
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceImproved polymerization and depolymerization kinetics of poly(ethylene terephthalate) by co-polymerization with 2,5-furandicarboxylic acid†
Anup S. Joshi ab,
Niloofar Alipourasiabiab,
Keerthi Vinnakotaab,
Maria R. Coleman*ab and
Joseph G. Lawrence*ab
ab,
Niloofar Alipourasiabiab,
Keerthi Vinnakotaab,
Maria R. Coleman*ab and
Joseph G. Lawrence*ab
aDepartment of Chemical Engineering, University of Toledo, OH 43606, USA. E-mail: joseph.lawrence@utoledo.edu; maria.coleman6@utoledo.edu
bPolymer Institute, University of Toledo, OH 43606, USA
First published on 5th July 2021
Abstract
Poly(ethylene terephthalate) (PET), known for its clarity, food safety, toughness, and barrier properties, is a preferred polymer for rigid packaging applications. PET is also one of the most recycled polymers worldwide. In light of climate change, significant efforts are underway to improve the carbon footprint of PET by synthesizing it from bio-based feedstocks. Often times, specific applications demand PET to be copolymerized with other monomers. This work focuses on copolymerization of PET with a bio-based co-monomer, 2,5-furandicarboxylic acid (FDCA) to produce the copolyester (PETF). We report the multifunction of FDCA to influence the esterification reaction kinetics and the depolymerization kinetics (via alkaline hydrolysis) of the copolyester PETF. NMR spectroscopy and titrimetric studies revealed that copolymerization of PET with different levels of FDCA improved the esterification reaction kinetics by enhancing the solubility of monomers. During the alkaline hydrolysis, the presence of FDCA units in the backbone almost doubled the PET conversion and monomer yield. Based on these findings, it is demonstrated that the FDCA facilitates the esterification, as well as depolymerization of PET, and potentially enables reduction of reaction temperatures or shortened reaction times to improve the carbon footprint of the PET synthesis and depolymerization process.
Introduction
In the new age of circular economy and heightened awareness of climate change, polyethylene terephthalate (PET) has gained greater acceptance as a circular packaging polymer due to its excellent properties and recyclability.1–6 Industrially, PET is synthesized either by transesterification of dimethyl terephthalate (DMT) with ethylene glycol (EG) (DMT process) or direct esterification of terephthalic acid (TPA) with EG (TPA process).7 With advancement in processes to obtain purified TPA, the capacity of PET produced from the direct esterification of TPA and EG has increased significantly in the last two decades. The esterification step of the TPA process is a dissolution limited process due to the extremely low solubility of TPA in EG.7,8 Higher reaction temperatures and pressures are required for the esterification step to dissolve the TPA in EG.Commercially available grades of PET used for packaging application generally contain small quantities of comonomers to modulate properties required for injection and stretch blow molding of PET.9 In the production of PET via TPA process, comonomers like isophthalic acid (IPA) are introduced in the free diacid form along with TPA. During the copolymerization, the comonomer diacid may compete with TPA for the esterification reaction with EG and may affect the dissolution and reaction kinetics, especially at higher molar contents. While detailed studies on the role of solubility in the esterification kinetics of PET have been reported,10–12 to the authors' knowledge, no published study on the effect of copolymerization on the esterification step of PET synthesis is available.
In the recent past, significant research effort has been devoted to develop bio-based alternatives to PET.13–17 Poly(ethylene 2,5-furandicarboxylate) (PEF), which can be produced from bio-based 2,5-furandicarboxylic acid (FDCA) and EG, has gained significant academic and industrial interest due to its superior properties and structural similarity to PET.18–27 Due to limited availability, high costs, and limited mechanical recycling compatibility of PEF with PET stream, copolymerization or blending of PET with PEF is being considered as an effective approach to avail the enhancements of PEF without affecting the cost or recyclability of the final packaging.27–30 In our previous work, we reported that FDCA has much higher solubility in EG compared to TPA.31 This difference in the solubility may play a role during the esterification step for synthesis of copolyesters of FDCA and TPA with EG (PETF copolyesters).
The primary focus of this work was to study the kinetics of co-esterification of TPA and FDCA with EG. The composition range of FDCA in the copolyesters was restricted to 20 mole percent due to the practical limitations associated with the cost of FDCA and recyclability of the copolyesters. Esterification kinetics were performed at two different temperatures of 250 °C and 225 °C. The temperature of 250 °C was chosen since the esterification step of PET synthesis is typically done at 250 °C commercially.7 The temperature of 225 °C was chosen to evaluate the possibility to perform the esterification of PETF copolyesters at milder conditions than those typically used for PET commercially. Since the advantage of higher solubility of FDCA in EG can play a crucial role only in the first step of the polymerization, this study was restricted to the analysis of the reaction conditions for esterification step of PET production. A previously developed NMR method32 was modified to track the end group conversion of hydroxyl and carboxyl end groups (for both diacids, TPA and FDCA) during the reaction. The end group conversion was also confirmed by titrimetry to validate the NMR method.
Lastly, proof-of-concept alkaline hydrolysis experiments were carried out with PET and PETF20 (copolyester with 20 mole% FDCA) to investigate the potential effect of the presence of FDCA on reaction kinetics for PET depolymerization. In summary, this work is focused on developing a mechanistic understanding of effect of FDCA on esterification and depolymerization kinetics of PET.
Experimental
Materials
2,5-Furandicarboxylic acid (99.27% by HPLC) was purchased from Chem-Impex International Inc. (Illinois, USA). Terephthalic acid (99+%) and dimethyl sulfoxide-d6 (99.5 + atom% D) were purchased from Acros Organics (New Jersey, USA). Ethylene glycol (certified ACS), dimethyl sulfoxide (certified ACS), sodium hydroxide pellets (CAS grade), and sulfuric acid (certified, 72% w/w, 24.0 N, ±0.1 N (12 M)) were purchased from Fisher Scientific (New Hampshire, USA). All the reagents were used as received without further purification.Methods
| δ2 = δd2 + δp2 + δh2 | (1) |
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 molar excess of glycol to diacid) were charged to a 4590 Parr micro bench top pressure reactor (Illinois, USA) equipped with a condenser assembly. No additional catalyst or DEG suppressor was added. Reaction mixture was stirred at 300 rpm under a nitrogen purge for 10 minutes to ensure inert atmosphere. After purging, the reactor was pressurized at 44 psi (3 atm) with industrial grade nitrogen. The mixture was heated to 100 °C over 15 minutes and held at that temperature for 25 minutes as a paste making step. NMR spectroscopy was performed to confirm that the paste making conditions were insufficient to initiate any esterification reaction and do not interfere with the kinetics experiments as shown in the ESI.† Following paste making, the mixture was heated to the esterification temperature over 15 minutes. The mixture was held at the esterification temperature for the desired time and quenched by immersing the reactor in a cold-water bath. This procedure was repeated at different times to study the kinetics of the reaction. For example, a reaction time of 0 minutes indicates that the mixture was immediately quenched following the ramp to esterification temperature without any hold.:1 molar excess of EG) using a synthesis protocol similar to the one described above. Following esterification at 180 °C for 120 minutes, the recovered oligomers were vacuum dried and milled prior to the solubility measurements. The PEF oligomers had a number average molecular weight of 930 Daltons and degree of polymerization of 5 as confirmed by the MALDI-MS (Fig. S10(a) in ESI†).
