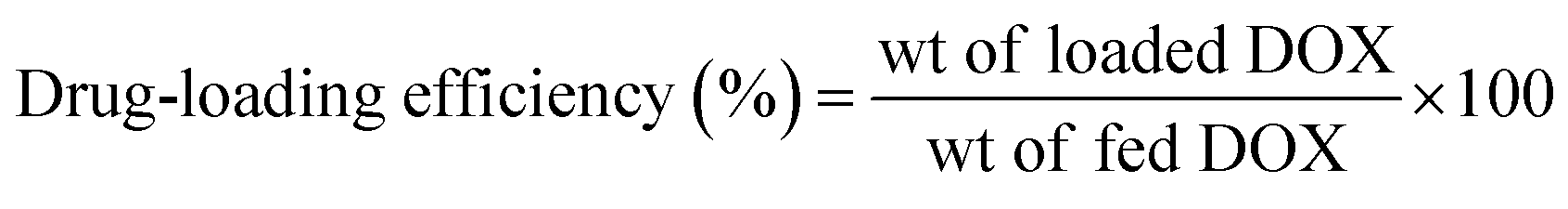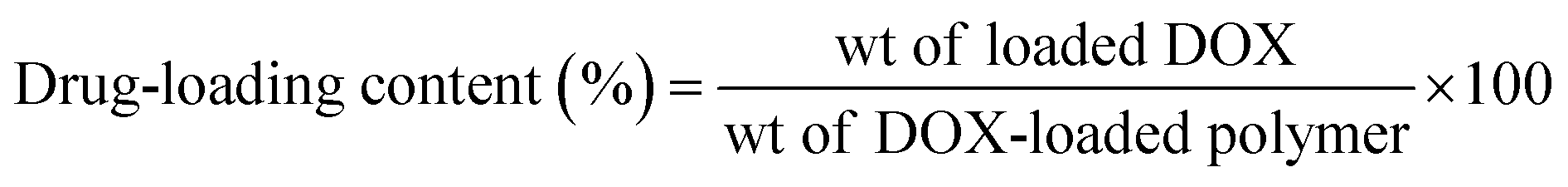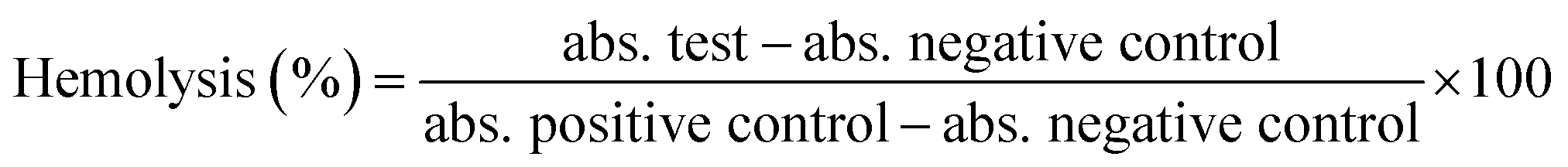DOI:
10.1039/D1RA04242D
(Paper)
RSC Adv., 2021,
11, 30532-30543
PEGylated ethyl cellulose micelles as a nanocarrier for drug delivery†
Received
1st June 2021
, Accepted 30th July 2021
First published on 8th November 2021
Abstract
Natural polymers provide a better alternative to synthetic polymers in the domain of drug delivery systems (DDSs) because of their renewability, biocompatibility, and low immunogenicity; therefore, they are being studied for the development of bulk/nanoformulations. Likewise, current methods for engineering natural polymers into micelles are in their infancy, and in-depth studies are required using natural polymers as controlled DDSs. Accordingly, in our present study, a new micellar DDS was synthesized using ethyl cellulose (EC) grafted with polyethylene glycol (PEG); it was characterized, its properties, cell toxicity, and hemocompatibility were evaluated, and its drug release kinetics were demonstrated using doxorubicin (DOX) as a model drug. Briefly, EC was grafted with PEG to form the amphiphilic copolymers EC-PEG1 and EC-PEG2 with varying PEG concentrations, and nano-micelles were prepared with and without the drug (DOX) via a dialysis method; the critical micelle concentrations (CMCs) were recorded to be 0.03 mg mL−1 and 0.00193 mg mL−1 for EC-PEG1 and EC-PEG2, respectively. The physicochemical properties of the respective nano-micelles were evaluated via various characterization techniques. The morphologies of the nano-micelles were analyzed via transmission electron microscopy (TEM), and the average size of the nano-micelles was recorded to be ∼80 nm. In vitro, drug release studies were done for 48 h, where 100% DOX release was recorded at pH 5.5 and 52% DOX release was recorded at pH 7.4 from the micelles. In addition, cytotoxicity studies suggested that DOX-loaded micelles were potent in killing MDA-MB-231 and MCF-7 cancer cells, and the blank micelles were non-toxic toward cancerous and normal cells. A cellular uptake study via fluorescence microscopy indicated the internalization of DOX-loaded micelles by cancer cells, delivering the DOX into the cellular compartments. Based on these studies, we concluded that the developed material should be studied further via in vivo studies to understand its potential as a controlled DDS to treat cancer.
