
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceReactive force fields for modeling oxidative degradation of organic matter in geological formations†
Jaewoong Hur a,
Younane N. Abousleimanb,
Katherine L. Hull*c and
Mohammad Javad Abdolhosseini Qomi*a
a,
Younane N. Abousleimanb,
Katherine L. Hull*c and
Mohammad Javad Abdolhosseini Qomi*a
aDepartment of Civil and Environmental Engineering, Henry Samueli School of Engineering, University of California, E4130 Engineering Gateway, Irvine, CA 92697-2175, USA. E-mail: mjaq@uci.edu
bIntegrated PoroMechanics Institute, School of Geosciences, The University of Oklahoma, Norman, Oklahoma 73019, USA
cAramco Services Company, Aramco Research Center – Houston, 16300 Park Row, Houston, Texas 77084, USA. E-mail: katherine.hull@aramcoamericas.com
First published on 1st September 2021
Abstract
In an attempt to better explore organic matter reaction and properties, at depth, to oxidative fluid additives, we have developed a new ReaxFF potential to model and describe the oxidative decompositions of aliphatic and aromatic hydrocarbons in the presence of the oxychlorine ClOn− oxidizers. By carefully adjusting the new H/C/O/Cl parameters, we show that the potential energies in both training and validation sets correlate well with calculated density functional theory (DFT) energies. Our parametrization yields a reliable empirical reactive force field with an RMS error of ∼1.57 eV, corresponding to a 1.70% average error. At this accuracy level, the reactive force field provides a reliable atomic-level picture of thermodynamically favorable reaction pathways governing oxidative degradation of H/C/O/Cl compounds. We demonstrate this capability by studying the structural degradation of small aromatic and aliphatic hydrocarbons in the presence of oxychlorine oxidizers in aqueous environments. We envision that such reactive force fields will be critical in understanding the oxidation processes of organic matter in geological reservoirs and the design of the next generation of reactive fluids for enhanced shale gas recovery and improved carbon dioxide adsorption and sequestration.
Introduction
Oxidation of natural organic matter (OM), whether in soil or geological formations, leads to changes in the chemical and physical properties of OM. Soil OM, when oxidized, undergoes changes in nutrient bioavailability, pollutant retention and its effects upon water quality, and soil productivity. Given that soils participate in half of the carbon that is recycled in the atmosphere,1 understanding the chemical redox processes that control CO2 release are of paramount concern for global climate initiatives. While soils contain more organic carbon than the combined totals of Earth's vegetation and atmosphere, the majority of organic carbon on earth is held in geological formations.2 Kerogen as the source of petroleum hydrocarbons3 is a crosslinked polymeric material with a disordered molecular structure intertwined with a nanogranular mineral matrix.4,5 It has the mechanical and physical characteristic signatures of polymers,6 though its chemical structure is not as readily oxidizable as synthetic polymers due to its compaction and thermal alteration over geological time. Since the 1950's, attempts to unravel the chemical structure of kerogen have utilized oxidation chemistry as a means of breaking the polymer structure into smaller fragments for analysis.7In unconventional source rock reservoirs, the hydrocarbon reserves are predominantly trapped in the pore space of the organic matter.8 Traditional hydraulic fracturing methods designed to fracture conventional reservoirs may not effectively crack and stimulate the ductile, polymeric kerogen. Chemical treatment of the polymeric properties of kerogen while taking into account its nanoscale porosities and ultra-low permeability, will introduce open flow channels, increase its permeability and eventually optimize recovery purposes. Recently, in an attempt to address the challenges associated with the kerogen's low permeability, Hull et al.9 designed fluid oxidizers as additives to the slick water in hydraulically fracking source rocks. The designed fluid additive has strong oxidizers with very high concentrations, and was successfully used recently. Oxidizing agents composed of oxyhalides such as bromate BrO3− and chlorate ClO3− are effective in breaking down shale's organic matter under reservoir temperatures.9
To further identify and develop oxidizers for various organic matter treatment, at depth, the earlier studies had to be generalized and optimized. In this work, we concentrate on oxychlorine ClOn− oxidizers as a class of effective oxidizing reagents to elucidate the oxidative degradation pathways likely to lead to organic matter cleavage, porosity enhancement, and increase in organic surface area. ClOn− oxidizers (n = 1–4), typically as alkali or alkaline earth metal salts, are stable, commercially available compounds with wide applicability. Hypochlorite ClO− is ubiquitously used for disinfection and water treatment, while chlorite ClO2− is one of the common oilfield oxidizers used in reservoir stimulation. Perchlorate ClO4−, though thermodynamically is a strong oxidant, is kinetically inert except under acidic conditions or in the presence of metal activator. ClO4− and chlorate ClO3− are also naturally occurring, coexisting on earth and on the Mars surface.10
The atomistic simulations of complex reactive processes at the organic matter–oxidizer interface require a numerically efficient framework to describe interatomic and intermolecular forces while faithfully describing the formation and breakage of covalent bonds. Although the first principle quantum chemistry approaches provide such information, their exorbitant computational cost still limits their application to short time and small length scales that render them unamenable to simulating multi-stage oxidative decomposition processes. Alternatively, adequately calibrated ReaxFF potentials can describe the formation, transition, and dissociation of chemical bonds with the density functional theory (DFT)-level accuracy at a reasonable computational cost.11,12 A well-calibrated ReaxFF can also simulate chemical reactions at the solid, liquid, and gas interfaces. This transferability gives ReaxFF a unique capability to simulate complex reactive processes involving multiple phases.13–16 These attributes make the ReaxFF potential an ideal simulation framework to unravel the mechanistic picture of oxidative reaction pathways at the kerogen-reactive fluid interface.
In this paper, we summarize ReaxFF's functional form and describe the new H/C/O/Cl ReaxFF parameterizations while comparing the calibrated ReaxFF's performance against DFT energetics. By performing ab initio molecular dynamics (MD) simulations, we propose the energetically-favorable oxidative decomposition pathways (reactant, transition, and product states) of small aromatic and aliphatic hydrocarbons in the presence of oxychlorine oxidizers (ClOn−: n = 1–4). Subsequently, we demonstrate that ReaxFF can quantitatively capture the energetics of proposed oxidation pathways. Finally, with the new ReaxFF parameterization at hand, we study the mechanistic picture of the oxidative degradations of small organic molecules with aromatic and aliphatic moieties representing the small unit of polymer-like kerogen frameworks with ClOn− in aqueous environments.
Model and methods
Density functional theory (DFT) calculations
We have systematically conducted a series of nonperiodic DFT energy calculations in establishing parameter optimization data sets used for the new ReaxFF parameterizations. The DFT calculations of the target molecules use B3LYP functional17,18 with different basis sets, as implemented in Gaussian16,19 such as 6-31G* (ref. 20) for minimum energy/single-point energy calculations, 6-31G** (ref. 21 and 22) for charge distribution calculations, 6-31G*(D3)/6-31++G**(D3)23 for van der Waals interatomic interaction energy calculations, and PBE functional24 for further charge evaluations by Bader analysis as considering adequate accuracy and computational time expense. For potential energy surfaces (PES) calculations, we employ the reliable B3LYP functional that has been extensively used and proven to fit the potential energies of various conformational H/C/O molecules.25–31 We performed the DFT (with B3LYP) calculations for closed-shell systems using the spin-restricted DFT (RDFT) approach to be consistent with the classical mechanics' concept employed in the ReaxFF parameterization procedure when reproducing the minimum and single-point energies. We note that it is essential to use a consistent DFT energy level across fitting and validation data sets to ensure that the newly calibrated ReaxFF properly represents the PES.Reactive force field (ReaxFF) parameterizations
In ReaxFF, the bond energy calculations are derived from a general energy expression like below;| ETOT = Eintra + Einter | (1) |
Eintra: this term represents all intramolecular interactions for bond order and bond energy estimations. It describes bond dissociations and formations based on bond distance, bond angle, and torsion angle-dependent terms. The parameters in these terms are adjusted to fit the minimized DFT energy curves for a predefined set of target molecules. The short-ranged bond order and bond energy result from the directional bonding environment surrounding atoms. The ReaxFF should be parameterized such that it reproduces a local bonding environment's energy and should be able to characterize the energy difference between conformational molecular structures. ReaxFF uses over- and under-coordination terms to penalize unphysical valence configurations that might occur during dynamic simulations in the cases of highly compressed and expanded systems. These terms ensure proper valency and allow a smooth transition from bonded to non-bonded configurations. Such transitions are usually defined by a cut-off tapering function as implemented in Tersoff-like bond order potentials such as REBO and AIREBO.32–34 It is noteworthy that the ReaxFF's short-ranged bond order ideas are inspired by the work of Pauling,35 Johnston,36 and others on chemical reactions.
