
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceNitration of aromatics with dinitrogen pentoxide in a liquefied 1,1,1,2-tetrafluoroethane medium†
Alexandr K. Kharchenko ,
Ruslan V. Fauziev,
Mikhail N. Zharkov,
Ilya V. Kuchurov and
Sergei G. Zlotin*
,
Ruslan V. Fauziev,
Mikhail N. Zharkov,
Ilya V. Kuchurov and
Sergei G. Zlotin*
N.D. Zelinsky Institute of Organic Chemistry Russian Academy of Sciences Leninsky Prospect, 47, Moscow, Russian Federation. E-mail: zlotion@ioc.ac.ru
First published on 27th July 2021
Abstract
Regardless of the sustainable development path, today, there are highly demanded chemical productions still operating that bear environmental and technological risks inherited from the previous century. The fabrication of nitro compounds, and nitroarenes in particular, is traditionally associated with acidic wastes formed in nitration reactions exploiting mixed acids. However, nitroarenes are indispensable for industrial and military applications. We faced the challenge and developed a greener, safer, and yet effective method for the production of nitroaromatics. The proposed approach comprises the application of an eco-friendly nitrating agent, namely dinitrogen pentoxide (DNP), in the medium of liquefied 1,1,1,2-tetrafluoroethane (TFE) – one of the most non-hazardous Freons. Importantly, the used TFE is not emitted into the atmosphere but is effortlessly recondensed and returned into the process. DNP is obtained via the oxidation of dinitrogen tetroxide with ozone. The elaborated method is characterized by high yields of the targeted nitro arenes, mild reaction conditions, and minimal amount of easy-to-utilize wastes.
Introduction
Today, the production of nitroaromatic compounds remains one of the largest among other industrial organic syntheses for being the basis of obtaining various amines,1 hydroxylamines,2 isocyanates,3 and annulated heterocyclic substances,4 which find their application in polymers, dyes, and drugs.5,6 Also, polynitroarenes, particularly trinitrotoluene, are well-known industrial and military explosives.7,8The conventional approach for the production of nitroarenes (both in laboratories and industry) employs the electrophilic nitration of arenes with a mixture of nitric and sulfuric acids.9,10 Its major disadvantage is the formation of large amounts of acidic waste often contaminated with nitric oxides (NOx).9 Moreover, the mixed acid cannot be used for the nitration of substrates bearing functional groups prone to acidic hydrolysis or oxidation.
Sulfuric acid acts as a protonating agent to generate active electrophilic species – nitronium cations [NO2]+.11 In some cases, it can be replaced with heterogeneous acidic catalysts, such as silica sulfuric acid,12 Nafion-H,13 or modified zeolites,14 though the efficacy of heterogeneous nitration is usually inferior to that of homogeneous nitration. The nitric acid–acetic anhydride nitrating system allows a more selective nitration of arenes;15,16 however, due to the formation of unstable acetyl nitrate in the reaction mass, risks of explosion rise dramatically.9 In some cases, metal nitrates and nitrites act as mild nitrating agents for arenes;17–26 however, the generation of stoichiometric quantities of metal salts in waste waters that require further disposal makes the scaling-up of such productions complicated. More rarely, metal-free nitrates,27 cyclic N-nitroimides28 and HNO3/HFIP system29 have been used as nitronium sources for the aromatic nitration.
A good alternative to the nitrating acid mixtures is dinitrogen pentoxide (DNP, N2O5), an effective and eco-friendly nitrating agent,10 obtained by chemical30–32 or electrochemical33,34 oxidation of N2O4, an industrial precursor of nitric acid. DNP can be used in nitration reactions almost stoichiometrically, thus significantly reducing the amounts of acidic wastes.10,35,36 Moreover, their composition becomes more narrow. The Kyodai nitration protocol using NO2/O3 mixtures, in which DNP can be in situ generated, also falls in this category of the nitration processes.37
DNP-based nitration reactions are dramatically influenced by the medium (solvent) used.38 For instance, DNP exhibits a prominent nitrating activity when combined with highly polar sulfuric acid taken in a far lesser amount than in the classic mixed acid nitrating system.39 The DNP-SO2 combination may also bring promising results.40 However, a disadvantage of such mixtures is the a priori presence of undesired sulfuric or sulfurous acid in the reaction wastes. By contrast, in low-polar media (e.g. in dichloromethane with a dipole moment of 1.6 D41) or in non-polar perfluorodecalin,42 DNP is less active, with the selectivity of the aromatic nitration being usually better.39 To increase the nitration efficacy for such cases, numerous Lewis and Bronsted acids were considered as heterogeneous catalysts, including zeolites,43,44 bismuth and rare-earth metal triflates,45,46 iron(II) and zirconium(IV) acetyl acetates.47,48
Recently, we have reported on N- and O-nitration reactions in the media of liquefied 1,1,1,2-tetrafluoroethane (TFE),36,49 an accessible and inexpensive refrigerant (R134a) widely used in different areas50,51 from industrial and household units to medical inhalers,52 where it is employed as gas-carrier due its complete nontoxicity.53 This gaseous, at normal conditions,54 resistant to strong oxidizers (including DNP) polar compound (DM = 2.1 D)54 can be easily turned to liquid under mild conditions. Importantly, TFE is characterized by zero ozone depletion potential and is not listed among the ozone-depleting substances.50,55 Although TFE carries some global warming potentials, we managed to render TFE-based technological processes to be environmentally safe by preventing its emission into the atmosphere. There are hardly any challenges in the TFE recycling since it is characterized by a relatively low saturated vapor pressure (0.6 MPa and 20 °C).35,54,56
Herein, we applied for the first time the DNP/TFE nitrating system for the clean, effective, and safe preparation of nitroarenes.
