H, J = 19.2, 3.8 Hz), 2.33 (s, 3H); 13C{1H} NMR (101 MHz, CDCl3) δ 206.2, 158.2, 140.7, 136.7, 136.6, 135.1, 129.6, 127.8, 127.5, 126.8, 123.4, 46.9, 44.1, 21.0; HRMS (EI, double focusing) m/z: [M]+ calcd for C16H14O 222.1045; found 222.1035.
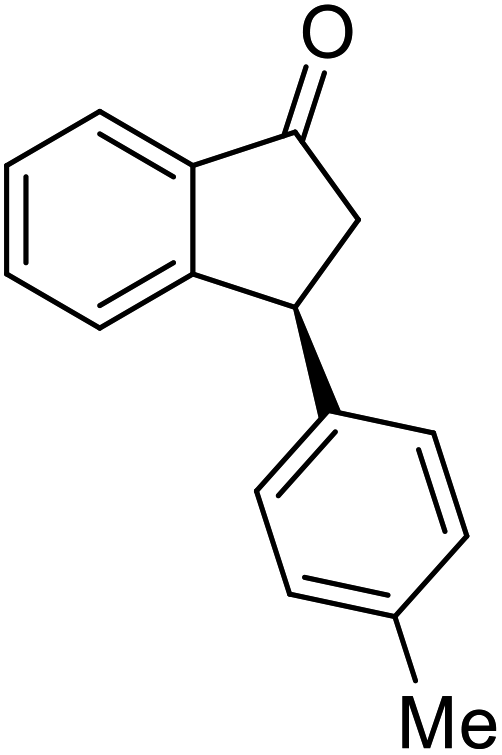
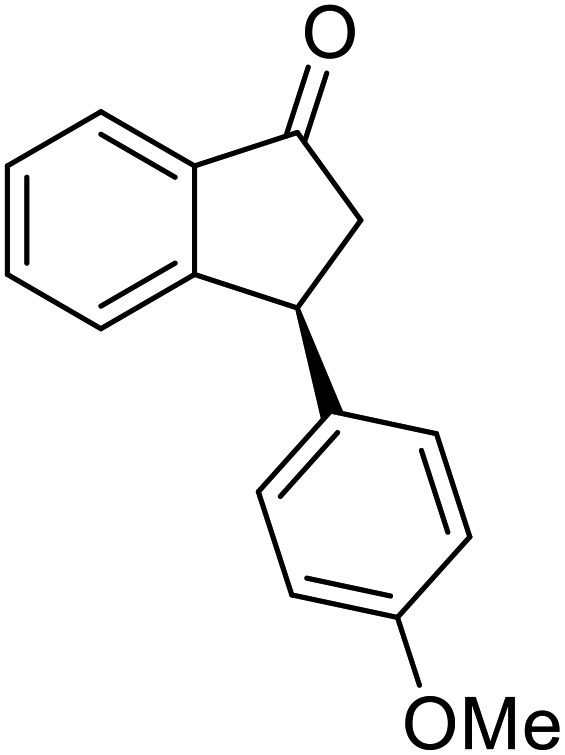
(1R,3R)-3-(4-Methoxyphenyl)-2,3-dihydro-1H-inden-1-ol (2n)
Yield 45.0% (53.6 mg as white solid); mp 110.8–111.3 °C; 99% ee (Chiralpak IB, 7% IPA in n-hexane, 1 mL min−1, 270 nm, tR(major) = 43.4 min, tR(minor) = 45.7 min); [α]28D = −20.6 (c 2.5, CH2Cl2); 1H NMR (CDCl3, 400 MHz): δ 7.5 (d, 1H, J = 7.4 Hz), 7.3–7.3 (m, 1H), 7.3–7.2 (m, 1H), 7.1 (d, 2H, J = 8.7 Hz), 6.9 (d, 1H, J = 7.4 Hz), 6.9 (d, 2H, J = 8.7 Hz), 5.3 (q, 1H, J = 6.9, 5.9 Hz), 4.1 (t, 1H, J = 8.4 Hz), 3.8 (s, 3H), 3.0 (dt, 1H, J = 12.9, 7.2 Hz), 2.0–1.8 (m, 2H) ppm; 13C{1H} NMR (101 MHz, CDCl3) δ 165.6, 160.9, 143.7, 130.2, 128.9, 127.0, 125.1, 116.0, 109.8, 55.7, 47.1, 44.5, 29.7; HRMS (EI, double focusing) m/z: [M]+ calcd for C16H16O2 240.1150; found 240.1161.
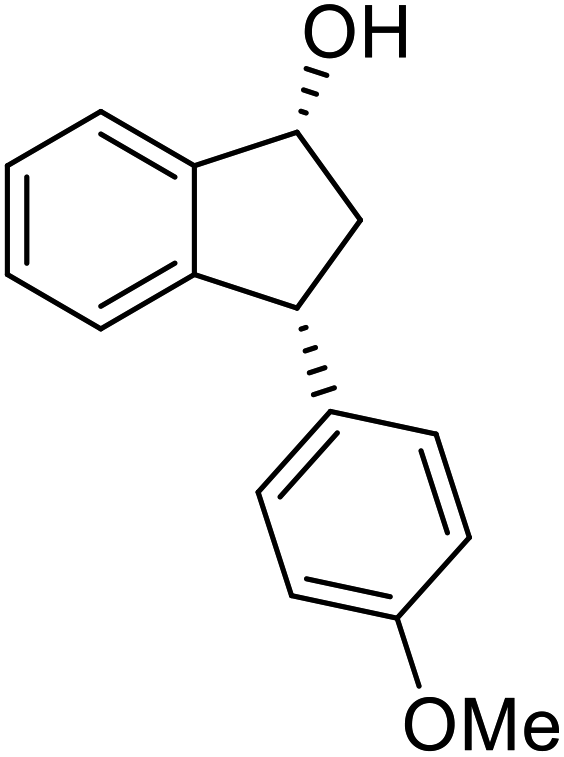
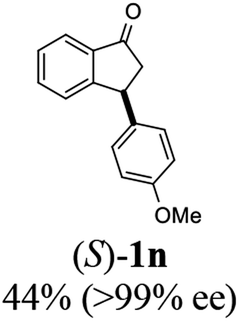
(S)-3-(4-Methoxyphenyl)-2,3-dihydro-1H-inden-1-one, (S)-1n
Yield 45.2% (53.8 mg, yellow solid); mp 72.7–73.1 °C; >99% (Chiralpak IB, 7% IPA in n-hexane, 1 mL min−1, 270 nm, tR(major) = 41.8 min, tR(minor) = 40.9 min); [α]20D = +69.7 (c 2.1, CH2Cl2). Literature values: [α]25D = +41.1 (c 0.6, CHCl3 for 70% ee).26 [α]20D = +59.1 (c 0.6, CH2Cl2 for 84% ee);23 1H NMR (CDCl3, 400 MHz): δ 7.80 (d, 1H, J = 7.7 Hz), 7.57 (t, 1H, J = 7.5 Hz), 7.41 (t, 1H, J = 7.4 Hz), 7.04 (d, 2H, J = 8.7 Hz), 6.85 (d, 2H, J = 8.7 Hz), 4.53 (d, 1H, J = 8.0 Hz), 3.79 (s, 3H), 3.21 (dd, 1H, J = 19.2, 8.0 Hz), 2.65 (dd, 1H, J = 19.2, 3.9 Hz); 13C{1H} NMR (101 MHz, CDCl3) δ 206.2, 158.6, 158.3, 136.7, 135.8, 135.1, 128.6, 127.8, 126.8, 123.3, 114.3, 55.3, 47.0, 43.7; HRMS (EI, double focusing) m/z: [M]+ calcd for C16H14O2 238.0994; found 238.1010.
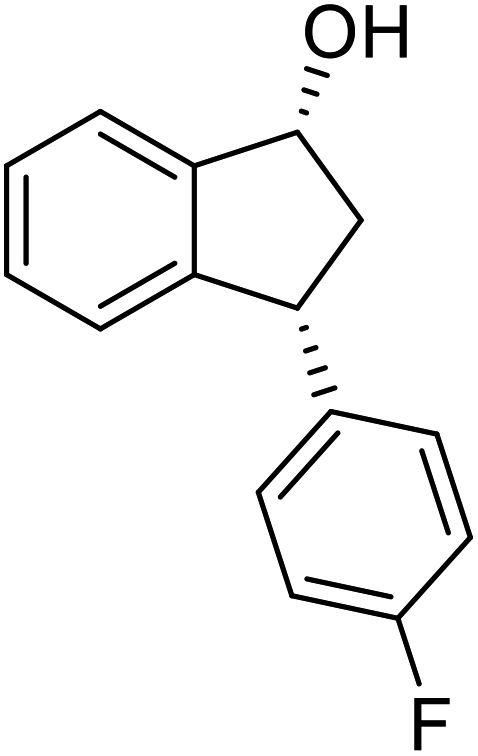
(1R,3R)-3-(4-Fluorophenyl)-2,3-dihydro-1H-inden-1-ol (2o)
Yield 48% (54 mg as white solid); mp 111.9–112.0 °C; 98.2% ee (Chiralpak IB, 0 to 4% IPA for 7 min in n-hexane, 1 mL min−1, 270 nm, tR(major) = 19.0 min, tR(minor) = 21.7 min); [α]28D = −14.4 (c 2.6, CH2Cl2); 1H NMR (CDCl3, 400 MHz): δ 7.47 (d, 1H, J = 7.4 Hz), 7.35–7.14 (m, 4H), 7.00 (t, 2H, J = 8.7 Hz), 6.93 (d, 1H, J = 7.4 Hz), 5.29 (t, 1H, J = 6.8 Hz), 4.17 (t, 1H, J = 8.4 Hz), 3.01 (dt, 1H, J = 13.0, 7.3 Hz), 2.02 (s, 1H), 1.90 (ddd, 1H, J = 12.9, 9.1, 7.6 Hz); 13C{1H} NMR (101 MHz, CDCl3) δ 161.7 (d, JC–F = 244.6 Hz), 145.5, 145.2, 140.0 (d, JC–F = 3.2 Hz), 129.7 (d, JC–F = 8.0 Hz), 128.5, 127.3, 125.0, 123.7, 115.4 (d, JC–F = 21.2 Hz), 75.0, 47.6, 47.2; HRMS (EI, double focusing) m/z: [M]+ calcd for C15H13FO 228.0950; found 228.0940.