:1 molar excess of glycol to diacid) were charged to a 4590 Parr micro bench top pressure reactor (Illinois, USA) equipped with a condenser assembly. No additional catalyst or DEG suppressor was added. Reaction mixture was stirred at 300 rpm under a nitrogen purge for 10 minutes to ensure inert atmosphere. After purging, the reactor was pressurized at 44 psi (3 atm) with industrial grade nitrogen. The mixture was heated to 100 °C over 15 minutes and held at that temperature for 25 minutes as a paste making step. NMR spectroscopy was performed to confirm that the paste making conditions were insufficient to initiate any esterification reaction and do not interfere with the kinetics experiments as shown in the ESI.† Following paste making, the mixture was heated to the esterification temperature over 15 minutes. The mixture was held at the esterification temperature for the desired time and quenched by immersing the reactor in a cold-water bath. This procedure was repeated at different times to study the kinetics of the reaction. For example, a reaction time of 0 minutes indicates that the mixture was immediately quenched following the ramp to esterification temperature without any hold.:1 molar excess of EG) using a synthesis protocol similar to the one described above. Following esterification at 180 °C for 120 minutes, the recovered oligomers were vacuum dried and milled prior to the solubility measurements. The PEF oligomers had a number average molecular weight of 930 Daltons and degree of polymerization of 5 as confirmed by the MALDI-MS (Fig. S10(a) in ESI†).Characterization techniques
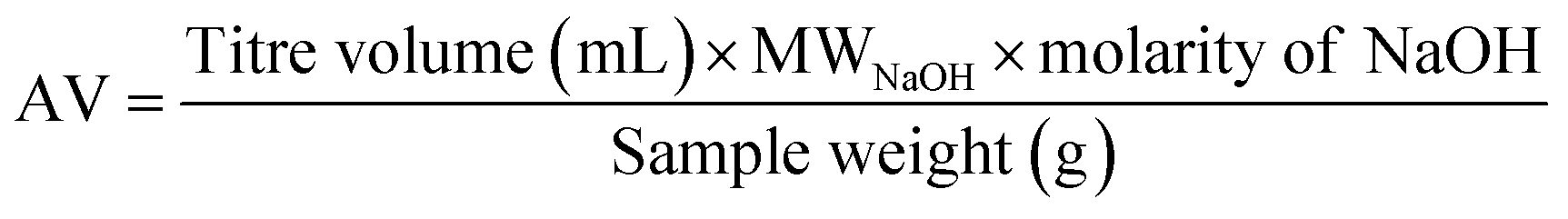 | (2) |
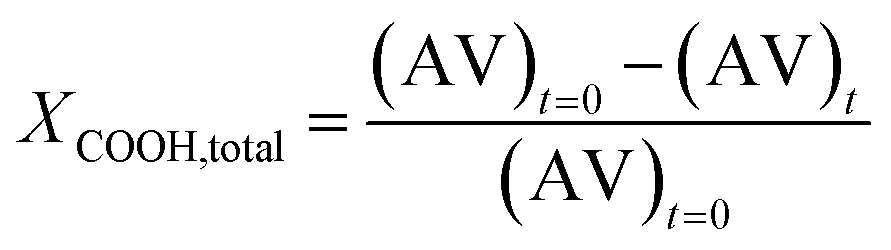 | (3) |
Results and discussion
As described in the Introduction section, the TPA process is a dissolution limited process due to limited solubility of terephthalic acid (TPA) in ethylene glycol (EG).7,8 Higher temperatures and pressures are required to dissolve the TPA in EG. In the case of synthesis of copolymers of PET via TPA process, the co-monomers like isophthalic acid are introduced in the diacid form for compatibility with the process. During the copolymerization, the co-monomer diacids are expected to compete with TPA for the esterification reaction with EG. Fig. 1 depicts a detailed reaction scheme for co-esterification of 2,5-furandicarboxylic acid (FDCA) and TPA with EG. Since this is a dissolution limited process,7,8 the difference in the solubilities between FDCA and TPA can potentially affect the esterification kinetics and copolymer microstructure. In our previous work, we reported that FDCA has much higher solubility in EG compared to TPA.31 Higher solubility of FDCA in EG could potentially translate into faster conversion of FDCA to bis-hydroxyethyl 2,5-furandicarboxylate (BHEF) and oligomers. During PET synthesis, oligomers of PET (bis-hydroxyethyl terephthalate or BHET) are often added to improve solubility of TPA in EG solution.7,8 The BHEF and FDCA-rich oligomers formed during the co-polymerization could provide an improved solubility effect similar to BHET.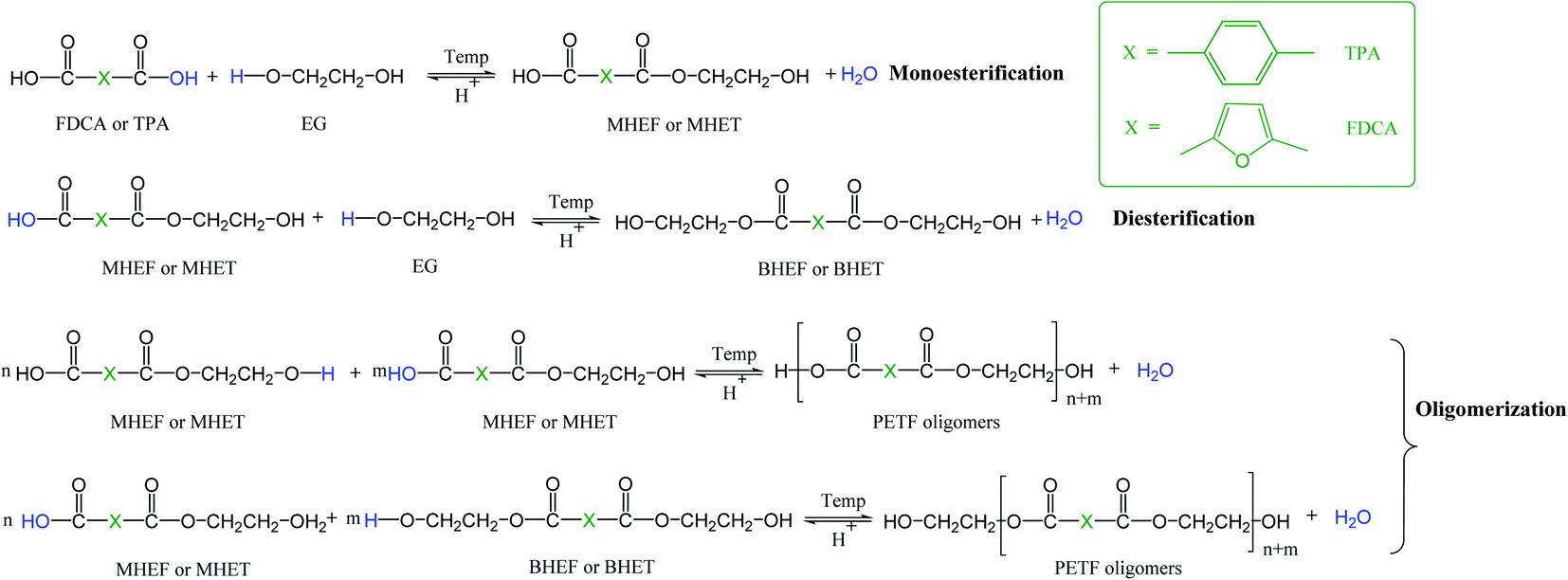 | ||
| Fig. 1 Series of chemical reactions involved during the co-esterification of diacid X (TPA and FDCA) with EG; MHEF = monohydroxyethyl 2,5-furandicarboxylate, MHET = monohydroxyethyl terephthalate, BHET = bishydroxyethyl terephthalate, BHEF = bishydroxyethyl 2,5-furandicarboxylate, PETF = poly(ethylene terephthalate-co-2,5-furandicarboxylate). | ||
To evaluate the effect of FDCA on dissolution and esterification kinetics of PET, direct esterification of copolymers of PET with FDCA was performed at 10 and 20 mole% of FDCA. As mentioned in the Experimental section, the protocol for industrial scale synthesis of PET was used for the esterification reactions for greater applicability of this work. The direct esterification was carried out at a commercially applicable temperature of 250 °C and a milder temperature of 225 °C to understand the effect of temperature on the kinetics. For simplicity, the copolyesters of PET with 10 and 20 mole% of FDCA are referred as PETF10 and PETF20, respectively. Note that the results and discussion are limited to the oligomers obtained during the esterification step.
End group conversion by NMR
1H and 13C NMR spectroscopy was used to track the hydroxyl and carboxyl end group conversion during the esterification of TPA and FDCA with EG. The details of the method are described in the ESI.† Fig. 2 shows the hydroxyl end group conversion (XOH) during the esterification of PETF co-polyesters at two esterification temperatures. As seen from Fig. 2(b), the reaction kinetics were much slower at 225 °C and the reaction had to be run for a longer time to achieve equilibrium conversions. At commercial synthesis conditions of 250 °C, equilibrium conversion was reached within one hour of reaction time for the co-polyesters. Please note that the esterification reaction conditions, and the reactor design were optimized to effectively remove the water generated during the esterification reaction without losing the EG in the condensate. Based on this assumption of no loss of EG, the equilibrium values of XOH were close to the theoretical conversion of 0.66 expected with 1.5:1 excess of hydroxyl end groups.
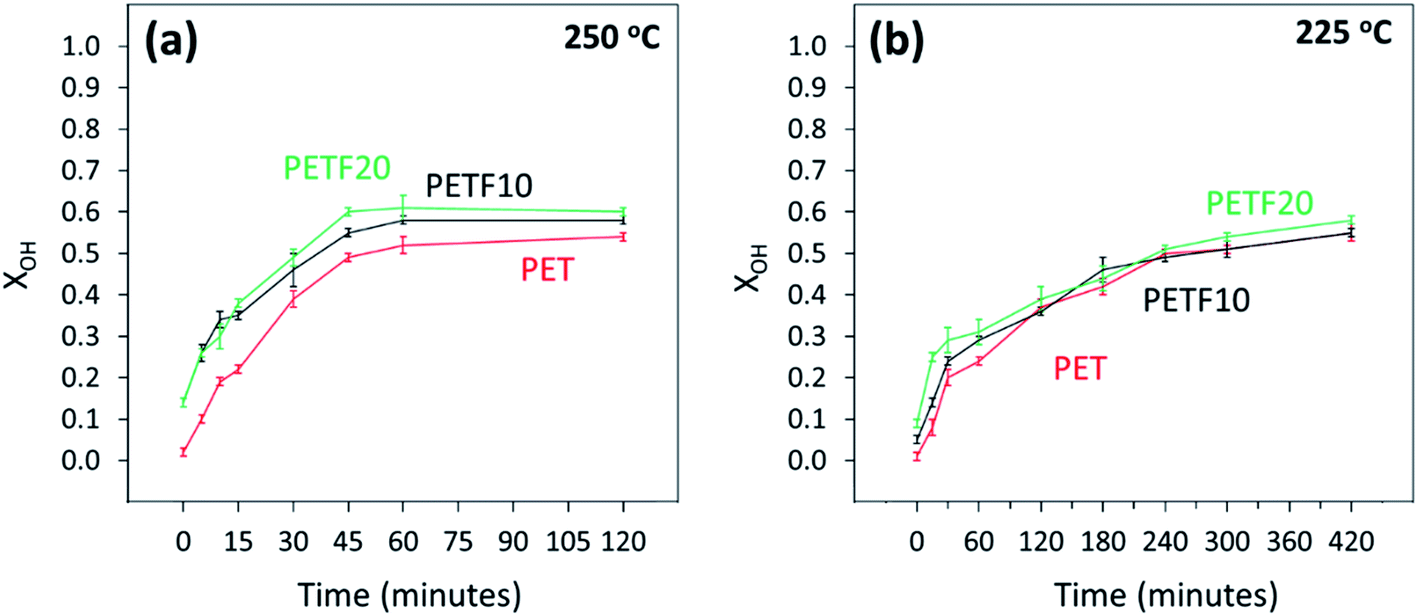 | ||
Fig. 2 Hydroxyl end group conversion determined by 1H NMR for PET ( ),PETF10 (-) and PETF20 ( ),PETF10 (-) and PETF20 ( ) for direct esterification performed at (a) 250 °C and (b) 225 °C. ) for direct esterification performed at (a) 250 °C and (b) 225 °C. | ||
The hydroxyl end group conversion increased significantly for the PETF co-polyesters compared to PET at 250 °C, especially at low reaction times. On the other hand, at 225 °C, the increase in XOH following copolymerization was much less evident possibly due to the reduced solubility of TPA in EG and slower reaction kinetics compared to 250 °C. The XOH values at both temperatures for the PETF10 and PETF20 samples at zero reaction time were greater than 0 indicating the onset of the reaction during the ramping step. However, the PET sample did not show any conversion of hydroxyl groups during the ramping step. This observation indicated that the FDCA starts reacting with the EG at temperatures much lower than the TPA confirming the results published in our previous work.31 The hydroxyl end group conversion was also employed to track the production of diethylene glycol, a known side reaction during the esterification step with a detailed discussion in the ESI.†
Acid end group conversion was determined for FDCA and TPA independently using the 13C NMR spectra as discussed in the ESI.† Total carboxyl end group conversion (XCOOH,total) was calculated from the FDCA (XCOOH,FDCA) and TPA (XCOOH,TPA) end group conversions using eqn (4).
| XCOOH,total = x × XCOOH,FDCA + (1 − x) × XCOOH,TPA | (4) |
As shown in Fig. 3, XCOOH,total followed a similar trend to that of XOH. Total acid end group conversion increased for the PETF copolyesters compared to PET. As was seen for XOH, the increase was significant especially at low reaction times. At 250 °C, the XCOOH,total values converged at higher times (Fig. 3(a)) to a value of 0.87. However, this convergence was not observed in the time scale studied at 225 °C (Fig. 3(b)) indicating the slower kinetics at 225 °C.