1. Introduction
Amphiphilic polymers have been explored as micelles for several decades and they have been extensively studied for various applications.1 In pharmaceutics, polymeric micelles (PMs) gained attention as DDSs because of their nanosize and their ability to solubilize hydrophobic molecules and shield them until the molecules are used. In vivo studies documented that these micelles have higher drug loading capacities and enhanced stability in comparison to other nanosystems like liposomes, nanoparticles, and dendrimers.2,3 They demonstrate prolonged circulation times in the bloodstream, avoiding rapid clearance by the renal system and reticuloendothelial system (RES).4 Micelles in an aqueous medium have an exposed hydrophilic corona, which is responsible for the protection of these systems from the RES; it also enhances the permeability of PMs and regulates the pharmacokinetic behavior. In addition, the inner hydrophobic domain supports and stabilizes hydrophobic drugs, allowing slow and sustained release.2 It has been well documented that nanosized PMs can extravasate and penetrate tumors and their extracellular matrix due to the loosely formed blood vessels and sluggish blood flow, thus generating the enhanced permeability and retention (EPR) effect.5,6 As a result, PMs become accumulated and deliver drugs at a controlled rate for a sustainable period, thereby decreasing drug resistance and increasing the effectiveness of therapy. Accordingly, anti-cancer formulations like Genexol-PM® and Nanoxel® have been developed and approved by the FDA for human treatment. In line with these studies, advanced PM formulations (e.g., NK105, NK911, and NC-6004) are being studied, and some are in the clinical trial stage for anti-cancer therapy.7
Generally, the designed PMs use synthetic block co-polymers and triblock copolymers and are extensively being studied for drug delivery applications.1 However, synthetic PMs are less compatible with human anatomy, showing high immunogenicity, as their interactions with human cells are less favorable. Further, to produce these PMs, complex protocols are involved, and the cost of manufacture is exorbitant; therefore, the application of these materials is unattainable for a wide range of the population. Currently, PM formulations designed from natural and renewable polymers are attracting much attention, because they are highly biocompatible with low immunogenicity. With proteins and polysaccharides being an integral part of humans, they are well tolerated and, further, developing these materials as nanoassemblies may not be expensive, as the design protocols would be less expensive and the materials are easily available from renewable resources and are safe to use. Accordingly, micelles are being investigated based on proteins (albumin, gelatin, zein, etc.) and polysaccharides (hyaluronic acid, chitosan, pullulan, dextran, alginate, xyloglucan, inulin, etc.) using functional modification to attain self-assembled nanocarriers.8
Polysaccharide-based DDSs are reported to form nano-micelles at very low concentrations in an aqueous medium, demonstrating the ability to enter cells and improve the drug pharmacokinetics, and providing a platform for sustained and controlled drug release.8 Ethyl cellulose (EC), a derivative of polysaccharide cellulose, is being explored for use in the fields of cosmetics, food additives, adhesives, and medicine owing to its chemically inert nature, stability, sustained drug release abilities, and good biocompatibility.9,10 In a report by Balzus et al., EC displayed extended-release behavior in comparison to polymers like Eudragit RS 100 and other lipids.11 However, there are very few reports about the application of EC in drug delivery systems, other than in oral and topical administrations. Nevertheless, S. Leitner et al. reported the development of EC nanoparticles as a new transfection tool for antisense oligonucleotide delivery.12 The hydrophobic nature of EC has benefits in terms of sustained release for oral administration; however, its hydrophobicity is a hurdle preventing its use in parenteral administration, as its hydrophobicity activates the macrophages of the immune system. To overcome this, the structure of EC needs to be modified without sacrificing its desirable properties. Therefore, the synthesis of EC-grafted polymers has attracted significant attention in recent years. For instance, Yuan et al. reported the synthesis, characterization, and in vitro degradation properties of EC-graft-poly(ε-caprolactone)-block-poly(L-lactide) copolymers obtained via sequential ring-opening polymerization.13 One of the best choices for drug delivery pharmaceutics is FDA-approved PEG because of its tunable properties and well-established safety profile in terms of biocompatibility and non-immunogenicity. Several polymers with different properties have been PEGylated to prolong their blood circulation times, shield them from the immune system, and allow them to complete their target function.14 For example, the PEGylation of cellulose or its derivatives has been explored to improve the dispersibility and colloidal stability. For instance, Lasseuguette grafted PEG with micro-fibrillated cellulose via EDC-coupling.15 T. Kaldéus et al. reported the PEGylation of TEMPO-oxidized cellulose and studied its colloidal stability.16 EC is a well-established material for oral delivery; however, there are few reports on the use of EC for systemic applications due to its drawbacks, as explained above. As the properties of EC are favorable for drug delivery, its potential application in invasive delivery has been explored via addressing its incompatibility and immunogenicity through grafting with PEG. For example, Huang et al. synthesized thermosensitive micelles from an amphiphilic graft copolymer of EC-poly(PEG methyl methacrylate), EC-PMMA, for systemic drug delivery applications.17 In their studies, they designed and developed EC-PMMA in the form of nano self-assemblies and characterized the thermosensitive behavior. However, a detailed study of EC-PMMA nano-assemblies for drug delivery and cytotoxicity and hemocompatibility studies have not been reported to support their potential for systemic applications. In accordance with these studies, we aimed to develop EC-PEG nano-micelles via a simple method, to evaluate their physicochemical properties, and to carry out in vitro studies, such as cytotoxicity, drug delivery, cell interaction, and hemolysis studies, so as to understand their potential for systemic applications. We have chosen doxorubicin (DOX) as a model drug for loading EC-PEG micelles for anti-cancer therapy in the form of a CDDS. DOX is a potent anti-cancer drug used in chemotherapy to treat solid tumors, lymphomas, soft tissue sarcomas, etc. However, its application at higher doses is limited because of its side effects, like cardiotoxicity, and its role in the development of tumor cell resistance.18 To mitigate these limitations, we designed amphiphilic copolymer-based micelles.
In this study, EC was converted to carboxylated EC (CEC) via TEMPO-mediated oxidation and this was subsequently grafted with mPEG(2000) via N,N′-dicyclohexylcarbodiimide (DCC) coupling. mPEG-grafted EC (EC-PEG) was developed in the form of micelles in an aqueous medium and loaded with hydrophobic DOX. The release pattern of the drug showed a sustained release profile. The in vitro effectivity of the system was also investigated toward the breast cancer cell lines MDA-MB-231 and MCF-7. Herein, the use of EC in the form of micelles as a DDS to deliver DOX is reported for the first time.