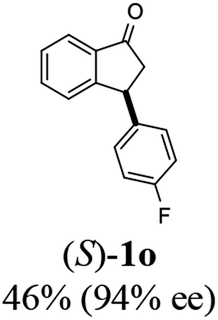
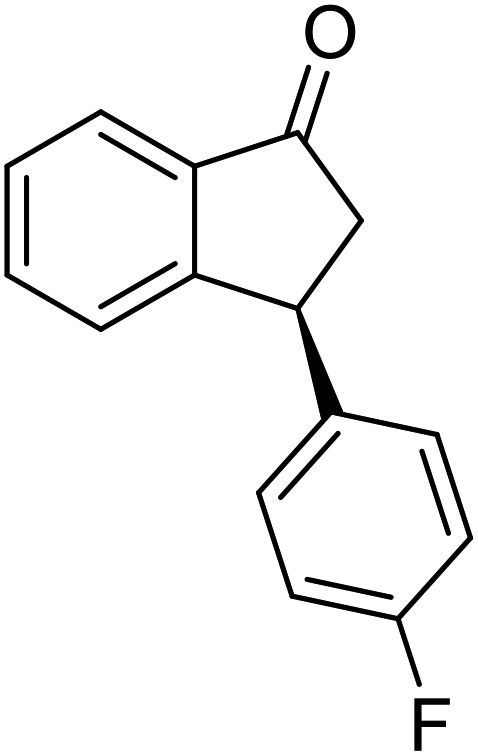
(S)-3-(4-Fluorophenyl)-2,3-dihydro-1H-inden-1-one, (S)-1o
Yield 49.9% (56.4 g, white solid); mp 116.5–117.1 °C; 94.2% ee (Chiralpak IB, 0 to 4% IPA for 7 min in n-hexane, 1 mL min−1, 270 nm, tR(major) = 16.3 min, tR(minor) = 17.4 min); [α]22D = +39.4 (c 2.6, CH2Cl2). Literature value [α]25D = +37.9 (c 0.3, CH2Cl2 for 90% ee);23 1H NMR (CDCl3, 400 MHz): δ 7.82 (d, 1H, J = 7.5 Hz), 7.58 (t, 1H, J = 7.5 Hz), 7.43 (t, 1H, J = 7.5 Hz), 7.24 (d, 1H), 7.12–7.05 (m, 2H), 7.00 (t, 2H, J = 8.6 Hz), 4.57 (dd, 1H, J = 8.0, 3.9 Hz), 3.23 (dd, 1H, J = 19.2, 8.0 Hz), 2.64 (dd, 1H, J = 19.2, 3.9 Hz); 13C{1H} NMR (101 MHz, CDCl3) δ 205.7, 163.0–160.6 (d, JC–F = 245.6 Hz), 157.7, 139.5–139.4 (d, JC–F = 3.4 Hz), 136.7, 135.2, 129.2–129.1 (d, JC–F = 8.0 Hz), 128.0, 126.8, 123.5, 115.9–115.7 (d, JC–F = 21.5 Hz), 46.9, 43.7; HRMS (EI, double focusing) m/z: [M]+ calcd for C15H11FO 226.0794; found 226.0791.
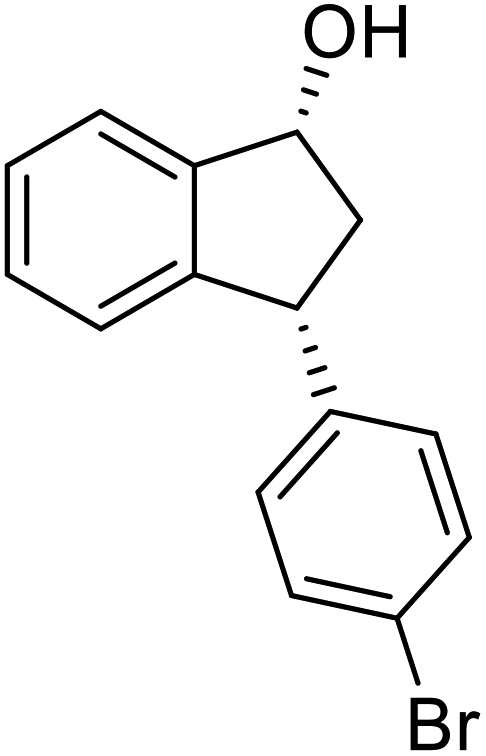
(1R,3R)-3-(4-Bromophenyl)-2,3-dihydro-1H-inden-1-ol (2p)
Yield 40% (58 mg as white solid); mp 132.2–132.7 °C; 99% ee (Chiralpak IB, 0 to 4% IPA for 7 min in n-hexane, 1 mL min−1, 270 nm, tR(major) = 20.2 min, tR(minor) = 21.5 min); [α]29D = −17.7 (c 1.4, CH2Cl2); 1H NMR (CDCl3, 400 MHz): δ 7.54–7.37 (m, 3H), 7.30 (t, 1H, J = 7.4 Hz), 7.27–7.20 (m, 1H), 7.10 (d, 2H, J = 8.4 Hz), 6.92 (d, 1H, J = 7.4 Hz), 5.36–5.20 (m, 1H), 4.15 (t, 1H, J = 8.3 Hz), 3.00 (dt, 1H, J = 13.0, 7.3 Hz), 2.08 (s, 1H), 1.89 (dt, 1H, J = 13.0, 8.3, 7.3 Hz); 13C{1H} NMR (101 MHz, CDCl3) δ 145.2, 145.0, 143.3, 131.7, 130.0, 128.5, 127.4, 125.0, 123.8, 120.4, 75.0, 47.8, 46.9; HRMS (EI, double focusing) m/z: [M]+ calcd for C15H13BrO 288.0150; found 288.0147.
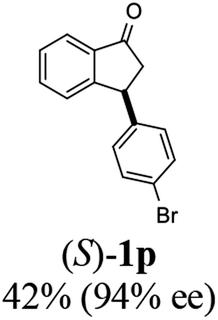
(S)-3-(4-Bromophenyl)-2,3-dihydro-1H-inden-1-one, (S)-1p
Yield 42.3% (60.7 mg, pale yellow solid); mp 60.1–60.5 °C; 94% ee (Chiralpak IB, 0 to 4% IPA for 7 min in n-hexane, 1 mL min−1, 270 nm, tR(major) = 17.3 min, tR(minor) = 18.2 min); [α]23D = +47.1 (c 2.9, CH2Cl2). Literature value: [α]25D = +44.0 (c 0.4, CH2Cl2 for 90% ee);23 1H NMR (CDCl3, 400 MHz): δ 7.82 (d, 1H, J = 7.8 Hz), 7.59 (d, 1H, J = 7.8 Hz), 7.49–7.39 (m, 3H), 7.24 (d, 1H, J = 7.8 Hz), 7.00 (d, 2H, J = 8.4 Hz), 4.55 (dd, 1H, J = 8.1, 3.8 Hz), 3.23 (dd, 1H, J = 19.2, 8.1 Hz), 2.63 (dd, 1H, J = 19.2, 3.8 Hz); 13C{1H} NMR (101 MHz, CDCl3) δ 205.4, 157.2, 142.7, 136.8, 135.2, 132.0, 129.4, 128.1, 126.8, 123.6, 120.9, 46.7, 43.9; HRMS (EI, double focusing) m/z: [M]+ calcd for C15H11BrO 285.9993; found 285.9991.
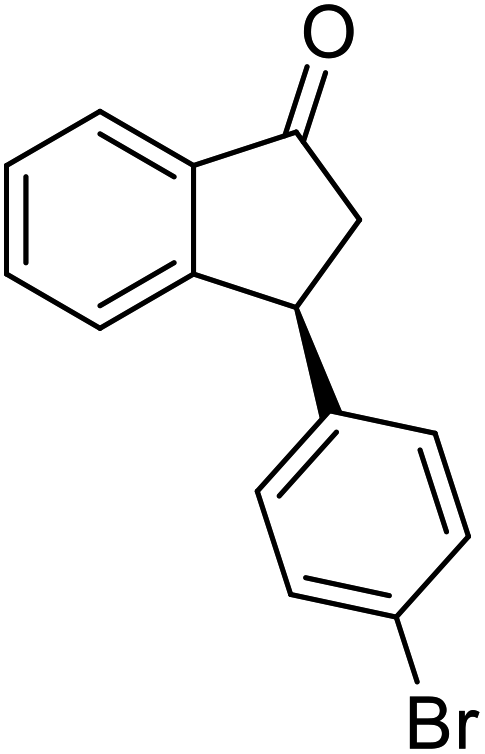
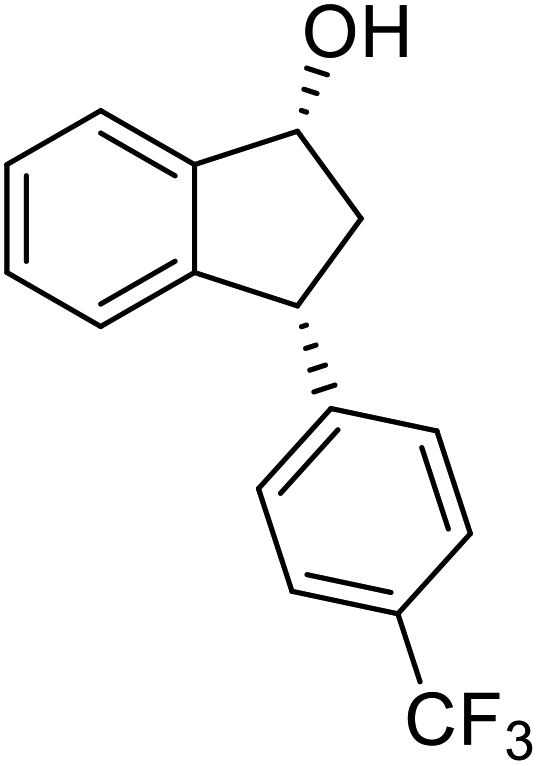
(1R,3R)-3-(4-(Trifluoromethyl)phenyl)-2,3-dihydro-1H-inden-1-ol (2q)
Yield 34% (47 mg as white solid); mp 98.5–99.7 °C; 97% ee (Chiralpak IB, 0 to 4% IPA for 7 min in n-hexane, 1 mL min−1, 270 nm, tR(major) = 20.7 min, tR(minor) = 18.8 min) [α]29D = −8.7 (c 3.8, CH2Cl2); 1H NMR (CDCl3, 400 MHz): δ 7.58 (d, 2H, J = 8.2 Hz), 7.49 (d, 1H, J = 7.5 Hz), 7.40–7.29 (m, 3H), 7.29–7.21 (m, 1H), 6.92 (d, 1H, J = 7.5 Hz), 5.37–5.29 (m, 1H), 4.26 (d, 1H, J = 8.9 Hz), 3.04 (dt, 1H, J = 13.0, 7.3 Hz), 2.04 (s, 1H), 1.94 (ddd, 1H, J = 13.0, 8.9, 7.3 Hz); 13C{1H} NMR (101 MHz, CDCl3) δ 148.5, 145.0 (d, JC–F = 48.4 Hz), 128.9 (q, JC–F = 32.4 Hz), 128.6, 127.6, 125.6 (t, JC–F = 3.7 Hz), 125.0, 123.9, 122.9, 75.0, 48.2, 46.8; HRMS (EI, double focusing) m/z: [M]+ calcd for C16H13F3O 278.0918; found 278.0915.
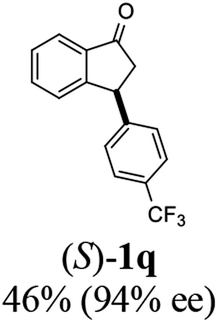
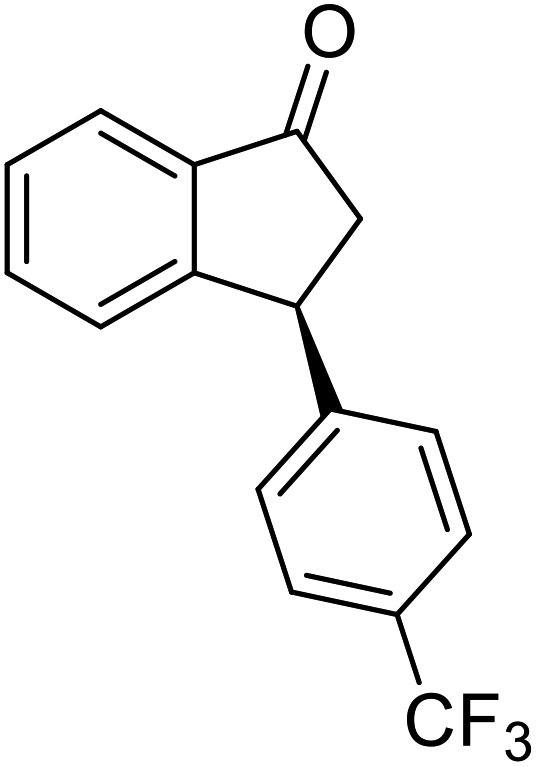
(S)-3-(4-(Trifluoromethyl)phenyl)-2,3-dihydro-1H-inden-1-one, (S)-1q
Yield 42.7% (58.9 mg, pale yellow solid); mp 84.2–84.5 °C; 94% ee (Chiralpak IB, 0 to 4% IPA for 7 min in n-hexane, 1 mL min−1, 270 nm, 270 nm, tR(major) = 16.9 min, tR(minor) = 20.9 min); [α]24D = +30.7 (c 3.2, CH2Cl2). Literature value: [α]25D = +32.0 (c 0.6 CH2Cl2 for 88% ee);23 1H NMR (CDCl3, 400 MHz): δ 7.84 (d, 1H, J = 7.5 Hz), 7.64–7.52 (m, 3H), 7.46 (d, 1H, J = 7.8 Hz), 7.25 (d, 3H, J = 8.1 Hz), 4.65 (dd, 1H, J = 8.1, 3.9 Hz), 3.26 (dd, 1H, J = 19.2, 8.1 Hz), 2.67 (dd, 1H, J = 19.2, 3.9 Hz); 13C{1H} NMR (101 MHz, CDCl3) δ 156.9, 147.8–147.8 (m), 136.8, 135.3, 129.4 (q, JC–F = 32.5 Hz), 128.3, 128.0, 126.8, 125.9 (q, JC–F = 3.8 Hz), 125.4–122.7 (d, JC–F = 272.0 Hz), 123.7, 46.5, 44.2; HRMS (EI, double focusing) m/z: [M]+ calcd for C16H11F3O 276.0762; found 276.0762.