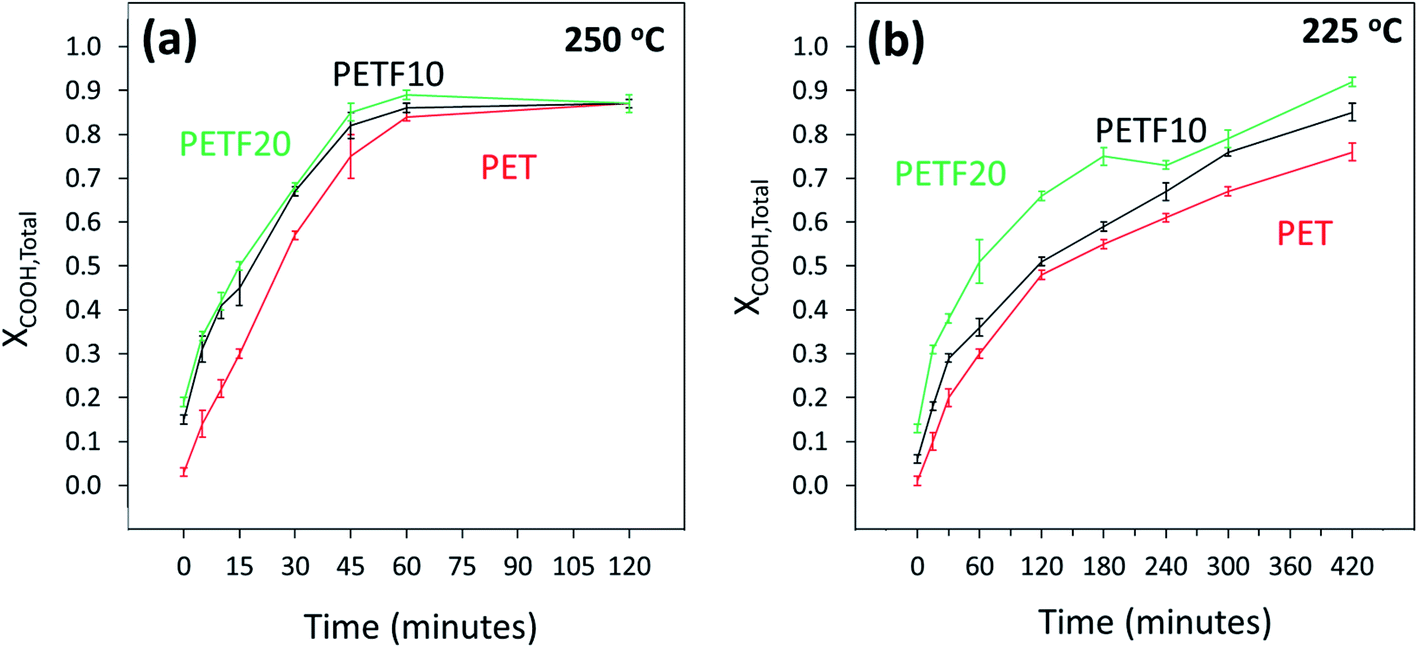 | ||
Fig. 3 Total carboxyl end group conversion determined by 13C NMR for PET ( ), PETF10 (-) and PETF20 ( ), PETF10 (-) and PETF20 ( ) for direct esterification performed at (a) 250 and (b) 225 °C. ) for direct esterification performed at (a) 250 and (b) 225 °C. | ||
Fig. 4(a) and (b) show the conversion of FDCA end groups at 250 and 225 °C respectively. The FDCA end groups reacted almost instantaneously at both reaction temperatures. Interestingly, almost half of the end groups were esterified during the ramping step (XCOOH,FDCA ∼ 0.5 at t = 0). As reported in our previous work, FDCA exhibits an order of magnitude higher solubility in EG compared to TPA at temperatures higher than 180 °C.31 Additionally, in the case of copolymers, FDCA was dissolved in a very large molar excess of EG (15:1 in case of 10 mole% FDCA since total molar ratio of EG: diacid was set to 1.5:1). High solubility and molar excess of EG to FDCA resulted in an instantaneous dissolution and esterification of FDCA end groups. This rapid esterification of FDCA end groups was primarily responsible for the increase in the XOH and XCOOH,total at low times.
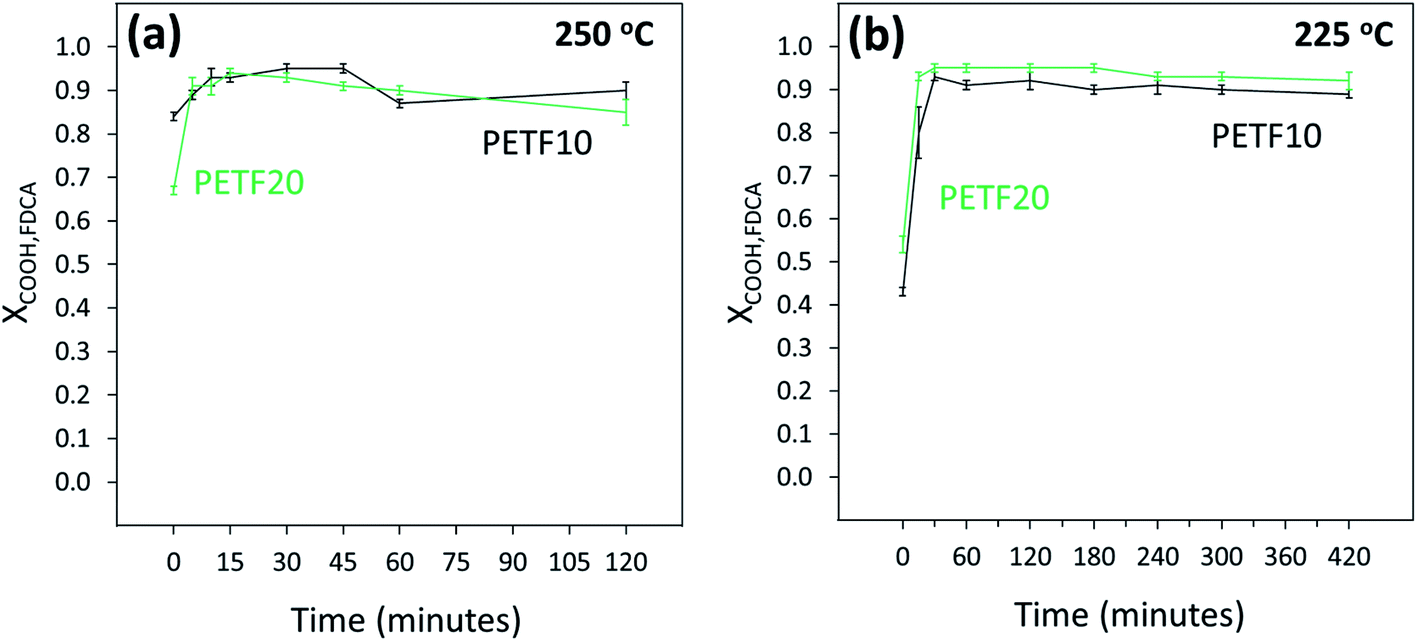 | ||
Fig. 4 FDCA end group conversion determined by 13C NMR for PETF10 (-) and PETF20 ( ) for direct esterification performed at (a) 250 °C and (b) 225 °C. ) for direct esterification performed at (a) 250 °C and (b) 225 °C. | ||
As shown in Fig. 1, esterification of TPA and FDCA with EG could be considered as competing parallel reactions. However, within 15 minutes of the reaction time at 250 and 225 °C, most of the FDCA was esterified (XCOOH,FDCA ∼ 0.9). On the other hand, most of the TPA was unreacted after 15 minutes (Fig. 5). Hence, due to the instantaneous esterification of FDCA, it is proposed that the reactions occurred in series rather than in parallel. Additionally, as shown in Fig. 5(a) and (b), TPA end group conversions increased for PETF10 and PETF20 samples compared to PET.
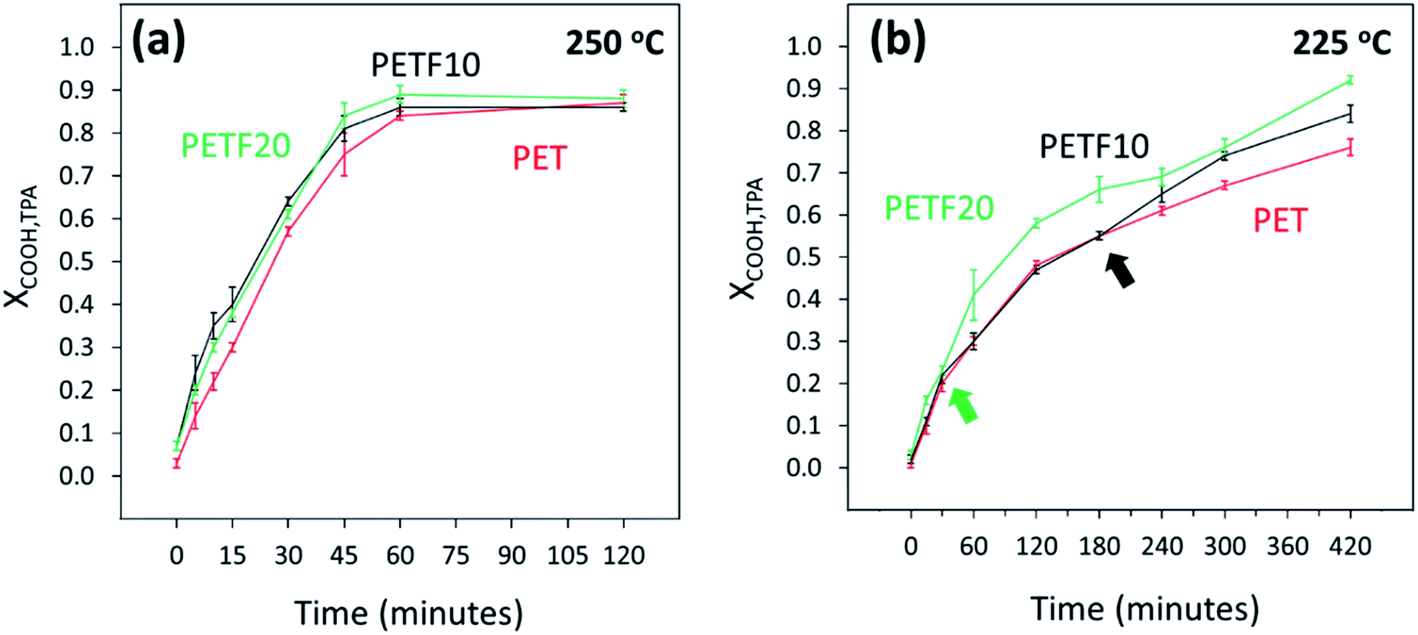 | ||
Fig. 5 TPA end group conversion determined by 13C NMR for PET ( ), PETF10 (-) and PETF20 ( ), PETF10 (-) and PETF20 ( ) for direct esterification performed at (a) 250 °C and (b) 225 °C. ) for direct esterification performed at (a) 250 °C and (b) 225 °C. | ||
At 250 °C, higher XCOOH,TPA was obtained in the case of PETF10 and PETF20 over the full reaction time. However, at 225 °C, the improvement in XCOOH,TPA for PETF samples occurred after a specific delay time as shown in Fig. 5(b) with arrows. This delay time observed at 225 °C was shorter for PETF20 (30 minutes) compared to PETF10 (180 minutes). Based on this observation, it is hypothesized that a certain concentration of esterified FDCA-rich oligomers in the reaction media was necessary to facilitate the conversion of TPA at 225 °C. This concentration was achieved at shorter times for PETF20 compared to PETF10 due to the higher molar ratio of FDCA in the feed for PETF20, and hence higher concentration of BHEF and FDCA-rich oligomers.
