
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMicrowave-assisted green construction of imidazole-fused hybrid scaffolds using 2-aminobenzimidazoles as building blocks†
Jung Pyo Kwaka,
Pham Duy Quang Dao a,
Nam Sik Yoonb and
Chan Sik Cho*a
a,
Nam Sik Yoonb and
Chan Sik Cho*a
aDepartment of Applied Chemistry, Kyungpook National University, 80 Daehakro, Bukgu, Daegu 41566, Republic of Korea. E-mail: cscho@knu.ac.kr
bDepartment of Textile System Engineering, Kyungpook National University, 80 Daehakro, Bukgu, Daegu 41566, Republic of Korea
First published on 17th June 2021
Abstract
A class of trinuclear imidazole-fused hybrid scaffolds was constructed by the reaction of 2-(2-bromoaryl)- and 2-(2-bromovinyl)imidazoles with 2-aminobenzimidazoles as building blocks in the presence of a base under microwave irradiation. A nucleophilic aromatic substitution followed by cyclization is proposed as a reaction pathway of this green process.
Introduction
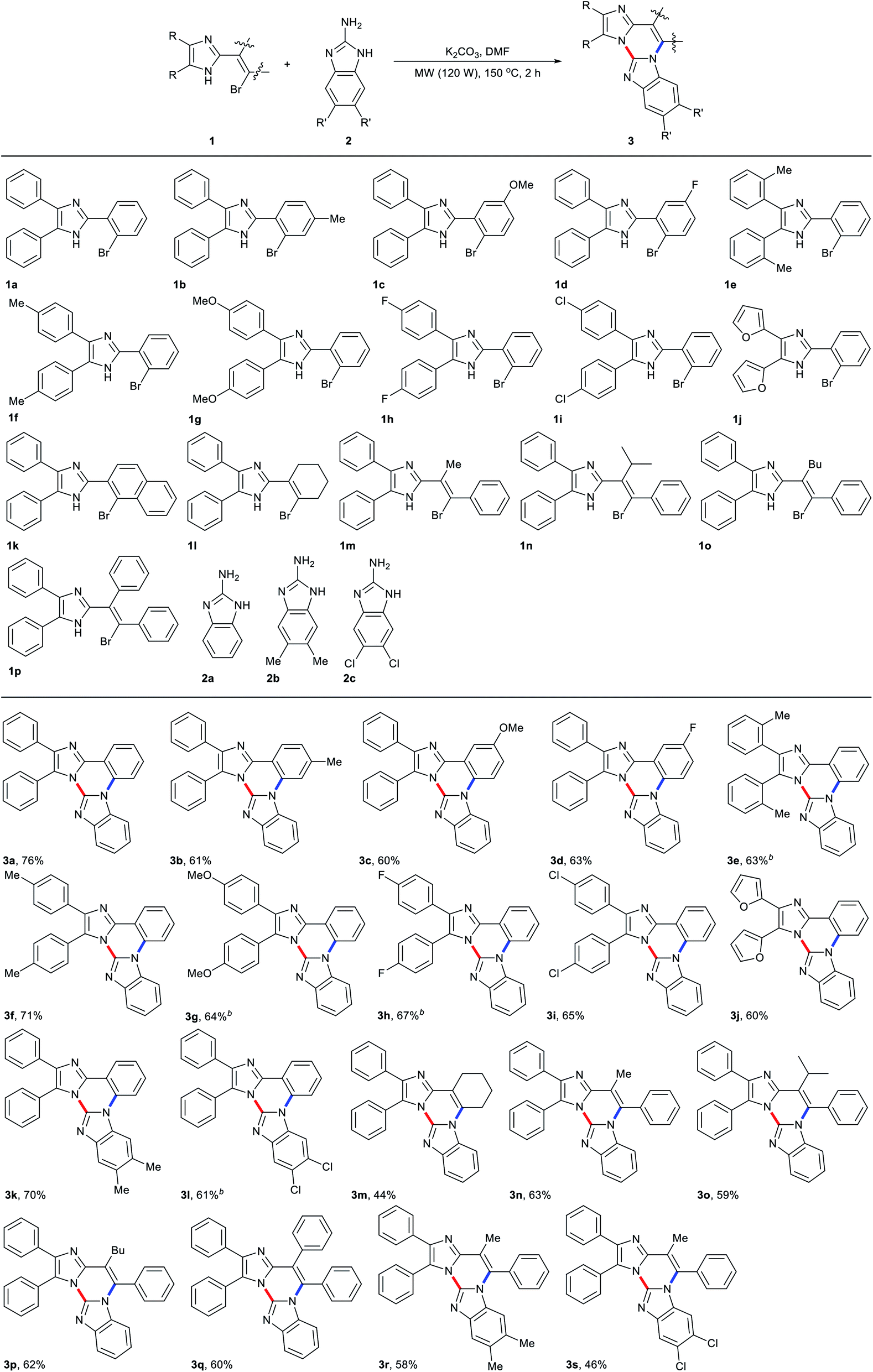
Diverse synthetic methods for the construction of N-fused hybrid scaffolds have been developed due to their characteristic properties that are not shown in each homonuclear heterocycle.1 In connection with this report, it is known that trinuclear N-fused hybrid structure A, benzo[4,5]imidazo[1,2-a]imidazo[1,2-c]quinazoline, exhibits biological activities and photoelectronic properties (Scheme 1).2,3 However, in contrast to well-known synthetic methods of each homonuclear quinazoline, imidazole and benzimidazole, several synthetic methods are reported for their trinuclear N-fused hybrid structures with a limited substrate scope.4 It is reported that bis(o-haloaryl)carbodiimides react with imidazole in the presence of CuI and 1,10-phenanthroline as a ligand along with a base to afford such a trinuclear compound via several sequential processes such as nucleophilic