Einter: this term can be decomposed to ECoulomb and EvdW. The electrostatic interactions between pairs of atoms are based on the charge populations reflected from charge transfer by electronegativity and polarization effects. The ECoulomb term uses a geometry-based charge calculation scheme37 similar to charge equilibration with the feature that Coulomb effects between bonded atoms are shielded to saturate for short distances. One of the most widely used methods for calculating atomic charges in DFT is the Mulliken charge population method. Mulliken charges do not fit the dipole and higher charge moments but do reasonably well in describing the local electronegativity and provide a self-consistent scheme to partition atomic charges. In other words, Mulliken is well suited to fit electronegativity and hardness parameters in the EEM37, similar to QEq38 schemes, employed in ReaxFF. The EvdW term includes all non-bonding interactions, which indicate short-ranged Pauli repulsion and long-ranged dispersion interactions.
We used the general H/C/O parameters31 and H/O parameters39 as initial parameter sets to optimize H/C/O/Cl parameters that are fully transferable with various conformational H/C/O/Cl molecules and aqueous environment described in this study. The optimized parameters for the new ReaxFF implemented in large-scale atomic/molecular massively parallel simulator (LAMMPS) code40 were determined by using continuous single-parameter search trial and error with in-house codes and fitting the minimum energies to the DFT energetics to minimize the root mean squared error (RMSE) expressed as below;
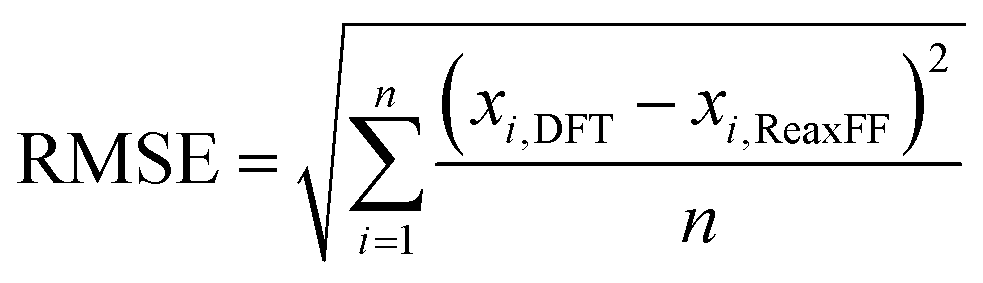 | (2) |
Transition state (TS) optimizations
To understand the local reaction mechanism of a small hydrocarbon oxidized by ClOn−, a transition state search is necessary to find a saddle-point of the potential energy landscape. This is a critical step in defining the minimum reaction path between the reactant and product states. To locate the transition state with one negative eigenvalue, we used the quadratic synchronous transit approach technique to get closer to the quadratic region of the transition state. We also used the eigenvector-following algorithm to complete the search and converge to the saddle point. These transition state optimizations