
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Methionine epimerization in cyclic peptides†
Pramodkumar D. Jadhav *a,
Jianheng Shena,
Peta-Gaye Burnetta,
Jian Yangb,
Ramaswami Sammynaikenc and
Martin J. T. Reaney*ade
*a,
Jianheng Shena,
Peta-Gaye Burnetta,
Jian Yangb,
Ramaswami Sammynaikenc and
Martin J. T. Reaney*ade
aDepartment of Plant Sciences, University of Saskatchewan, Saskatoon, SK S7N 5A8, Canada. E-mail: pramodkumar.jadhav@usask.ca; martin.reaney@usask.ca
bDrug Discovery and Development Research Group, College of Pharmacy and Nutrition, University of Saskatchewan, 107 Wiggins Road, Saskatoon, SK S7N 5E5, Canada
cSaskatchewan Structural Sciences Centre, University of Saskatchewan, 110 Science Place, Saskatoon, SK S7N 5C9, Canada
dPrairie Tide Diversified Inc., 102 Melville Street, Saskatoon, Saskatchewan S7J 0R1, Canada
eGuangdong Saskatchewan Oilseed Joint Laboratory, Department of Food Science and Engineering, Jinan University, 601, Huangpu Avenue West, Guangzhou, Guangdong 510632, China
First published on 11th June 2021
Abstract
Bioactive flax cyclic octa- and nona-peptides containing single methionine (Met) and its oxidized forms were treated under mild alkaline conditions to perform regio-selective epimerization. Cyclic peptide epimerization at the Met α-proton in a single chemical step has not been reported previously. The epimerization rate varies among Met oxidation states and ring size. These D-amino isomers along with the developed Met alkylation strategy will enable an approach to novel chemical functionalization of biomolecules. The amino acid configurations were confirmed by Marfey derivatizations, and cytotoxicity studies show the difference among the isomers. These D-amino analogs can act as a potential biomarker in plant protein processing and biomedical applications.
1. Introduction
Cyclic peptides belong to an important class of naturally occurring biologically active compounds. For example, tyrocidine A1 and gramicidin S2 are used as antibiotics whereas cyclosporine A3 is used as an immunosuppressant drug. Cyclic peptides are more stable than linear peptides and resist protease degradation, and cyclic Gαi binding peptide [NaC-(MITWYEFVAGTK)] has shown proteolytic stability compared to the parent linear molecule.4 Furthermore, the presence of D-amino acids enhances structural/conformational flexibility as compared to the L-isomers. Incorporation of D-amino acids can increase biological activity by stabilizing specific molecular conformations. For example, cyclopentapeptide, astin-C analogs containing D-amino acid residues show a better immunosuppressive activity than its isomers.5 In the case of omega-agatoxins IVB and IVC, the peptide with D-serine in position 46 is about four times more potent than the L-serine isomer in its inhibitory action on P-type Ca channels in rat cerebellar Purkinje cells.6 Another interesting property of D-amino acid peptides is improved resistance to protease hydrolysis that leads to greater stability and improved therapeutic applications. For example, the incorporation of single D-amino acid in a cyclic decapeptide increases stability and maintains breast cancer cell targeting.7Selective transformation of functional groups on biomolecules such as carbohydrates and peptides is important in biomedical research to prepare adducts and study structure–function relationships. Post-translational epimerization of selective amino acids in the peptide chain is usually a formidable challenge and requires enzymes with discriminating competence.8 Some examples include S-adenosyl-L-methionine radical epimerase family catalyzes L to D-amino acids into ribosomally synthesized peptides.9–11 Enzymes have been used to generate D-alanine from L-serine through a two-step process of the dehydration–hydrogenation process.12 Also, a peptide epimerase, BotH, helps isomerization of L-Asp to D-Asp during bottromycin biosynthesis.13 However, the use of chemical synthesis for selective epimerization on cyclic peptides is very rare. A selective epimerization at Leu of the cyclic depsiheptapeptide via 5-aminooxazole intermediate was performed in four synthetic steps.14 Another example includes epimerization of bicyclic hexapeptide RA-VII via oxazole intermediate.15 Classically, incorporation of D-amino acids into peptides has been performed by solid-phase synthesis, where amino acids are sequentially added to solid phase support to prepare linear peptides, and then release them by a reaction that results in cyclic products (e.g. Merrifield resin). These technologies consist of specialized resin, hazardous solvents, reagents, and intensive chromatography that can lower the yield of a peptide synthesis.5,16–18 In addition, site-directed chemical modification without affecting other functional groups is often a formidable challenge. Cyclic peptides provide a unique opportunity for selective functionalization of identical functional groups. For example, vitamin B12 biomimetic synthesis, the nucleotide component selectively reacts with only one ester group of cobyrinic acid heptakis(cyanomethylester).19,20
Plant cyclic peptides lacking disulfide bonds with head-to-tail cyclization from proteinogenic amino acids were termed orbitides.21 These generally contain amino acids in L-form with exceptions including, schnabepeptide (cyclooctapeptide) found in Schnabelia oligophylla containing D-Trp.22 Flaxseed orbitide LO 1 (cyclolinopeptide A), first discovered in a precipitated flaxseed oil slime, possesses immunosuppressive activity (ESI, Table S1†).23,24 About 39 flax orbitides containing proteinogenic amino acids (such as Gly, Pro, Phe, Val, Leu, Ile, Met, and Trp) were reported and have various biological activities including acting as immunosuppressants, antioxidants, anti-inflammatories, anticancer agents and osteoclast differentiation inhibitors.25 The aim of the study is to exploit conformational differentiation in a cyclic peptide system for selective Met epimerization. The Met containing orbitides are shown in Table S1 (ESI†).