addition, tautomerization, intramolecular Ullmann-type C–N coupling, and intramolecular sp2 C–H arylation (Scheme 2, route a).5 Kumar and co-workers demonstrated that 2-(2-bromophenyl)imidazoles are C–N coupled and oxidatively cyclized with benzimidazole by subsequent treatment of CuI and Pd(OAc)2/Cu(OAc)2 (or Cu(OAc)2) to give such a trinuclear compound (Scheme 2, route b).2,6 Such a similar coupling and cyclization for the construction of such a trinuclear compound was also exemplified by multistep processes starting from 1-benzyl-4,5-diphenyl-1H-imidazole/2-chlorobenzimidazole and 2-fluorobenzonitrile/benzimidazole, respectively (Scheme 2, routes c and d).3 However, these precedents still have some drawbacks such as limitation of substrate scope, the requirement of transition metal catalysts and contamination of residual metals in products, and tedious multistep processes. As part of our continuing studies directed toward transition metal-catalyzed and transition metal-free synthesis of polynuclear N-fused hybrid scaffolds,7 we recently reported that compounds B–D can be formed by double nucleophilic aromatic substitution of 2-(2-bromoaryl)- and 2-(2-bromovinyl)benzimidazoles with 2-aminoazoles in the presence of a base (Scheme 1).8,9 This work started during the course of the extension of such a transition metal-free double nucleophilic aromatic substitution to synthesize polynuclear N-fused hybrid scaffolds. This report shows an example of the green construction of trinuclear N-fused hybrid structure A by transition metal-free double C(sp2)–N coupling between 2-(2-bromoaryl)-4,5-diaryl-1H-imidazoles and 2-aminobenzimidazoles under microwave irradiation (Scheme 2, route e).10,11 The present protocol can also be extended to the reaction of 2-(2-bromovinyl)imidazoles with 2-aminobenzimidazole to produce trinuclear N-fused hybrid structure E (Scheme 2, route f).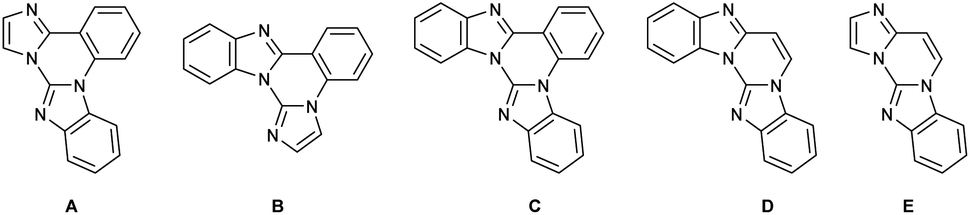 | ||