2. Experimental methods
2.1 Materials
Ethyl cellulose (ethoxy content: 44–51%, 18–24 mPa s) was procured from S.D. Fine Chemicals, India. Doxorubicin HCl (hydrochloride) and 4-dimethylaminopyridine (DMAP) were obtained from Himedia Laboratories, India. Poly(ethylene glycol) methyl ether (Mw: 2000; mPEG) and (2,2,6,6-tetramethylpiperidin-1-yl)oxidanyl (TEMPO) were procured from Aldrich, USA. N,N′-Dicyclohexylcarbodiimide (DCC) was procured from Spectrochem Pvt. Ltd, India. DMEM (Dulbecco's modified Eagle's medium), MEM (minimal essential medium), and MTT (3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide) were purchased from Invitrogen, India. All solvents and salts were of analytical grade and used without any further purification. Cell lines were procured from the National Centre for Cell Science (NCCS) Pune.
2.2 Synthesis and characterization of the EC-PEG graft polymer
2.2.1 TEMPO-mediated oxidation of EC. The oxidation of the hydroxy groups of EC (–CH2OH) was done according to a reported literature method by J. Araki et al.,19 as shown in Fig. 1. Briefly, TEMPO (0.4 g) and sodium bromide (4 g) were added to 400 mL of an aqueous suspension of EC (10 mg mL−1, 4 g), and this was stirred at room temperature (RT) for 15 min. The oxidation was initiated via the addition of 60 mL of NaClO (sodium hypochlorite) solution, where the concentration of NaClO was maintained at 25 wt% with respect to EC (1.25 g of NaClO for 5 g of EC). The solution pH was maintained between 10 and 11 while stirring at RT for 4 h. After 4 h, NaCl (30 g) was added to the reaction mixture and the EC suspension was precipitated. The product was filtered through a pore-fritted glass filter and washed with 0.5–1.0 M NaCl. The product was dispersed in NaCl of the same concentration and centrifuged at 12![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 rpm for 30 min at 25 °C. This washing procedure was repeated 3 times to remove unreacted NaClO. The EC sodium salt was converted to the free acid form via washing it twice with 0.1 N HCl. The obtained product was dialyzed against deionized water for 3 days with frequent water changes. The colloidal carboxylic EC obtained was dried, quantified, and characterized to confirm the product (yield: 2.32 g, 58%).
000 rpm for 30 min at 25 °C. This washing procedure was repeated 3 times to remove unreacted NaClO. The EC sodium salt was converted to the free acid form via washing it twice with 0.1 N HCl. The obtained product was dialyzed against deionized water for 3 days with frequent water changes. The colloidal carboxylic EC obtained was dried, quantified, and characterized to confirm the product (yield: 2.32 g, 58%).
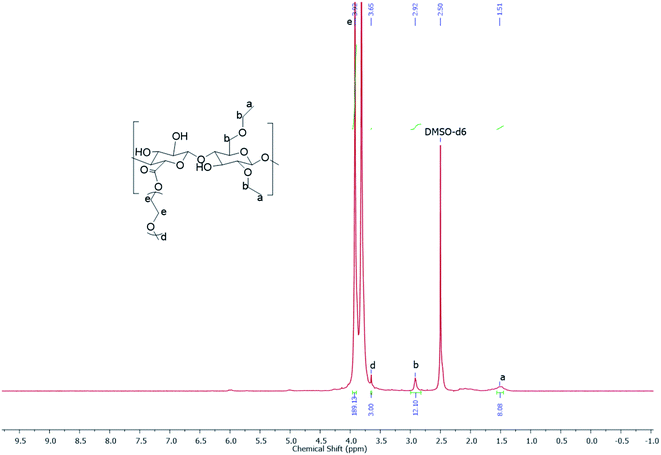 |
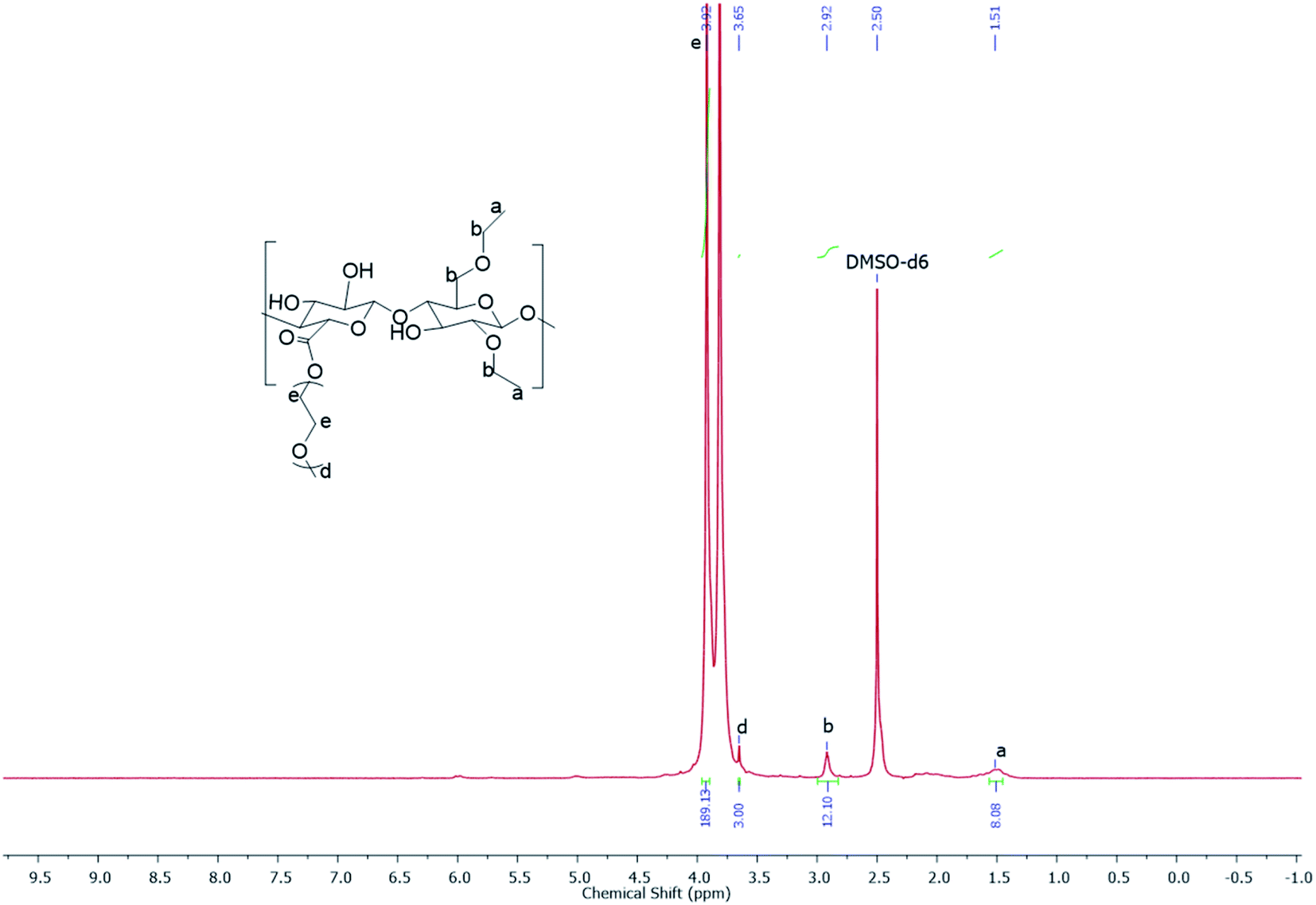
| | Fig. 1 The 1H NMR spectrum of EC-PEG1 recorded using a Bruker AV-200 NMR spectrometer operating at a frequency of 200 MHz with DMSO-d6 as the solvent. | |