can be readily performed using the Gaussian16 program.19 In this work, we employed ab initio molecular dynamics (MD) to determine a suitable initial geometry for the constrained transition state calculations. For these MD simulations, we used the generalized gradient approximation and Perdew–Burke–Ernzerhof functional (GGA–PBE24), implemented in the Vienna Ab Initio Simulation Package (VASP).41–43 We used the projector-augmented wave (PAW) method with a plane-wave basis set to describe the interaction between the core and valence electrons. We performed single point ReaxFF energy calculations with the DFT-optimized geometries to check whether the ReaxFF's TS energy profile matches that of DFT.Molecular dynamics (MD) simulations
The system configurations for the new ReaxFF MD simulations are composed of the following components: (a) 23 perchlorate (ClO4−) oxidizers as benchmarking the same concentration (0.013 M) of bromate BrO3− oxidizer used in experiments to break down the polymeric structure of organic matter,9 (b) 23 hydronium ions (H3+O) which are added to ensure charge-neutrality in the model for each simulation box, and (c) relaxed water box at ∼1.0 g cm−3 density placed in a periodic box. The MD simulations of the new ReaxFF implemented in the LAMMPS code40 are performed in the NVT ensemble for 100 ps. The equations of motion are integrated via the velocity Verlet algorithm with a time step of 0.25 fs at 450 K using a thermostat with a 100 fs of temperature rescaling constant. We used DFT-optimized geometry for all molecules, including the relaxed aliphatic and aromatic molecules. To initialize MD configurations, we placed the DFT-relaxed molecules randomly in the simulation box.Results and discussion
Potential energy surface (PES) of the new ReaxFF parameterization
Since the new ReaxFF parameterization is to provide a chemical and physical means of describing the oxidative decompositions of the small aromatic and aliphatic hydrocarbons with chlorate oxidizers (ClOn−: n = 1–4), the reliable DFT training and testing data sets should include a wide range of reactive and non-reactive H/C/O/Cl bonding environments. The extensive DFT data set is needed to ensure accurate PES descriptions. The H/C/O/Cl compound data sets used herein are as follows: (a) thirty-five small