| Scheme 1 Trinuclear N-fused hybrid scaffolds. | ||
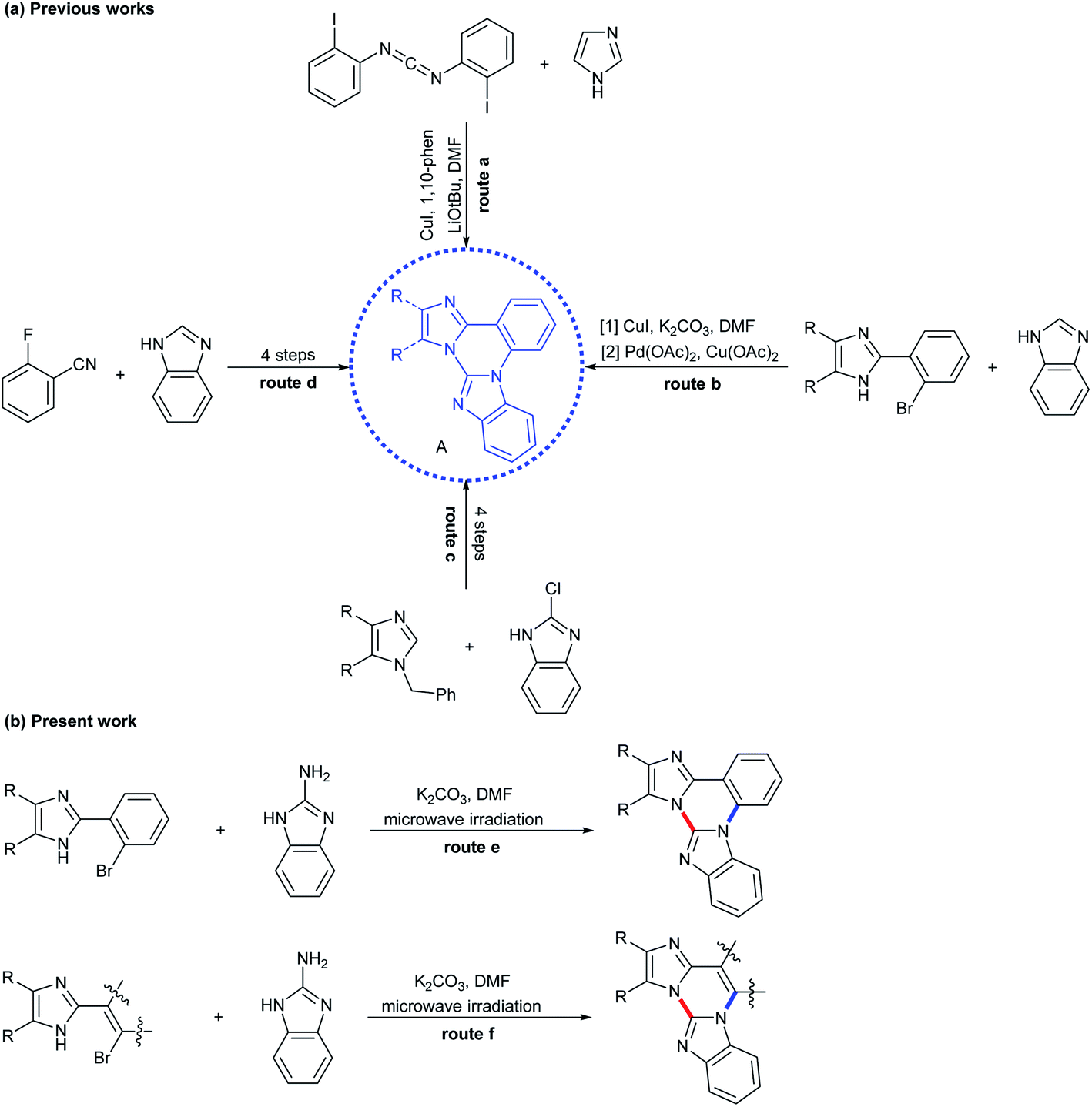 | ||
| Scheme 2 Synthetic routes for trinuclear N-fused hybrid scaffolds. | ||
Results and discussion
Based on our recent report on transition metal-free coupling and cyclization of 2-(2-bromoaryl)- and 2-(2-bromovinyl)benzimidazoles with 2-aminoazoles leading to trinuclear N-fused hybrid scaffolds, we optimized the reaction conditions with 2-(2-bromophenyl)-4,5-diphenyl-1H-imidazole (1a) and 2-aminobenzimidazole (2a) as model substrates to produce trinuclear N-fused hybrid compound 3a effectively (Table 1).8 Treatment of 1a with 1.5 equiv. of 2a in DMF in the presence of K3PO4 (1 equiv.) at 150 °C for 2 h under microwave irradiation (120 W of initial power) produced 3a in 39% isolated yield with complete conversion of 1a (Table 1, entry 1). Although it is still obscure why such an excess amount of base is needed for the effective formation of 3a, the yield of 3a gradually increased with an increase of the molar ratio of K3PO4 to 1a up to 3 (Table 1, entries 2–4).9a The yield of 3a also increases on prolonging the reaction time up to 2 h and a lower yield of 3a was observed with a short reaction time nevertheless complete conversion of 1a (Table 1, entries 3, 5, and 6). Lower yield of 3a was observed with incomplete conversion of 1a under lower reaction temperature, and the yield increased with the increase in temperature up to 150 °C (Table 1, entries 3, 7, and 8). Among solvents examined in combination with K3PO4 under the employed conditions, DMF was found to be of choice in terms of both the yield of 3a and complete conversion of 1a (Table 1, entries 3, 9, and 10). No significant change of the yield of 3a was observed with higher molar ratio of [2a]/[1a] (Table 1, entry 11). The