2.2.2 Determination of the carboxylate charge density of EC. The carboxylate charge density of EC after the oxidation reaction was estimated via the conductometric titration method. Briefly, TEMPO-oxidized EC (240 mg) was dispersed in 30 mL of de-ionized (DI) water. This dispersion was stirred and sonicated for 30 min in a bath sonicator (Bio-Technics, India) and left standing overnight. Later, 10 mL of the dispersion was added to 80 mL of DI water. Subsequently, 50 μL of concentrated HCl was added to the carboxylated EC (CEC) dispersion and this was titrated against 0.5 M NaOH. The conductivity of the solution was measured (Orion Star, Thermo Scientific Conductometer, India) upon each addition of 20 μL of 0.5 M NaOH to the CEC-HCl dispersion. The degree of carboxylated EC was estimated via plotting a graph of the conductivity of the CEC-HCl dispersion against the volume of NaOH.
2.2.3 PEGylation of carboxylated EC (CEC). mPEG (Mw 2000) was grafted to CEC using DCC reagent and the catalyst DMAP. For example, in a two-necked RB flask, CEC [400 mg, (0.33 mmol (calculated using the titration eqn (S1)))] was dispersed in 10 mL of dry tetrahydrofuran (THF) and stirred for 45 min under a N2 atmosphere at RT. A solution of DCC (0.99 mmol) and DMAP (0.165 mmol) prepared in dry THF was added to the CEC solution and stirred for 2 h at RT. Later, to the solution mixture, mPEG (0.33 mmol) dissolved in 5 mL of dry THF was added dropwise using a syringe. Following this, the reaction mixture was stirred under a nitrogen atmosphere at 40 °C for 48 h. After 48 h, the reaction was stopped and the solution was filtered through Whatman's filter paper to collect the filtrate. This filtrate was concentrated using a rotary evaporator at 50 °C and then precipitated in diethyl ether. The diethyl ether was evaporated and the precipitate was dispersed in DI water and dialyzed (10 kDa cut-off) against DI water for 48 h to remove the unreacted mPEG and reagents, obtaining an EC to PEG ration of 1:1. Similarly, in another set of reactions, mPEG (0.66 mmol) was reacted with EC following the same procedure and reaction conditions to obtain an EC to PEG ratio of 1:2. The final EC-PEG compounds (1:1 and 1:2), labeled EC-PEG1 and EC-PEG2, respectively, were characterized via 1H NMR, 13C NMR, and FTIR studies. The yield of this reaction was 320 mg (80%).
2.2.4 Critical micellar concentration (CMC) of EC-PEG. Following a protocol reported by J. Chen et al.,20 the CMCs of EC-PEG1 and EC-PEG2 were determined. The hydrophobic probe pyrene was used for estimating the CMCs of the respective EC-PEG materials in water via fluorescence spectroscopy. A total of 25 μL of pyrene in acetone solvent (20 μg mL−1) was added to individual 5 mL vials, and the acetone was allowed to evaporate to obtain dry pyrene. To these respective vials, 5 mL of an aqueous solution of EC-PEG copolymer was added, with varying concentrations from 0.00025 to 0.09 mg mL−1. The final concentration of pyrene in each sample solution was 0.1 μg mL−1. The excitation spectra (300–360 nm) of the solutions were recorded at an emission wavelength of 395 nm. The ratios of the peak intensity at 338 nm to the intensity at 334 nm (I338/I334) of the excitation spectra were recorded and plotted as a function of the polymer concentration (logC (mg mL−1)).