The proposed mechanism of epimerization is shown below in Scheme 1. Generally, racemases and epimerases are enzymes that catalyze the epimerization of biological molecules. Enzymes utilize a one- or two-step mechanism to subtract the α-H proton of an amino acid in a protein or peptide by a deprotonation/reprotonation mechanism.8,26 In the current study, the base (potassium ethoxide) abstracts protons from one face and solvent protonation occurs through another face to produce a D-amino isomer. The acidity of α-H of different amino acid residues such as Leu, Ile, Phe, Val, and Met are quite similar27,28 and hence the Met epimerization was not considered kinetically favored over other amino acids in this cyclic system. To afford selective epimerization at the Met site there might be two possible mechanisms. The orbitides cyclic structure imposes a steric constraint allowing base to have selective access to α-H of Met or there is a polar effect of sulfur, where solvents such as butanol and methanol were bonded close to MetO2 and Met in LOs 4 and 2 respectively. This is observed in the crystal structure as shown in Fig. 1.29,30 Hence solvent can play a major role in resonance stabilization of the carbanion intermediate generated by base thus allowing base access for proton abstraction. Also, there were reports of oxazolone mechanism to support the effect of sidechain on Met epimerization. The proximity of a Lewis base in the structure of Met and its oxidized products might stabilize isomerization intermediates.31,32 We have found experimentally that eight-membered rings were more prone to epimerization as compare to nine membered (Table 1). Hence steric factors and ring orientation potentially affect alpha proton abstraction.
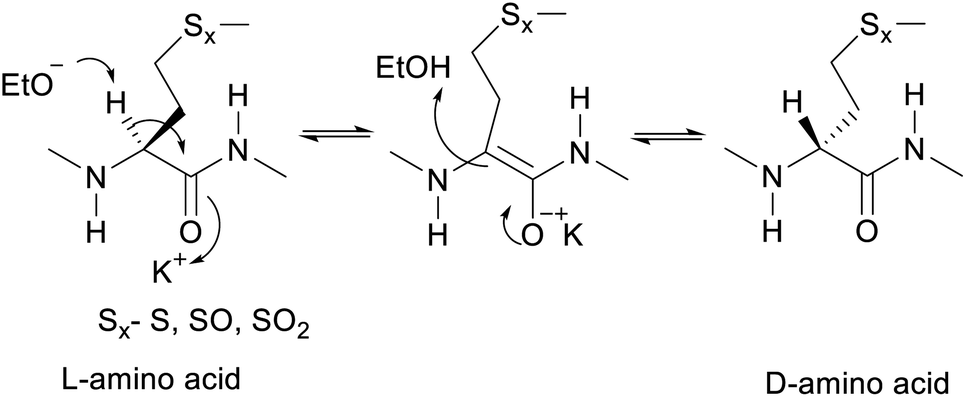 | ||
| Scheme 1 Proposed mechanism of base catalyzed epimerization. | ||
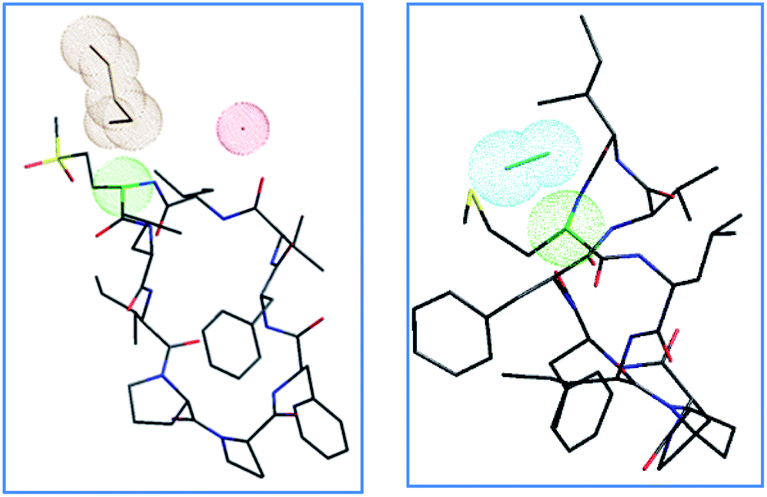 | ||
| Fig. 1 Crystal structures of LO 4 butanol solvate29 and LO 2 methanol solvate,30 with color-coded atoms sulfur – yellow, oxygen – red, nitrogen – blue, carbon – black, α carbon Met/MetO2 – green, solvent clouds: butanol – brown, water – red, methanol – cyan. | ||
| LO | Area under curve | LO | Area under curve | % L to D conversion |
|---|---|---|---|---|
| 2 | 574.834 | 8 | 31.1244 | 5.14 |
| 3 | 2314.17 | 9 | 964.877 | 29.43 |
| 4 | 1251.1 | 10 | 322.223 | 20.48 |
| 5 | 767.894 | 11 | 227.048 | 22.82 |
| 6 | 411.138 | 12 | 508.19 | 55.28 |
| 7 | 319.425 | 13 | 502.883 | 61.16 |
2. Materials and methods
2.1 Reagents
D-Isomer of Met LOs 8, 9, 10, and 12 were custom synthesized by Chempeptide Limited (Shanghai, China) and other chemicals were obtained from Sigma-Aldrich Canada Ltd., Oakville, ON, Canada. HPLC grade solvents were used for LC and MS analysis. Prairie Tide Diversified Inc. (Saskatoon, SK Canada) supplied LOs for the study. LOs stock solutions for cytotoxicity assay were prepared in dimethyl sulfoxide (DMSO). Human breast cancer triple-negative-subtype cell line MDA-MB-231 and HER2-subtype cell line Sk-Br-3 were obtained from American Type Culture Collection (ATCC, Manassas, VA, USA). McCoy's 5A Modified Medium, Leibovitz's L-15 Medium and cell culture media, were obtained from Thermo Fisher Scientific (Ottawa, ON, Canada). CytoTox Green Cytotoxicity Assay was acquired from the Promega Corporation (Madison, WI, USA).2.2 Cell culture