reaction also proceeded with other bases such as KOtBu, K2CO3, and Cs2CO3 with complete conversion of 1a and similar activities to that of K3PO4 under the employed conditions, and K2CO3 was shown to be the base of choice (Table 1, entries 3, 12–14). However, as is the case for our recent report on the reaction of 2-(2-bromoaryl)- and 2-(2-bromovinyl)benzimidazoles with 2-aminoazoles, 3a was not produced at all in the absence of base and no other spots were observed except for both substrates on TLC (Table 1, entry 15).8
|
|||||||
|---|---|---|---|---|---|---|---|
| Entry | [Base]/[1a] | Base | Solvent | Temp. (°C) | Time (h) | Conv. of 1a (%) | Yieldb (%) |
| a Reaction conditions: 1a (0.3 mmol), 2a (0.45 mmol), base, DMF (3 mL), under microwave irradiation (120 W of initial power).b Isolated yield.c [2a]/[1a] = 2.d Usual heating (5 mL screw-capped vial). | |||||||
| 1 | 1 | K3PO4 | DMF | 150 | 2 | 100 | 39 |
| 2 | 2 | K3PO4 | DMF | 150 | 2 | 100 | 54 |
| 3 | 3 | K3PO4 | DMF | 150 | 2 | 100 | 65 |
| 4 | 4 | K3PO4 | DMF | 150 | 2 | 100 | 66 |
| 5 | 3 | K3PO4 | DMF | 150 | 3 | 100 | 66 |
| 6 | 3 | K3PO4 | DMF | 150 | 1 | 100 | 29 |
| 7 | 3 | K3PO4 | DMF | 100 | 2 | 66 | 34 |
| 8 | 3 | K3PO4 | DMF | 125 | 2 | 85 | 41 |
| 9 | 3 | K3PO4 | HMPA | 150 | 2 | 100 | 47 |
| 10 | 3 | K3PO4 | DMSO | 150 | 2 | 97 | 0 |
| 11c | 3 | K3PO4 | DMF | 150 | 2 | 100 | 66 |
| 12 | 3 | KOtBu | DMF | 150 | 2 | 100 | 58 |
| 13 | 3 | K2CO3 | DMF | 150 | 2 | 100 | 76 |
| 14 | 3 | Cs2CO3 | DMF | 150 | 2 | 100 | 68 |
| 15 | 3 | — | DMF | 150 | 2 | 0 | 0 |
| 16d | 3 | K2CO3 | DMF | 150 | 20 | 100 | 33 |
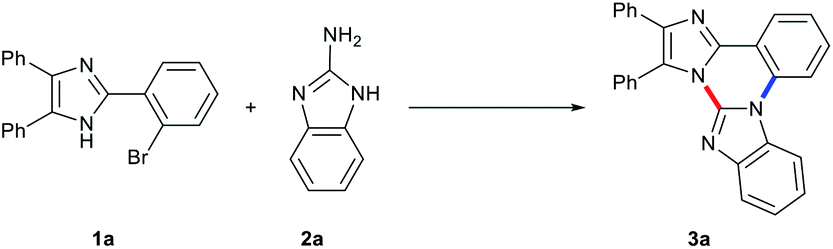
Similar treatment of 1a with 2a under usual heating conditions screw-capped vial, 150 °C for 20 h resulted in only 33% yield of 3a along with several unidentifiable side products with complete conversion of 1a (Table 1, entry 16).