2.3 Preparation and characterization of micelles
2.3.1 Preparation of EC-PEG micelles. The dialysis method was used for fabricating micelles of EC-PEG (1:1 and 1:2). Briefly, EC-PEG1 (20 mg) was dissolved in 1 mL of DMF, and 10 mL of PBS buffer (pH 7.4) was added dropwise while sonicating at a 20% amplitude (Vibracell, VCX 500, USA). Later this solution was dialyzed against DI water for 24 h using a dialysis membrane with a 10 kDa cut-off to obtain micelles. These micelles were lyophilized to obtain dry powder, which was used for characterization. In a similar fashion, EC-PEG2 micelles were also prepared and characterized.
2.3.2 Preparation of DOX-loaded EC-PEG micelles. EC-PEG micelles loaded with DOX were prepared at 0.1% (w/v) following the described procedure. Briefly, DOX (1 mg) was dissolved in 1 mL of DMF, 1.5 eq. of triethylamine was added, and the mixture was stirred at RT. Later, 20 mg of EC-PEG1 was added to this DOX solution and this was stirred for 1 h while maintaining RT conditions. To this, PBS buffer (10 mL) was added dropwise under sonication at a 20% amplitude. Further, this solution was dialyzed against DI water for 24 h using a membrane with a 10 kDa cut-off. After dialysis, the obtained DOX-loaded micelles were lyophilized to give DOX-EC-PEG1 powder, which was red in color. The same protocol was followed to obtain DOX-loaded micelles of EC-PEG2. The developed formulations are shown in Table 1. The micelles with and without DOX were characterized via various physicochemical characterization methods.
Table 1 Formulations and their characteristics
| Batch |
EC-PEG1% (w/v) |
DOX% (w/v) |
Hydrodynamic diameter (nm) |
PDI |
Drug-loading efficiency (%) |
Drug-loading content (%) |
| B1 |
2 |
— |
153.5 |
0.293 |
— |
— |
| B2 |
2 |
0.1 |
204.9 |
0.122 |
36.73 |
1.73 |
| B3 |
2 |
0.15 |
257.7 |
0.297 |
52.73 |
4.11 |
| Batch |
EC-PEG2% (w/v) |
DOX% (w/v) |
Hydrodynamic diameter (nm) |
PDI |
Drug-loading efficiency (%) |
Drug-loading content (%) |
| B4 |
2 |
— |
249.9 |
0.191 |
— |
— |
| B5 |
2 |
0.1 |
338.6 |
0.180 |
32.93 |
1.55 |
| B6 |
2 |
0.15 |
350.5 |
0.357 |
52.51 |
3.66 |
2.4 Characterizations
2.4.1 NMR spectroscopy. 1H NMR spectra for the characterization of mPEG and EC-PEG were recorded using a Bruker AV-200 NMR spectrometer operating at a 1H frequency of 200 MHz. 1H spectra of all compounds were recorded in DMSO-d6 (concentration of polymer = 50 mg mL−1). The samples were homogenized before recording the NMR spectra. Similarly, 13C NMR spectra of mPEG and EC-PEG were also recorded using a Bruker AV-500 NMR spectrometer operating at a 13C frequency of 125 MHz.
2.5 Physicochemical characterization of the micelles
The size distribution, hydrodynamic diameter, and charge density data for blank and DOX-loaded micelles were determined via DLS (90 Plus Brookhaven Instruments Corp, PALS zeta potential analyzer, USA). The morphologies of the micelles were investigated via TEM (FEI Tecnai TF20, 200 kV FEG high-resolution transmission electron microscope, USA). For TEM analysis, samples were diluted (5×) in Millipore water, stained with 1% (w/v) uranyl acetate dye, and drop-cast on a copper grid mesh.
The drug loading efficiency and content percentages were determined using a UV-vis spectrophotometer (UV 1601 PC UV spectrophotometer, Shimadzu, Japan). As an example, 1.5 mg of DOX-loaded micelles were dissolved in 1 mL of DMF and filtered, and the DOX content was estimated using the standard DOX calibration curve recorded at 480 nm. The drug loading efficiency and loading content percentages were calculated using the following equations:
| |
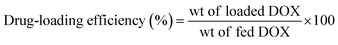 | (1) |
| |
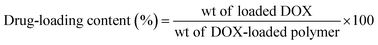 | (2) |
FTIR spectroscopy analysis was conducted on DOX, bare polymers (EC and mPEG), grafted copolymers (EC-PEG1 and EC-PEG2), and DOX-loaded micelles (batches 3 and 6) to confirm the grafting of mPEG with EC and the functional interactions with DOX. For FTIR, samples (2–3 mg) were ground with anhydrous potassium bromide, KBr, (97 mg) to form a pellet, which was used for analysis (PerkinElmer spectrometer I, FT-IR diffuse reflectance (DRIFT) mode, USA). The scans were recorded from 400 cm−1 to 4000 cm−1 with an average of 10 scans per sample.
Thermal analysis was carried out using DSC (Model Q100 DSC, TA instruments, Newcastle, DE, USA) to investigate the thermal properties of EC in its pristine form, after grafting, and after DOX loading (batches 3 and 6). The samples were crimped in aluminum pans and the thermal properties were analyzed in cycles. In the first heating cycle, the temperature was ramped from −70 °C to 100 °C at a rate of 10 °C min−1. The sample was then cooled to −70 °C in the 2nd cycle at a rate of 10 °C min−1, and in the third heating cycle, the temperature was ramped from −70 °C to 100 °C at a rate of 10 °C min−1. The entire experiment was done under nitrogen gas (50 mL min−1). An empty aluminum pan was used as a reference pan.