A Forma Series II 3110 Water-Jacketed CO2 Incubator (Thermo Fisher Scientific) containing T-75 cell culture flasks at 37 °C under a humidified atmosphere was used to culture human breast cancer cell lines MDA-MB-231 and Sk-Br-3. 10% fetal bovine serum (FBS) and 1% penicillin under 5% CO2 were used to supplement McCoy's 5A Modified Medium to culture Sk-Br-3 cell line, and 10% FBS and 1% penicillin under 0% CO2 were used to supplement Leibovitz's L-15 Medium to culture MDA-MB-231 cell line. Each cell line's culture medium was changed every 2–3 days.2.3 Instrumentation
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :15 for 3 min, 85:15 to 60:40 in 18 min, to 10:90 in 0.5 min, 10:90 for 1 min, to 85:15 in 0.5 min, to equilibration for 2 min) with column compartment at 30 °C. Chromolith® High Resolution RP-18e columns 50 mm × 4.6 mm i.d. (Merck KGaA, Darmstadt, Germany) equipped with in-line filters were used for LO analytical characterization. H2O and CH3CN linear HPLC gradient with a flow rate of 2 mL min−1 (50:50 to 20:80 for 1.8 min, 20:80 to 10:90 in 0.1 min, to 50:50 in 0.1 min, to equilibration for 1 min), and a column compartment at 32 °C was used.
:15 for 3 min, 85:15 to 60:40 in 18 min, to 10:90 in 0.5 min, 10:90 for 1 min, to 85:15 in 0.5 min, to equilibration for 2 min) with column compartment at 30 °C. Chromolith® High Resolution RP-18e columns 50 mm × 4.6 mm i.d. (Merck KGaA, Darmstadt, Germany) equipped with in-line filters were used for LO analytical characterization. H2O and CH3CN linear HPLC gradient with a flow rate of 2 mL min−1 (50:50 to 20:80 for 1.8 min, 20:80 to 10:90 in 0.1 min, to 50:50 in 0.1 min, to equilibration for 1 min), and a column compartment at 32 °C was used.Preparative reversed phase chromatography for separation of natural and KOH treated LOs 4 and 7 was performed using Chromolith® High Resolution RP-18e column (100 × 4.6 mm i.d., Merck KGaA, Darmstadt, Germany) at 214 nm. The mobile phase consisted of H2O–CH3CN (50:50 for 6.5 min at 1.2 mL min−1, 50:50 to 5:95 in 0.25 min at 1.2 mL min−1, 5:95 for 0.5 min at 2 mL min−1, 5:95 to 50:50 in 0.25 min at 2 mL min−1, to equilibration for 2.5 min at a flow rate of 2 mL min−1) with column compartment at 32 °C. Separation of natural and KOH treated LOs 3 and 6 was performed with same column. The mobile phase consisted of H2O–CH3CN (50:50 for 7.0 min at 0.8 mL min−1, 50:50 to 10:90 in 0.25 min at 0.8 mL min−1, 10:90 to 50:50 in 0.25 min at 2 mL min−1, to equilibration for 2.5 min at a flow rate of 2 mL min−1) with the column compartment at 32 °C.