Results and discussion
We started with the nitration of benzene (1) in a liquefied TFE medium as a model reaction (Scheme 1, Table 1). The reactions were carried out under similar conditions (20 °C, 0.6 MPa). The use of nearly stoichiometric quantity of DNP (1.1 equiv.) inevitably resulted in the formation of nitrobenzene (2). The yield of 2 increased with the reaction time and reached the maximum at 30 min (Table 1, entries 1–3). When using 2.2 equiv. DNP, only a small amount of dinitration product 3 was observed over 0.5 h (entry 4). A significant increase in the amount of DNP (up to 10 equiv.) and the reaction time (up to 8 h) were required for introducing the second nitro group. In this case, meta-dinitrobenzene (3) was selectively formed (rr meta/para >20![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) and no traces of trinitrobenzene were recorded.
:1) and no traces of trinitrobenzene were recorded.
 | ||
| Scheme 1 Model reaction. | ||
| Entry | DNP, equiv. | Time, min | Conversionb, % | 2/3 Ratiob, % |
|---|---|---|---|---|
| a Reaction conditions, unless otherwise specified: 1 (5 mmol), DNP (5.5 mmol), 20 °C, 0.6 MPa.b 1H NMR data.c Compound 3 is not registered.d Compound 2 is not registered. | ||||
| 1 | 1.1 | 10 | 56 | —c |
| 2 | 1.1 | 20 | 78 | —c |
| 3 | 1.1 | 30 | 98 | —c |
| 4 | 2.2 | 30 | 97 | 98:2 |
| 5 | 10.0 | 480 | 95 | —d |
The nitration of toluene (4) with 1.1 equiv. of DNP under similar conditions (0.6 MPa and 20 °C) led to the formation of an almost equimolar mixture of para- and ortho-nitrotoluenes 5a and 5b at full conversion in 30 min (Scheme 2). In the presence of 5 equiv. of DNP, isomeric dinitrotoluenes 6a and 6b were obtained in a >4:1 ratio. These results generally correspond to the data reported for the perfluorodecalin medium;42 however, the reaction in TFE went much faster (0.5 h vs. 12 h). The noticeable activation of DNP in liq. TFE was also proved by the easy nitration of ortho-nitrotoluene (5b) to dinitrotoluenes (6a) and (6b). The reaction proceeded in 89% total yield by the action of only 1.1 equiv. of DNP (Scheme 3), while a similar reaction in CH2Cl2 was characterized by the poor conversion of substrate 5b (<6%).57
 | ||
| Scheme 2 Mono- and dinitration of toluene (4) with DNP in liq. TFE. | ||
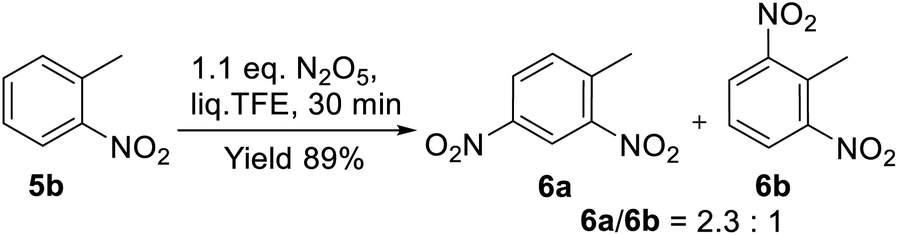 | ||
| Scheme 3 Nitration of o-nitrotoluene (5b) with DNP in liq. TFE. | ||
A wide range of arenes underwent the nitration reaction under proposed conditions (DNP/liq. TFE, 0.6 MPa, 20 °C). The nitration of meta-xylene (7) with 1 equiv. of DNP proceeded quantitatively with the formation of 4-nitro isomer 8a as the major product (Scheme 4).
 | ||
| Scheme 4 Nitration of m-xylene (7) with DNP in liq. TFE. | ||
The nitration of the activated substrates went faster in the liq. TFE medium, and the number of nitro groups introduced into the aromatic ring depends on the nitrating agent amount. Indeed, anisole (9) reacted with an equimolar amount of DNP affording a mixture of isomeric nitroanisoles 10a and 10b in a quantitative total yield over 5 min (Scheme 5). The main isomer was ortho-nitroanisole (10b), which is typical in aromatic nitration processes employing nitronium salts as nitrating agents and differs significantly from reactions with mixed acids.58 This result indirectly indicates that DNP molecules reversibly dissociated in liq. TFE with the formation of the nitronium cation [NO2]+, which determines the nitration mechanism and the ratio of isomers. Simultaneously, with a larger amount of the nitrating agent (2.2 equiv.), the reaction afforded isomeric dinitroanisoles 11a and 11b in a 4:1 ratio with the total yield of 93% over 0.5 h.