In summary, hydroxyl and total carboxyl end group conversions increased with the presence of FDCA in the reaction media. The FDCA carboxyl end groups were almost instantaneously converted to the esterified products. These esterified FDCA-rich oligomers accelerated the conversion of TPA carboxyl end group in PETF copolyesters compared to PET. At 250 °C, XCOOH,TPA of 0.84 was achieved for PET sample after 60 minutes of esterification reaction. A similar value of conversion was achieved in 45 minutes for PETF20 reducing the reaction time by 15 minutes or 25%. Esterification performed at 225 °C showed significantly slower reaction kinetics compared to 250 °C. Surprisingly, even at 225 °C, almost all of the FDCA was esterified within the first 15 minutes of reaction. TPA carboxyl end group conversions at 225 °C showed a delay time before the improvement due to esterified FDCA products was observed.
Composition and degree of randomness by NMR
1H NMR was also employed to investigate the composition and the degree of randomness of the growing chains as a function of esterification time and temperature. The oxyethylene proton peak corresponding to the in-chain EG unit showed peak splitting depending on the acid units surrounding it. Based on the area under the peaks, the mole fractions and degree of randomness of PEF and PET blocks was calculated (as explained in the ESI†) and summarized in Table 1.| Time (min) | 250 °C | 225 °C | ||
|---|---|---|---|---|
| PETF10 | PETF20 | PETF10 | PETF20 | |
| 0 | 49 ± 01 (1.04) | 69 ± 01 (0.56) | 66 ± 04 (0.83) | 85 ± 05 (1.23) |
| 5 | 22 ± 07 (1.03) | 47 ± 04 (0.85) | — | — |
| 10 | 19 ± 01 (1.50) | 33 ± 01 (1.00) | — | — |
| 15 | 15 ± 03 (1.53) | 26 ± 01 (0.99) | 35 ± 03 (1.37) | 56 ± 03 (0.86) |
| 30 | 11 ± 02 (1.26) | 25 ± 03 (1.00) | 24 ± 01 (1.10) | 41 ± 01 (1.01) |
| 45 | — | 21 ± 02 (1.33) | — | — |
| 60 | — | 26 ± 02 (1.13) | 20 ± 02 (1.22) | 39 ± 01 (0.95) |
| 120 | — | 22 ± 04 (1.10) | 18 ± 01 (1.12) | 31 ± 03 (0.95) |
| 180 | — | — | 19 ± 01 (1.24) | 24 ± 01 (0.97) |
| 240 | — | — | 19 ± 01 (1.21) | 26 ± 01 (0.97) |
| 300 | — | — | 16 ± 03 (1.39) | 24 ± 01 (1.08) |
Fig. 6 shows the composition of the growing copolyester chains as a function of the reaction time and temperature. At low reaction times, the values of the ratio of PEF to PET unit were much higher confirming the observation that the FDCA reacted very rapidly to produce ‘FDCA-rich oligomers’ followed by incorporation of the TPA into the growing chain. As time progressed, TPA units were incorporated in the growing chains, and eventually expected mole% values were achieved in the case of 250 °C. At 225 °C, the mole% of furan units was higher than the expected values for the time scale studied. This indicated that the incorporation of TPA units was not complete after 300 minutes of esterification reaction time as observed from the Fig. 3(b).
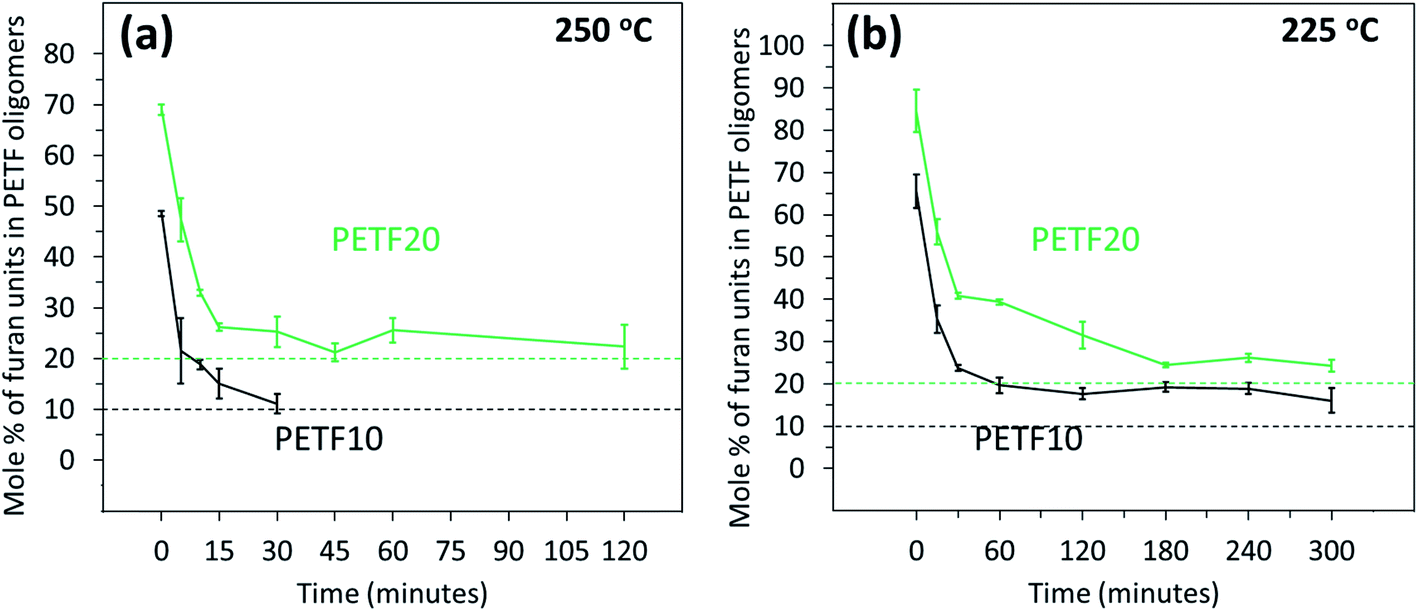 | ||
Fig. 6 Mole% of furan in PETF copolyesters determined by 1H NMR for PETF10 (-) and PETF20 ( ) for direct esterification performed at (a) 250 °C and (b) 225 °C. Dashed lines indicate the expected mole% values (10% for PETF10 and 20% for PETF20). ) for direct esterification performed at (a) 250 °C and (b) 225 °C. Dashed lines indicate the expected mole% values (10% for PETF10 and 20% for PETF20). | ||
The values of degree of randomness (R) at very low times were lower than 1 in some cases indicating a block character for the copolymers. This could be due to the blocks of rapidly esterified FDCA-rich oligomers and absence of PET oligomers formed at low times. However, for most of the reaction time, the R values remained between 1.0 to 1.5 indicating a random microstructure. These R values suggested that even though FDCA reacted faster than TPA, the reversible nature of the esterification reaction caused a redistribution of the repeat units at higher times and randomized the FDCA units in the backbone.
Based on the hydroxyl and carboxyl end group conversions and the degree of randomness analysis, it was concluded that the FDCA dissolved in EG almost instantaneously and reacted to form bis-hydroxyethyl 2,5-furandicarboxylate (BHEF) and FDCA-rich oligomers which promoted the dissolution-rate-limited-esterification of TPA with EG most likely by acting as a co-solvent and enhancing the solubility of the TPA in the mixture of EG and oligomers. Note that, the observed improvement in the reaction kinetics of TPA was different for 250 °C compared to 225 °C. This difference was primarily attributed to the temperature effect on the solubility of TPA in EG. At 250 °C, the solubility of TPA in EG was sufficient to promote the reaction and the presence of FDCA-rich oligomers enhanced the dissolution of TPA only marginally. On the other hand, at 225 °C, the esterification kinetics of FDCA was slower compared to 250 °C (Fig. 4). This resulted in a delay time to obtain sufficient concentration of the BHEF and FDCA-rich oligomers to promote the esterification of TPA with EG. Another possible explanation for the observed improvement in TPA conversion was that the dissolved FDCA improved reaction kinetics with catalytic effect since the esterification is an acid-catalyzed reaction.8 As explained in the following sections, detailed solubility studies and pKa measurements were performed to further investigate the observed improvement in the reaction kinetics of esterification of TPA with EG in the presence of FDCA.