:40 for 2 min, to 10:90 in 8 min, to 60:40 in 0.5 min, to equilibration for 5.5 min) was used for chromatography.2.4 Synthesis
2.5 Cytotoxicity assay
MDA-MB-231 and Sk-Br-3 cells were plated in 96-well plates at 8000 cells per well (final culture volume – 300 μL) and allowed to grow overnight for attachment before being treated with LO. LO stock solutions were prepared in DMSO at various concentrations of 200 μM; 100 μM; 50 μM, 25 μM, 12.5 μM. CytoTox Green reagent was added to wells during treatments. DMSO treated cells (final concentration at 2%) were used as a negative control. Following the manufacturer's recommended protocols, LOs cytotoxicity was continuously monitored at 24 h intervals for 120 h and calculated using the equation.| Cytotoxicity (%) = [OD (experiment) − OD (vehicle control)]/[OD (maximum cell death) − OD (vehicle control)] × 100% |
2.6 Statistical analysis
Statistical analyses of the results were performed using GraphPad Prism 6 (GraphPad Software, La Jolla, CA, USA) using a one-way ANOVA. Significance differences are indicated by asterisks: *p ≤ 0.05, **p ≤ 0.01, ***p ≤ 0.001, and ****p ≤ 0.0001.3. Results and discussion
Orbitides containing one Met or their oxidized forms with eight or nine amino acids in the chain were selected. L-forms were naturally available from flax (isolated) while the D-forms were chemically synthesized. These cyclic peptides contain other hydrophobic amino acids such as Pro, Phe, Val, Leu, and Ile. Initially, LOs 1, 4 and 7 mixtures treated with KOH in 70% aqueous ethanol (Scheme 2). But we observed two additional peaks in the HPLC chromatogram at 214 nm and 280 nm (Fig. 2). It suggests that there is a change in the structure of these peptides. Hence MS/MS of these samples was performed to characterize the new compounds and similar molecular masses of the new peaks like 4 and 7 and similar fragmentation patterns were observed. However, no extra peak was observed for non-Met containing LO 1. Hence, this suggests that amino acid isomers might have formed to give the same molecular mass. The LO 4 (L-MetO2) was compared with synthetic analog LO 10 (D-MetO2), as these LO analogs are diastereomers in the chiral environment, they were resolved in reversed phase HPLC with LO 4 elutes first and KOH treated LO 4 shows an additional peak (Fig. 2). We performed the NMR study to monitor alkaline treatment of LOs 3 and 6 mixture with multiple acquisitions and time delay experiment. 50 NMR acquisitions were performed on a single sample with 16 scans and 10 s delays between each acquisition (total time: 64.1 min). The epimerization reaction happens quickly with few differences in the spectra. Spectral changes were observed on the MetO methyl residues (2.70 ppm) (ESI, Fig. S21†).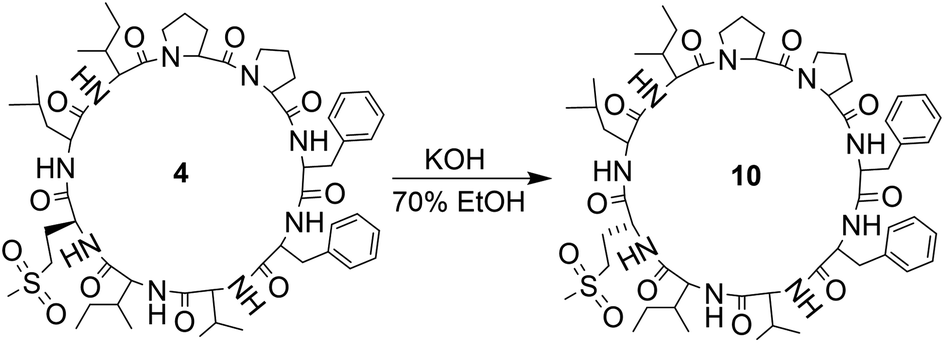 | ||