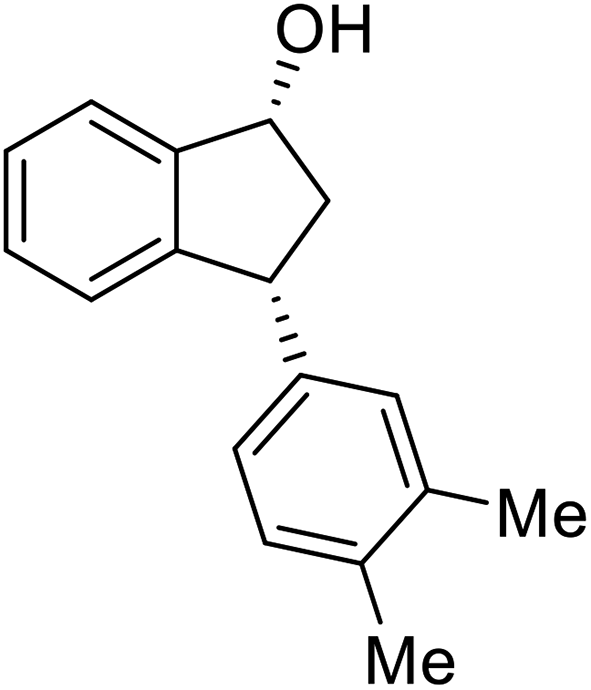
(1R,3R)-3-(3,4-Dimethylphenyl)-2,3-dihydro-1H-inden-1-ol (2r)
Yield 44% (52 mg as white solid); mp 89.8–90.8 °C; 99% ee (Chiralpak IB, 0 to 2% IPA for 7 min in n-hexane, 1 mL min−1, 270 nm, tR(major) = 29.7 min, tR(minor) = 24.3 min); [α]29D = −21.3 (c 2.8, CH2Cl2); 1H NMR (CDCl3, 400 MHz): δ 7.47 (d, 1H, J = 7.4 Hz), 7.29 (t, 1H, J = 7.4 Hz), 7.22 (d, 1H, J = 7.2 Hz), 7.09 (d, 1H, J = 7.7 Hz), 7.00 (s, 1H), 6.96 (d, 2H, J = 7.5 Hz), 5.27 (d, 1H, J = 7.2 Hz), 4.12 (d, 1H, J = 8.3 Hz), 3.00 (dt, 1H, J = 12.8, 7.2 Hz), 2.25 (s, 3H), 2.24 (s, 3H), 2.01–1.86 (m, 2H); 13C{1H} NMR (101 MHz, CDCl3) δ 145.9, 145.2, 141.7, 136.7, 134.8, 129.8, 129.5, 128.3, 127.1, 125.6, 125.1, 123.6, 75.1, 47.9, 47.3, 19.8, 19.4; HRMS (EI, double focusing) m/z: [M]+ calcd for C17H18O 238.1358; found 238.1358.
(S)-3-(3,4-Dimethylphenyl)-2,3-dihydro-1H-inden-1-one, (S)-1r
Yield 49.6% (58.6 mg, pale yellow solid); mp 103.9–105.0 °C; >99% ee (Chiralpak IB, 0 to 2% IPA for 7 min in n-hexane, 1 mL min−1, 270 nm, tR(major) = 17.0 min, tR(minor) = 17.8 min); [α]24D = +58.7 (c 1.1, CH2Cl2); 1H NMR (CDCl3, 400 MHz): δ 7.80 (d, 1H, J = 7.7 Hz), 7.55 (t, 1H, J = 7.5 Hz), 7.40 (t, 1H, J = 7.4 Hz), 7.27 (d, 1H, J = 7.7 Hz), 7.07 (d, 1H, J = 7.6 Hz), 6.93–6.81 (m, 2H), 4.50 (dd, 1H, J = 8.0, 3.8 Hz), 3.20 (dd, 1H, J = 19.2, 8.0 Hz), 2.67 (dd, 1H, J = 19.2, 3.8 Hz), 2.23 (s, 3H), 2.21 (s, 3H); 13C{1H} NMR (101 MHz, CDCl3) δ 206.3, 158.3, 141.1, 137.1, 136.7, 135.2, 135.0, 130.1, 128.8, 127.7, 126.9, 125.0, 123.3, 46.9, 44.1, 19.8, 19.3; HRMS (EI, double focusing) m/z: [M]+ calcd for C17H16O 236.1201; found 236.1201.
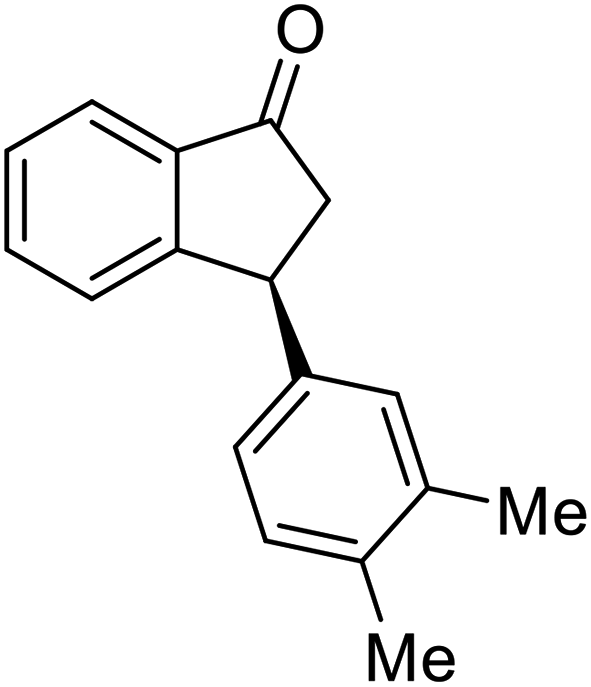
(1R,3R)-3-(3,4-Dichlorophenyl)-2,3-dihydro-1H-inden-1-ol (2s)
Yield 42% (58 mg as white solid); mp 91.7–93.1 °C; 99% ee (Chiralpak IB, 0 to 4.5% IPA for 10 min in n-hexane, 0.8 mL min−1, 270 nm, tR(major) = 20.9 min, tR(minor) = 22.0 min); [α]29D = −18.3 (c 2.0, CH2Cl2); 1H NMR (CDCl3, 400 MHz): δ 7.48 (d, 1H, J = 7.4 Hz), 7.38 (d, 1H, J = 8.3 Hz), 7.35–7.25 (m, 3H), 7.07 (dd, 1H, J = 8.3, 2.0 Hz), 6.94 (d, 1H, J = 7.4 Hz), 5.30 (s, 1H), 4.15 (t, 1H, J = 8.3 Hz), 3.01 (dt, 1H, J = 13.1, 7.3 Hz), 2.01 (s, 1H), 1.89 (ddd, 1H, J = 13.0, 8.8, 7.3 Hz); 13C{1H} NMR (101 MHz, CDCl3) δ 145.1, 144.7, 144.5, 132.6, 130.6, 130.6, 130.2, 128.7, 127.7, 127.7, 124.9, 123.9, 74.9, 47.6, 46.7; HRMS (EI, double focusing) m/z: [M]+ calcd for C15H12Cl2O 278.0265; found 278.0263.
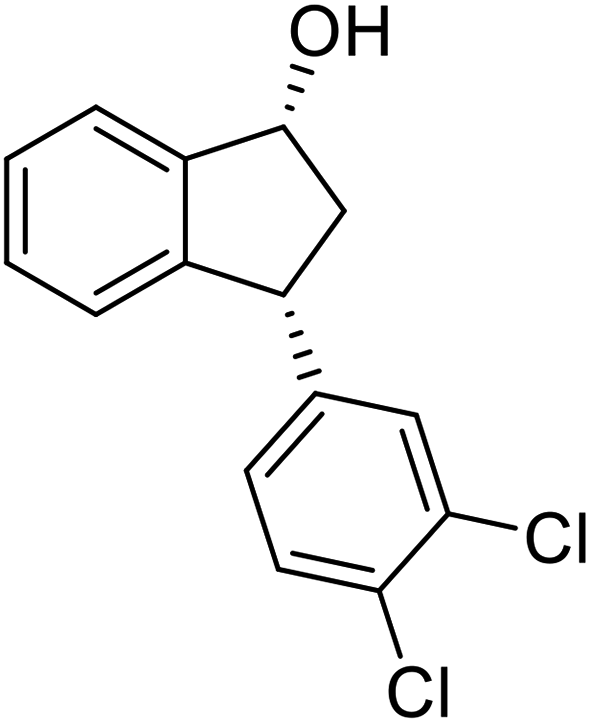
(S)-3-(3,4-Dichlorophenyl)-2,3-dihydro-1H-inden-1-one, (S)-1s
Yield 46.8% (64.8 g, white solid); mp 114.1–114.5 °C; >99% ee (Chiralpak IB, 0 to 4.5% IPA for 10 min in n-hexane, 0.8 mL min−1, 270 nm, tR(major) = 23.7 min, tR(minor) = 22.8 min); [α]25D = +35.5 (c 2.4, CH2Cl2). Literature values: [α]23D = +48 (c 1.0, CHCl3 for 92% ee).7 [α]25D = +38.2 (c 0.5, CH2Cl2 for 90% ee).23 [α]24D = +49.5 (c 1.33, CHCl3, for 98% ee);19 1H NMR (CDCl3, 400 MHz): δ 7.83 (d, 1H, J = 7.7 Hz), 7.61 (t, 1H, J = 7.4 Hz), 7.46 (t, 1H, J = 7.4 Hz), 7.38 (d, 1H, J = 8.3 Hz), 7.26 (d, 1H, J = 7.7 Hz), 7.23 (d, 1H, J = 2.1 Hz), 6.95 (dd, 1H, J = 8.3, 2.1 Hz), 4.55 (dd, 1H, J = 8.1, 3.8 Hz), 3.23 (dd, 1H, J = 19.2, 8.1 Hz), 2.62 (dd, 1H, J = 19.2, 3.8 Hz); 13C{1H} NMR (101 MHz, CDCl3) δ 204.9, 156.5, 144.0, 136.8, 135.4, 133.0, 131.1, 130.9, 129.7, 128.4, 127.0, 126.7, 123.7, 46.5, 43.6; HRMS (EI, double focusing) m/z: [M]+ calcd for C15H10Cl2O 276.0109; found 276.0104.