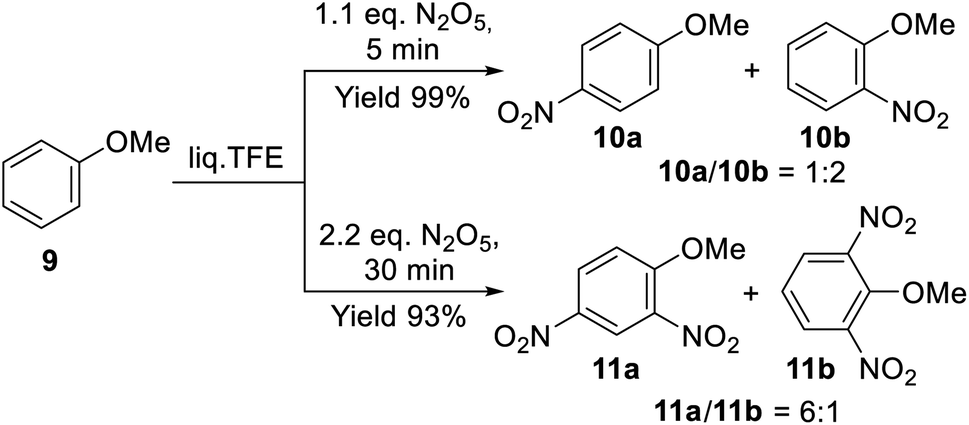 | ||
| Scheme 5 Mono- and dinitration of anisole (9) with DNP in liq. TFE. | ||
The reaction of phenol (12) with 3.3 equiv. of DNP under proposed conditions afforded 2,4,6-trinitrophenol (13), the exhaustive nitration product, in 82% yield over 0.5 h (Scheme 6). Accessing a comparable yield of 13 in a non-polar perfluorodecalin medium required a significant excess of DNP (7 equiv.) and a longer reaction time (2 h).42
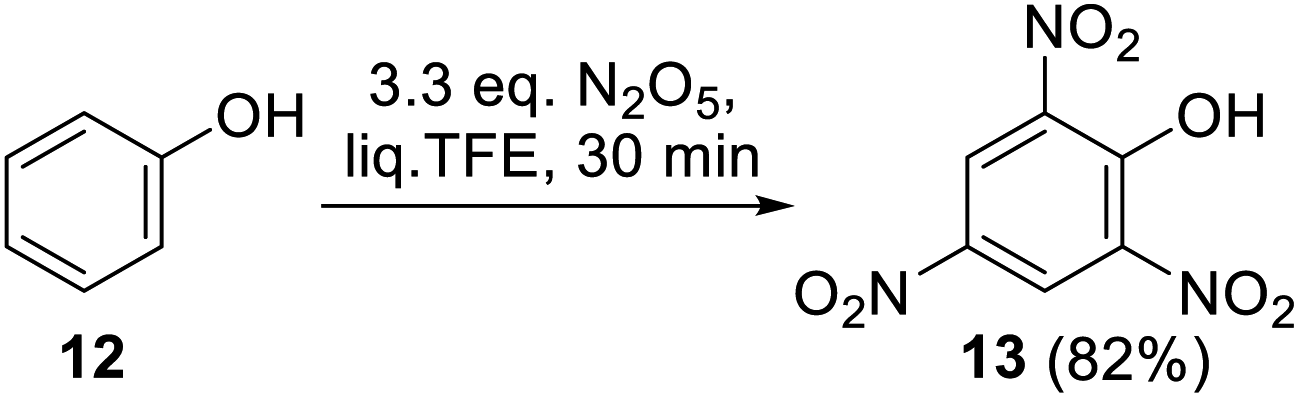 | ||
| Scheme 6 Exhaustive nitration of phenol (12) with DNP in liq. TFE. | ||
As noted above, the nitration of aromatic substrates deactivated with electron-withdrawing groups under the action of DNP is usually conducted in the presence of catalysts or in strongly acidic media.39,44,48 We managed to carry out the non-catalytic nitration of similar compounds in the liq. TFE medium. Therefore, the reactions of halogenated benzenes 14–16 or methyl benzoate (20) with the equimolar amount of DNP afforded isomeric mononitro products 17–19 or 21 in high yields under mild conditions (Scheme 7). Notably, para-isomers 17a–19a prevailed in products 17–19, which could be isolated as mixtures of isomers a and b by simple filtration. To achieve a high conversion for 20, the reaction time was increased from 30 to 45 min, and the isomers 21a and 21b were isolated in this case by flash chromatography in 19% and 72% yield, respectively.
 | ||
| Scheme 7 Nitration of deactivated arenes 14–16 and 20 with DNP in liq. TFE. | ||
Meta- and para-dichlorobenzenes 22 and 24 readily underwent the reaction with DNP under optimal conditions (Scheme 8). The nitration products 23 and 25 were obtained here as single isomers, and their structures were unambiguously confirmed by 1H NMR spectroscopy.