molecules that have mostly aliphatic-like structures as the PES fitting data set and (b) thirty-five larger molecules that are predominantly aromatic-like structures as the PES validation data set.Fig. 1 shows bond formation energies that are evaluated at energy-minimized geometries for seventy different molecules in the training and validation data sets. We compare the new ReaxFF energies against the DFT energies at B3LYP/6-31G* level of theory. The bond formation energies were calculated by
| Ef = Ecomplex − nHEH − nCEC − nOEO − nClECl | (3) |
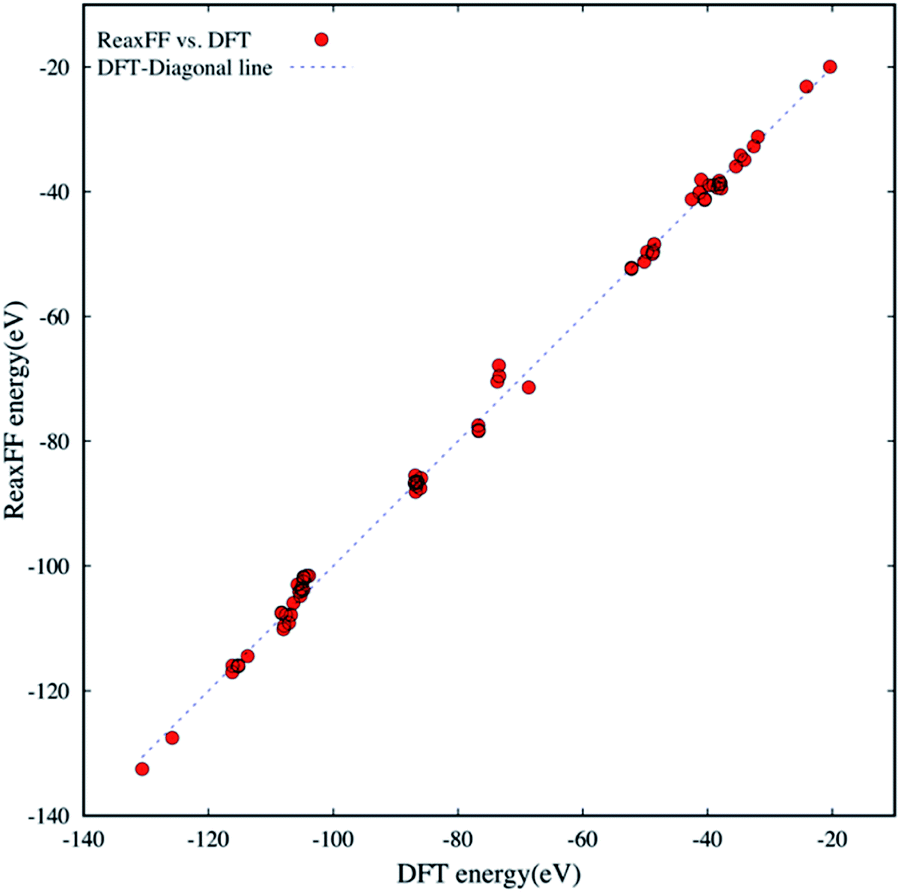 | ||
| Fig. 1 The comparison of DFT and ReaxFF bond formation energies for seventy H/C/O/Cl compounds in the fitting and validation data sets. The dashed diagonal line serves as a guide for eyes. | ||
PES using the new ReaxFF for reactive bonding interactions
As explained earlier for Eintra term, all the directional bonding interactions for empirical bond order estimations in describing the bond environment are based on bond distance, bond angle, and torsion angle. We calibrated the ReaxFF to fit the energy-minimized curves for the target molecular systems when compared to DFT (B3LYP/6-31G*) energies.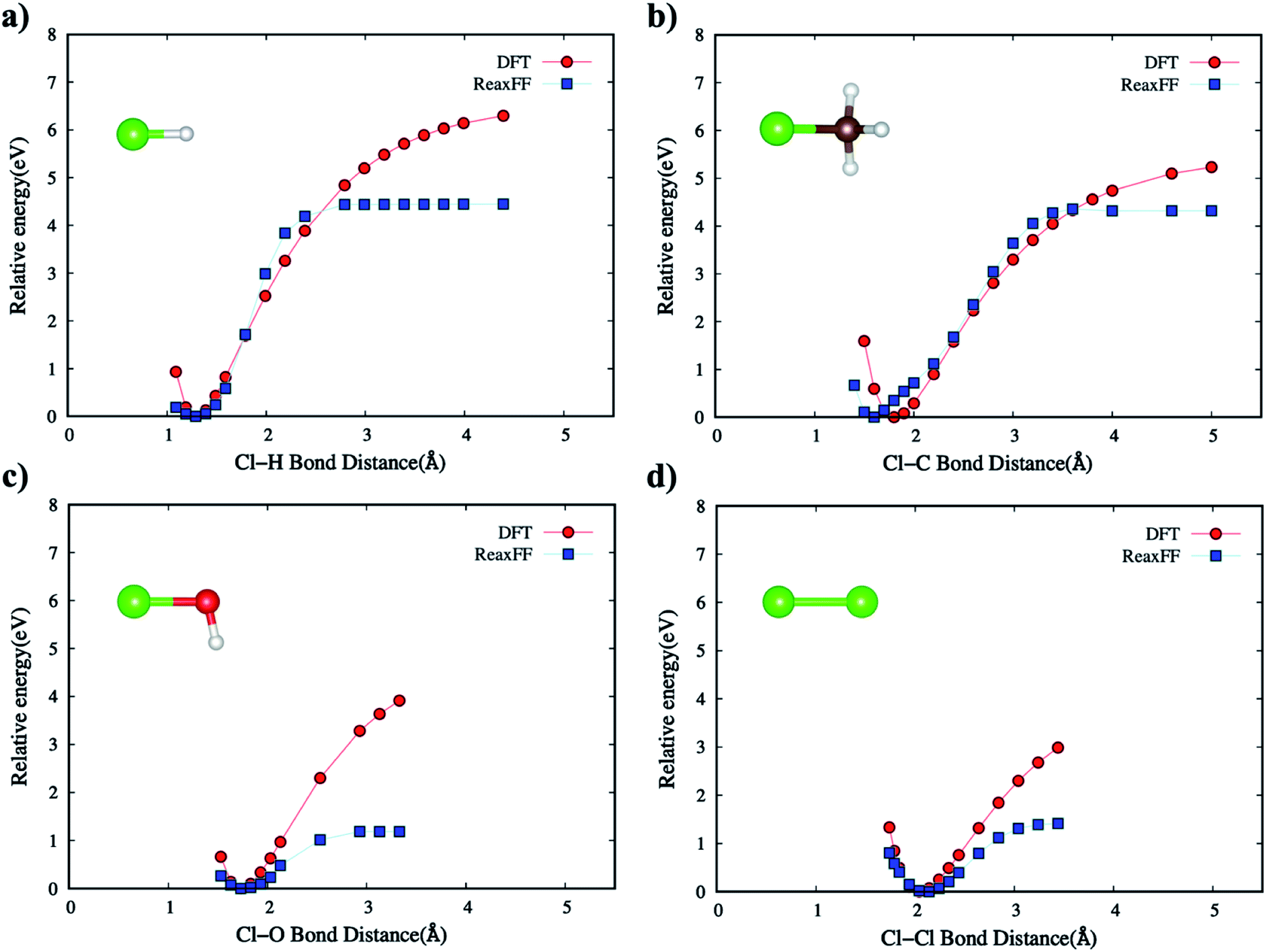 | ||
| Fig. 2 The comparison of DFT and ReaxFF bond dissociation energies by changing interatomic bond distances for (a) Cl–H bond in HCl, (b) Cl–C bond in CH3Cl, (c) Cl–O bond in ClOH, and (d) Cl–Cl bond in Cl2. The brown, red, white, and green-colored balls indicate C, O, H, and Cl atoms, respectively. | ||
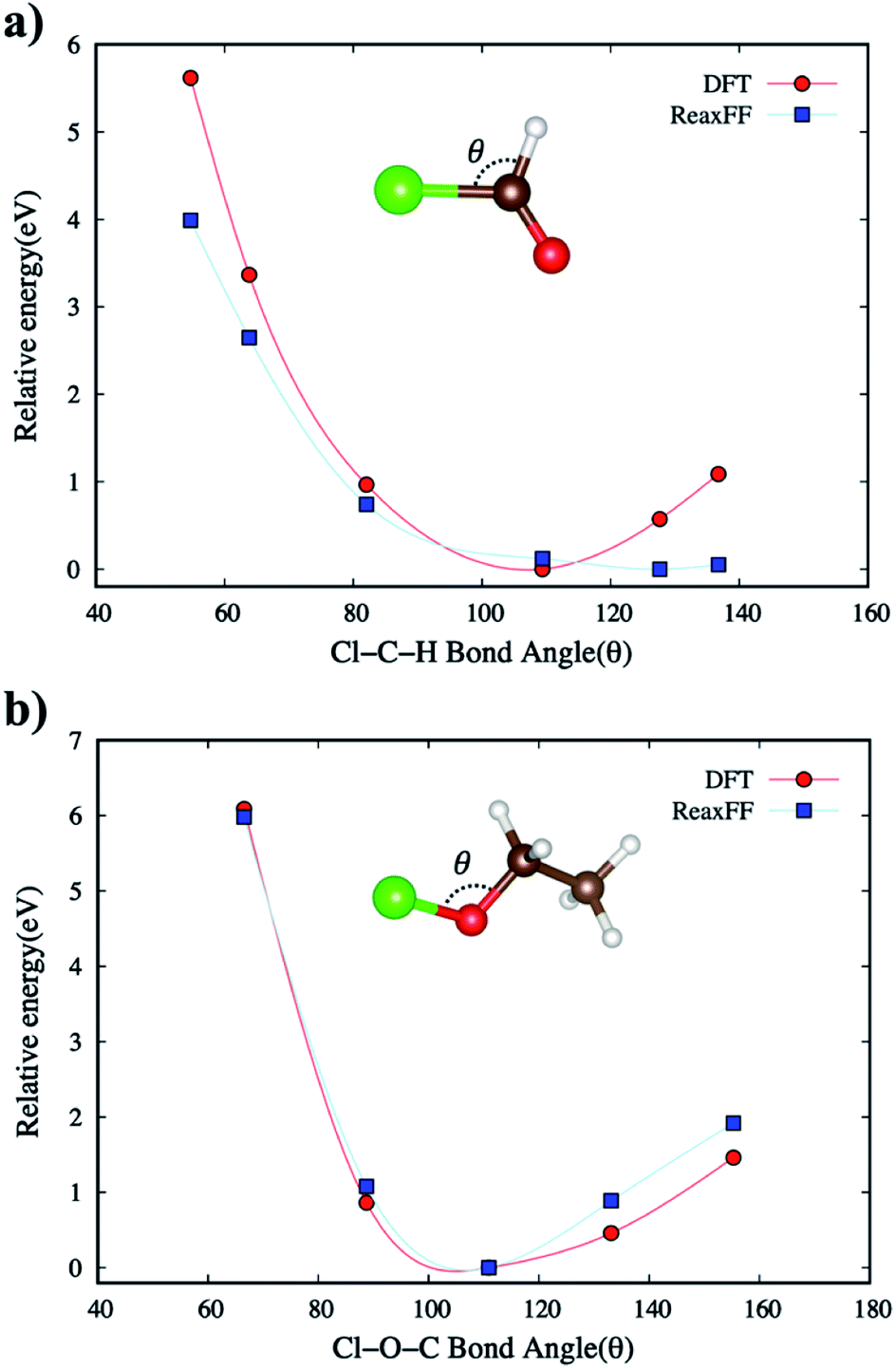 | ||
| Fig. 3 The comparison of relative energies of DFT and ReaxFF for (a) Cl–C–H bond angle in CHClO and (b) Cl–O–C bond angle in C2H5ClO. The brown, red, white, and green-colored balls indicate C, O, H, and Cl atoms, respectively. | ||
PES of the new ReaxFF for non-bonded interactions
We have also parametrized the non-bonded terms in the new ReaxFF force field. This parameterization includes fitting both charge distribution parameters, also known as the EEM method,37 and dispersion interaction coefficients. Following electronegativity rules, the new ReaxFF includes parameters that allow charge distributions, correlated with charge transfers, which are dependent on the local bonding environment of a particular atom.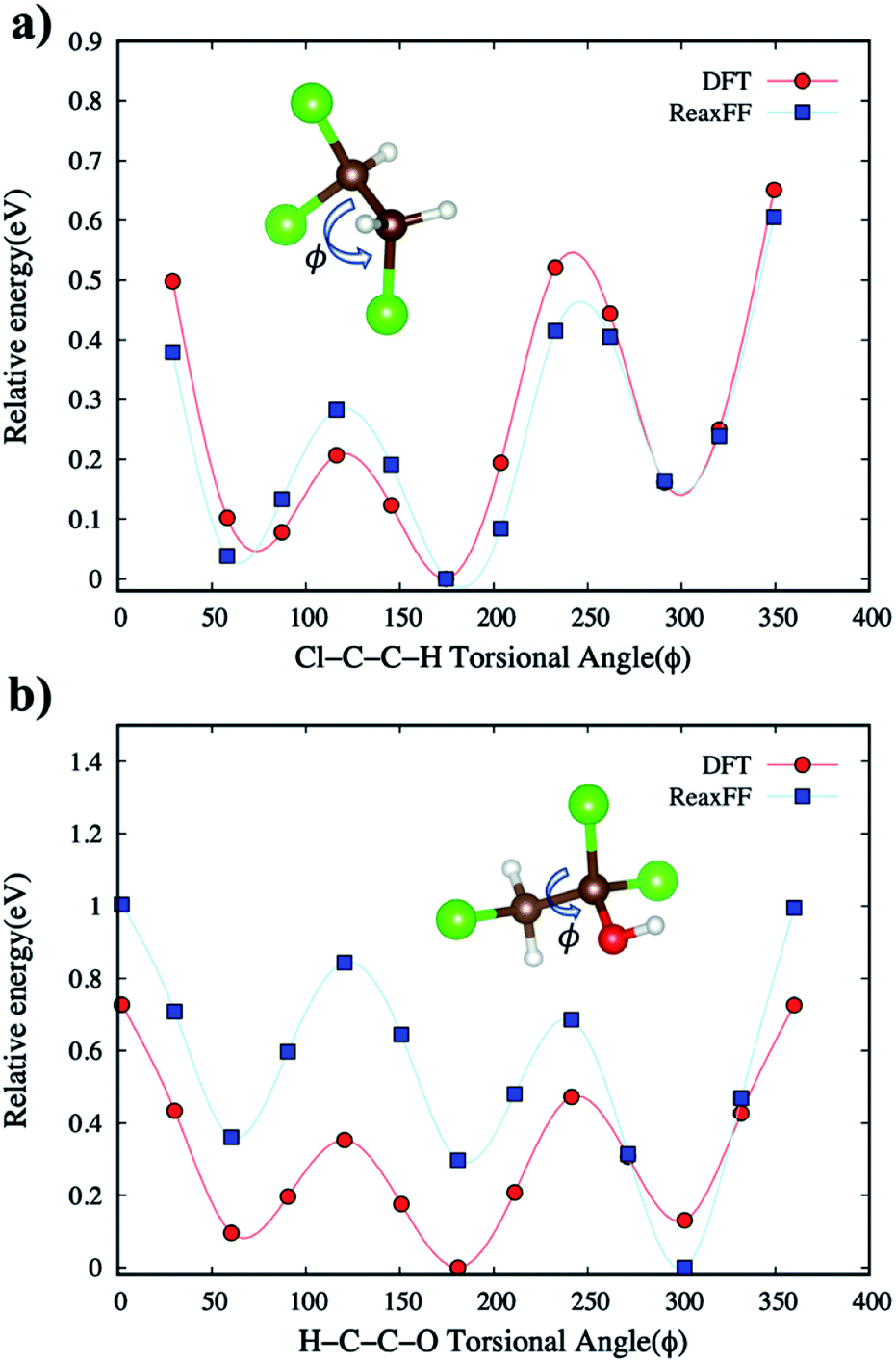 | ||