Effect of FDCA and pre-synthesized PEF oligomers on the solubility of TPA in EG
In a previous study, we reported that the solubility of FDCA in EG is an order of magnitude higher than that of TPA at the esterification temperatures.31 For PET synthesis, the solubility of TPA in EG is often enhanced by adding small quantities of PET oligomers.7,8 Solubility experiments were carried out to confirm the proposed hypothesis that BHEF and PEF rich oligomers acted as co-solvents and improved TPA solubility in EG. A fixed quantity of TPA was mixed with different quantities of EG in the presence of FDCA or pre-synthesized PEF oligomers.Fig. 7 shows the effect of FDCA and PEF oligomers on the solubility of TPA in moles per kg of EG. Please note that for the experiments with FDCA and PEF oligomers, the weight of TPA was kept constant and additional 20 mole% of FDCA or PEF oligomers was added. The solubility curve of TPA shifted to lower temperatures by ∼10 °C in the presence of 20 mole% pre-synthesized PEF oligomers throughout the temperature range studied. As mentioned previously, similar effect of enhanced solubility of TPA in EG has been reported when PET oligomers were mixed with EG.8
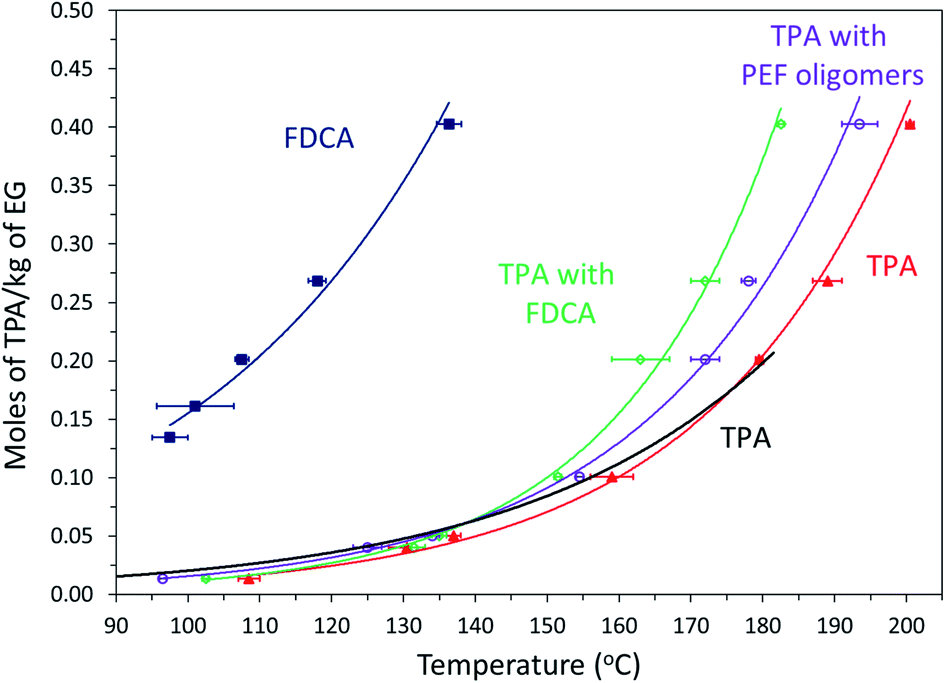 | ||
Fig. 7 Solubility of TPA in moles per kg of EG determined using clear point method for TPA alone (filled triangle,  ), TPA (black line) reported by Yamada et al.,12 TPA with 20 mole% pre-synthesized PEF oligomers (empty circle, ), TPA (black line) reported by Yamada et al.,12 TPA with 20 mole% pre-synthesized PEF oligomers (empty circle,  ), TPA with 20 mole% FDCA (empty diamond, ), TPA with 20 mole% FDCA (empty diamond,  ), and FDCA reproduced from previous work31 (filled square, ), and FDCA reproduced from previous work31 (filled square,  ). ). | ||
The trend in the improvement of solubility was different for the samples with the 20 mole% FDCA. The FDCA did not impact the solubility of TPA at lower temperatures (90 to 140 °C). However, the solubility curve of TPA improved significantly in the presence of FDCA at temperatures higher than 140 °C. The previous work on esterification of FDCA with EG revealed that the onset of esterification reaction of FDCA occurred at 140 °C.31 Hence, this significant deviation in the solubility of TPA in presence of FDCA at temperatures higher than 140 °C was primarily attributed to the in situ esterified PEF oligomers. However, the improvement in solubility of TPA by in situ esterified PEF oligomers was greater than the improvement observed in the case of TPA with pre-synthesized PEF oligomers. MALDI-MS was employed to investigate the difference in the in situ esterified PEF oligomers and the pre-synthesized PEF oligomers. As confirmed from the MALDI-MS spectrum in Fig. S10 in ESI,† the primary difference was the degree of polymerization. In the case of in situ esterification, most of FDCA was only converted to BHEF and PEF dimer due to the huge excess of EG. Hence, it was hypothesized that the presence of the BHEF formed during the heating step had a greater effect on solubility of TPA in EG compared to the pre-synthesized PEF oligomers with higher degree of polymerization. As previously discussed, during the copolymerization reaction with 10 and 20 mole% FDCA, the instantaneously dissolved FDCA reacted with large molar excess of EG (relative to moles of FDCA) most likely will form BHEF at shorter reaction times (oligomers are expected to form as the reaction progresses since the molar excess of EG relative to moles of FDCA is reduced due to reaction with TPA) which resulted in the observed improvement in TPA conversions.
Solubility parameters for the monomers and oligomers were calculated using the group contribution method devised by Hoftyzer and Van Krevelen33 to support the hypothesis of difference in solubility of TPA in EG in the presence of BHEF vs. PEF oligomers. For consistency, molar volumes of the monomers and oligomers were predicted using the group contribution method by Fedors.33 Table S2 from ESI† shows the calculated solubility parameters for different chemical species involved in the esterification reaction. Based on the calculated solubility parameters, potential of mutual solubility of two species can be determined from the individual components using eqn (5).
| Δδ1,2 = [(δd,1 − δd,2)2 + (δp,1 − δp,2)2 + (δh,1 − δh,2)2] | (5) |
The lower the value of Δδ1,2, the greater the solubility of two components with each other. ΔδEG,x and ΔδTPA,x were calculated to understand the solubility of EG and TPA with the different chemical species.37 Fig. 8(a) and (b) show the calculated Δδ values for EG and TPA, respectively. As shown in Fig. 8(a), lower value of Δδ for the EG,FDCA pair compared to EG,TPA pair indicates that FDCA is expected to have higher solubility in EG compared to TPA. These results are consistent with the observed solubility values reported in Fig. 7. Similarly, the solubility of BHEF in EG was predicted to be higher than the PEF pentamer based on the Δδ values calculated for the respective pairs. Even though the BHEF and PEF pentamer have similar δd and δp values, the hydrogen bonding contribution (δh) was higher for BHEF compared to PEF tetramer (Table S2†). This difference in the hydrogen bonding contribution should result in improved solubility of the EG,BHEF pair relative to the EG,PEF tetramer pair. Additionally, TPA is predicted to have much higher solubility in the BHEF and PEF pentamer compared to EG (Fig. 8(b)). Hence, based on the solubility parameter analysis, it was concluded that the sequential nature of the co-esterification on PETF copolyesters with FDCA reacting prior to TPA resulted in improved solubility of TPA in EG due to the presence of BHEF and PEF oligomers in the reaction mixture. The solubility studies revealed that, for the temperature range considered, copolymerization with FDCA would result in a better solubility enhancement than the direct addition of pre-synthesized PEF oligomers primarily due to better solubility of BHEF in EG over PEF oligomers.