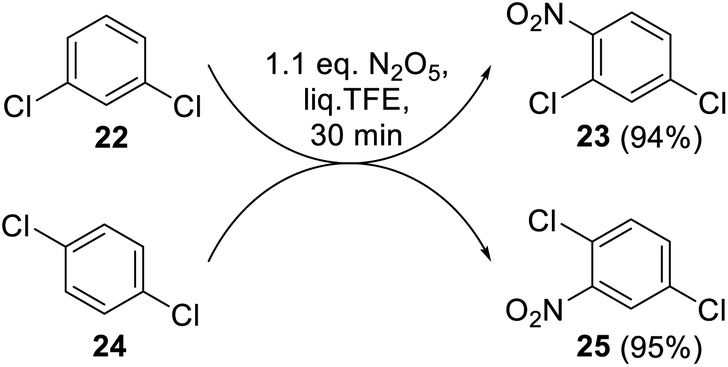 | ||
| Scheme 8 Nitration of dichlorobenzenes 22 and 24 with DNP in liq. TFE. | ||
Functionalized anizoles 26–28 containing electron-withdrawing substituents (cyano-, acetyl, or formyl groups) in the para-position reacted with DNP regioselectively in the liq. TFE medium. Nitro compounds 29 and 30 bearing cyano- and acetyl groups were generated in these reactions in nearly quantitative yields over 45 min and did not require further purification (Scheme 9). The yield of para-methoxybenzaldehyde (31) was somewhat lower (75%), which may be attributed to oxidative by-side processes with the participation of the formyl group under the reaction conditions.
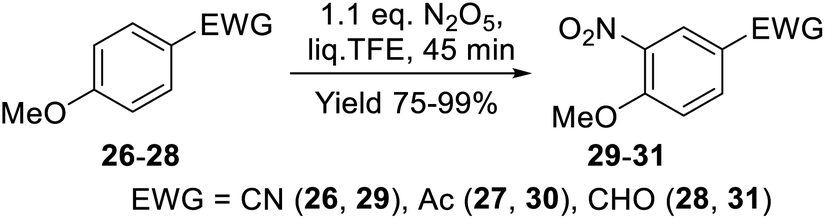 | ||
| Scheme 9 Nitration of anisole derivatives 26–28 with DNP in liq. TFE. | ||
Along with electronic factors, the solubility of starting substrates and reaction products in liq. TFE can exert a significant impact on the nitration reaction outcome. Indeed, the poor solubility of naphthalene (32) in liq. TFE made the nitration heterogeneous at the substrate load of 5 mmol. This led to the situation when the first formed 1-nitronaphthalene (33), which is better soluble in liq. TFE than 32, and competitively underwent further nitration in the solution. As a result, a mixture of unchanged naphthalene (32) and its 1-nitro- (33) and 1,8-dinitro- (34) derivatives were produced (Scheme 10, reaction A). The problem was fixed by reducing the naphthalene load by half, thus transferring it to the solution before the DNP addition (Scheme 10, reaction B). Under these homogeneous conditions, 1-nitronaphthalene (33) was formed exclusively in a near quantitative yield. Therefore, the solubility phenomenon should be considered when planning the further synthesis of nitroarenes in the liq. TFE medium.
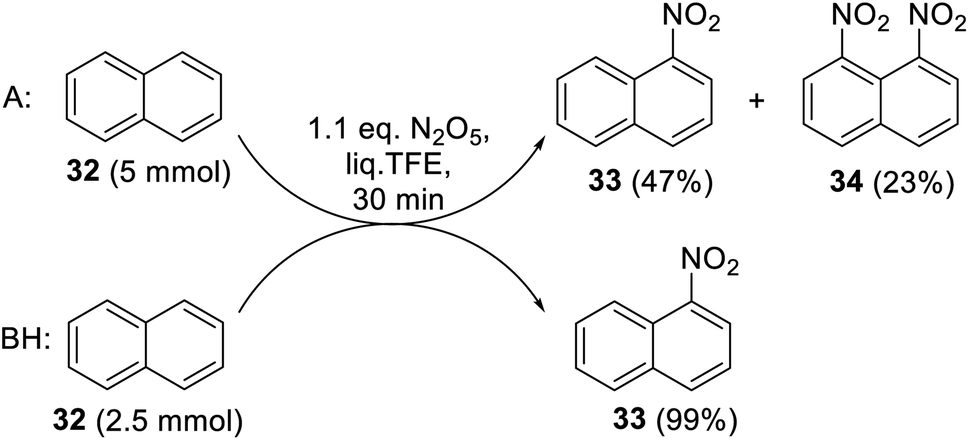 | ||
| Scheme 10 Nitration of naphthalene (32) with DNP in liq. TFE under heterogeneous (A) and homogenous (B) conditions. | ||
The developed nitration procedure was simple and environmentally benign. Due to the low pressure in the reactor (≤0.6 MPa), the process did not require an expensive high-pressure equipment. In most cases, pure nitro products could be isolated in high yields right after the final decompression and water flushing. Importantly, the exhaust TFE gas was not vented into the atmosphere, but passed through a desiccant tube, and then liquefied in a cooled auxiliary vessel for further reuse. The simplicity of the by-product nitric acid utilization and the affordability of the TFE recycling make the developed procedure attractive both from industrial and green chemistry viewpoints.