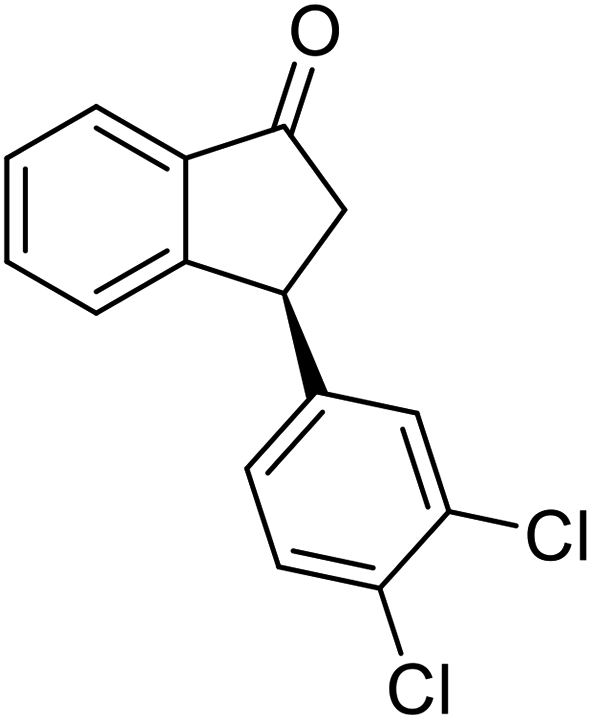
(1R,3R)-5-Fluoro-3-(p-tolyl)-2,3-dihydro-1H-inden-1-ol (2t)
Yield 42% (50 mg as white solid); mp 87.3–88.1 °C; 99% ee (Chiralpak IB, 0 to 3% IPA for 8 min in n-hexane, 1 mL min−1, 270 nm, tR(major) = 25.2 min, tR(minor) = 22.4 min); [α]29D = −33.7 (c 1.4, CH2Cl2); 1H NMR (CDCl3, 400 MHz): δ 7.40 (dd, 1H, J = 8.3, 5.2 Hz), 7.14 (d, 2H, J = 8.1 Hz), 7.10 (d, 2H, J = 8.1 Hz), 6.96 (t, 1H, J = 8.7 Hz), 6.62 (d, 1H, J = 9.0 Hz), 5.23 (t, 1H, J = 7.2 Hz), 4.11 (t, 1H, J = 8.4 Hz), 3.01 (dt, 1H, J = 12.9, 7.3 Hz), 2.34 (s, 3H), 2.13–1.88 (m, 2H); 13C{1H} NMR (101 MHz, CDCl3) δ 163.4 (d, JC–F = 245.5 Hz), 148.2 (d, JC–F = 8.0 Hz), 140.8 (d, JC–F = 2.3 Hz), 140.5, 136.4, 129.4, 128.0, 125.0 (d, JC–F = 9.0 Hz), 114.4 (d, JC–F = 22.9 Hz), 111.9 (d, JC–F = 22.3 Hz), 74.4, 47.8, 47.5, 21.0; HRMS (EI, double focusing) m/z: [M]+ calcd for C16H15FO 242.1107; found 242.1116.
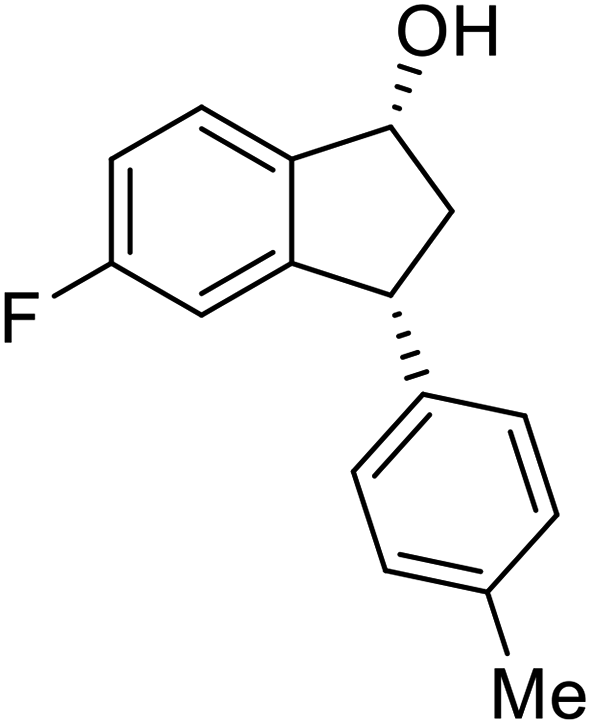
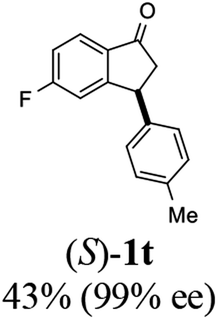
(S)-5-Fluoro-3-(p-tolyl)-2,3-dihydro-1H-inden-1-one, (S)-1t
Yield 43% (51.6 mg, pale brown solid); mp 82.7–83.3 °C; 99% ee (Chiralpak IB, 0 to 3% IPA for 8 min in n-hexane, 1 mL min−1, 270 nm, tR(major) = 16.4 min, tR(minor) = 17.2 min); [α]26D = +60.2 (c 2.3, CH2Cl2); 1H NMR (CDCl3, 400 MHz): δ 7.80 (dd, 1H, J = 8.5, 5.3 Hz), 7.19–7.06 (m, 3H), 7.01 (d, 2H, J = 8.1 Hz), 6.91 (d, 1H, J = 8.3 Hz), 4.51 (dd, 1H, J = 8.1, 3.9 Hz), 3.23 (dd, 1H, J = 19.2, 8.1 Hz), 2.70 (dd, 1H, J = 19.2, 3.9 Hz), 2.34 (s, 3H); 13C{1H} NMR (101 MHz, CDCl3) δ 204.1, 167.4 (d, JC–F = 256.8 Hz), 161.1 (d, JC–F = 9.6 Hz), 139.9, 137.0, 133.1 (d, JC–F = 1.8 Hz), 129.7, 127.4, 125.7 (d, JC–F = 10.4 Hz), 116.3 (d, JC–F = 24.0 Hz), 113.4 (d, JC–F = 22.4 Hz), 47.0, 43.9, 21.0; HRMS (EI, double focusing) m/z: [M]+ calcd for C16H13FO 240.0950; found 240.0961.
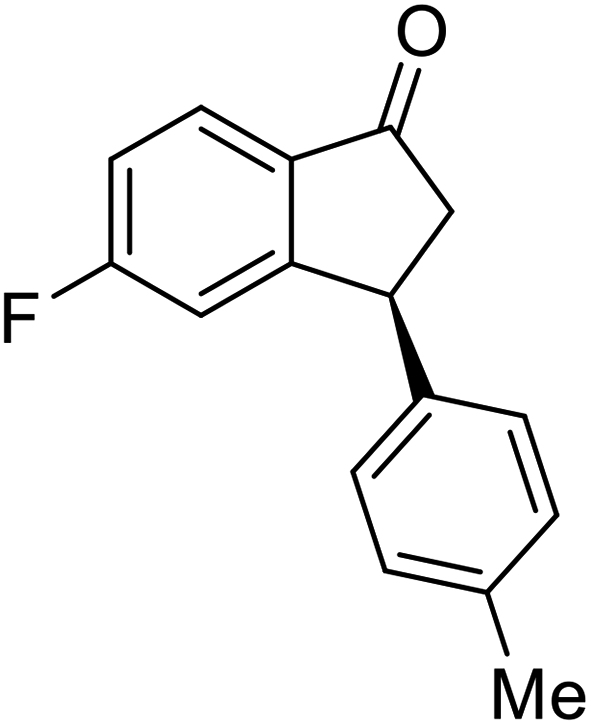
(1R,3R)-5-Fluoro-3-(4-methoxyphenyl)-2,3-dihydro-1H-inden-1-ol (2u)
Yield 42% (53 mg as white solid); mp 86.5–87.0 °C; 95% ee (Chiralpak IB, 0 to 6% IPA for 7 min in n-hexane, 1 mL min−1, 270 nm, tR(major) = 23.7, tR(minor) = 21.2 min); [α]29D = −34.1 (c 1.9, CH2Cl2); 1H NMR (CDCl3, 400 MHz): δ 7.40 (dd, 1H, J = 8.3, 5.2 Hz), 7.14 (d, 2H, J = 8.7 Hz), 7.04–6.92 (m, 1H), 6.87 (d, 2H, J = 8.7 Hz), 6.66–6.59 (m, 1H), 5.24 (t, 1H, J = 6.8 Hz), 4.11 (t, 1H, J = 8.4 Hz), 3.81 (s, 3H), 3.02 (dt, 1H, J = 12.9, 7.2 Hz), 2.02–1.88 (m, 2H); 13C{1H} NMR (101 MHz, CDCl3) δ 163.4 (d, JC–F = 245.5 Hz), 158.5, 148.4 (d, JC–F = 7.9 Hz), 140.7 (d, JC–F = 2.2 Hz), 135.6, 129.1, 125.0 (d, JC–F = 9.1 Hz), 114.4 (d, JC–F = 22.8 Hz), 114.1, 111.8 (d, JC–F = 22.3 Hz), 74.4, 55.3, 47.6, 47.4; HRMS (EI, double focusing) m/z: [M]+ calcd for C16H15FO2 258.1056; found 258.1056.
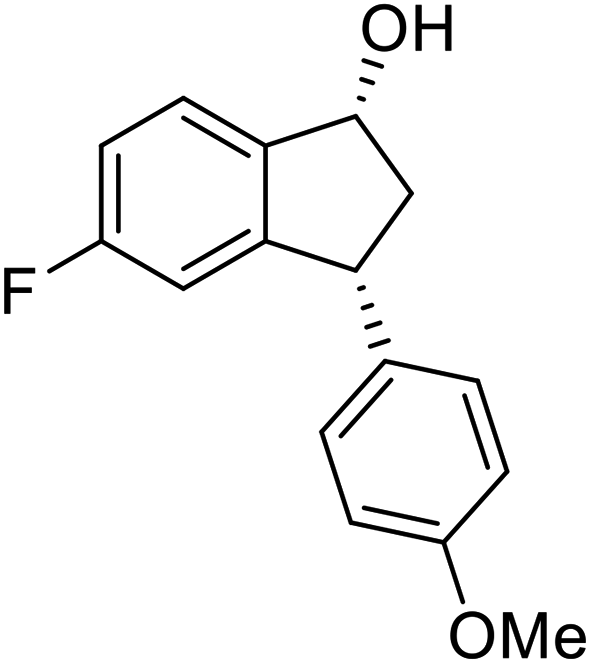
(S)-5-Fluoro-3-(4-methoxyphenyl)-2,3-dihydro-1H-inden-1-one, (S)-1u
Yield 44.8% (57.4 mg, pale brown solid); mp 112.3–112.9 °C; 98% ee (Chiralpak IB, 0 to 6% IPA for 7 min in n-hexane, 1 mL min−1, 270 nm, tR(major) = 18.2 min, tR(minor) = 17.7 min); [α]27D = +55.5 (c 2.5, CH2Cl2); 1H NMR (CDCl3, 400 MHz): δ 7.80 (dd, 1H, J = 8.5, 5.3 Hz), 7.10 (td, 1H, J = 8.5, 1.8 Hz), 7.04 (d, 2H, J = 8.7 Hz), 6.91 (dd, 1H, J = 8.5, 1.8 Hz), 6.86 (d, 2H, J = 8.7 Hz), 4.50 (dd, 1H, J = 8.0, 3.9 Hz), 3.80 (s, 3H), 3.23 (dd, 1H, J = 19.2, 8.1 Hz), 2.68 (dd, 1H, J = 19.2, 3.9 Hz); 13C{1H} NMR (101 MHz, CDCl3) δ 204.1, 167.4 (d, JC–F = 256.8 Hz), 161.2 (d, JC–F = 9.6 Hz), 158.8, 134.9, 133.1 (d, JC–F = 1.8 Hz), 128.6, 125.7 (d, JC–F = 10.3 Hz), 116.3 (d, JC–F = 24.0 Hz), 114.4, 113.4 (d, JC–F = 22.4 Hz), 55.3, 47.1, 43.6; HRMS (EI, double focusing) m/z: [M]+ calcd for C16H13FO2 256.0900; found 256.0903.