2.5.1 In vitro DOX release. The release of DOX from the micelles was studied via dispersing 5 mg of DOX-loaded micelles (B3 and B6) in 10 mL of buffer and enclosing them in a dialysis membrane tube (Mw cut-off: 12 kDa). This dialysis bag was kept in 20 mL of phosphate buffer (100 mM, pH 7.4) and incubated at 37 °C in a shaker bath (Julabo SW23) at 100 rpm. At regular intervals of time, 1 mL was withdrawn from the buffer and 1 mL of fresh buffer was added to maintain the sink conditions. The withdrawn buffer was analyzed for the amount of DOX released using a UV-vis spectrophotometer at 480 nm. Similarly, release studies were performed in phosphate buffer at pH 5.5.
2.5.2 Hemolysis assays. To study the biocompatibility of the developed graft polymer toward RBCs (red blood cells), hemolysis assays were conducted. Briefly, blood was collected in tubes containing EDTA from healthy volunteers at the National Chemical Laboratory, Pune, India. The RBCs were separated from the whole blood by density gradient centrifugation. As an example, 5 mL of whole blood was added slowly to 5 mL of PBS and centrifuged at 2000 rpm for 30 min. The supernatant, which was devoid of RBCs, was discarded and the pellet was washed thrice with PBS and centrifuged again for 30 min at 2000 rpm. Later, the cells were dispersed in PBS to prepare a 2% (v/v) stock dispersion. Further, 2 mL of this stock dispersion was dispensed in 2 mL vials in duplicate, and test samples, such as EC, CEC, B1, B3, B4, and B6, at concentrations of 0.5 mg mL−1 and 1 mg mL−1 were added to the respective vials. Respective controls, like a RBC suspension in PBS (negative) and a RBC suspension in DI water (positive), were prepared. All respective test samples and controls were incubated at 37 °C for 2 h under gentle shaking every 30 min to re-suspend precipitated RBCs. After the incubation period, the suspensions were centrifuged at 1500g for 10 min at RT. The obtained supernatant was dispensed in a 96-well plate, and the hemoglobin (Hb) release was obtained spectrophotometrically using a microtiter plate reader at 540 nm (Multiskan Ex, (51118170 (200–240 V) Thermo Scientific, Finland)). Considering 100% cell lysis in DI water and 0% lysis in PBS, the hemolysis percentages for test samples were calculated using the following equation.| |
 | (3) |
2.6 Cell studies
The fibroblast cell line L929 and breast cancer cell line MDA-MB-231 were maintained in DMEM supplemented with 10% FBS under standard conditions at 37 °C in a humidified CO2 incubator. Under similar incubation conditions, the breast cancer cell line MCF-7 was also cultured in MEM supplemented with 10% FBS. Cells were routinely grown as monolayer cultures in a 25 cm2 flask and passaged once a week using trypsin/EDTA at 80% confluence.
2.6.1 Cytotoxicity assays. Blank micelles, B1, and B4 were investigated for their cellular toxicity toward L929 fibroblast cells via MTT assays. A confluent flask of L929 cells was trypsinized to harvest cells, which were further seeded in a 96-well plate at 10000 cells per well. The plate was incubated at 37 °C under a 5% CO2 humidified atmosphere for 16 h to allow cells to attach and form a monolayer. Later, the media from the wells was flicked off, and B1 and B4 micelles were added, respectively. Stock concentrations of micelles at 1 mg mL−1 were prepared in serum-free media. From these, a series of concentrations ranging from 0 to 400 μg mL−1 was added to the respective wells, which were incubated for 48 h. Post incubation, the media was removed from the wells and MTT solution prepared in DMEM-FBS medium was added to the wells. Following this, the plate was incubated in the dark at 37 °C in a humidified CO2 incubator for 4 h. Finally, the MTT media in the wells was replaced with 100 μL of DMSO and the plate was read at 550 nm using a plate reader (Multiskan Ex, (51118170 (200–240 V) Thermo Scientific, Finland)). Cells grown in wells devoid of any test sample were considered as a positive control and cells incubated in media with 30% DMSO (v/v) were the negative control. The relative cell viabilities were calculated via comparing the absorbance read from test samples to the positive control [(abs. sample/abs. positive control) × 100]. Data are presented as average ± SD (n = 3). The cytotoxicity of B1 and B4 micelles was also evaluated toward MDA-MB-231 cells via this assay using the same parameters. Similarly, the MTT assay was also carried out for DOX-loaded micelles (B3 and B6) in MDA-MB-231 and MCF-7 cells for 72 h.
2.6.2 In vitro cellular uptake. The uptake of the B3 and B6 micelles was studied in MDA-MB-231 cells. 5 × 104 cells were seeded on sterile 12 mm round coverslips placed in a 24 well plate, and these were incubated for 24 h at 37 °C under a 5% CO2 atmosphere. After incubation, the media were aspirated from the wells followed by washing thrice with PBS. Free DOX at a concentration of 4 μg mL−1 and DOX-loaded micelles (B3 and B6) at 100 μg mL−1 containing an equivalent amount of DOX (calculated based on the loading efficiency) were dispersed in serum-free media and incubated for 4 and 8 h separately. Later, the cells were washed thrice with PBS and fixed via adding 300 μL of 4% paraformaldehyde and incubating for 15 min at RT. After the fixing step, the cells were washed extensively with PBS and incubated with Alexa Fluor 488 phalloidin (60 nM) for 30 min in the dark to colorize the actin filaments, so as to allow the identification of the cytoplasm and cell boundaries. The nuclei were stained with DAPI (300 nM) for 15 min in the dark, which was washed off with PBS. Finally, the coverslips were mounted on a clean slide with the application of mounting media (Fluoroshield). Excess mounting media was dabbed off with tissue, and the cells were visualized using an epifluorescence microscope (Carl Zeiss, model: Axio Observer Z1, oil-emersion objective, 63×). The nuclei stained with DAPI were observed under the blue channel, the cytoskeletons stained with Alexa Fluor 488 under the green channel, and DOX under the red channel.