| Fig. 4 The comparison of relative energies of DFT and ReaxFF for (a) Cl–C–C–H torsional angle in C2H3Cl3 and (b) H–C–C–O torsional angle in C2H3Cl3O. The brown, red, white, and green-colored balls indicate C, O, H, and Cl atoms, respectively. | ||
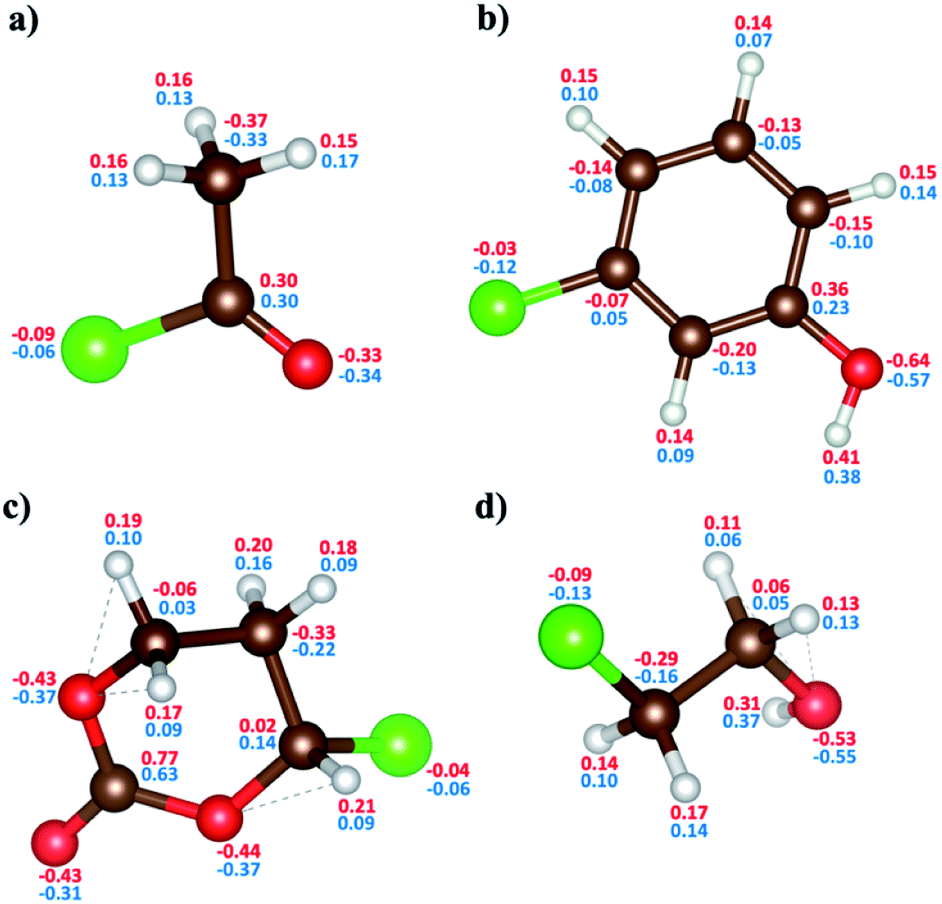 | ||
| Fig. 5 The comparison of charge distributions of DFT and ReaxFF in (a) C2H3ClO, (b) C6H5ClO, (c) C4H5ClO3, and (d) C2H5ClO. The red-colored values are for DFT-calculated Mulliken charges and the blue-colored values are for the ReaxFF-calculated EEM charges based on electronegativity. The brown, red, white, and green-colored balls indicate C, O, H, and Cl atoms, respectively. | ||
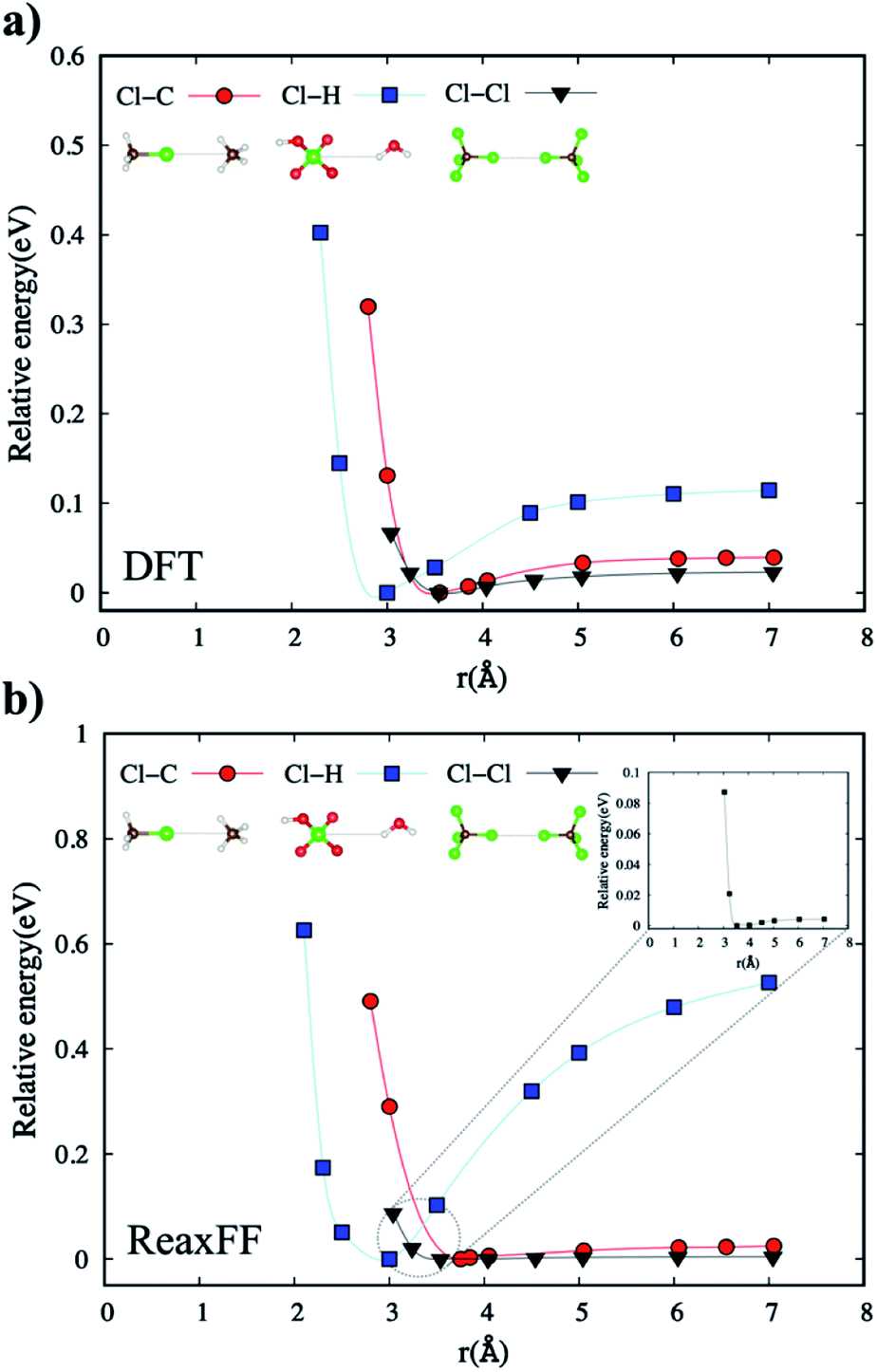 | ||
| Fig. 6 The comparison of (a) DFT and (b) the new ReaxFF non-bonded energies for Cl–C in between CH3Cl and CH4, Cl–H in between H2O and H-terminated ClO4− (perchloric acid), and Cl–Cl in between two CCl4 molecules. The inset in part (b) shows the non-bonded interactions of Cl–Cl atom pair in CCl4–CCl4. | ||
The non-bonded energies of the Cl–Cl and Cl–H calculated by the DFT and new ReaxFF are consistent with regard to the position of energy minimum (equilibrium distance) and the energy curve pattern. The equilibrium Cl–C distance predicted by the new ReaxFF is slightly longer (∼0.2 Å) than that calculated by DFT. However, the non-bonded energy pattern for the Cl–Cl and Cl–H cases remain consistent between the new ReaxFF potential and DFT calculations. We also note that the depth of the non-bonded attractive energy well is less than 0.55 eV, especially for the Cl–H pair interaction, Fig. 6b. The majority of this attractive interaction might arise from coulombic interactions, as shown in the inset of Fig. S5b provided in ESI.† The aforementioned discrepancy in the magnitude of potential well depth might be due in part to the previous H/C/O parameterizations with regard to hydrocarbon oxidation31 on which we based our H/C/O/Cl parameter set. Having shown such slight differences between DFT and the new ReaxFF results, we note that the attractive energy well's depth is still smaller than the 1.57 eV threshold satisfied in the parametrization of the new ReaxFF to formation energy of the H/C/O/Cl compound data set.