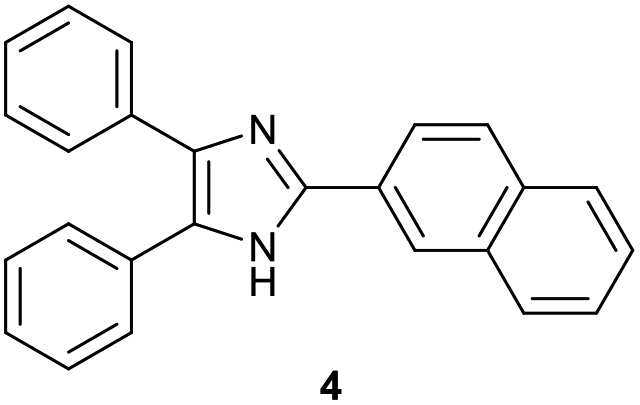
After the reaction conditions had been optimized, various 2-(2-bromoaryl)- and 2-(2-bromovinyl)imidazoles 1 were subjected to the reaction with 2-aminobenzimidazoles 2, and the representative are shown in Table 2.12–14 2-(2-Bromoaryl)imidazoles 1b–d having electron-donating and withdrawing substituents on the bromoaryl moiety were also coupled and cyclized with 2a to give the corresponding trinuclear N-fused hybrid scaffolds 3b–d in 60–63% yields, irrespective of the electronic nature of such substituents. With 2-(2-bromophenyl)-4,5-diaryl-1H-imidazoles 1e–i having electron-donating and withdrawing substituents on the phenyl moiety attached to imidazole ring, the corresponding trinuclear N-fused hybrid scaffolds 3e–i were also formed in 63–71% yields. Prolonging the reaction time up to 4 h was needed for an allowable yield of 3e, 3g and 3h from the reaction with the corresponding 2-(2-bromophenyl)imidazoles (1e, 1g and 1h). The reaction of 2-(2-bromophenyl)-4,5-diheteroarylimidazole 1j with 2a also proceeded to give the corresponding trinuclear N-fused compound 3j in similar yield. Performing the reaction of 1a with dimethyl- and dichloro-substituted 2-aminobenzimidazoles (2b and 2c) also afforded the corresponding trinuclear N-fused hybrid scaffolds (3k and 3l). Here again, prolonging the reaction time was necessary for the effective formation of 3l from the reaction of 1a and 2c. However, no expected cyclized product was observed with the reaction of 2-(1-bromonaphthalen-2-yl)-4,5-diphenyl-1H-imidazole (1k) and 2a, a considerable amount of 2-(naphthalen-2-yl)-4,5-diphenyl-1H-imidazole 4 by debromination of 1k being produced in 57% yield with 59% conversion of 1k. It is known that aryl bromides were reduced to the corresponding arenes in the presence of a base by DMF as hydrogen source.15 Similar treatment of 2-(2-bromovinyl)imidazoles 1l–p having alkyl and phenyl substituents on vinyl moiety with 2-aminobenzimidazoles 2a–c under the employed conditions also afforded the corresponding imidazole-fused hybrid compounds 3m–s irrespective of the identity of such substituents. To the best of our knowledge, there are no reports on the synthetic method and biological activity of such a scaffold.
On the other hand, a one-pot reaction starting from 2-bromobenzaldehyde (5) and benzil (6) under microwave irradiation renders the synthesis of 3a practical.13 Treatment of 5 with equimolar amount of 6 in the presence of NH4OAc and AcOH in EtOH at 70 °C under microwave irradiation followed by removal of solvent afforded a white solid. Subsequent addition of 2a, K2CO3 and DMF to the microwave reaction tube containing a white solid and heating at 150 °C for 3 h produced 3a in 63% yield (eqn (1)).