| Scheme 2 Alkaline treatment of LO 4. | ||
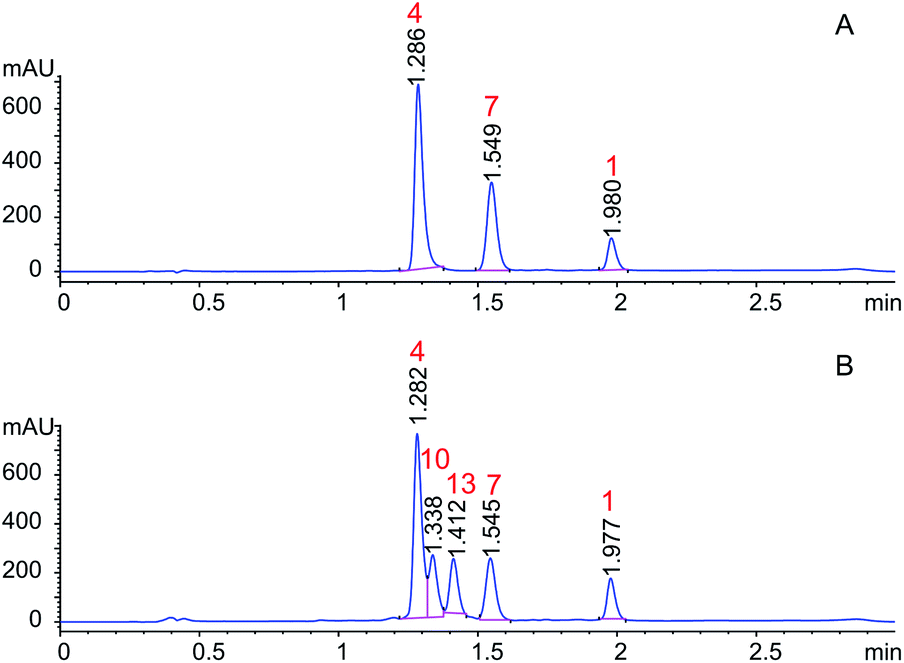 | ||
| Fig. 2 HPLC chromatogram of (A) LOs 1, 4 and 7; (B) KOH treated LOs 1, 4 and 7. | ||
To understand the alkaline treatment effect, we must determine all amino acid configurations present in the cyclic peptide. Hence these new peaks were purified using preparative LC techniques. Finally, isolated new cyclic peptides were hydrolyzed and Marfey's derivatives of the amino acids were synthesized to determine amino acid chirality. Marfey's reagent has been used to determine the optical purity of amino acids and peptides. This strategy involves the treatment of racemic analytes with an optical reagent to prepare pairs of diastereomers. These diastereomers were separated using reversed phase chromatography.
LOs were hydrolyzed by heating in 1 M HCl and derivatizing with Marfey's reagent. The optimized HPLC conditions were developed to separate Marfey's derivatives of 8 amino acids (Met, MetO, MetO2, Pro, Val, Phe, Leu, IIe) using a single chromatographic gradient (Fig. 3). L-Amino acid Marfey's derivatives eluted before the corresponding D-isomer. The optimized chromatographic conditions were used to compared hydrolyzed LO 4 and purified KOH treated LO 4 (LO 10) after derivatizing with Marfey's reagent. HPLC analysis showed selective epimerization at α-H of MetO2 in LO 10 with the formation of D-MetO2. These results confirm that the alkali catalyzed reaction proceeded efficiently to produce the D-isomer product. Similar, results were obtained with KOH treated LO 7 (LO 13) shown in Fig. 3. Small additional peaks indicated that side-reactions occurred where non-Met residues (D-Phe/D-Ile/D-Leu) were also isomerized. Alkaline treatment was also performed on reduced LOs of 4 (2 and 3) and 7 (5 and 6), and the products analyzed. However, in this case other LOs 2, 3, 5 and 6 were not purified after KOH treatment but their isomers were confirmed by comparison with available synthetic D-isomers (ESI, Fig. S12–S20†). It was found that LOs 2, 3, 5, and 6 show racemization of Met and MetO amino acids. KOH-treated LO 2 shows the formation of an additional peak in HPLC with similar MS/MS fragmentation (ESI, Fig. S1†), and KOH-treated LO 5 showed 3 peaks in the chromatogram with same molecular mass. It is difficult to characterize the third peak, it might be proline isomerization (cis–trans) of D-Met or another amino acid isomerization. We are claiming that Met isomerization is kinetically favored over other amino acids due to its orientation in cyclic peptide/reactivity, though other amino acids might also isomerize (ESI, Fig. S2†). The eight membered ring (LOs 5, 6 and 7) shows a higher epimerization rate in the order of MetO2 > MetO > Met residues while in nine membered (LOs 2, 3 and 4) the order was MetO > MetO2 > Met (Table 1).