Transition state (TS) simulations of hydrocarbon oxidation
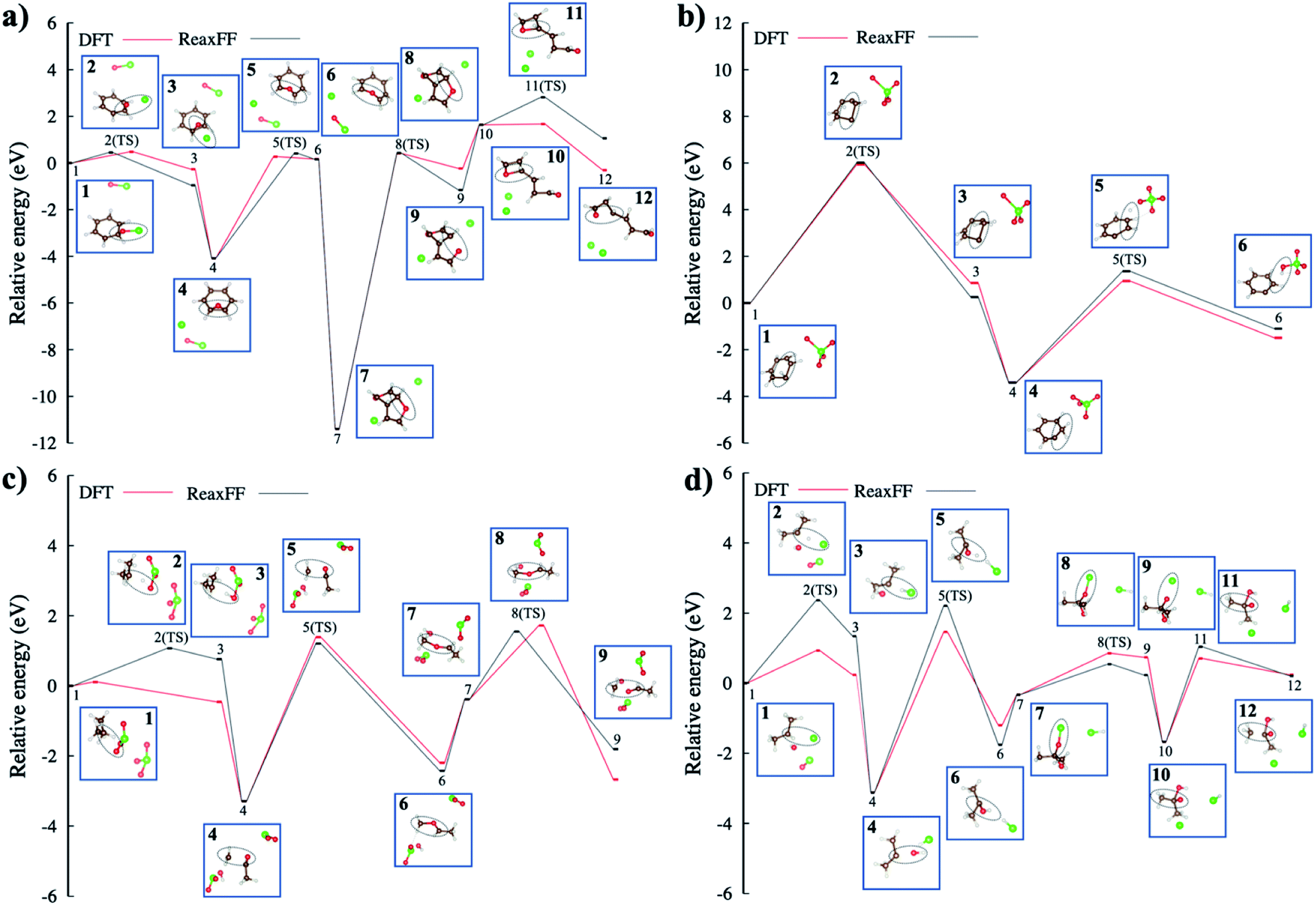 | ||
| Fig. 7 The proposed reaction pathways and potential energy surfaces in the oxidative decompositions of small aromatic and aliphatic hydrocarbons with oxychlorine oxidizers (ClOn−; 1 ≤ n ≤ 4), as compared between the DFT and ReaxFF energetics; (a) oxygen transfer to the benzene forms a seven-membered benzene oxide ring and C–O bond breakings with the addition of ClO− produces a linear molecules, (b) proton transfers in the internal benzene and to the oxygen of ClO4−, (c) proton transfer to the oxygen of ClO3−, bond transition from a C–C bond of the oxygen-combined isopropyl backbone to a C–O bond with the ˙CH3 radical, and the C–O bond breaking to take apart the ˙CH3 radical that produce an acetaldehyde, (d) proton transfer to ˙Cl radical to produce HCl and oxygen transfer with the addition of ClO− that generates acetic acid. The brown, red, white, and green-colored balls indicate C, O, H, and Cl atoms, respectively. | ||
Fig. 7b shows a proton transfer within the benzene itself and then, its transfer to the oxygen of ClO4−. This reaction coordinate shows how the presence of ClO4− affects the structural deformation of the benzene, induced by strong vibrations. The proton transfer energy barrier is shown to be ∼6.0 eV, which is well correlated to DFT energy. In the meantime, the energy barrier of the other proton transfer to the oxygen of ClO4− molecule is ∼2.5 eV indicating that the unsaturated benzene configuration with a carbon radical requires less energy to be endothermic for the proton hopping. The proposed proton transfer reaction steps are detailed in Scheme 1 presented in ESI.†
In Fig. 7d, the radical–radical combination, i.e., the combination of ˙H and ˙Cl radicals to form hydrochloric acid, gives rise to an energy barrier of ∼2.36 eV. We also note that Cl's strong electronegativity (χCl = 3.16) may play a role in the proton transfer process. In the meantime, hydroxyl radical approaches the unsaturated isopropyl configuration to form a 2-propanol radical. Finally, an additional ClO− oxygenates the 2-propanol radical site, with an energy barrier of ∼0.87 eV, forming a carboxyl functional group. At the end, the released ˙CH3 radical yields acetic acid in this reaction profile.