3. Results and discussion
EC, a natural polymer derivative, was grafted covalently with mPEG to develop an amphiphilic copolymer that assembles into micellar structures in an aqueous medium. The EC core of the micellar architecture was used for loading the anti-cancer drug DOX. The purpose of the mPEG graft on the outer layer is to protect the drug carrier from being recognized by the immune system and to prolong the micelle circulation time in the blood for effective therapy.
3.1 TEMPO-mediated oxidation and the determination of the carboxylate charge density on EC
TEMPO-mediated oxidation has been explored over the last few decades for the conversion of polysaccharide alcoholic hydroxyls to carboxyls under aqueous alkaline conditions.21 It is a highly selective reaction and oxidizes only the primary hydroxyl group at carbon (C-6) on the EC backbone. TEMPO, in combination with NaBr and NaClO, efficiently converts the hydroxyl groups to carboxylates via aldehydes. Under alkaline conditions, the selectivity of TEMPO is further enhanced; hence, the oxidation of EC was performed at pH 10–11.22–24 The degree of substitution of primary hydroxyl groups of EC with carboxylate groups after TEMPO oxidation was calculated via conductometric titration, as described in the ESI.† From the graph in Fig. S2 and eqn (S1),† the density of EC carboxylate groups was calculated to be 625 mmol kg−1. It was observed that with an increase in the volume of NaClO, the degree of carboxylation was increased, which is similar to the reported literature.19 The concentration of NaClO was optimized at 25% relative to the dry weight of EC to obtain a carboxylate content of ∼600 mmol kg−1 in EC. The reproducible yield of this reaction was 58%. The carboxylate groups are available for further modification with mPEG so as to increase the hydrophilicity of EC.
3.2 PEGylation of carboxylated EC (CEC)
After the confirmation of the oxidation of EC based on conductometric titration, the product, CEC, was used for synthesizing graft copolymers of EC-PEG via esterification. The reaction was carried out in THF; the carboxylic acid of CEC was deprotonated via DMAP, and DCC was nucleophilically added to give the intermediate O-acylisourea, a good leaving group. The hydroxyl groups of mPEG were nucleophilically reacted with this intermediate to form the desired ester bond between the carboxyl group of CEC and the hydroxyl group of mPEG.25 Along with the grafted copolymer, the side product dicyclohexyl urea (DCU) was obtained, which was removed during the filtration steps. The grafting of mPEG onto CEC was performed with 1 and 2 equivalents, and the products were labeled as EC-PEG1 and EC-PEG2, respectively. The final products were dialyzed against DI water to remove unreacted mPEG and reagents, and the products were finally confirmed via 1H NMR and 13C NMR spectroscopy analysis (see the ESI†) in DMSO-d6. The yield of this reaction was optimized to 80%. Fig. 1 shows the 1H NMR spectrum (DMSO-d6) of the EC-PEG1 copolymer with the following peaks (ppm): δ 3.92 (–OCH2CH2O–), δ 3.65 (CH3OCH2CH2OH), δ 1.51 (–CH2CH3 of ethyl groups), and δ 2.92 (CH3CH2OCH–, CH3CH2OCH2CH). Integration of the peaks corresponding to the terminal methyl (–CH3) of mPEG at 3.65 ppm and the methylene (–CH2CH2–) group of ethylene glycol at 3.92 ppm suggests 46 repeating units, corresponding to mPEG2000. From the reported literature,25–29 in the 1H NMR spectrum of ethyl cellulose (EC), the protons of the EC backbone appear between 3.4 and 4.5 ppm. The peaks of mPEG also appear in a similar region, which resulted in the merging of the peaks of protons of EC with the protons of mPEG.