The synthetic potential of the method was demonstrated by the facile nitration of ibuprofen methyl ester 35 with DNP in the liq. TFE medium (Scheme 11). Isomeric nitration products 36a and 36b were obtained in a total yield of 93% and in a 2:1 ratio. Isomer 36a is a precursor of bromide 37, which has a higher anti-inflammatory activity than the parent drug.59
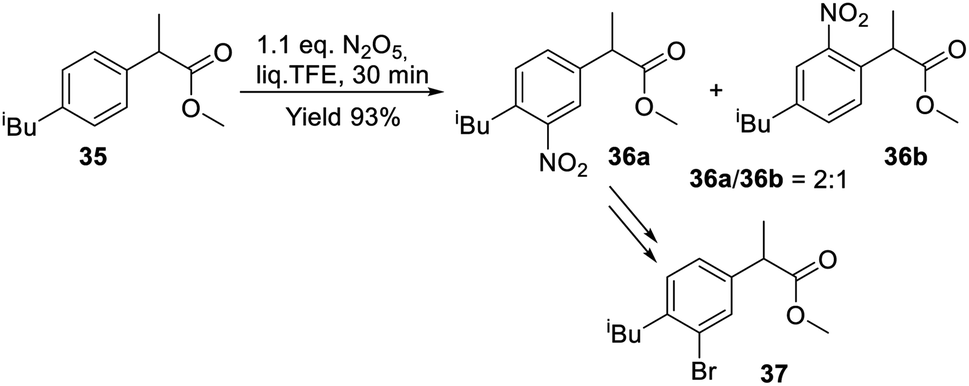 | ||
| Scheme 11 Nitration of ibuprofen methyl ester in liq. TFE. | ||
Moreover, the nitration of naproxen ester 38 and its analog 39 also proceeded smoothly under the proposed conditions affording corresponding nitro derivatives 40 and 41 as single regioisomers in 83% or 79% yield, respectively (Scheme 12). The mild reaction conditions (1.1 eq. of DNP, 0.6 MPa, 20 °C) along with the selectivity of aromatic nitration higher than that of the classical methods make our approach useful for selective modifications of biologically active substances or their precursors at the later stages of production.
 | ||
| Scheme 12 Nitration of naproxen ester 38 and its analog 39 with DNP in liq. TFE. | ||
Conclusion
In summary, we have developed an effective and safe method for the nitration of aromatic compounds with dinitrogen pentoxide in the medium of liquefied 1,1,1,2-tetrafluoroethane (TFE). The reactions were conducted under mild conditions (0.6 MPa, 20 °C), which made it possible to embrace functionally substituted arenes and, hence, synthesize valuable precursors for biologically active substances. The method eliminated the use of toxic organic solvents typical of traditional nitration, and significantly reduced the acidic waste amount. Upon the synthesis completion, the solvent (TFE) was easily separated from the reaction products by decompression, and then re-condensed to be subsequently re-used. This prevented its emission into the atmosphere and made the proposed method sustainable and environmentally friendly.Experimental section
Materials
The reactions were carried out in a 22.4 cm3 steel autoclave equipped with sapphire windows, magnetic stirrer, and pressure and temperature sensors. An auxiliary 12 cm3 steel dosing vessel with sapphire windows and a magnetic stirrer was used for the preparation of nitrating agent solutions. Melting points were obtained on Stuart® SMP40. 1H and 13C NMR spectra were recorded on a Bruker® AM-300 (300.13 and 75.47 MHz, respectively). The high-resolution mass spectra (HRMS) were measured on a Bruker microTOF II spectrometer via electrospray ionization (ESI).TFE, O2 (Grade 3.5), N2 (Grade 5) and N2O4 were obtained from Linde Gas Rus. Unless otherwise mentioned, all substrates were purchased from Acros Organics. Substrates 35 and 38 were prepared via a routine etherification procedure.60 Substrate 39 was synthesized from anisole, oxalyl chloride and methanol (see ESI†).