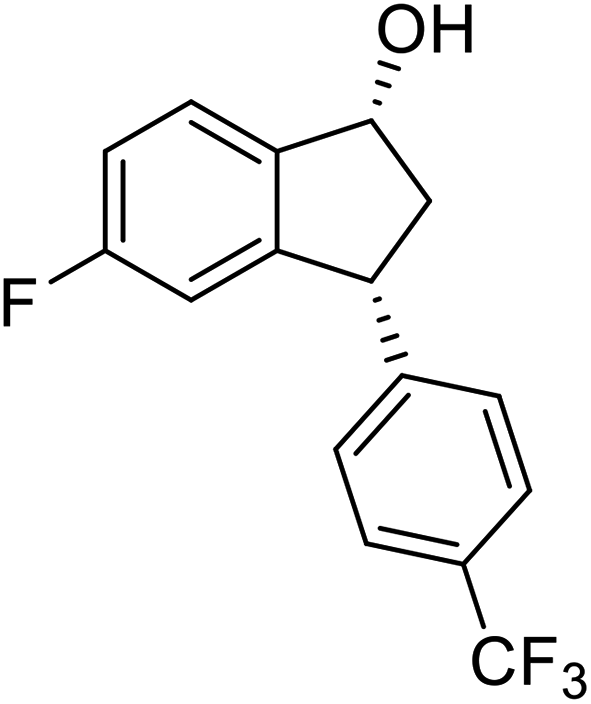
(1R,3R)-5-Fluoro-3-(4-(trifluoromethyl)phenyl)-2,3-dihydro-1H-inden-1-ol (2v)
Yield 43% (48 mg as white solid); mp 86.1–86.5 °C; 92% ee (Chiralpak IB, 0 to 2% IPA for 60 min in n-hexane, 0.6 mL min−1, 270 nm, tR(major) = 48.3 min, tR(minor) = 46.1 min); [α]29D = −21.9 (c 2.6, CH2Cl2); 1H NMR (CDCl3, 400 MHz): δ 7.59 (d, 2H, J = 8.1 Hz), 7.44 (dd, 1H, J = 8.3, 5.2 Hz), 7.35 (d, 2H, J = 8.1 Hz), 7.01 (t, 1H, J = 8.6 Hz), 6.60 (d, 1H, J = 8.8 Hz), 5.29 (s, 1H), 4.24 (t, 1H, J = 8.3 Hz), 3.06 (dt, 1H, J = 13.1, 7.3 Hz), 1.98 (ddd, 2H, J = 13.1, 8.9, 7.1 Hz); 13C{1H} NMR (101 MHz, CDCl3) δ 163.4 (d, JC–F = 246.4 Hz), 147.7–147.6 (m), 147.0 (d, JC–F = 7.9 Hz), 140.8 (d, JC–F = 2.4 Hz), 129.2 (d, JC–F = 32.4 Hz), 128.6, 125.7 (q, JC–F = 3.8 Hz), 125.4, 125.3, 114.9 (d, JC–F = 22.9 Hz), 111.8 (d, JC–F = 22.4 Hz), 74.3, 48.1, 47.1; HRMS (EI, double focusing) m/z: [M]+ calcd for C16H12F4O 296.0824; found 296.0824.
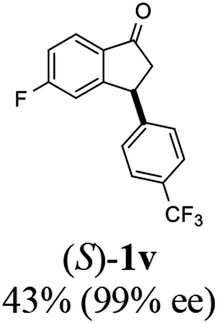
(S)-5-Fluoro-3-(4-(trifluoromethyl)phenyl)-2,3-dihydro-1H-inden-1-one, (S)-1v
Yield 43.2% (63.5 mg, pale brown solid); mp 112.0–112.4 °C; 99% ee (Chiralpak IB, 0 to 2% IPA for 60 min in n-hexane, 0.6 mL min−1, 270 nm, tR(major) = 44.6 min, tR(minor) = 43.7 min); [α]27D = +29.9 (c 2.9, CH2Cl2); 1H NMR (CDCl3, 400 MHz): δ 7.84 (dd, 1H, J = 8.5, 5.3 Hz), 7.60 (d, 2H, J = 8.1 Hz), 7.25 (d, 2H, J = 8.1 Hz), 7.15 (td, 1H, J = 8.6, 2.1 Hz), 6.89 (dd, 1H, J = 8.4, 1.7 Hz), 4.62 (dd, 1H, J = 8.2, 3.9 Hz), 3.28 (dd, 1H, J = 19.2, 8.2 Hz), 2.69 (dd, 1H, J = 19.2, 3.9 Hz); 13C{1H} NMR (101 MHz, CDCl3) δ 203.1, 168.7, 166.2, 159.7 (d, JC–F = 9.5 Hz), 146.9, 133.2 (d, JC–F = 1.9 Hz), 129.7 (q, JC–F = 32.6 Hz), 128.0, 126.1 (q, JC–F = 3.8 Hz), 124.0 (d, JC–F = 271.9 Hz), 116.8 (d, JC–F = 23.9 Hz), 113.4 (d, JC–F = 22.5 Hz), 46.7, 44.0; HRMS (EI, double focusing) m/z: [M]+ calcd for C16H10F4O 294.0668; found 294.0674.
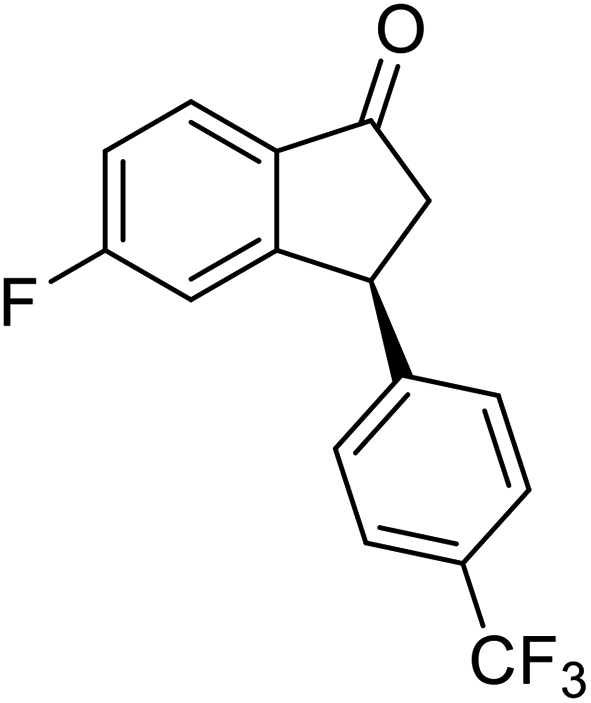
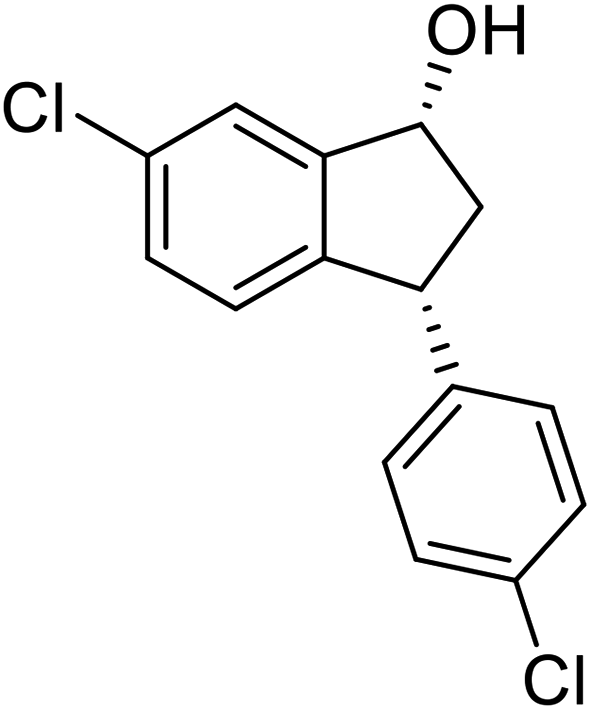
(1R,3R)-6-Chloro-3-(4-chlorophenyl)-2,3-dihydro-1H-inden-1-ol (2w)
Yield 49.6% (68.7 mg as white solid); mp 100.5–101.1 °C; 98% ee (Chiralpak IB, 0 to 4% IPA for 7 min in n-hexane, 1 mL min−1, 270 nm, tR(major) = 21.8 min, tR(minor) = 20.1 min); [α]29D = −42.4 (c 3.4, CH2Cl2); 1H NMR (CDCl3, 400 MHz): δ 7.45 (d, 1H, J = 1.8 Hz), 7.30 (d, 2H, J = 8.4 Hz), 7.21 (dd, 1H, J = 8.1, 1.8 Hz), 7.14 (d, 2H, J = 8.4 Hz), 6.84 (d, 1H, J = 8.1 Hz), 5.26 (q, 1H, J = 7.0 Hz), 4.12 (d, 1H, J = 8.3 Hz), 3.03 (dt, 1H, J = 13.0, 7.2 Hz), 2.0–1.8 (m, 2H) ppm; 13C{1H} NMR (CDCl3, 101 MHz): δ 147.1, 143.5, 142.2, 133.3, 132.6, 129.5, 128.9, 128.7, 126.2, 124.2, 47.3, 47.3; HRMS (EI, double focusing) m/z: [M]+ calcd for C15H12Cl2O 278.0265; found 296.0268.
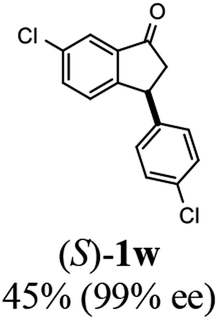
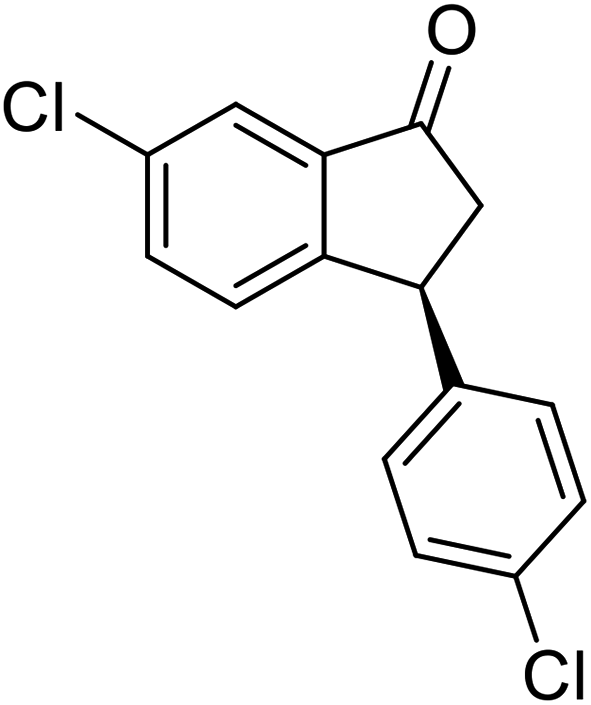
(S)-6-Chloro-3-(4-chlorophenyl)-2,3-dihydro-1H-inden-1-one, (S)-1w
Yield: 45.3% (62.7 mg, white solid); mp 81.6–82.1 °C; >99% ee (Chiralpak IB, 0 to 4% IPA for 7 min in n-hexane, 1 mL min−1, 270 nm, tR(major) = 17.9 min, tR(minor) = 16.9 min); [α]28D = +44.9 (c 2.9, CH2Cl2); 1H NMR (CDCl3, 400 MHz): δ 7.77 (d, 1H, J = 2.0 Hz), 7.54 (dd, 1H, J = 8.2, 2.0 Hz), 7.29 (d, 2H, J = 8.4 Hz), 7.19 (d, 1H, J = 8.2 Hz), 7.04 (d, 2H, J = 8.4 Hz), 4.53 (dd, 1H, J = 8.1, 3.9 Hz), 3.26 (dd, 1H, J = 19.3, 8.1 Hz), 2.66 (dd, 1H, J = 19.3, 3.9 Hz) ppm; 13C{1H} NMR (CDCl3, 101 MHz): δ 203.9, 155.3, 141.6, 138.2, 135.2, 134.7, 133.1, 129.2, 128.9, 128.0, 123.4, 47.0, 43.4 ppm; HRMS (EI, double focusing) m/z: [M]+ calcd for C15H10Cl2O 276.0109; found 276.0108.