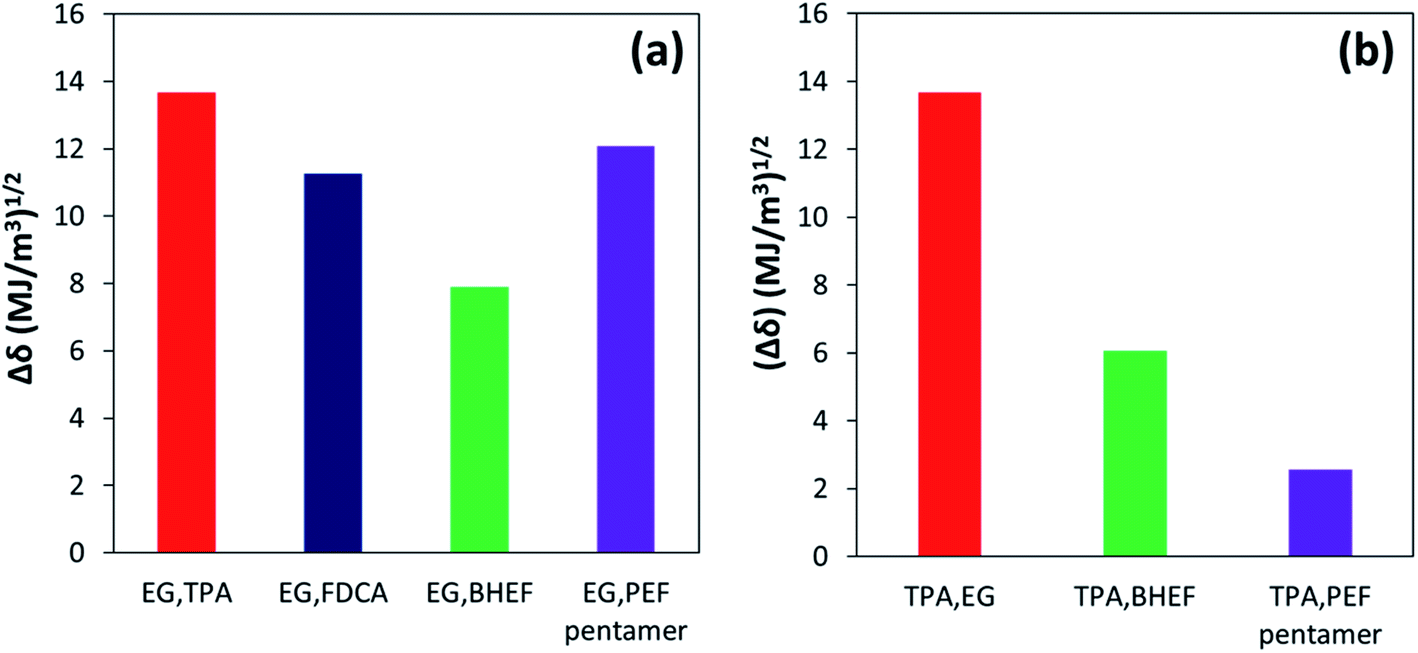 | ||
| Fig. 8 Δδ values calculated using eqn (1) to predict solubility of different reaction species with (a) EG and (b) TPA. | ||
Prediction of pKa of FDCA and TPA
Direct esterification of carboxylic acids with glycols is an acid-catalyzed Fischer esterification reaction.8 The carboxylic acid can auto-catalyze the reaction depending on the strength or the pKa of the acid. The pKa value determines the equilibrium between the acid and its conjugate base with lower values of pKa indicating that the acid will be present in the deprotonated form at neutral pH. These free protons can auto catalyze the esterification reaction.8 Hence, pKa of the diacid can influence the esterification reaction kinetics. In the case of PET, direct esterification of TPA with EG has been reported to be auto-catalyzed by TPA.7,8 Otton and Ratton reported that the esterification reaction rate increased linearly with decreasing the pKa of the carboxylic acid.38 The objective of this section was to predict the pKa for FDCA and compare it with the reported values of pKa for TPA. Different predictive tools are available for calculating the pKas based on the structure. The accuracy of the predictions has been reported to be in good agreement with the experimental values in the case of carboxylic acids.39 Chemicalize tool provided by ChemAxon was used for pKa predictions for TPA and FDCA. The predicted pKa values for TPA were very close to the experimental values reported in the literature as shown in Table 2. The predicted values for FDCA were lower than TPA indicating that FDCA is a stronger diacid compared to TPA. Similar trend was observed in the case of monoesterified TPA and FDCA. Based on these predicted pKa values and extending the observation by Otton and Ratton,38 FDCA should exhibit higher esterification rates than TPA. In conjunction with the reported esterification kinetics data, these results indicate that the improved solubility and faster esterification kinetics are responsible for instantaneous dissolution and conversion of FDCA for PETF copolyesters. The dissolved esterified products of FDCA increase the solubility and potentially reactivity of the dissolved TPA and result in improved TPA conversions at the reaction temperatures studied. A detailed modeling of the dissolution and reaction kinetics should be used to fit the experimental data and obtain the esterification rates to confirm the effect of pKa of FDCA on the esterification rates. However, due to the complexity of the copolymerization process, modeling of the reactions was considered outside of the scope of the current study.| Species | Structure | pKa | Source |
|---|---|---|---|
| TPA | 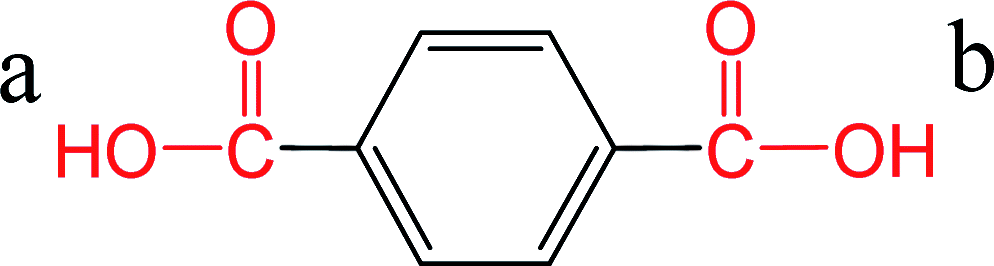 |
a: 3.51 | 38 |
| b: 4.82 | |||
| a: 3.32 | ChemAxon | ||
| b: 4.56 | |||
| Monoesterified TPA | 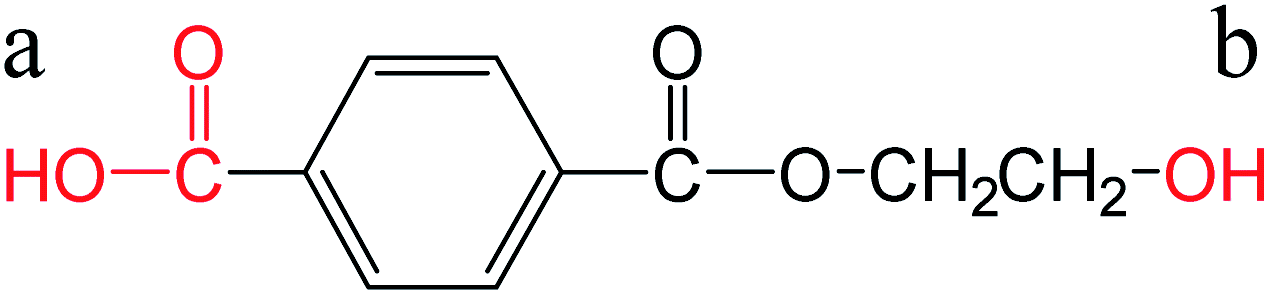 |
a: 3.61 | ChemAxon |
| b: 15.10 | |||
| FDCA | 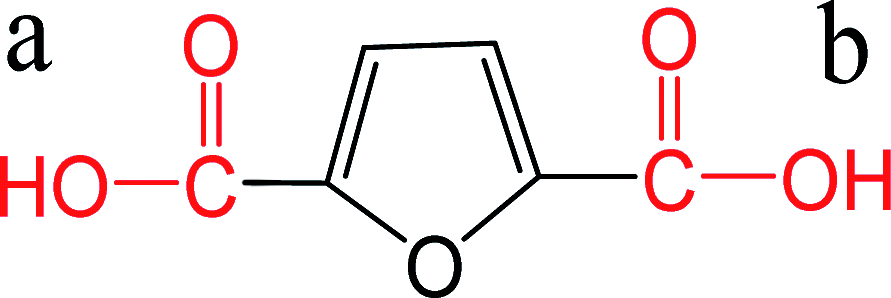 |
a: 2.76 | ChemAxon |
| b: 3.47 | |||
| Monoesterified FDCA | 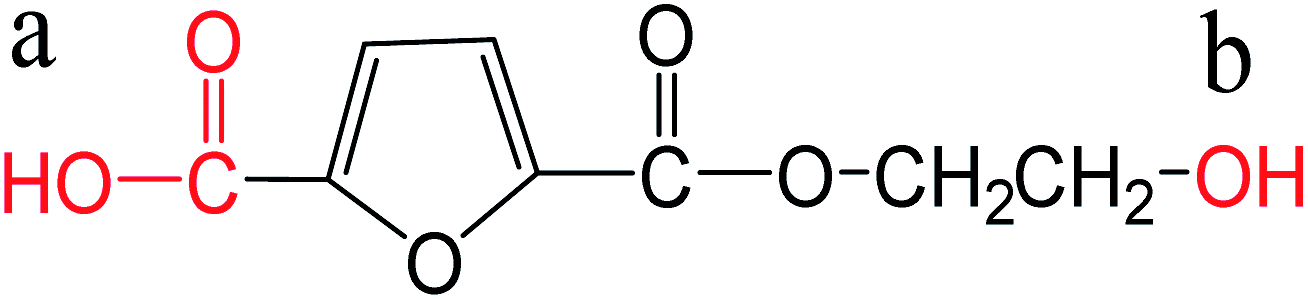 |
a: 3.06 | ChemAxon |
| b: 15.10 |
End group conversion by titration
To confirm the NMR methods used above, the carboxyl end group conversions were determined by titration and compared with the values obtained by NMR as shown in Table 3. The percent difference between the methods calculated using the eqn (6) was ±13% validating the end group analysis studies performed based on the NMR method.
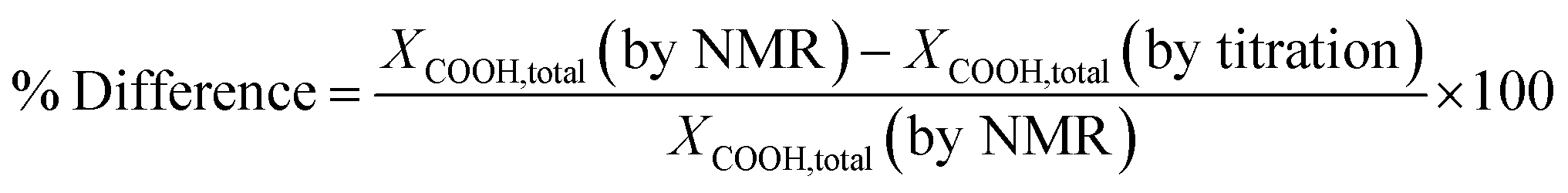 | (6) |
| T (°C) | t (min) | Sample | Mn (Da) | Đ | XCOOH,total by NMR | XCOOH,total by titration | % difference in XCOOH,total |
|---|---|---|---|---|---|---|---|
| 250 | 30 | PET | 900 | 1.08 | 0.57 ± 0.01 | 0.51 ± 0.01 | 11.1 |
| PETF10 | 960 | 1.07 | 0.67 ± 0.01 | 0.64 ± 0.01 | 5.2 | ||
| PETF20 | 960 | 1.08 | 0.68 ± 0.01 | 0.59 ± 0.01 | 12.6 | ||
| 60 | PET | 890 | 1.08 | 0.84 ± 0.01 | 0.84 ± 0.04 | 0 | |
| PETF10 | 970 | 1.08 | 0.86 ± 0.01 | 0.80 ± 0.03 | 7.3 | ||
| PETF20 | 960 | 1.09 | 0.89 ± 0.01 | 0.80 ± 0.03 | 10.4 | ||
| 225 | 180 | PET | 870 | 1.07 | 0.55 ± 0.01 | 0.49 ± 0.03 | 11.5 |
| PETF10 | 880 | 1.09 | 0.59 ± 0.01 | 0.66 ± 0.01 | −11.1 | ||
| PETF20 | 970 | 1.07 | 0.75 ± 0.02 | 0.68 ± 0.01 | 9.9 | ||
| 420 | PET | 920 | 1.08 | 0.76 ± 0.02 | 0.81 ± 0.04 | −6.7 | |
| PETF10 | 950 | 1.09 | 0.85 ± 0.02 | 0.81 ± 0.01 | 4.6 | ||
| PETF20 | 970 | 1.08 | 0.92 ± 0.01 | 0.86 ± 0.02 | 6.7 |
The trend of increase in the end group conversion for PETF samples was consistent in the end group data by titration. The observed variability between the two methods was consistent with the literature reports40 and was primarily attributed to the random error arising from the inherent differences in the techniques.