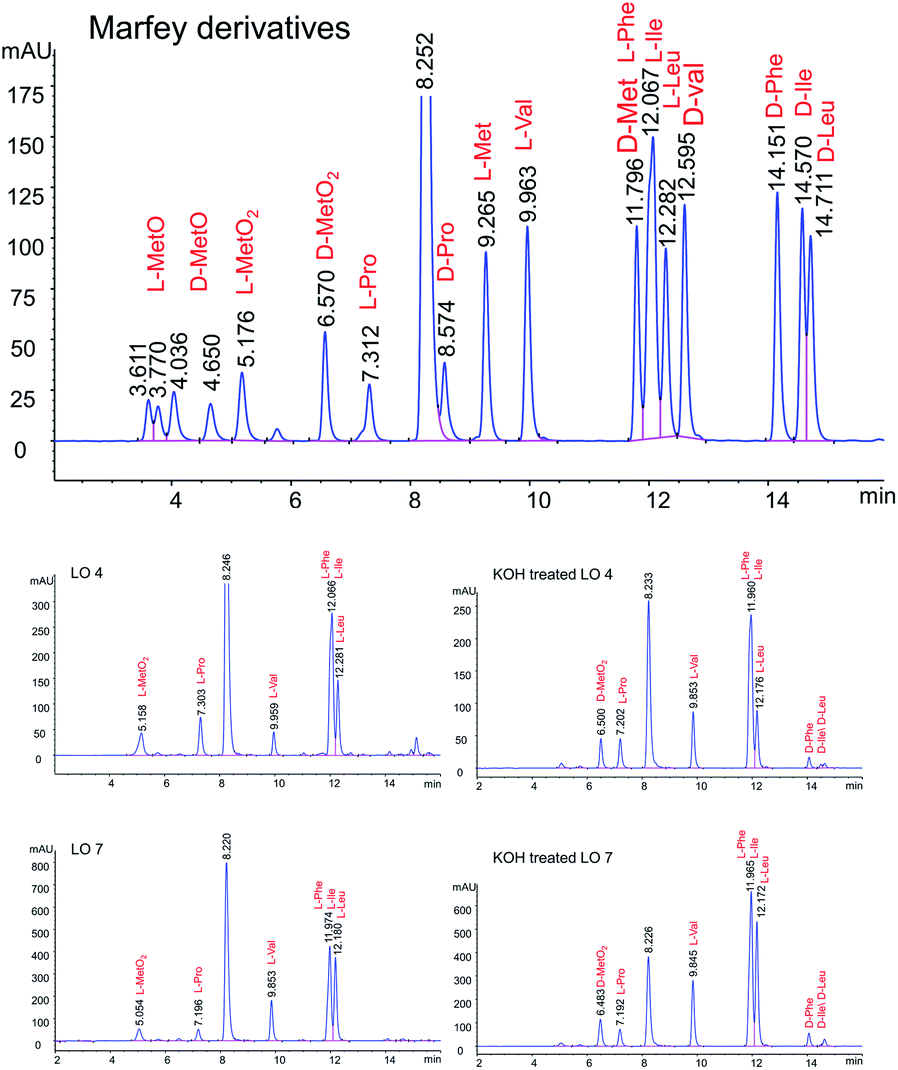 | ||
| Fig. 3 Separation of Marfey derivatized hydrolysate LO 4, LO 7, KOH treated LO 4 and KOH treated LO 7. | ||
Cytotoxicity studies were designed to understand the effect of isomerization on biological activity. Both natural and synthesized D-analogs were studied using MDA-MB-231 and Sk-Br-3 cell lines (ESI, Fig. S3–S11†). There were cytotoxicity differences observed between L- and D-pair and a higher concentration of 200 μM was required for full inhibition in these cell lines. MetO pairs show low cytotoxicity and the most notable difference was found in D-Met LO 8 with cytotoxicity of ∼100% at 48 h at 100 μM concentration against the MDA-MB-231 cells compared to ∼40% for L-Met-LO 2 (Fig. 4).
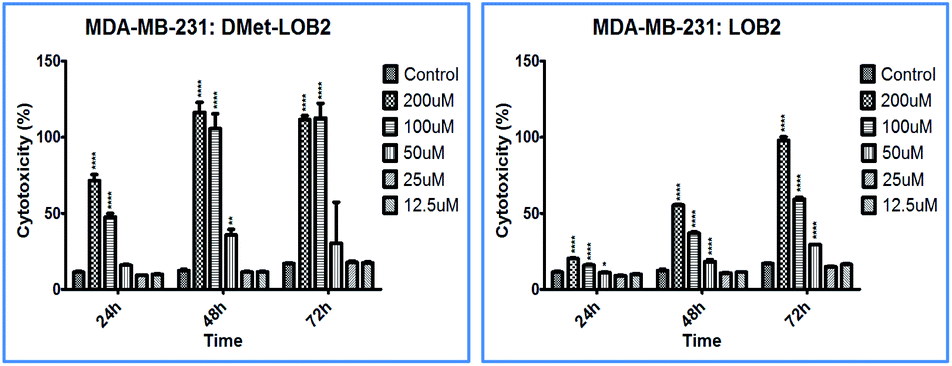 | ||
| Fig. 4 % cytotoxicity of D-Met-LO 8 and L-Met LO 2 against MDA-MB-231 cell lines. | ||
We have developed selective chemistry for isomerizing Met, MetO, and MetO2. Hence, this ability followed by modification is a route to more chemical novelty. For example, these Met side chains have been successfully coupled with dye, affinity matrix and protein for use as fluorescent tags, protein purification and antibodies production.33–35 Click type chemistry was also developed to alkylate Met to prepare multifunctional polypeptides.36 Also, ionic copolypeptide vesicles and sulfonium tethered peptides were also prepared.37,38
4. Conclusions
In summary, orbitides containing one Met and its oxidized forms produce D-analogs under alkaline conditions by selective epimerization of Met, MetO, and MetO2. Both D and L-isomers isomers were isolated, hydrolyzed, and derivatized with Marfey's reagent to confirm the amino acid configuration. The eight membered ring and MetO/MetO2 were more prone to epimerization. The selective epimerization on cyclic peptides will help to generate rapid structural analogs for future SAR studies. Cyclic peptide conformational orientation was utilized for selective functionalization and have potential to similar cyclic peptides systems. Cytotoxicity studies show the difference between D and L-isomers at higher concentrations. These orbitides can act as a potential biomarkers for \food processing and biomedical applications.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors thank the financial support from the Agriculture Development Fund (ADF) from the Saskatchewan Ministry of Agriculture (ADF-20080205, 20120099 and 20120146). Genome Canada, Total Utilization Flax Genomics (TUFGEN-1309), and Canada Foundation for Innovation (CFI-23426) also provided financial support.References
- H. D. Mootz and M. A. Marahiel, J. Bacteriol., 1997, 179, 6843–6850 CrossRef CAS PubMed.