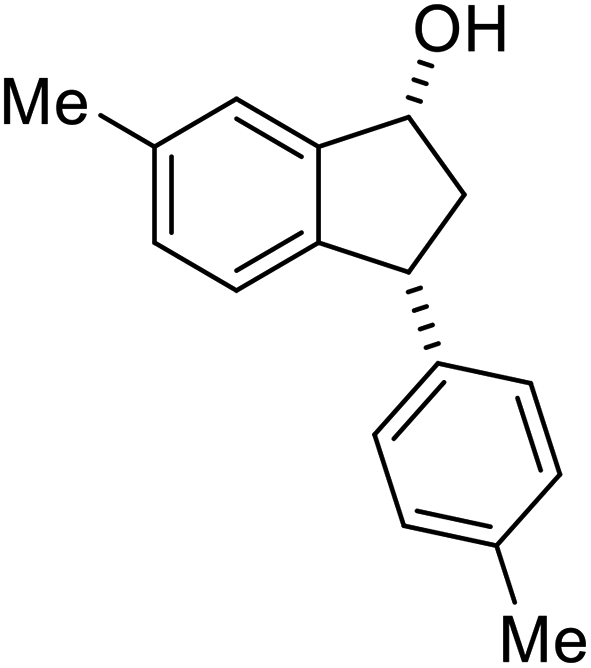
(1R,3R)-6-Methyl-3-(p-tolyl)-2,3-dihydro-1H-inden-1-ol (2x)
Yield: 42.3% (50.0 mg, white solid); mp 100.8–101.8 °C; 98% ee (Chiralpak IB, 0 to 3% IPA for 8 min in n-hexane, 1 mL min−1, 270 nm, tR(major) = 31.6 min, tR(minor) = 21.6 min); [α]29D = −32.4 (c 3.4, CH2Cl2); 1H NMR (CDCl3, 400 MHz): δ 7.29 (s, 1H), 7.17–7.08 (m, 4H), 7.05 (d, 1H, J = 7.8 Hz), 6.84 (d, 1H, J = 7.8 Hz), 5.25 (d, 1H, J = 7.2 Hz), 4.12 (t, 1H, J = 8.4 Hz), 3.00 (dt, 1H, J = 12.8, 7.2 Hz), 2.38 (s, 3H), 2.34 (s, 3H), 1.95–1.83 (m, 2H) ppm; 13C{1H} NMR (CDCl3, 101 MHz): δ = 145.4, 142.9, 141.5, 136.9, 136.1, 129.3, 129.3, 128.1, 124.8, 124.2, 75.1, 47.6, 47.5, 21.3, 21.0 ppm; HRMS (EI, double focusing) m/z: [M]+ calcd for C17H18O 238.1358; found 238.1371.
(S)-6-Methyl-3-(p-tolyl)-2,3-dihydro-1H-inden-1-one, (S)-1x
Yield: 46.1% (54.4 mg, white solid); mp 77.7–78.4 °C; 99% ee (Chiralpak IB, 0 to 3% IPA for 8 min in n-hexane, 1 mL min−1, 270 nm, tR(major) = 17.6 min, tR(minor) = 17.3 min); [α]28D = +56.9 (c 2.5, CH2Cl2); 1H NMR (CDCl3, 400 MHz): δ = 7.60 (s, 1H), 7.38 (d, 1H, J = 8.0 Hz), 7.15 (d, 1H, J = 8.0 Hz), 7.11 (d, 2H, J = 8.0 Hz), 7.00 (d, 2H, J = 8.0 Hz), 4.50 (dd, 1H, J = 8.0, 3.8 Hz), 3.21 (dd, 1H, J = 19.2, 8.0 Hz), 2.65 (dd, 1H, J = 19.2, 3.8 Hz), 2.42 (s, 3H), 2.32 (s, 3H) ppm; 13C{1H} (CDCl3, 101 MHz): δ 206.3, 155.6, 140.9, 137.8, 136.9, 136.5, 136.3, 129.5, 127.5, 126.5, 123.2, 76.7, 47.3, 43.7, 21.1, 21.0 ppm; HRMS (EI, double focusing) m/z: [M]+ calcd for C17H16O 236.1201; found 236.1206.
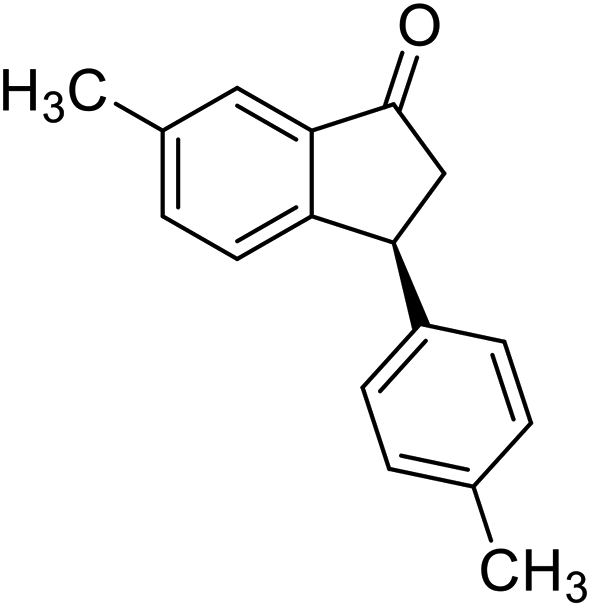
(1R,3S)-3-(Furan-2-yl)-2,3-dihydro-1H-inden-1-ol (2y)
Yield 40% (40 mg, pale brown solid); mp 109.9–111.0 °C; 98% ee (Chiralpak IB, 0 to 5% EtOH for 3 min in n-hexane, 0.8 mL min−1, 270 nm, tR(major) = 18.8 min, tR(minor) = 15.4 min); [α]20D = −39.6 (c 1.8, CH2Cl2); 1H NMR (CDCl3, 400 MHz): δ 7.48 (d, 1H, J = 6.8 Hz), 7.36–7.26 (m, 3H), 7.23–7.17 (m, 1H), 6.31 (t, 1H, J = 3.0, 1.8 Hz), 6.13 (d, 1H, J = 3.1 Hz), 5.24 (t, 1H, J = 6.3 Hz), 4.34 (t, 1H, J = 7.5 Hz), 2.91 (ddt, 1H, J = 1350.7, 13.4, 7.9, 6.9 Hz), 2.21–2.03 (m, 2H); 13C{1H} NMR (101 MHz, CDCl3) δ 156.9, 144.7, 143.0, 141.7, 128.6, 127.7, 124.9, 124.4, 110.2, 105.3, 75.1, 42.3, 41.5; HRMS (EI, double focusing) m/z: [M]+ calcd for C13H12O2 200.0837; found 200.0846.
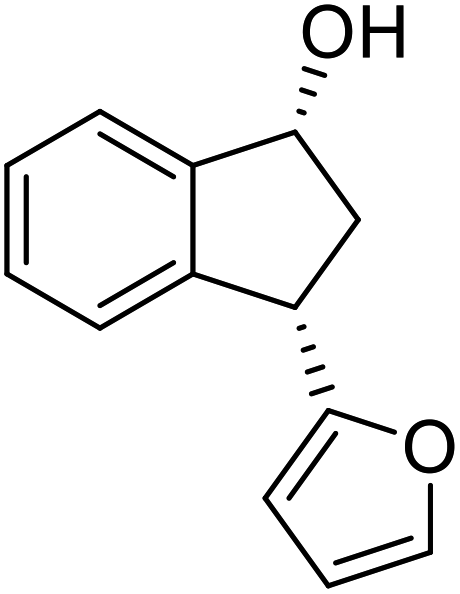
(R)-3-(Furan-2-yl)-2,3-dihydro-1H-inden-1-one, (R)-1y
tYield 41.3% (41.0 mg, brown oil); 95% ee (Chiralpak IB, 0 to 5% EtOH for 3 min in n-hexane, 0.8 mL min−1, 270 nm, tR(major) = 14.1 min, tR(minor) = 14.4 min); [α]19D = −7.8 (c 1.5, CH2Cl2) Literature values: [α]25D = −4.3 (c 0.6, CHCl3 for 50% ee).26 [α]25D = −7.4 (c 0.3, CHCl3 for 58% ee);23 1H NMR (CDCl3, 400 MHz): δ 7.80 (d, 1H, J = 7.6 Hz), 7.65–7.59 (m, 2H), 7.52 (d, 1H, J = 7.8 Hz), 7.44 (t, 1H, J = 7.4 Hz), 7.35 (d, 1H, J = 1.8 Hz), 6.31 (t, 1H, J = 3.2, 1.8 Hz), 6.11 (d, 1H, J = 3.2 Hz), 4.69 (dd, 1H, J = 8.1, 4.1 Hz), 3.13 (dd, 1H, J = 19.0, 8.1 Hz), 2.88 (dd, 1H, J = 19.0, 4.1 Hz); 13C{1H} NMR (101 MHz, CDCl3) δ 205.0, 155.2, 154.7, 142.2, 136.4, 135.0, 128.3, 126.6, 123.7, 110.3, 105.8, 42.8, 37.7; HRMS (EI, double focusing) m/z: [M]+ calcd for C13H10O2 198.0681; found 198.0677.
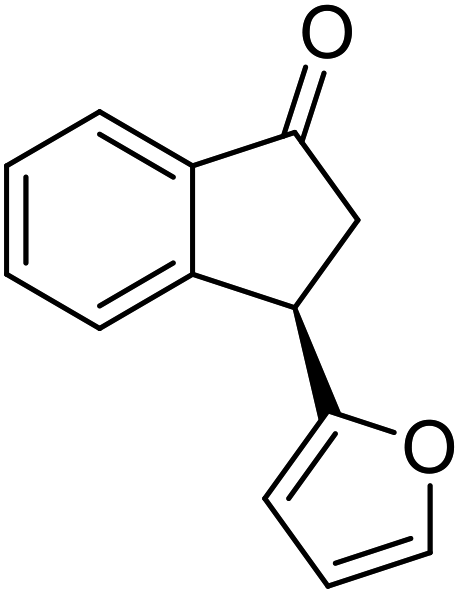
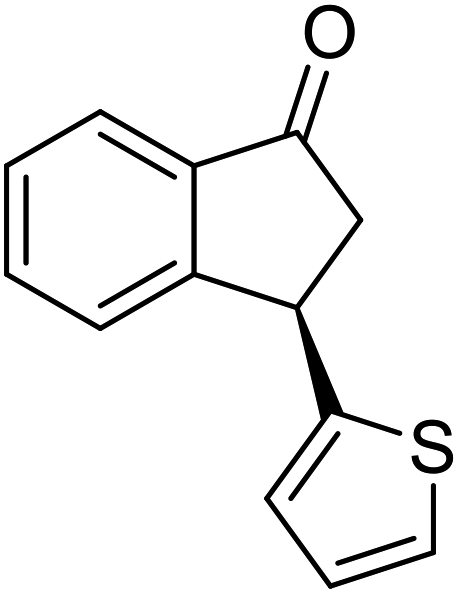
(1R,3S)-3-(Thiophen-2-yl)-2,3-dihydro-1H-inden-1-ol (2z)
Yield 42% (50 mg, pale brown solid); mp 75.9–76.2 °C; 98% ee (Chiralpak IB, 0 to 5% EtOH for 7 min in n-hexane, 1 mL min−1, 270 nm, tR(major) = 14.7 min, tR(minor) = 14.2 min); [α]22D = −5.2 (c 2.3, CH2Cl2); 1H NMR (CDCl3, 400 MHz): δ 7.46 (d, 1H, J = 7.3 Hz), 7.34–7.24 (m, 2H), 7.18 (dd, 1H, J = 5.1, 1.2 Hz), 7.14 (d, 1H, J = 7.4 Hz), 6.96 (dd, 1H, J = 5.1, 3.5 Hz), 6.92 (d, 1H, J = 3.1 Hz), 5.23 (t, 1H, J = 7.0 Hz), 4.49 (t, 1H, J = 8.2 Hz), 3.06 (dt, 1H, J = 12.9, 7.2 Hz), 2.15–1.96 (m, 2H); 13C{1H} NMR (101 MHz, CDCl3) δ 148.0, 144.9, 144.6, 128.5, 127.6, 126.8, 124.9, 124.6, 123.9, 123.9, 74.8, 47.5, 43.1; HRMS (EI, double focusing) m/z: [M]+ calcd for C13H12OS 216.0609; found 216.0617.