Molecular weight evolution by MALDI-MS
MALDI-MS spectra were recorded to monitor the effect of copolymerization on the number average molecular weight (Mn) and dispersity or polydispersity index of the samples (Đ). Two different reaction times corresponding to XCOOH,total for PET of approximately 0.5 and 0.8 were chosen for each reaction temperature as shown in Table 3. The recorded MALDI-MS spectra are shown in the ESI.† At both reaction temperatures, copolymerization with FDCA marginally increased the number average molecular weight of the oligomers after a reaction time corresponding to XCOOH,total ∼ 0.5. The Mn remained unchanged at higher reaction times. The dispersity (Đ) was close to 1.08 and was unchanged by the reaction time, temperature, or presence of FDCA. The low values of Đ indicated the effective redistribution of repeat units in growing chains as expected in the case of low molecular weight oligomers. It has been reported that the esterification step produces oligomers with 4 to 5 repeat units.7 Any further increase in the molecular weight needs an effective catalyst system and removal of EG which only takes place in the polycondensation step. The degree of polymerization for the samples was between 4 and 5 (repeat unit molecular weight = 192) consistent with the reports.7 Hence this data confirmed that the presence of FDCA simply improved the kinetics of the esterification reaction without affecting the degree of polymerization of the oligomers.Alkaline hydrolysis of PETF20 vs. PET
This work demonstrated that the solubility and pKa differences in FDCA and TPA result in improved esterification kinetics. Considering the reversible nature of condensation reactions, it was hypothesized that a similar improvement in kinetics may be observed in the depolymerization of PETF copolymers via an alkaline hydrolysis pathway. As a proof-of-concept, a simple alkaline hydrolysis experiment was carried out on polymer (PET and PETF20) film flakes in a glass vial as described in the Experimental section. Due to the nature of the glass vial set up, the reactions were carried out at low temperatures (lower than 100 °C to avoid boiling of the water). To achieve equilibrium conversions at lower temperatures, reaction was run for an extended period of time (3 days). After the workup, the solid residue was weighed to determine monomer molar yield and conversion of PET using eqn (7) and (8).
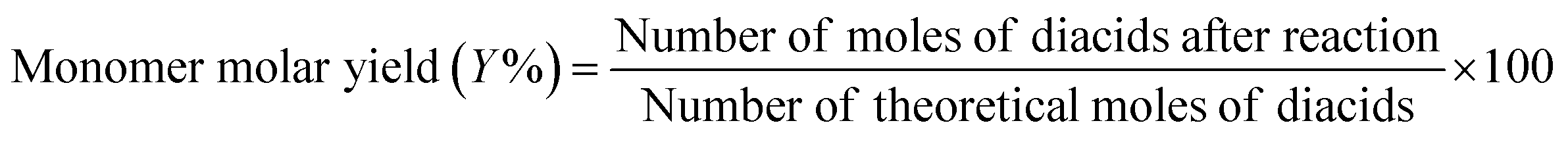 | (7) |
 | (8) |
The % conversion and monomer yield of PET and PETF20 depolymerization reactions are shown in Fig. 9. Both, the conversion and yield almost doubled for PETF20 flake samples compared to PET. 1H NMR spectroscopy was employed to confirm the purity of the diacid monomers recovered after the depolymerization (Fig. S13 in ESI†). NMR spectrum confirms that depolymerization was complete and both, FDCA and TPA were recovered as a mixture.
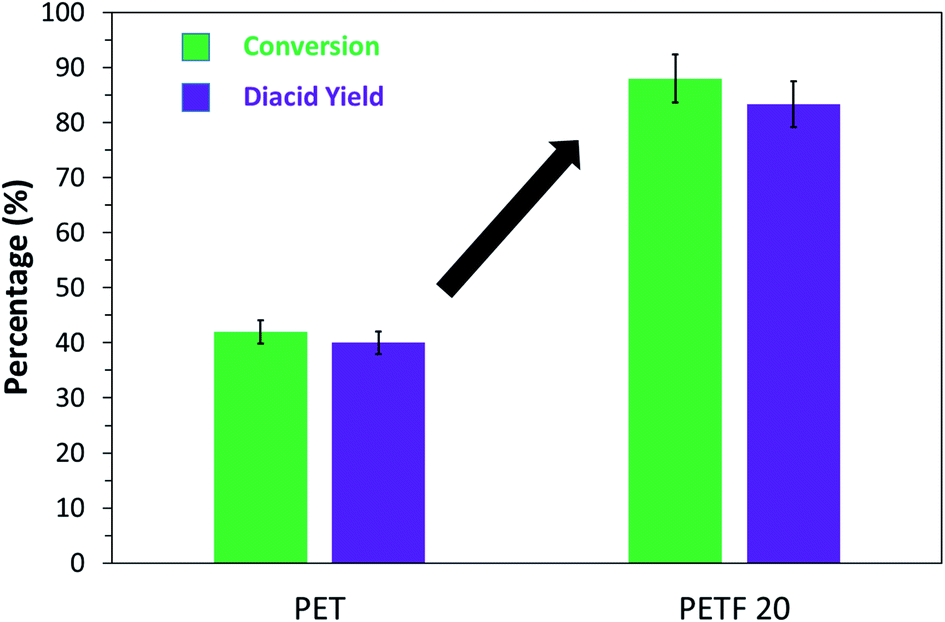 | ||
Fig. 9 % Conversion ( ) and % diacid yield ( ) and % diacid yield ( ) of PET vs. PETF20 flakes after alkaline hydrolysis with 1.1 M NaOH solution for 3 days at 90 °C. ) of PET vs. PETF20 flakes after alkaline hydrolysis with 1.1 M NaOH solution for 3 days at 90 °C. | ||
PET depolymerization reactions are known to be surface reactions, where the alkali, in this case, NaOH, reacts with the surface ester linkages and continues to scrape away the surface of the flakes.41 Based on the well-proven surface mechanism of depolymerization and the solubility work done in this study we propose the following possible explanations for the observed increase in depolymerization kinetics in case of PETF20, (1) higher solubility parameter of PETF20 enhances the interaction of solvent molecules with the polymer flakes, swelling the polymer matrix resulting in faster reaction kinetics; (2) the ester linkage with FDCA may be more labile to hydrolytic degradation resulting in more rapid depolymerization. Similar effect of higher hydrolytic degradation has been reported in the case of other bio-based co-monomers like isosorbide.42 A more detailed study of alkaline hydrolysis of copolyester of PET and PEF will be presented in a future paper.
Conclusions
The effect of copolymerization with FDCA on the dissolution and esterification of TPA with EG was studied in detail in this work. End group conversion analysis based on NMR revealed that the co-esterification reactions occurred ‘in series’ rather than ‘in parallel’ as FDCA reacted with EG almost instantaneously to form FDCA-rich oligomers followed by esterification of TPA with EG. The presence of FDCA-rich oligomers promoted the dissolution of TPA in the reaction mixture and improved the TPA esterification reaction kinetics for PETF copolyester samples compared to PET homopolyester. The carboxyl end group conversions obtained from the NMR were confirmed by titrimetry validating the method used. MALDI-MS results showed that the copolymerization did not affect the degree of polymerization, however, the number average molecular weight increased marginally at shorter reaction times by copolymerization with FDCA. This increase was attributed to the improved kinetics at shorter times as shown by the NMR results. FDCA was predicted to be more acidic than TPA based on the calculated values of pKa. Since esterification is an acid-catalyzed reaction, the lower pKa and thus, the higher acidity of FDCA could also contribute to the enhancement of kinetics in addition to the co-solvent effect of FDCA-rich oligomers. As demonstrated in the ESI,† the higher acidity of FDCA also resulted in increased DEG production, a side product of the esterification step. Based on the improved esterification kinetics, it is proposed that the esterification step of PETF copolyesters can be performed at lower temperatures or times and reduce the energy consumption during the synthesis step.Due to the reversible nature of the condensation reactions, the effect of copolymerization with FDCA on depolymerization kinetics of PET was also investigated through a proof-of concept alkaline hydrolysis reaction. PETF20 copolymers showed almost double equilibrium conversion and monomer yield compared to PET homopolymer. A more comprehensive study on depolymerization of PETF copolymers at industrially relevant temperatures and recovery of FDCA from the diacid mixture will be published in a subsequent article.
Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
Authors would like to acknowledge the Polyesters and Barrier Materials Consortia at the Polymer Institute of the University of Toledo for their financial support and guidance with this project. The authors would like to express thanks to Dr B. Leif Hanson and Dr Yong-Wah Kim for their help with MALDI-ToF-MS and NMR studies, respectively.References
- R. Rizvi, E. P. Nguyen, M. D. Kowal, W. H. Mak, S. Rasel, M. A. Islam, A. Abdelaal, A. S. Joshi, S. Zekriardehani, M. R. Coleman and R. B. Kaner, High-Throughput Continuous Production of Shear-Exfoliated 2D Layered Materials using Compressible Flows, Adv. Mater., 2018, 30, 1800200 CrossRef.
- A. Polyakova, R. Y. F. Liu, D. A. Schiraldi, A. Hiltner and E. Baer, Oxygen-barrier properties of copolymers based on ethylene terephthalate, J. Polym. Sci., Part B: Polym. Phys., 2001, 39(16), 1889–1899 CrossRef CAS.
- K. Majdzadeh-Ardakani, S. Zekriardehani, M. R. Coleman and S. A. Jabarin, A Novel Approach to Improve the Barrier Properties of PET/Clay Nanocomposites, Int. J. Polym. Sci., 2017, 2017, 10 Search PubMed.
- S. Zekriardehani, S. A. Jabarin, D. R. Gidley and M. R. Coleman, Effect of Chain Dynamics, Crystallinity, and Free Volume on the Barrier Properties of Poly(ethylene terephthalate) Biaxially Oriented Films, Macromolecules, 2017, 50(7), 2845–2855 CrossRef CAS.
- S. Zekriardehani, A. S. Joshi, S. A. Jabarin and M. R. Coleman, Combined effect of small molecule antiplasticizers and strain induced crystallization on properties of polyethylene terephthalate, Polym. Cryst., 2018, 1(3), e10016 Search PubMed.
- S. Zekriardehani, A. S. Joshi, S. A. Jabarin, D. W. Gidley and M. R. Coleman, Effect of Dimethyl Terephthalate and Dimethyl Isophthalate on the Free Volume and Barrier Properties of Poly(ethylene terephthalate) (PET): Amorphous PET, Macromolecules, 2018, 51(2), 456–467 CrossRef CAS.
- S. M. Aharoni, Industrial-Scale Production of Polyesters, Especially Poly(Ethylene Terephthalate), in Handbook of Thermoplastic Polyesters, Wiley-VCH Verlag GmbH & Co. KGaA, 2005, pp. 59–103 Search PubMed.
- T. Rieckmann and S. Völker, Poly(Ethylene Terephthalate) Polymerization – Mechanism, Catalysis, Kinetics, Mass Transfer and Reactor Design, in Modern Polyesters: Chemistry and Technology of Polyesters and Copolyesters, John Wiley & Sons, Ltd, 2004, pp. 29–115 Search PubMed.