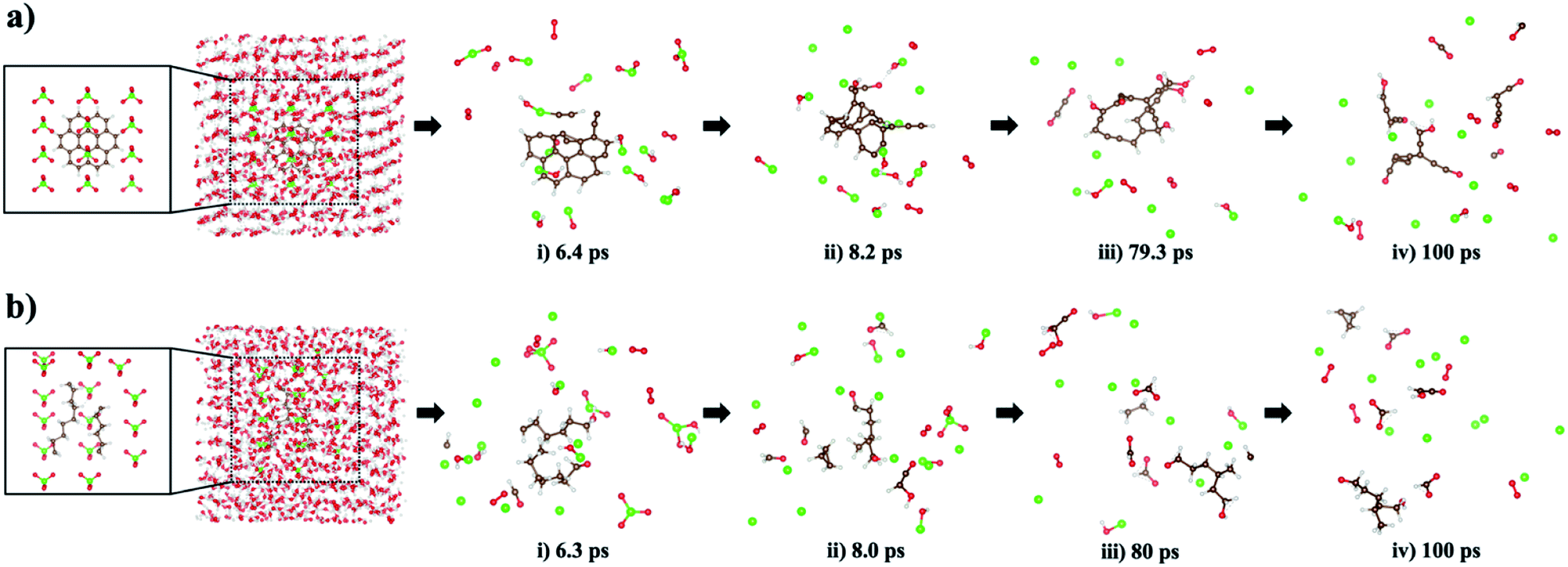 | ||
| Fig. 8 Trajectory snapshots of the new ReaxFF-MD simulations: (a) coronene (C24H12) as an aromatic hydrocarbon and (b) two relaxed polyethylenes (–(CH2–CH2)n–, one for n = 9 and the other for n = 6) as aliphatic hydrocarbons with ClO4− oxidizers and H3+O ions dissolved in water. The brown, red, white, and green-colored balls indicate C, O, H, and Cl atoms, respectively. In time (i)–(iv) snapshots, water molecules are not displayed to clearly show the oxidative fragmentation process. | ||
Molecular dynamics (MD) simulations of oxidative decomposition
Using this newly parameterized ReaxFF, we carried out MD simulations to observe the mechanistic picture of oxidative decomposition of small hydrocarbon molecules at 450 K using ClO4− oxidizers, shown in Fig. 8. For these MD simulations, we selected coronene (C24H12) as an aromatic hydrocarbon, and two relaxed polyethylenes (–(CH2–CH2)n–, where n = 9 and n = 6) as aliphatic hydrocarbons. During MD simulations, the decompositions of ClO4−, which were experimentally confirmed,15 produce O2 and ˙Cl radical, which are the main source for generating the H/C/O/Cl compounds, whenever H and C atoms detach from the deformed hydrocarbon structures. As seen in the stage (i) snapshot taken from the MD trajectory at 6.4 ps, the coronene structure degrades to produce a small H/C/O/Cl compound and O2/ClOH/ClOn− (n: 1–3). In stage (ii) at 8.2 ps, all but one of the remaining aromatic rings in the parent structure are broken to form a hydrocarbon chain terminated with a hydrophilic H/C/O. Complete degradation of the fused ring matrix takes place at stage (iii) of the 79.3 ps snapshot while a small H/C/O molecule is separated. A new H/C/O/Cl compound and other H/C/O species appear at 100 ps stage (iv) verifying the irreversible nature of the ring breaking.Oxidative degradation of the two aliphatic polyethylenes occurs in stage (i) at 6.3 ps where small molecules containing H/C/O elements are formed. In stage (ii), the curved –(CH2–CH2)n– domain found in the snapshot at 6.3 ps predominantly loses its chain characteristics while gaining a terminal H/O. Also, more C1 and H atoms released from the curved –(CH2–CH2)n– domain combine with H/O/Cl elements to form H/C/O and H/C/O/Cl compounds. In stage (iii) and (iv) snapshots, short hydrocarbon fragments (i.e., HnCn: n = 1–3) and small H/C/O compounds are observed. The time period of 100 ps is enough to see a variety of H/C/O/Cl compounds derived from aliphatic hydrocarbon fragmentation, though no chlorocarbons are observed at the end of the simulations.
Conclusions
In this paper, we developed a new ReaxFF parameter set to model the oxidative degradation of small aromatic and aliphatic hydrocarbons in response to oxychlorine oxidizers (ClOn−: n = 1–4). The potential energy surface in the calibrated ReaxFF correlates well with that of DFT (R2 = 0.997). The new H/C/O/Cl parameterization shows the RMS error of only ∼1.57 eV, corresponding to the average percent error of 1.70%. The energy patterns estimated by the new ReaxFF for covalent bonding interactions provide a good description of the DFT energies with regards to the bond distance, bond angle, and torsional angle changes. Furthermore, the new ReaxFF adequately reproduces the non-bonding interactions such as coulombic, and intermolecular dispersions as well as H-bonding interactions. Through ab initio MD simulations, we benchmarked thermodynamically-favorable oxidation paths for small hydrocarbon molecules by ClOn−. We showed that the ReaxFF potential could quantitatively reproduce the energetics of proposed reaction paths. We subsequently performed MD simulations to study the effect of perchlorate on the degradation of small aromatic and aliphatic hydrocarbons. The MD trajectories describe the unfolding of hydrocarbon fragmentation, delineating how H/C/O/Cl species are formed and broken during complex oxidative processes.This reactive force field serves only as a proof of concept that the properly parameterized ReaxFF potentials can be employed to obtain the atomic-level understanding of the oxidative degradation processes at the kerogen-oxidizing fluid interface. We have already geared our parameterization efforts toward developing Na/H/C/O/Cl and Na/H/C/O/Br parameter sets to resolve the oxidative reaction pathways responsible for kerogen fragmentation and degradation reported in benchtop experiments.9 We envision that such transferable reactive force fields will pave the path for designing the next generation of reactive oxidizing fluids for improving the efficiency of stimulation operations in unconventional reservoirs and pretreatment of geological reservoirs for enhanced carbon dioxide sequestration.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors gratefully acknowledge computational support by the UCI HPC staff.References
- J. Lehmann and M. Kleber, Nature, 2015, 528, 60–68 CrossRef CAS PubMed.
- M. Vandenbroucke and C. Largeau, Org. Geochem., 2007, 38, 719–833 CrossRef CAS.
- H. Sanei, Sci. Rep., 2020, 10, 15595 CrossRef CAS PubMed.
- F.-J. Ulm and Y. Abousleiman, Acta Geotechnica, 2006, 2, 77–88 CrossRef.
- K. L. Hull, Y. N. Abousleiman, Y. Han, G. A. Al-Muntasheri, P. Hosemann, S. S. Parker and C. B. Howard, SPE J., 2017, 22, 1024–1033 CrossRef.
- Y. N. Abousleiman, K. L. Hull, Y. Han, G. Al-Muntasheri, P. Hosemann, S. Parker and C. B. Howard, Acta Geotechnica, 2016, 11, 573–594 CrossRef.
- W. E. Robinson, H. H. Heady and A. B. Hubbard, Ind. Eng. Chem., 1953, 45, 788–791 CrossRef CAS.