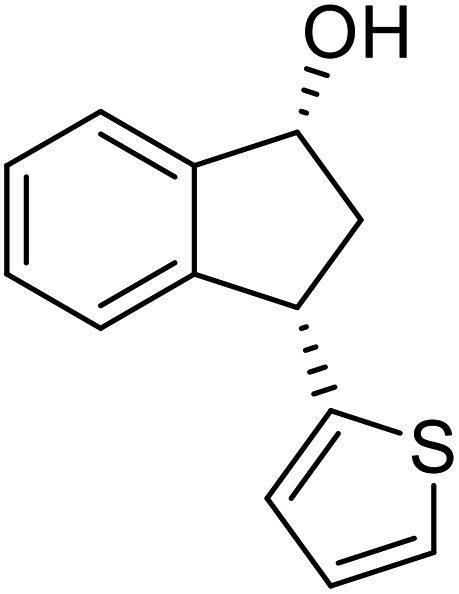
(R)-3-(Thiophen-2-yl)-2,3-dihydro-1H-inden-1-one, (R)-1z
Yield 49.7% (53.2 mg, brown solid); mp 54.8–55.0 °C; 94% ee (Chiralpak IB, 0 to 5% EtOH for 7 min in n-hexane, 1 mL min−1, 270 nm, tR(major) = 34.5 min, tR(minor) = 16.4 min); [α]21D = −3.0 (c 2.4, CH2Cl2). Literature value for (S)-1z: [α]23D = +8 (c 1.0, CHCl3 for 93% ee);27 1H NMR (CDCl3, 400 MHz): δ 7.80 (d, 1H, J = 7.6 Hz), 7.61 (t, 1H, J = 7.5 Hz), 7.51–7.40 (m, 2H), 7.19 (d, 1H, J = 5.1 Hz), 6.95 (dd, 1H, J = 5.1, 3.5 Hz), 6.88 (d, 1H, J = 3.5 Hz), 4.89 (dd, 1H, J = 8.0, 4.0 Hz), 3.27 (dd, 1H, J = 19.1, 8.0 Hz), 2.80 (dd, 1H, J = 19.1, 4.0 Hz); 13C{1H} NMR (101 MHz, CDCl3) δ 204.8, 156.7, 146.8, 136.1, 135.1, 128.3, 126.9, 126.7, 124.7, 124.3, 123.5, 47.2, 39.4; HRMS (EI, double focusing) m/z: [M]+ calcd for C13H10OS 214.0452; found 214.0457.
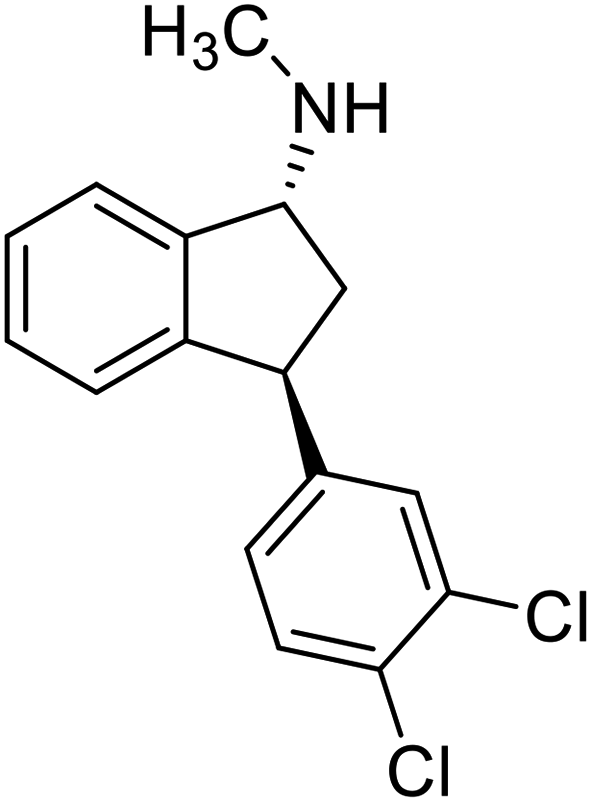
Synthesis of (1R,3S)-3-(3,4-dichlorophenyl)-N-methyl-2,3-dihydro-1H-inden-1-amine, (+)-indatraline7
A solution of (1S,3S)-3-(3,4-dichlorophenyl)-2,3-dihydro-1H-inden-1-ol (2s) (87 mg, 0.3 mmol) and triethylamine (210 μL, 1.5 mmol) dissolved in anhydrous THF (3.0 mL) was cooled to −20 °C and methananesulfonyl chloride (70 μL, 0.9 mmol) was added dropwise. The reaction mixture was stirred at −20 °C for 1 h. Then 2 M solution of methylamine in THF (3.75 mL 7.5 mmol) was added slowly over 30 min. The reaction mixture was allowed to warm to rt and stirred 18 hours. The solvent was removed by rotary evaporation, and EtOAc (10 mL) and water (10 mL) were added. The phases were separated and the aqueous layer was re-extracted with EtOAc (20 mL × 3) and the combined organic layers were washed with brine (40 mL), dried over anhydrous MgSO4, concentrated by rotary evaporation. The crude residue was purified by silica-gel column chromatography (EtOAc:Et3N = 95:5) to give (+)-indatraline (56.6 mg, 65%) as a yellow oil.
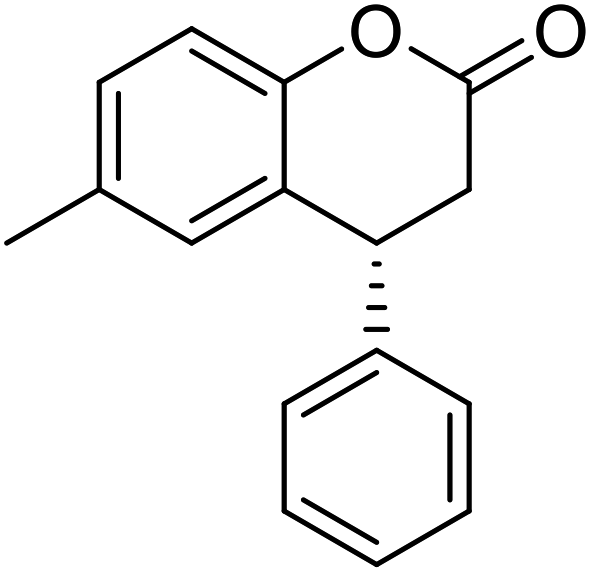
Synthesis of (R)-6-methyl-4-phenylchroman-2-one [(R)-5]27
To a solution of (R)-1c (90 mg, 0.4 mmol) and p-toluenesulfonic acid (19.5 mg, 0.09 mmol) in CH2Cl2 (6 mL) was added m-CPBA (443 mg 1.76 mmol) portionwise. The solution was heated to reflux for 24 hours. After cooling to room temperature, the reaction mixture was quenched with saturated aqueous NaHCO3 (10 mL) and Na2S2O3 (10 mL) and extracted with EtOAc (20 mL × 3). The combined organic layers were dried over anhydrous MgSO4 and concentrated by rotary evaporator. The crude residue was purified by flash column chromatography on silica-gel (EtOAc: n-hexane 1:10) to provide (R)-5 as a white solid (65%, 65 mg).
Synthesis54 of (S)-7 and (S)-8
A suspension of hydroxylamine hydrochloride (144 mg, 2.1 mmol) and NaOAc (212 mg, 2.6 mmol) in 80% aqueous EtOH (20 mL) was stirred at rt for 30 min. (S)-3-(3,4-dichlorophenyl)-2,3-dihydro-1H-inden-1-one ((S)-1s) (360 mg 1.3 mmol) was added and the reaction mixture was heated gently to reflux for 2 h. After cooling to rt, the solvent was evaporated under vacuum and the residue was diluted with EtOAc (60 mL) and washed with water (30 mL) and brine successively. The organic layer was dried over anhydrous MgSO4, and concentrated by rotary evaporation. The crude oxime product (ca. 353 mg) was used in the next step without further purification. To a stirred solution of 1-indanone oxime (300 mg, ca.1.0 mmol) and p-toluenesulfonyl chloride (225 mg, 1.1 mmol) in 3 mL of acetone was added 4 N NaOH (0.5 mL) solution dropwise at −10 °C. After 5 min, the cooling bath was removed and the reaction mixture was stirred for 2 h at rt. Then the reaction mixture was poured into 50 g of ice and extracted with EtOAc (30 mL × 3). The combined extracts were dried over anhydrous MgSO4 and concentrated by rotary and the resulting residue was purified by flash chromatography (EtOAc:n-hexane 1:5) on silica-gel to give (E)-indanone oxime-O-tosylate ((S)-6, 360 mg, 78%) as a white solid. To a solution of (E)-indanone oxime-O-tosylate ((S)-6, 100 mg, 0.22 mmol) in CH2Cl2 (3 mL) at −40 °C was added AlCl3 (49 mg, 0.34 mmol) portionwise. The reaction mixture was stirred for 30 minutes at that temperature and allow to warm to rt. The reaction mixture was stirred for additional 2 h. Then, water (15 mL) was added carefully for quenching, and extracted with EtOAc (30 mL × 3). The combined organic layers were washed with brine (40 mL), dried over anhydrous MgSO4 and concentrated by rotary evaporation. The crude residue was purified by flash column chromatography on silica-gel (EtOAc:n-hexane 1:3) to give (S)-7 (26.8 mg, 41% yield), and (S)-8 (23.1 mg, 35% yield). Rf for (S)-7 = 0.2 and Rf for (S)-8 = 0.1 (EtOAc:n-hexane 1:2).
Yield 41% (26.4 mg, as white solid); mp 131.1–131.9 °C; 99% ee (Chiralpak IB, 6% EtOH in n-hexane, 1 mL min−1, 270 nm, tR(major) = 17.5 min, tR(minor) = 19.7 min); [α]26D = −86.8 (c 0.5, CH2Cl2); 1H NMR (CDCl3, 400 MHz): δ = 9.5 (s, 1H), 7.4 (d, 1H, J = 8.3 Hz), 7.3–7.2 (m, 1H), 7.1–7.0 (m, 2H), 6.9 (d, 2H, J = 7.9 Hz), 4.3 (t, 1H, J = 7.1 Hz), 3.0 (dd, 1H, J = 16.2, 6.3 Hz), 2.9 (dd, 1H, J = 16.2, 7.9 Hz); 13C{H} NMR (CDCl3, 101 MHz): δ = 170.5, 141.9, 137.0, 132.9, 131.3, 130.9, 129.8, 128.6, 128.2, 127.1, 125.2, 123.6, 116.1, 41.2, 38.2; HRMS (EI, double focusing) m/z: [M]+ calcd for C15H11Cl2NO 291.0218; found 291.0210.