Synthesis of N2O5 (DNP)
DNP was obtained via NO2 oxidation with ozone (O3). The process was arranged in a 2 l gas-flow reactor with an inner-coiled tube cooler operating in continuous mode. NO2 was fed into the reactor from the cylinder heated up to 35–40 °C at a flow rate of 22.4 ml min−1 (1 mmol min−1). O3 was delivered by an ozone generation unit in the form of its 20 wt% mixture with O2. The flow rate was sufficient to have the brownish NO2 fully engaged in the oxidation reaction (the discoloration was determined visually). The gases entered the reactor through the coaxial nozzle for better mixing and more uniform distribution throughout the vessel. The process temperature was maintained by the cooling water and did not exceed 25 °C in the first third of the reactor according to the thermocouple readings. The resultant gaseous DNP was condensed in two successive traps cooled at −80 °C giving practically a quantitative yield of nearly 7 g h−1. After the desired DNP quantity was obtained, the NO2 feed and O3 generation were stopped, the system was insufflated with pure O2, and the product was weighed and used as intended.More details and the experimental setup scheme are given in ESI.†
General nitration procedure
The principal scheme of the experimental setup for the nitration of aromatics is given in Fig. 1 (for details also see ESI†). | ||
| Fig. 1 Laboratory setup for the nitration of aromatics with DNP in the TFE medium: 1 is cylinder with TFE; 2 is the piston pump with a gas condensation chamber; 3 is the autoclave-reactor equipped with two look-through windows, stir-bar and sensors of temperature and pressure; 4 and 10 are thermostats; 5 is the auxiliary dosing vessel; 6 is the magnetic stirrer; 7 is the syringe pump; 8 is the beaker with aqueous NaHCO3; 9 is the cylinder for TFE re-condensation; 11 is the desiccant tube. | ||
A steel autoclave-reactor containing 5.0 mmol of the substrate was filled with liquefied TFE at room temperature by one third of the volume and cooled to 5 °C. The auxiliary dosing vessel was charged with 5.5 mmol of N2O5, and then filled with TFE fluid by half at room temperature. Using the pressure difference (ΔP = ∼0.3 MPa) between the vessels, the N2O5 solution was slowly moved into the reactor (quick temperature growth should be avoided) at constant stirring. The dosing vessel was refilled with TFE by one third to dissolve and wash residuals of the nitrating agent into the reactor. The reaction mixture was stirred at room temperature and 0.6 MPa for the time shown in Schemes 1–11. Then, 5 ml of 2 N aqueous NaHCO3 was added by a syringe pump to the reactor to neutralize the nitrating agent excess and the formed nitric acid. From the decompressed reactor, TFE was streamed into the recondensation line for its further re-use. The autoclave was opened, and the nitration products were filtered off, washed with distilled water (2 × 3 ml), dried in air to afford corresponding nitro compounds as individual substances or mixtures of isomers. For compounds 2; 5a,b; 8a,b; 15a,b; 19a,b; 29; 34a,b; 38 and 39 another work-up procedure was applied. The products were extracted from the aqueous suspension with ethyl acetate (2 × 25 ml). The combined organic extracts were washed with distilled water (2 × 25 ml), and then dried over anhydrous MgSO4. The solvent was evaporated under a reduced pressure to afford the targeted nitro compounds. Compounds 19a,b; 29; 34a,b; 38 and 39 were further purified by column chromatography (silica gel, eluent EtOAc/petroleum ether, and gradient). The structures and isomer ratios of all nitro compounds were determined by 1H NMR spectroscopy.
TFE recuperation procedure
After the completion of each nitration process, TFE was re-condensed from gradually decompressing reactor 3 (see Fig. 1) to cylinder 9 cooled to −10 °C. When cylinder 9 was fully filled with the exhausted TFE (V = 1.0 L, about 50 experiments), it was switched by a three-way valve to desiccant tube 11 filled with a mixture of activated molecular sieves and granulated alkali. Then, the cylinder was warmed up to 30–35 °C for efficient TFE evaporation. After passing through tube 11, the gaseous TFE was re-condensed in empty cylinder 1 cooled to −10 °C. The collected moisture-free TFE (>95% of the initial amount by weight) was reused in the nitration reactions, thereby significantly improving the process performance. The TFE recuperation procedure was repeated at least five times, and the same results were attained in control experiments (99% yield of the 10a:10b = 2:1 mixture in the quick (5 min) nitration of anisole 9).
Author contributions
Alexandr K. Kharchenko: conceptualization, investigation, methodology, writing – original draft. Ruslan V. Fauziev: investigation. Mikhail N. Zharkov: methodology, writing – original draft, writing – review & editing. Ilya V. Kuchurov: conceptualization, funding acquisition, project administration, writing – original draft. Sergei G. Zlotin: supervision, writing – review & editing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors are grateful to Dr Lyubov N. Mitireva for the thorough English language editing. This research was supported by the Russian Science Foundation (Project no. 18-73-10207).Notes and references
- N. Daems, J. Wouters, C. Van Goethem, K. Baert, C. Poleunis, A. Delcorte, A. Hubin, I. F. J. Vankelecom and P. P. Pescarmona, Appl. Catal., B, 2018, 226, 509–522 CrossRef CAS
.