- J. Kratzschmar, M. Krause and M. A. Marahiel, J. Bacteriol., 1989, 171, 5422–5429 CrossRef CAS PubMed.
- E. A. Emmel, C. L. Verweij, D. B. Durand, K. M. Higgins, E. Lacy and G. R. Crabtree, Science, 1989, 246, 1617–1620 CrossRef CAS PubMed.
- S. W. Millward, S. Fiacco, R. J. Austin and R. W. Roberts, ACS Chem. Biol., 2007, 2, 625–634 CrossRef CAS PubMed.
- F. Li, X.-X. Guo, G.-Z. Zeng, W.-W. Qin, B. Zhang and N.-H. Tan, Bioorg. Med. Chem. Lett., 2018, 28, 2523–2527 CrossRef CAS PubMed.
- S. D. Heck, C. J. Siok, K. J. Krapcho, P. R. Kelbaugh, P. F. Thadeio, M. J. Welch, R. D. Williams, A. H. Ganong, M. E. Kelly and A. J. Lanzetti, Science, 1994, 266, 1065–1068 CrossRef CAS PubMed.
- Y. Raghuwanshi, H. Etayash, R. Soudy, I. Paiva, A. Lavasanifar and K. Kaur, J. Med. Chem., 2017, 60, 4893–4903 CrossRef CAS PubMed.
- S. D. Heck, W. S. Faraci, P. R. Kelbaugh, N. A. Saccomano, P. F. Thadeio and R. A. Volkmann, Proc. Natl. Acad. Sci. U. S. A., 1996, 93, 4036–4039 CrossRef CAS PubMed.
- A. L. Vagstad, T. Kuranaga, S. Püntener, V. R. Pattabiraman, J. W. Bode and J. Piel, Angew. Chem., Int. Ed., 2019, 58, 2246–2250 CrossRef CAS PubMed.
- B. I. Morinaka, A. L. Vagstad, M. J. Helf, M. Gugger, C. Kegler, M. F. Freeman, H. B. Bode and J. Piel, Angew. Chem., 2014, 126, 8643–8647 CrossRef.
- B. I. Morinaka, A. L. Vagstad and J. Piel, in Methods Enzymol, Elsevier, 2018, pp. 237–257 Search PubMed.
- P. D. Cotter, P. M. O'Connor, L. A. Draper, E. M. Lawton, L. H. Deegan, C. Hill and R. P. Ross, Proc. Natl. Acad. Sci. U. S. A., 2005, 102, 18584–18589 CrossRef CAS PubMed.
- A. Sikandar, L. Franz, S. Adam, J. Santos-Aberturas, L. Horbal, A. Luzhetskyy, A. W. Truman, O. V. Kalinina and J. Koehnke, Nat. Chem. Biol., 2020, 16, 1013–1018 CrossRef PubMed.
- B. Oberhauser, K. Baumann, B. Grohmann and H. Sperner, Synlett, 1999, 893–896 CrossRef CAS.
- Y. Hitotsuyanagi, Y. Matsumoto, S.-I. Sasaki, K. Yamaguchi, H. Itokawa and K. Takeya, Tetrahedron Lett., 2001, 42, 1535–1537 CrossRef CAS.
- R. B. Merrifield, J. Am. Chem. Soc., 1963, 85, 2149–2154 CrossRef CAS.
- R. B. Merrifield, Science, 1965, 150, 178–185 CrossRef CAS PubMed.
- R. B. Merrifield, Adv. Enzymol. Relat. Areas Mol. Biol., 1969, 32, 221–296 CAS.