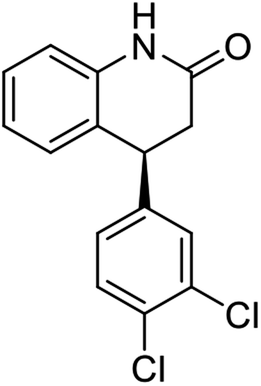
Yield 35% (23.1 mg, as white solid); mp 176.8–177.2 °C; 98% ee (Chiralpak IA, 10% EtOH in n-hexane, 1 mL min−1, 270 nm, tR(major) = 19.6 min, tR(minor) = 25.1 min); [α]26D = +21.2 (c 0.9, CH2Cl2); 1H NMR (CDCl3, 400 MHz): δ = 8.2 (d, 1H, J = 7.1 Hz), 7.5–7.4 (m, 3H), 7.0–7.0 (m, 3H), 6.7 (s, 1H), 4.3–4.2 (m, 1H), 3.8 (ddd, 1H, J = 12.5, 5.2, 2.7 Hz), 3.7 (ddd, 1H, J = 12.5, 7.1, 3.1 Hz); 13C{H} NMR (CDCl3, 101 MHz): δ = 166.0, 141.0, 140.0, 132.9, 132.8, 131.6, 130.7, 130.4, 128.8, 128.4, 128.0, 127.9, 127.6, 46.9, 43.3; HRMS (EI, double focusing) m/z: [M]+ calcd for C15H11Cl2NO 291.0218; found 291.0218.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research was financially supported by grants from the National Research Foundation of Korea (2017M3A9A5051181) and Korea Research Institute of Chemical Technology (SI2151-20).References
- B. Gabriele, R. Mancuso and L. Veltri, Chem.–Eur. J., 2016, 22, 5056–5094 CrossRef CAS PubMed.
- C. Borie, L. Ackermann and M. Nechab, Chem. Soc. Rev., 2016, 45, 1368–1386 RSC.
- M. Turek, D. Szczęsna, M. Koprowski and P. Bałczewski, Beilstein J. Org. Chem., 2017, 13, 451–494 CrossRef CAS PubMed.
- S. Faiz, M. Yousaf, A. F. Zahoor, S. A. R. Naqvi, A. Irfan and G. Zaman, Synth. Commun., 2017, 47, 1121–1135 CrossRef CAS.
- S. A. Patil, R. Patil and S. A. Patil, Eur. J. Med. Chem., 2017, 138, 182–198 CrossRef CAS PubMed.
- M.-L. Tang, P. Peng, Z.-Y. Liu, J. Zhang, J.-M. Yu and X. Sun, Chem.–Eur. J., 2016, 22, 14535–14539 CrossRef PubMed.
- X. Qin, M. W. Yao Lee and J. S. Zhou, Org. Lett., 2019, 21, 5990–5994 CrossRef CAS PubMed.
- H. Yu, I. J. Kim, J. E. Folk, X. Tian, R. B. Rothman, M. H. Baumann, C. M. Dersch, J. L. Flippen-Anderson, D. Parrish, A. E. Jacobson and K. C. Rice, J. Med. Chem., 2004, 47, 2624–2634 CrossRef CAS PubMed.
- M. Froimowitz, K.-M. Wu, A. Moussa, R. M. Haidar, J. Jurayj, C. George and E. L. Gardner, J. Med. Chem., 2000, 43, 4981–4992 CrossRef CAS PubMed.
- K. P. Bogeso, A. V. Christensen, J. Hyttel and T. Liljefors, J. Med. Chem., 1985, 28, 1817–1828 CrossRef CAS PubMed.
- K. P. Boegesoe, J. Arnt, V. Boeck, A. V. Christensen, J. Hyttel and K. G. Jensen, J. Med. Chem., 1988, 31, 2247–2256 CrossRef CAS PubMed.
- X.-D. Hao, J. Chang, B.-Y. Qin, C. Zhong, Z.-B. Chu, J. Huang, W.-J. Zhou and X. Sun, Eur. J. Med. Chem., 2015, 102, 26–38 CrossRef CAS PubMed.
- T. Atsumi, Y. Murakami, K. Shibuya, K. Tonosaki and S. Fujisawa, Anticancer Res., 2005, 25, 4029 CAS.
- G. G. Bianco, H. M. C. Ferraz, A. M. Costa, L. V. Costa-Lotufo, C. Pessoa, M. O. de Moraes, M. G. Schrems, A. Pfaltz and L. F. Silva, J. Org. Chem., 2009, 74, 2561–2566 CrossRef CAS PubMed.
- S. B. Bhorkade, K. B. Gavhane and V. S. Shinde, Tetrahedron, 2016, 72, 1954–1959 CrossRef CAS.
- S. H. Lee, S. J. Park, I. S. Kim and Y. H. Jung, Tetrahedron, 2013, 69, 1877–1880 CrossRef CAS.
- G. Wang, C. Zheng and G. Zhao, Tetrahedron: Asymmetry, 2006, 17, 2074–2081 CrossRef CAS.
- N. G. Nørager, L. L. R. Lorentz-Petersen, L. O. Lyngsø, J. Kehler and K. Juhl, Synlett, 2011, 1753–1755 Search PubMed.
- S. Roesner, J. M. Casatejada, T. G. Elford, R. P. Sonawane and V. K. Aggarwal, Org. Lett., 2011, 13, 5740–5743 CrossRef CAS PubMed.
- Q. Yan, D. Kong, M. Li, G. Hou and G. Zi, J. Am. Chem. Soc., 2015, 137, 10177–10181 CrossRef CAS PubMed.
- K. Yoo, H. Kim and J. Yun, Chem.–Eur. J., 2009, 15, 11134–11138 CrossRef CAS PubMed.
- W.-T. Wei, J.-Y. Yeh, T.-S. Kuo and H.-L. Wu, Chem.–Eur. J., 2011, 17, 11405–11409 CrossRef CAS PubMed.
- J. Yan, Y. Nie, F. Gao, Q. Yuan, F. Xie and W. Zhang, Tetrahedron, 2021, 84, 132003 CrossRef CAS.
- W. M. Clark, A. J. Kassick, M. A. Plotkin, A. M. Eldridge and I. Lantos, Org. Lett., 1999, 1, 1839–1842 CrossRef CAS PubMed.
- B. H. Lee, Y. L. Choi, S. Shin and J.-N. Heo, J. Org. Chem., 2011, 76, 6611–6618 CrossRef CAS PubMed.
- A. Minatti, X. Zheng and S. L. Buchwald, J. Org. Chem., 2007, 72, 9253–9258 CrossRef CAS PubMed.
- G. Yue, K. Lei, H. Hirao and J. Zhou, Angew. Chem., Int. Ed., 2015, 54, 6531–6535 CrossRef CAS PubMed.
- S. Mannathan, S. Raoufmoghaddam, J. N. H. Reek, J. G. de Vries and A. J. Minnaard, ChemCatChem, 2017, 9, 551–554 CrossRef CAS.
- Y.-N. Yu and M.-H. Xu, J. Org. Chem., 2013, 78, 2736–2741 CrossRef CAS PubMed.
- T. Ikariya and A. J. Blacker, Acc. Chem. Res., 2007, 40, 1300–1308 CrossRef CAS PubMed.
- T. Ikariya, K. Murata and R. Noyori, Org. Biomol. Chem., 2006, 4, 393–406 RSC.
- R. Noyori, M. Yamakawa and S. Hashiguchi, J. Org. Chem., 2001, 66, 7931–7944 CrossRef CAS PubMed.
- R. Noyori and S. Hashiguchi, Acc. Chem. Res., 1997, 30, 97–102 CrossRef CAS.
- G. N. Hans, A. Zanotti-Gerosa and M. Wills, Chem. Rec., 2016, 16, 2623–2643 CrossRef PubMed.
- F. Foubelo, C. Nájera and M. Yus, Tetrahedron: Asymmetry, 2015, 26, 769–790 CrossRef CAS.
- T. Touge, T. Hakamata, H. Nara, T. Kobayashi, N. Sayo, T. Saito, Y. Kayaki and T. Ikariya, J. Am. Chem. Soc., 2011, 133, 14960–14963 CrossRef CAS PubMed.
- L.-S. Zheng, Q. Llopis, P.-G. Echeverria, C. Férard, G. Guillamot, P. Phansavath and V. Ratovelomanana-Vidal, J. Org. Chem., 2017, 82, 5607–5615 CrossRef CAS PubMed.
- A. E. Cotman, M. Lozinšek, B. Wang, M. Stephan and B. Mohar, Org. Lett., 2019, 21, 3644–3648 CrossRef CAS PubMed.
- S. Rast, B. Modec, M. Stephan and B. Mohar, Org. Biomol. Chem., 2016, 14, 2112–2120 RSC.
- A. E. Cotman, B. Modec and B. Mohar, Org. Lett., 2018, 20, 2921–2924 CrossRef CAS PubMed.
- T. Touge, H. Nara, M. Kida, K. Matsumura and Y. Kayaki, Org. Lett., 2021, 23, 3070–3075 CrossRef CAS PubMed.
- I. Kaljurand, A. Kütt, L. Sooväli, T. Rodima, V. Mäemets, I. Leito and I. A. Koppel, J. Org. Chem., 2005, 70, 1019–1028 CrossRef CAS PubMed.
- R. Rendy, Y. Zhang, A. McElrea, A. Gomez and D. A. Klumpp, J. Org. Chem., 2004, 69, 2340–2347 CrossRef CAS PubMed.
- B. Venkat Ramulu, A. Gopi Krishna Reddy and G. Satyanarayana, Synlett, 2013, 24, 868–872 CrossRef.
- B. V. Ramulu, P. Niharika and G. Satyanarayana, Synthesis, 2015, 47, 1255–1268 CrossRef CAS.
- A. Püschl, H. C. Rudbeck, A. Faldt, A. Confante and J. Kehler, Synthesis, 2005, 291–295 CrossRef.
- N. Parveen and G. Sekar, Adv. Synth. Catal., 2019, 361, 4581–4595 CrossRef CAS.
- P. A. Dub and J. C. Gordon, Dalton Trans., 2016, 45, 6756–6781 RSC.
- P. A. Dub and T. Ikariya, J. Am. Chem. Soc., 2013, 135, 2604–2619 CrossRef CAS PubMed.
- P. Van Kerrebroeck, K. Kreder, U. Jonas, N. Zinner and A. Wein, Urology, 2001, 57, 414–421 CrossRef CAS PubMed.
- F. Ulgheri, M. Marchetti and O. Piccolo, J. Org. Chem., 2007, 72, 6056–6059 CrossRef CAS PubMed.
- B. D. Gallagher, B. R. Taft and B. H. Lipshutz, Org. Lett., 2009, 11, 5374–5377 CrossRef CAS PubMed.
- G. Chen, N. Tokunaga and T. Hayashi, Org. Lett., 2005, 7, 2285–2288 CrossRef CAS PubMed.
- B. S. Lee, I. Y. Lee, B.-S. Lee, C. E. Song and D. Y. Chi, Bull. Korean Chem. Soc., 2000, 21, 860 CAS.
- T. Korenaga, R. Sasaki, T. Takemoto, T. Yasuda and M. Watanabe, Adv. Synth. Catal., 2018, 360, 322–333 CrossRef CAS.
- Y. Luo and A. J. Carnell, Angew. Chem., Int. Ed., 2010, 49, 2750–2754 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures for the synthesis of starting racemic 3-arylindanones, copies of 1H-, 13C-NMR, and chiral HPLC chromatograms for all new compounds. See DOI: 10.1039/d1ra04538e |
| This journal is © The Royal Society of Chemistry 2021 |