- S. Doherty, J. G. Knight, T. Backhouse, R. J. Summers, E. Abood, W. Simpson, W. Paget, R. A. Bourne, T. W. Chamberlain, R. Stones, K. R. J. Lovelock, J. M. Seymour, M. A. Isaacs, C. Hardacre, H. Daly and N. H. Rees, ACS Catal., 2019, 9, 4777–4791 CrossRef CAS
- S. B. Halligudi, R. V. Chaudharl and L. K. Doralswamy, Ind. Eng. Chem. Process Des. Dev., 1984, 23, 794–801 CrossRef CAS
- A. D. Kotov, M. A. Prokaznikov, E. A. Antonova and A. I. Rusakov, Chem. Heterocycl. Compd., 2014, 50, 647–657 CrossRef CAS
- G. Booth, in Ullmann's Encyclopedia of Industrial Chemistry, Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim, Germany, 2000 Search PubMed
- K. Nepali, H. Y. Lee and J. P. Liou, J. Med. Chem., 2019, 62, 2851–2893 CrossRef CAS PubMed
- R. Meyer, J. Köhler and A. Homburg, Explosives, Wiley-VCH Verlag GmbH&Co. KGaA, Weinheim, Germany, 7th edn, 2016 Search PubMed
- T. M. Klapötke, Energetic Materials Encyclopedia, De Gruyter, 1st edn, 2018 Search PubMed
- G. A. Olah, R. Malhotra and S. C. Narang, NITRATION: Methods and Mechanisms, Wiley-VCH, 1989 Search PubMed
- J. P. Agrawal and R. Hodgson, Organic Chemistry of Explosives, John Wiley & Sons, Ltd, Chichester, UK, 2007 Search PubMed
- B. Galabov, D. Nalbantova, P. V. R. Schleyer and H. F. Schaefer, Acc. Chem. Res., 2016, 49, 1191–1199 CrossRef CAS PubMed
- M. A. Zolfigol, B. B. F. Mirjalili, A. Bamoniri, M. A. K. Zarchi, A. Zarei, L. Khazdooz and J. Noei, Bull. Korean Chem. Soc., 2004, 25, 1414–1416 CrossRef CAS
- G. A. Olah, R. Malhotra and S. C. Narang, J. Org. Chem., 1978, 43, 4628–4630 CrossRef CAS
- B. M. Choudary, M. Sateesh, M. Lakshmi Kantam, K. Koteswara Rao, K. V. Ram Prasad, K. V. Raghavan and J. A. R. P. Sarma, Chem. Commun., 2000, 25–26 RSC
- K. Smith, A. Musson and G. A. DeBoos, J. Org. Chem., 1998, 63, 8448–8454 CrossRef CAS
- R. O. Norman and G. K. Radda, J. Am. Chem. Soc., 1961, 3030–3037 RSC
- S. Samajdar, F. F. Becker and B. K. Banik, Tetrahedron Lett., 2000, 41, 8017–8020 CrossRef CAS
- V. Anuradha, P. V. Srinivas, P. Aparna and J. M. Rao, Tetrahedron Lett., 2006, 47, 4933–4935 CrossRef CAS
- A. K. Bose, S. N. Ganguly, M. S. Manhas, S. Rao, J. Speck, U. Pekelny and E. Pombo-Villars, Tetrahedron Lett., 2006, 47, 1885–1888 CrossRef CAS
- R. Rajagopal and K. V. Srinivasan, Synth. Commun., 2003, 33, 961–966 CrossRef CAS
- M. F. A. Dove, B. Manz, J. Montgomery, G. Pattenden and S. A. Wood, J. Chem. Soc., Perkin Trans. 1, 1998, 1589–1590 RSC
- X. Yang, C. Xi and Y. Jiang, Tetrahedron
Lett., 2005, 46, 8781–8783 CrossRef CAS
- M. A. Zolfigol, E. Ghaemi and E. Madrakian, Synlett, 2003, 2003, 191–194 CrossRef
- M. A. Zolfigol, E. Ghaemi and E. Madrakian, Synth. Commun., 2000, 30, 1689–1694 CrossRef CAS
- N. Nowrouzi, A. M. Mehranpour, E. Bashiri and Z. Shayan, Tetrahedron Lett., 2012, 53, 4841–4842 CrossRef CAS
- G. Yan and M. Yang, Org. Biomol. Chem., 2013, 11, 2554–2566 RSC
- H. Sepehrmansourie, M. Zarei, M. A. Zolfigol, A. Mehrzad and H. R. Hafizi-Atabak, RSC Adv., 2021, 11, 8367–8374 RSC
- R. Calvo, K. Zhang, A. Passera and D. Katayev, Nat. Commun., 2019, 10, 1–8 CrossRef CAS PubMed
- L. Lu, H. Liu and R. Hua, Org. Lett., 2018, 20, 3197–3201 CrossRef CAS PubMed
- N. C. Paul, in Nitration. Recent Laboratory and Industrial Development. ACS Symposium Series 623, ed. L. F. Albright, R. V. C. Carr and R. J. Schmitt, American Chemical Society, Washington, DC, 1996, vol. 623, pp. 165–173 Search PubMed
- T. E. Devendorf and J. R. Stacy, ACS Symp. Ser., 1996, 623, 68–77 CrossRef CAS