- A. Eschenmoser, Angew. Chem., Int. Ed. Engl., 1988, 27, 5–39 CrossRef.
- A. Eschenmoser, Angew. Chem., 1988, 100, 5–40 CrossRef CAS.
- P. G. Arnison, M. J. Bibb, G. Bierbaum, A. A. Bowers, T. S. Bugni, G. Bulaj, J. A. Camarero, D. J. Campopiano, G. L. Challis, J. Clardy, P. D. Cotter, D. J. Craik, M. Dawson, E. Dittmann, S. Donadio, P. C. Dorrestein, K.-D. Entian, M. A. Fischbach, J. S. Garavelli, U. Göransson, C. W. Gruber, D. H. Haft, T. K. Hemscheidt, C. Hertweck, C. Hill, A. R. Horswill, M. Jaspars, W. L. Kelly, J. P. Klinman, O. P. Kuipers, A. J. Link, W. Liu, M. A. Marahiel, D. A. Mitchell, G. N. Moll, B. S. Moore, R. Müller, S. K. Nair, I. F. Nes, G. E. Norris, B. M. Olivera, H. Onaka, M. L. Patchett, J. Piel, M. J. T. Reaney, S. Rebuffat, R. P. Ross, H.-G. Sahl, E. W. Schmidt, M. E. Selsted, K. Severinov, B. Shen, K. Sivonen, L. Smith, T. Stein, R. D. Süssmuth, J. R. Tagg, G.-L. Tang, A. W. Truman, J. C. Vederas, C. T. Walsh, J. D. Walton, S. C. Wenzel, J. M. Willey and W. A. Van Der Donk, Nat. Prod. Rep., 2013, 30, 108–160 RSC.
- Z. Yan, W. Ming-Kui, P. Shu-Lin and D. Li-Sheng, Acta Bot. Sin., 2001, 43, 431–434 Search PubMed.
- H. P. Kaufmann and A. Tobschirbel, Chem. Ber., 1959, 92, 2805–2809 CrossRef CAS.
- Z. Wieczorek, B. Bengtsson, J. Trojnar and I. Z. Siemion, Pept. Res., 1991, 4, 275–283 CAS.
- Y. Y. Shim, Z. Song, P. D. Jadhav and M. J. T. Reaney, Trends Food Sci. Technol., 2019, 93, 197–211 CrossRef CAS.
- M. E. Tanner, Acc. Chem. Res., 2002, 35, 237–246 CrossRef CAS PubMed.
- S. Yamada, C. Hongo, R. Yoshioka and I. Chibata, J. Org. Chem., 1983, 48, 843–846 CrossRef CAS.
- B. Chan, A. Karton, C. J. Easton and L. Radom, J. Chem. Theory Comput., 2016, 12, 1606–1613 CrossRef CAS PubMed.
- P. Jadhav, G. Schatte, S. Labiuk, P.-G. Burnett, B. Li, D. Okinyo-Owiti, M. Reaney, P. Grochulski, M. Fodje and R. Sammynaiken, Acta Crystallogr., Sect. E: Struct. Rep. Online, 2011, 67, O2360–O2361 CrossRef CAS PubMed.
- G. Schatte, S. Labiuk, B. Li, P.-G. Burnett, M. Reaney, P. Grochulski, M. Fodje, J. Yang and R. Sammynaiken, Acta Crystallogr., Sect. E: Struct. Rep. Online, 2012, 68, o50–o51 CrossRef CAS PubMed.
- J. Kovacs, E. M. Holleran and K. Y. Hui, J. Org. Chem., 1980, 45, 1060–1065 CrossRef CAS.
- G. G. Smith and G. V. Reddy, J. Org. Chem., 1989, 54, 4529–4535 CrossRef CAS.
- P. D. Jadhav, J. Shen, R. Sammynaiken and M. J. T. Reaney, Chem.–Eur. J., 2015, 21, 17023–17034 CrossRef CAS PubMed.
- P. D. Jadhav, Y. Y. Shim and M. J. T. Reaney, RSC Adv., 2018, 8, 17702–17709 RSC.
- P. D. Jadhav, Y. Y. Shim and M. J. T. Reaney, Bioconjugate Chem., 2016, 27, 2346–2358 CrossRef CAS PubMed.
- J. R. Kramer and T. J. Deming, Biomacromolecules, 2012, 13, 1719–1723 CrossRef CAS PubMed.
- A. R. Rodriguez, U.-J. Choe, D. T. Kamei and T. J. Deming, Isr. J. Chem., 2016, 56, 607–613 CrossRef CAS.
- Y. Li, C. Lian, Z. Hou, D. Wang, R. Wang, C. Wan, W. Zhong, R. Zhao, Y. Wang, S. Li, F. Yin and Z. Li, Chem. Commun., 2020, 56, 3741–3744 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1ra04260b |
| This journal is © The Royal Society of Chemistry 2021 |