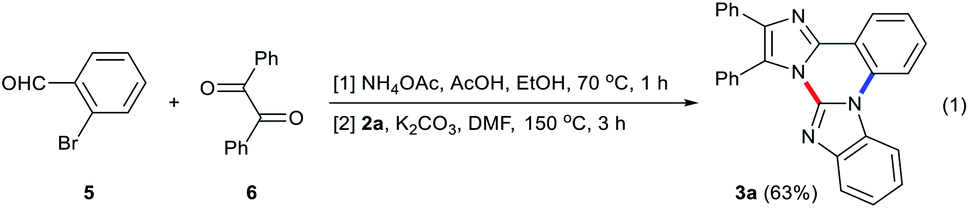
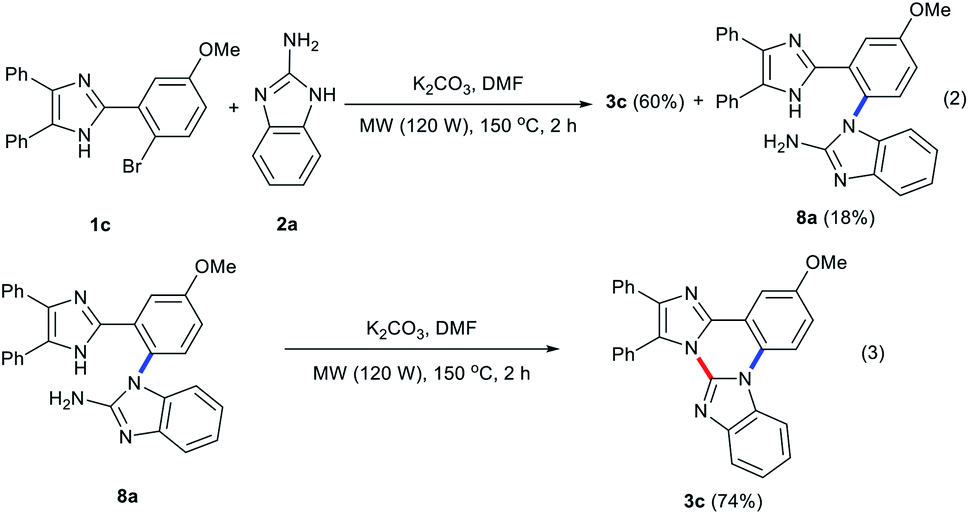
The present reaction seems to proceed via an initial formation of a C–N coupled intermediate 8 by an addition–elimination nucleophilic aromatic substitution by way of a resonance-stabilized carbanion, Meisenheimer complex 7 generated by a nucleophilic attack of 2a to the carbon attached to Br in 1 (Scheme 3).16 The ortho-substituted imidazole group of 1 facilitates the formation of such a Meisenheimer complex 7 by further electron delocalization over phenyl and imidazole rings. Fortunately, a C–N coupled intermediate 8a could be isolated in 18% yield from the reaction of 1c with 2a under the optimized conditions (eqn (2)). Performing the reaction of 1c with 2a for 4 h under the employed conditions resulted in 68% yield of 3c with complete disappearance of 8a. The intermediate 8a thus isolated was readily cyclized under the optimized conditions to give 3c in 74% yield (eqn (3)). These results indicate a doubtless evidence for the formation of such a C–N coupled intermediate 8 during the reaction course between 1 and 2. Subsequent intramolecular nucleophilic addition in intermediate 8 produces an intermediate 9, which triggers β-elimination to give 3 with the evolution of ammonia (Scheme 3).17 The evolution of ammonia was testified by the color change of Nessler's reagent, an alkali solution of K2HgI4.18
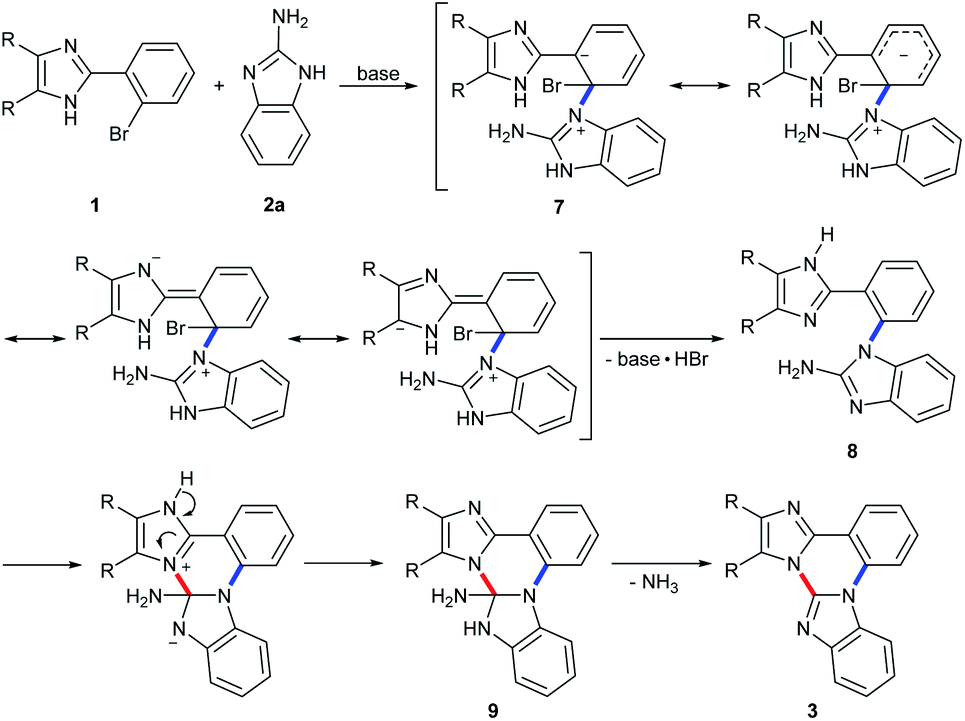 | ||
| Scheme 3 A plausible reaction pathway. | ||
Conclusions
In summary, we developed a transition metal-free synthesis of trinuclear imidazole-fused hybrid scaffolds by K2CO3-mediated coupling and cyclization of 2-(2-bromoaryl)- and 2-(2-bromovinyl)imidazoles with 2-aminobenzimidazoles as building blocks under microwave irradiation. The present protocol provides a green synthetic method for such trinuclear N-fused hybrid scaffolds. Further challenges on the green construction of polynuclear N-fused hybrid scaffolds via transition metal-free C(sp2)–N coupling are in progress.Experimental
General information