- M. B. Talawar, R. Sivabalan, B. G. Polke, U. R. Nair, G. M. Gore and S. N. Asthana, J. Hazard. Mater., 2005, 124, 153–164 CrossRef CAS PubMed
- Q. Wang, M. Su, X. Zhang, L. Wang, J. Wang and Z. Mi, Electrochim. Acta, 2007, 52, 3667–3672 CrossRef CAS
- R. D. Chapman and G. D. Smith, in Nitration. Recent Laboratory and Industrial Development. ACS Symposium Series 623, ed. L. F. Albright, R. V. C. Carr and R. J. Schmitt, American Chemical Society, Washington, DC, 1996, vol. 623, pp. 78–96 Search PubMed
- M. N. Zharkov, S. S. Arabadzhi, I. V. Kuchurov and S. G. Zlotin, React. Chem. Eng., 2019, 4, 1303–1308 RSC
- M. Zharkov, I. Kuchurov, I. Fomenkov, V. Tartakovsky, I. Fedyanin and S. Zlotin, Synthesis, 2017, 49, 1103–1108 CAS
- H. Suzuki, K. Hisatome and N. Nonoyama, Synthesis, 1999, 1291–1293 CrossRef CAS
- R. W. Millar, M. E. Colclough, P. Golding, P. J. Honey, N. C. Paul, M. J. Stewart and A. J. Sanderson, Philos. Trans. R. Soc., A, 1992, 339, 305–319 CrossRef CAS
- R. W. Millar, A. W. Arber, R. M. Endsor, J. Hamid and M. E. Colclough, J. Energ. Mater., 2011, 29, 88–114 CrossRef CAS
- J. M. Bakke and I. Hegbom, Acta Chem. Scand., 1994, 48, 181–182 CrossRef CAS
- M. Sansotera, S. Geran Malek Kheyli, A. Baggioli, C. L. Bianchi, M. P. Pedeferri, M. V. Diamanti and W. Navarrini, Chem. Eng. J., 2019, 361, 885–896 CrossRef CAS
- M. R. Crampton, E. L. Cropper, L. M. Gibbons and R. W. Millar, Green Chem., 2002, 4, 275–278 RSC
- R. Damavarapu, K. Jayasuriya and T. J. Kwok, US Pat. US5977418A, 1998
- X. M. Ma, B. D. Li, L. Chen, M. Lu and C. X. Lv, Chin. Chem. Lett., 2012, 23, 809–812 CrossRef CAS
- H. Qian, Y. Wang, D. Liu and C. Lv, Lett. Org. Chem., 2014, 11, 509–512 CrossRef CAS
- X. M. Ma, B. D. Li, M. Lu and C. X. Lv, Chin. Chem. Lett., 2012, 23, 73–76 CrossRef CAS
- A. J. Hill, R. W. Millar and J. P. B. Sandall, Org. Biomol. Chem., 2004, 2, 90–92 RSC
- R. R. Bak and A. J. Smallridge, Tetrahedron Lett., 2001, 42, 6767–6769 CrossRef CAS
- I. V. Kuchurov, S. S. Arabadzhi, M. N. Zharkov, L. L. Fershtat and S. G. Zlotin, ACS Sustainable Chem. Eng., 2018, 6, 2535–2540 CrossRef CAS
- S. Corr, J. Fluorine Chem., 2002, 118, 55–67 CrossRef CAS
- M. Capeans, I. Glushkov, R. Guida, F. Hahn and S. Haider, Nucl. Instrum. Methods Phys. Res., Sect. A, 2012, 661, S214–S221 CrossRef CAS
- W. F. S. Sellers, Allergy, Asthma, Clin. Immunol., 2017, 13, 30 CrossRef PubMed
- H. H. Emmen, E. M. G. Hoogendijk, W. A. A. Klöpping-Ketelaars, H. Muijser, E. Duistermaat, J. C. Ravensberg, D. J. Alexander, D. Borkhataria, G. M. Rusch and B. Schmit, Regul. Toxicol. Pharmacol., 2000, 32, 22–35 CrossRef CAS PubMed
- Thermophysical Properties of Fluid Systems, accessed 21 April 2021, https://webbook.nist.gov/chemistry/fluid/ Search PubMed.
- P. Barker, R. Cary and S. Dobson, Concise International Chemical Assessment Document 11. 1,1,1,2-Tetrafluoroethane, Geneva, 1998 Search PubMed.
- M. N. Zharkov, I. V. Kuchurov and S. G. Zlotin, CrystEngComm, 2020, 22, 7549–7555 RSC
- R. P. Claridge, N. L. Lancaster, R. W. Millar, R. B. Moodie and J. P. B. Sandall, J. Chem. Soc., Perkin Trans. 2, 1999, 1815–1818 RSC
- G. A. Olah, H. C. Lin, J. A. Olah and S. C. Narang, Proc. Natl. Acad. Sci. U. S. A., 1978, 75, 545–548 CrossRef CAS PubMed
- K. Gupta, C. J. Kaub, K. N. Carey, E. G. Casillas, B. S. Selinsky and P. J. Loll, Bioorg. Med. Chem. Lett., 2004, 14, 667–671 CrossRef CAS PubMed
- D. Chavez-Flores and J. M. Salvador, Tetrahedron: Asymmetry, 2012, 23, 237–239 CrossRef CAS
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1ra04536a |
| This journal is © The Royal Society of Chemistry 2021 |