- V. B. Gupta and Z. Bashir, PET Fibers, Films, and Bottles: Sections 1–4.13, in Handbook of Thermoplastic Polyesters, Wiley-VCH Verlag GmbH & Co. KGaA, 2005, pp. 317–361 Search PubMed.
- C.-K. Kang, B. C. Lee, D. W. Ihm and D. A. Tremblay, A simulation study on continuous direct esterification process for poly(ethylene terephthalate) synthesis, J. Appl. Polym. Sci., 1997, 63(2), 163–174 CrossRef CAS.
- K. S. Yang, K. H. An, C. N. Choi, S. R. Jin and C. Y. Kim, Solubility and esterification kinetics of terephthalic acid in ethylene glycol III. The effects of functional groups, J. Appl. Polym. Sci., 1996, 60(7), 1033–1039 CrossRef CAS.
- T. Yamada, Y. Imamura and O. Makimura, A mathematical model for computer simulation of a direct continuous esterification process between terephthalic acid and ethylene glycol: part 1. Model development, Polym. Eng. Sci., 1985, 25(12), 788–795 CrossRef CAS.
- A. Gandini, D. Coelho, M. Gomes, B. Reis and A. Silvestre, Materials from renewable resources based on furan monomers and furan chemistry: work in progress, J. Mater. Chem., 2009, 19(45), 8656–8664 RSC.
- A. F. Sousa, C. Vilela, A. C. Fonseca, M. Matos, C. S. R. Freire, G.-J. M. Gruter, J. F. J. Coelho and A. J. D. Silvestre, Biobased polyesters and other polymers from 2,5-furandicarboxylic acid: a tribute to furan excellency, Polym. Chem., 2015, 6(33), 5961–5983 RSC.
- C. Vilela, A. F. Sousa, A. C. Fonseca, A. C. Serra, J. F. J. Coelho, C. S. R. Freire and A. J. D. Silvestre, The quest for sustainable polyesters – insights into the future, Polym. Chem., 2014, 5(9), 3119–3141 RSC.
- K. M. Zia, A. Noreen, M. Zuber, S. Tabasum and M. Mujahid, Recent developments and future prospects on bio-based polyesters derived from renewable resources: a review, Int. J. Biol. Macromol., 2016, 82, 1028–1040 CrossRef CAS PubMed.
- X. Fei, J. Wang, J. Zhu, X. Wang and X. Liu, Biobased Poly(ethylene 2,5-furancoate): No Longer an Alternative, but an Irreplaceable Polyester in the Polymer Industry, ACS Sustainable Chem. Eng., 2020, 8(23), 8471–8485 CrossRef CAS.
- A. Gandini, A. J. D. Silvestre, C. P. Neto, A. F. Sousa and M. Gomes, The furan counterpart of poly(ethylene terephthalate): an alternative material based on renewable resources, J. Polym. Sci., Part A: Polym. Chem., 2009, 47(1), 295–298 CrossRef CAS.
- M. Gomes, A. Gandini, A. J. D. Silvestre and B. Reis, Synthesis and characterization of poly(2,5-furan dicarboxylate)s based on a variety of diols, J. Polym. Sci., Part A: Polym. Chem., 2011, 49(17), 3759–3768 CrossRef CAS.
- E. de Jong, M. A. Dam, L. Sipos and G. J. M. Gruter, Furandicarboxylic Acid (FDCA), A Versatile Building Block for a Very Interesting Class of Polyesters, in Biobased Monomers, Polymers, and Materials, American Chemical Society, 2012, vol. 1105, pp. 1–13 Search PubMed.
- A. J. J. E. Eerhart, A. P. C. Faaij and M. K. Patel, Replacing fossil based PET with biobased PEF; process analysis, energy and GHG balance, Energy Environ. Sci., 2012, 5(4), 6407–6422 RSC.
- G.-J. M. Gruter, L. Sipos and M. A. Dam, Accelerating Research into Bio-Based FDCA-Polyesters by Using Small Scale Parallel Film Reactors, Comb. Chem. High Throughput Screening, 2012, 15(2), 180–188 CrossRef CAS.
- M. Jiang, Q. Liu, Q. Zhang, C. Ye and G. Zhou, A series of furan-aromatic polyesters synthesized via direct esterification method based on renewable resources, J. Polym. Sci., Part A: Polym. Chem., 2012, 50(5), 1026–1036 CrossRef CAS.
- P. Gopalakrishnan, S. Narayan-Sarathy, T. Ghosh, K. Mahajan and M. N. Belgacem, Synthesis and characterization of bio-based furanic polyesters, J. Polym. Res., 2013, 21(1), 340 CrossRef.
- S. K. Burgess, O. Karvan, J. R. Johnson, R. M. Kriegel and W. J. Koros, Oxygen sorption and transport in amorphous poly(ethylene furanoate), Polymer, 2014, 55(18), 4748–4756 CrossRef CAS.
- S. K. Burgess, J. E. Leisen, B. E. Kraftschik, C. R. Mubarak, R. M. Kriegel and W. J. Koros, Chain Mobility, Thermal, and Mechanical Properties of Poly(ethylene furanoate) Compared to Poly(ethylene terephthalate), Macromolecules, 2014, 47(4), 1383–1391 CrossRef CAS.
- S. K. Burgess, Fundamentals of Transport in Poly(ethylene Terephthalate) and Poly(ethylene Furanoate) Barrier Materials, Georgia Institute of Technology, Atlanta, 2015 Search PubMed.
- A. F. Sousa, M. Matos, C. S. R. Freire, A. J. D. Silvestre and J. F. J. Coelho, New copolyesters derived from terephthalic and 2,5-furandicarboxylic acids: a step forward in the development of biobased polyesters, Polymer, 2013, 54(2), 513–519 CrossRef CAS.
- M. Konstantopoulou, Z. Terzopoulou, M. Nerantzaki, J. Tsagkalias, D. S. Achilias, D. N. Bikiaris, S. Exarhopoulos, D. G. Papageorgiou and G. Z. Papageorgiou, Poly(ethylene furanoate-co-ethylene terephthalate) biobased copolymers: synthesis, thermal properties and cocrystallization behavior, Eur. Polym. J., 2017, 89, 349–366 CrossRef CAS.
- J. Min, L. Tingting, Z. Qiang, C. Ying and Z. Guangyuan, From Fossil Resources to Renewable Resources: Synthesis, Structure, Properties and Comparison of Terephthalic Acid-2,5-Furandicarboxylic Acid-Diol Copolyesters, J. Renewable Mater., 2015, 3(2), 120–141 CrossRef CAS.
- A. S. Joshi, N. Alipourasiabi, Y.-W. Kim, M. R. Coleman and J. G. Lawrence, Role of enhanced solubility in esterification of 2,5-furandicarboxylic acid with ethylene glycol at reduced temperatures: energy efficient synthesis of poly(ethylene 2,5-furandicarboxylate), React. Chem. Eng., 2018, 3(4), 447–453 RSC.
- R. Pétiaud, H. Waton and Q.-T. Pham, A 1H and 13C NMR study of the products from direct polyesterification of ethylene glycol and terephthalic acid, Polymer, 1992, 33(15), 3155–3161 CrossRef.
- D. W. Van Krevelen and K. Te Nijenhuis, Chapter 7 - Cohesive Properties and Solubility, in Properties of Polymers, Elsevier, Amsterdam, 4th edn, 2009, pp. 189–227 Search PubMed.
- A. S. Joshi, J. G. Lawrence and M. R. Coleman, Effect of Biaxial Orientation on Microstructure and Properties of Renewable Copolyesters of Poly(ethylene terephthalate) with 2,5-Furandicarboxylic Acid for Packaging Application, ACS Appl. Polym. Mater., 2019, 1(7), 1798–1810 CrossRef CAS.
- G. P. Karayannidis, A. P. Chatziavgoustis and D. S. Achilias, Poly(ethylene terephthalate) recycling and recovery of pure terephthalic acid by alkaline hydrolysis, Adv. Polym. Technol., 2002, 21(4), 250–259 CrossRef CAS.
- R. Yamadera and M. Murano, The determination of randomness in copolyesters by high resolution nuclear magnetic resonance, J. Polym. Sci., Part A-1: Polym. Chem., 1967, 5(9), 2259–2268 CrossRef CAS.
- A. V. Raghu, G. S. Gadaginamath, S. S. Jawalkar, S. B. Halligudi and T. M. Aminabhavi, Synthesis, characterization, and molecular modeling studies of novel polyurethanes based on 2,2′-[ethane-1,2-diylbis(nitrilomethylylidene)]diphenol and 2,2′-[hexane-1,6-diylbis(nitrilomethylylidene)]diphenol hard segments, J. Polym. Sci., Part A: Polym. Chem., 2006, 44(20), 6032–6046 CrossRef CAS.
- J. Otton and S. Ratton, Investigation of the formation of poly(ethylene terephthalate) with model molecules: kinetics and mechanism of the catalytic esterification and alcoholysis reactions. I. Carboxylic acid catalysis (monofunctional reactants), J. Polym. Sci., Part A: Polym. Chem., 1988, 26(8), 2183–2197 CrossRef CAS.
- L. Settimo, K. Bellman and R. M. A. Knegtel, Comparison of the Accuracy of Experimental and Predicted pKa Values of Basic and Acidic Compounds, Pharm. Res., 2014, 31(4), 1082–1095 CrossRef CAS PubMed.
- M. Lomelí-Rodríguez, M. Martín-Molina, M. Jiménez-Pardo, Z. Nasim-Afzal, S. I. Cauët, T. E. Davies, M. Rivera-Toledo and J. A. Lopez-Sanchez, Synthesis and kinetic modeling of biomass-derived renewable polyesters, J. Polym. Sci., Part A: Polym. Chem., 2016, 54(18), 2876–2887 CrossRef.
- B.-Z. Wan, C.-Y. Kao and W.-H. Cheng, Kinetics of Depolymerization of Poly(ethylene terephthalate) in a Potassium Hydroxide Solution, Ind. Eng. Chem. Res., 2001, 40(2), 509–514 CrossRef CAS.
- J. Chen, J. Wu, J. Qi and H. Wang, Systematic Study of Thermal and (Bio)Degradable Properties of Semiaromatic Copolyesters Based on Naturally Occurring Isosorbide, ACS Sustainable Chem. Eng., 2019, 7(1), 1061–1071 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1ra04359e |
| This journal is © The Royal Society of Chemistry 2021 |