- B. J. Katz and I. Arango, Org. Geochem., 2018, 123, 1–16 CrossRef CAS.
- K. L. Hull, D. Jacobi and Y. N. Abousleiman, Energy Fuels, 2019, 33, 4758–4766 CrossRef CAS.
- K. Mitra and J. G. Catalano, J. Geophys. Res.: Planets, 2019, 124, 2893–2916 CAS.
- M. Zheng, X. Li, J. Liu, Z. Wang, X. Gong, L. Guo and W. Song, Energy Fuels, 2014, 28, 522–534 CrossRef CAS.
- K. Nomura, R. K. Kalia, A. Nakano and P. Vashishta, Comput. Phys. Commun., 2008, 178, 73–87 CrossRef CAS.
- A. C. T. van Duin, S. Dasgupta, F. Lorant and W. A. Goddard, J. Phys. Chem. A, 2001, 105, 9396–9409 CrossRef CAS.
- T. P. Senftle, S. Hong, M. M. Islam, S. B. Kylasa, Y. Zheng, Y. K. Shin, C. Junkermeier, R. Engel-Herbert, M. J. Janik, H. M. Aktulga, T. Verstraelen, A. Grama and A. C. T. van Duin, npj Comput. Mater., 2016, 2, 15011 CrossRef CAS.
- S. Masoumi, S. Zare, H. Valipour and M. J. Abdolhosseini Qomi, J. Phys. Chem. C, 2019, 123, 4755–4766 CrossRef CAS.
- M. J. A. Qomi, M. Bauchy, F.-J. Ulm and R. J.-M. Pellenq, J. Chem. Phys., 2014, 140, 054515 CrossRef PubMed.
- A. D. Becke, J. Chem. Phys., 1993, 98, 5648–5652 CrossRef CAS.
- C. Lee, W. Yang and R. G. Parr, Phys. Rev. B: Condens. Matter Mater. Phys., 1988, 37, 785–789 CrossRef CAS PubMed.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, X. Li, M. Caricato, A. V. Marenich, J. Bloino, B. G. Janesko, R. Gomperts, B. Mennucci, H. P. Hratchian, J. V. Ortiz, A. F. Izmaylov, J. L. Sonnenberg, D. Williams-Young, F. Ding, F. Lipparini, F. Egidi, J. Goings, B. Peng, A. Petrone, T. Henderson, D. Ranasinghe, V. G. Zakrzewski, J. Gao, N. Rega, G. Zheng, W. Liang, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell, J. A. Montgomery Jr, J. E. Peralta, F. Ogliaro, M. J. Bearpark, J. J. Heyd, E. N. Brothers, K. N. Kudin, V. N. Staroverov, T. A. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. P. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, J. M. Millam, M. Klene, C. Adamo, R. Cammi, J. W. Ochterski, R. L. Martin, K. Morokuma, O. Farkas, J. B. Foresman and D. J. Fox, Gaussian 16 Rev. C.01, Wallingford, CT, 2016 Search PubMed.
- W. J. Hehre, R. Ditchfield and J. A. Pople, J. Chem. Phys., 1972, 56, 2257–2261 CrossRef CAS.
- P. C. Hariharan and J. A. Pople, Chem. Phys. Lett., 1972, 16, 217 CrossRef CAS.
- M. M. Francl, W. J. Pietro, W. J. Hehre, J. S. Binkley, M. S. Gordon, D. J. DeFrees and J. A. Pople, J. Chem. Phys., 1982, 77, 3654–3665 CrossRef CAS.
- S. Grimme, J. Antony, S. Ehrlich and H. Krieg, J. Chem. Phys., 2010, 132, 154104 CrossRef PubMed.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865–3868 CrossRef CAS PubMed.
- M. L. Coote, J. Phys. Chem. A, 2004, 108, 3865–3872 CrossRef CAS.
- D. Chakraborty, R. P. Muller, S. Dasgupta and W. A. Goddard, J. Phys. Chem. A, 2000, 104, 2261–2272 CrossRef CAS.
- K. N. Rogstad, Y. H. Jang, L. C. Sowers and W. A. Goddard, Chem. Res. Toxicol., 2003, 16, 1455–1462 Search PubMed.
- X. Xu, R. P. Muller and W. A. Goddard, Proc. Natl. Acad. Sci. U. S. A., 2002, 99, 3376–3381 CrossRef CAS PubMed.
- M. T. Feldmann, S. L. Widicus, G. A. Blake, D. R. Kent and W. A. Goddard, J. Chem. Phys., 2005, 123, 034034 CrossRef PubMed.
- M. S. Rudner, D. R. Kent, W. A. Goddard and J. D. Roberts, J. Phys. Chem. A, 2005, 109, 9083–9088 CrossRef CAS PubMed.
- K. Chenoweth, A. C. T. van Duin and W. A. Goddard, J. Phys. Chem. A, 2008, 112, 1040–1053 CrossRef CAS PubMed.
- J. Tersoff, Phys. Rev. B: Condens. Matter Mater. Phys., 1988, 37, 6991–7000 CrossRef PubMed.
- D. W. Brenner, O. A. Shenderova, J. A. Harrison, S. J. Stuart, B. Ni and S. B. Sinnott, J. Phys.: Condens. Matter, 2002, 14, 783–802 CrossRef CAS.
- S. J. Stuart, A. B. Tutein and J. A. Harrison, J. Chem. Phys., 2000, 112, 6472–6486 CrossRef CAS.
- L. Pauling, The nature of the chemical bond, Cornell University Press, Ithaca, NY, 3rd edn, 1960 Search PubMed.
- H. S. Johnston and C. Parr, J. Am. Chem. Soc., 1963, 85, 2544–2551 CrossRef CAS.
- W. J. Mortier, S. K. Ghosh and S. Shankar, J. Am. Chem. Soc., 1986, 108, 4315–4320 CrossRef CAS.
- A. K. Rappe and W. A. Goddard, J. Phys. Chem., 1991, 95, 3358–3363 CrossRef CAS.
- W. Zhang and A. C. T. van Duin, J. Phys. Chem. B, 2017, 121, 6021–6032 CrossRef CAS PubMed.
- S. Plimpton, J. Comput. Phys., 1995, 117, 1–19 CrossRef CAS http://lammps.sandia.gov.
- G. Kresse and J. Hafner, Phys. Rev. B: Condens. Matter Mater. Phys., 1993, 47, 558–561 CrossRef CAS PubMed.
- G. Kresse and J. Furthmüller, Comput. Mater. Sci., 1996, 6, 15–50 CrossRef CAS.
- G. Kresse and J. Furthmüller, Phys. Rev. B: Condens. Matter Mater. Phys., 1996, 54, 11169–11186 CrossRef CAS PubMed.
- L. Latriano, B. D. Goldstein and G. Witz, Proc. Natl. Acad. Sci. U. S. A., 1986, 83, 8356–8360 CrossRef CAS PubMed.
- A. Karich, M. Kluge, R. Ullrich and M. Hofrichter, AMB Express, 2013, 3, 5 CrossRef PubMed.
- M.-J. Cheng, K. Chenoweth, J. Oxgaard, A. van Duin and W. A. Goddard, J. Phys. Chem. C, 2007, 111, 5115–5127 CrossRef CAS.
- M. D. Argyle, K. Chen, A. T. Bell and E. Iglesia, J. Phys. Chem. B, 2002, 106, 5421–5427 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Bond formation energies, PES data for reactive and non-reactive bonding, atomic charges, TS schemes, the new ReaxFF parameters, and parameterization flow chart. See DOI: 10.1039/d1ra04397h |
| This journal is © The Royal Society of Chemistry 2021 |