1H (500 MHz) and 13C NMR (125 MHz) spectra were recorded on a Bruker Avance Digital 500 spectrometer. Melting points were determined on a microscopic melting point apparatus (Stanford Research Inc. MPA 100 automated melting point apparatus). High-resolution mass data were recorded using electronic ionization (HRMS-EI, magnetic sector-electric sector double focusing mass analyzer) at Korea Basic Science Institute, Daegu Center, Korea. All reactions were carried out in a sealed tube under microwave irradiation (CEM, Discover LabMate) and the reaction temperature was maintained by an external infrared sensor. The products were isolated by TLC (a glass plate coated with Kieselgel 60 GF254, Merck). 2-(2-Bromoaryl)- and 2-(2-bromovinyl)-4,5-diaryl-1H-imidazoles 1 were synthesized from the corresponding aldehydes and 1,2-diarylethane-1,2-diones (see ESI†).12,13 2-Aminobenzimidazoles 2 were prepared from 1,2-phenylenediamines and cyanogen bromide by known methods.19 Other commercially available organic and inorganic reagents were used without further purification.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Research Foundation of Korea (NRF) grant funded by the Korea government (Ministry of Science and ICT) (2020R1F1A1054806).Notes and references
- For reviews, see: (a) A. R. Katritzky, C. A. Ramsden, J. A. Joule and V. V. Zhdankin, Handbook of Heterocyclic Chemistry, Elsevier, 2010 Search PubMed; (b) J. Alvarez-Builla and J. J. Vaquero, in Modern Heterocyclic Chemistry, ed. J. Alvarez-Builla, J. J. Vaquero and J. Barluenga, Wiley-VHC, Weinheim, 2011, ch. 22, pp. 1989–2070 CrossRef; (c) X.-X. Guo, D.-W. Gu, Z. Wu and W. Zhang, Chem. Rev., 2015, 115, 1622 CrossRef CAS PubMed; (d) S. K. Manna, T. Das and S. Samanta, ChemistrySelect, 2019, 4, 8781 CrossRef CAS.
- N. K. Nandwana, R. P. Singh, O. P. S. Patel, S. Dhiman, H. K. Saini, P. N. Jha and A. Kumar, ACS Omega, 2018, 3, 16338 CrossRef CAS PubMed.
- (a) D. B. Knowles, C. Lin, P. B. Mackenzie, J.-Y. Tsai, R. Walters, S. A. Beers, C. S. Brown, W. H. Yeager and E. Barron, Utility Pat., 2008156879, 2008; Chem. Abstr., 2008, 150, 86349 Search PubMed; (b) T. Y. Park, J. S. Bae, S. G. Hong, D. H. Lee, D. U. Lee, S. Y. Jeon and S. S. Kim, Korean patent 2010097797, 2010; Chem. Abstr., 2010, 153, 456673 Search PubMed.
- For the synthesis of scaffold B, another N-fused mode of scaffold A (Scheme 1), see: (a) D. Chen, L. Huang, J. Yang, J. Ma, Y. Zheng, Y. Luo, Y. Shen, J. Wu, C. Feng and X. Lv, Tetrahedron Lett., 2018, 59, 2005 CrossRef CAS; (b) X. Guo, J. Hu, M. Zhang and L. Wang, Asian J. Org. Chem., 2019, 8, 417 CrossRef CAS.
- G. Yuan, H. Liu, J. Gao, K. Yang, Q. Niu, H. Mao, X. Wang and X. Lv, J. Org. Chem., 2014, 79, 1749 CrossRef CAS PubMed.
- N. K. Nandwana, K. Pericherla, P. Kaswan and A. Kumar, Org. Biomol. Chem., 2015, 13, 2947 RSC.
- (a) P. D. Q. Dao, H. K. Lee, H.-S. Sohn, N. S. Yoon and C. S. Cho, ACS Omega, 2017, 2, 2953 CrossRef CAS PubMed; (b) H. K. Lee, P. D. Q. Dao, Y. Kim and C. S. Cho, Synthesis, 2018, 50, 3243 CrossRef CAS; (c) P. D. Q. Dao, S. L. Ho and C. S. Cho, ACS Omega, 2018, 3, 5643 CrossRef CAS PubMed; (d) D. Y. Kim, P. D. Q. Dao and C. S. Cho, ACS Omega, 2018, 3, 17456 CrossRef CAS PubMed; (e) T. D. Diep, P. D. Q. Dao and C. S. Cho, Eur. J. Org. Chem., 2019, 4071 CrossRef CAS; (f) D. Y. Kim, P. D. Q. Dao, N. S. Yoon and C. S. Cho, Asian J. Org. Chem., 2019, 8, 1726 CrossRef CAS; (g) P. D. Q. Dao and C. S. Cho, Eur. J. Org. Chem., 2020, 330 CrossRef CAS; (h) T. D. Diep, P. D. Q. Dao, S. L. Ho and C. S. Cho, Eur. J. Org. Chem., 2020, 2807 CrossRef CAS; (i) J. P. Kwak, P. D. Q. Dao and C. S. Cho, Eur. J. Org. Chem., 2020, 3468 CrossRef CAS.
- P. D. Q. Dao, H.-J. Lim and C. S. Cho, Green Chem., 2019, 21, 6590 RSC.
- For our recent reports for transition metal-free cyclizations via homolytic aromatic substitution (HAS) type pathway, see: (a) P. D. Q. Dao, S. L. Ho, H.-J. Lim and C. S. Cho, J. Org. Chem., 2018, 83, 4140 CrossRef CAS PubMed; (b) P. D. Q. Dao, H.-J. Lim and C. S. Cho, ACS Omega, 2018, 3, 12114 CrossRef CAS PubMed.
- For recent reports on transition metal-free C(sp2)–N bond formation, see: (a) C.-L. Sun and Z.-J. Shi, Chem. Rev., 2014, 114, 9219 CrossRef CAS PubMed; (b) W. Liu, J. Li, P. Querard and C.-J. Li, J. Am. Chem. Soc., 2019, 141, 6755 CrossRef CAS PubMed; (c) F. Diness and D. P. Fairlie, Angew. Chem., Int. Ed., 2012, 51, 8012 CrossRef CAS PubMed; (d) F. Diness and M. Begtrup, Org. Lett., 2014, 16, 3130 CrossRef CAS PubMed; (e) A. H. Sandtorv and D. R. Stuart, Angew. Chem., Int. Ed., 2016, 55, 15812 CrossRef CAS PubMed; (f) C. B. Jacobson, M. Meldal and F. Diness, Chem.–Eur. J., 2017, 23, 846 CrossRef PubMed.
- P. D. Q. Dao and C. S. Cho, J. Org. Chem., 2020, 85, 13354 CrossRef PubMed.
- Y. Yuan and H. Zhu, Eur. J. Org. Chem., 2012, 329 CrossRef CAS.
- M.-Y. Gang, J.-Q. Liu and X.-S. Wang, Tetrahedron, 2017, 73, 4698 CrossRef CAS.
- For biological activities of imidazo[1,2-c]quinazolines, see: (a) G. Domány, T. Gizur, A. Gere, K. Takács-Novák, G. Farsang, G. G. Ferenczy, G. Tárkányi and M. Demeter, Eur. J. Med. Chem., 1998, 33, 181 CrossRef; (b) E. E. Korshin, L. A. Sabirova and Y. A. Levin, Synthesis, 2012, 44, 3512 CrossRef CAS.
- H.-X. Zheng, X.-H. Shan, J.-P. Qu and Y.-B. Kang, Org. Lett., 2017, 19, 5114 CrossRef CAS PubMed.
- M. B. Smith, March's Advanced Organic Chemistry: Reactions, Mechanisms, and Structure, John Wiley & Sons, New Jersey, 2020, pp. 767–838 Search PubMed.
- Y. Ashida, K. Arashiba, K. Nakajima and Y. Nishibayashi, Nature, 2019, 568, 536 CrossRef CAS PubMed.
- W. M. Haynes, CRC Handbook of Chemistry and Physics, CRC Press, Boca Raton, FL, 97th edn, 2017, p. 91, section 8 Search PubMed.
- (a) M. Beesu, S. S. Malladi, L. M. Fox, C. D. Jones, A. Dixit and S. A. David, J. Med. Chem., 2014, 57, 7325 CrossRef CAS PubMed; (b) X. Guida, H. Jianhua and L. Xiaomin, Eur. J. Med. Chem., 2006, 41, 1080 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1ra04396j |
| This journal is © The Royal Society of Chemistry 2021 |