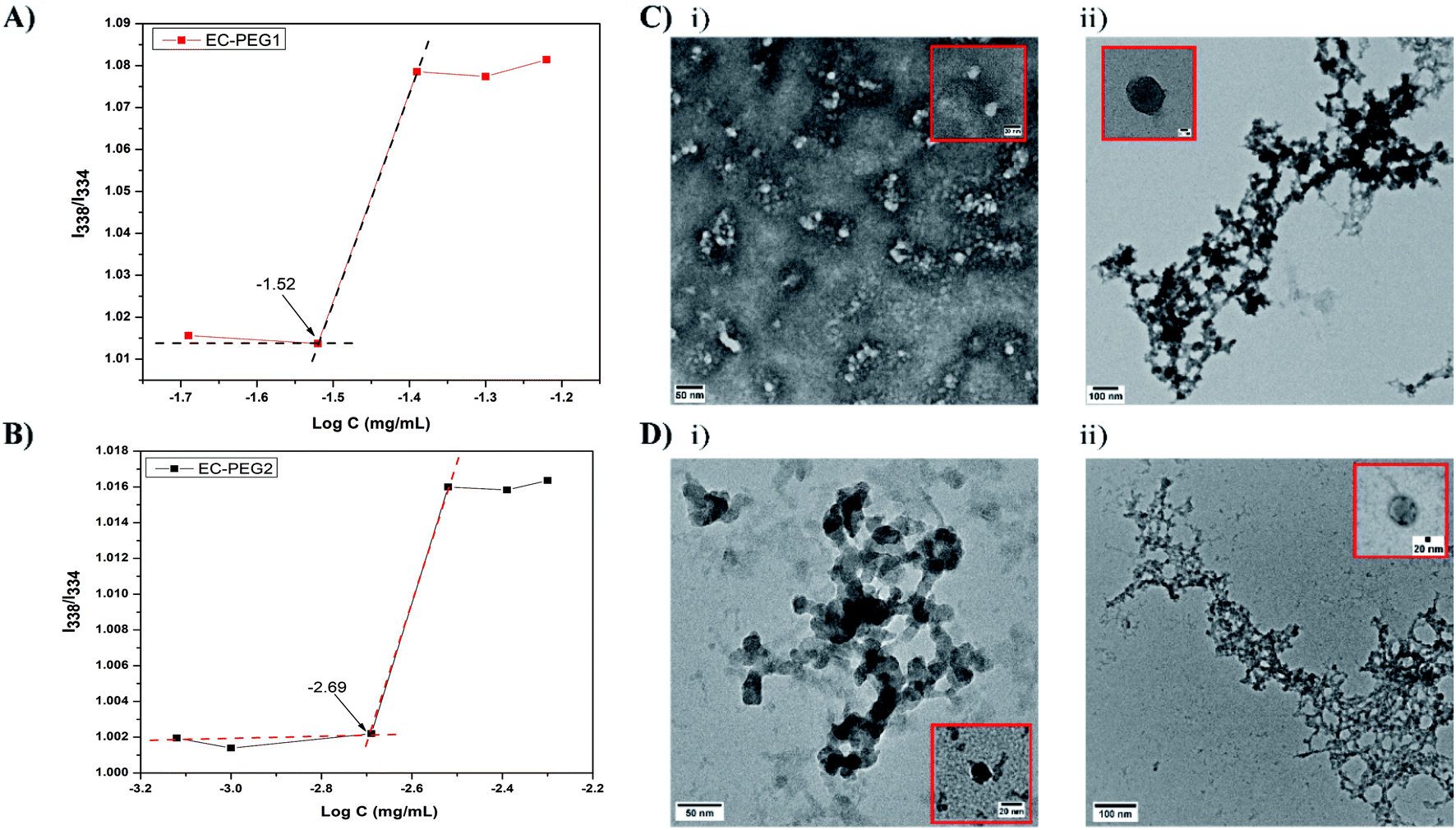
The CMC is an important factor deciding the micelle formation capacity of any amphiphilic polymer. To determine the CMCs of the developed graft copolymers of EC-PEG, the well-documented method of fluorometry was used with the hydrophobic probe pyrene. The excitation spectra of the probes with increasing concentrations of polymer were monitored at an emission wavelength of 395 nm. Fig. 2A and B shows the I338/I334 intensity ratios plotted against the log of concentration, varying from 0.00025 mg mL−1 to 0.09 mg mL−1. The CMC value was taken from the intersection point of the tangent to the curve at high concentrations with a horizontal line passing through the point at low concentrations. From the graphs, the CMCs of EC-PEG1 (Fig. 2A) and EC-PEG2 (Fig. 2B) were observed to be 0.03 mg mL−1 and 0.00193 mg mL−1, respectively. Therefore, with an increase in the amount of mPEG grafting to EC, the CMC value dropped. This suggested that in an aqueous medium, EC-PEG2 formed micelles at a much lower concentration compared to EC-PEG1. The grafting of 2 equivalents of mPEG to EC made the molecules more amphiphilic compared to 1 equivalent of mPEG. This observation was similar to that reported in the literature by Chen et al., where a polymer modified with PEG with a Mw of 5000 had a lower CMC value than that modified with PEG with a Mw of 2000.20 A lower CMC value signifies the ability of a polymer to maintain a stable micellar structure under diluted conditions, like in the bloodstream.30
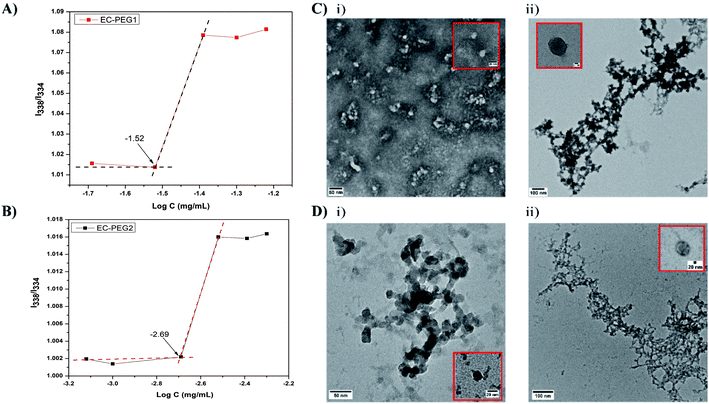 |
| | Fig. 2 (A) The critical micelle concentration (CMC) of EC-PEG1 derived from a plot of the I338/I334 ratio vs. the copolymer concentration. (B) The CMC of EC-PEG2 derived from a plot of the I338/I334 ratio vs. the copolymer concentration. (C) (i) A TEM image of B1 micelles (scale bar: 50 nm) and (ii) a TEM image of B3 micelles (scale bar: 100 nm). (D) (i) A TEM image of B4 micelles (scale bar: 50 nm) and (ii) a TEM image of B6 micelles (scale bar: 100 nm). | |
3.3 The preparation and physicochemical characterization of micelles
Table 1 shows the various formulations of EC-PEG with and without DOX at various concentrations, which were abbreviated as B1, B2, B3, B4, B5, and B6. Micelles of EC-PEG (B1 and B4) were prepared via dissolving the polymer in DMF and dialyzing these against DI water. Similarly, DOX-loaded micelles were also fabricated via dissolving the copolymer along with DOX in DMF and dialyzing against DI water. Fig. 2C and D shows the morphologies and size distributions of the prepared micelles, which were analyzed via DLS and TEM. From the DLS measurements, as shown in Table 1, it was seen that the sizes (B1 = 153.5 and B4 = 249.9 nm) of the micelles increased with the addition of DOX; this confirmed the interaction and entrapment of DOX within the hydrophobic cores of the micelles. TEM images of blank and DOX-loaded micelles shown in Fig. 2C and D present average micelle sizes below 150 nm, which are lower than the mean diameters measured via the DLS method. It should be noted that the mean diameters of the micelles determined via the DLS method depend on the size of the biggest micelle. Further, DLS measures the hydrodynamic diameters of solvated micelles whereas TEM measures dried micelles.12 The PEG chains in the outer domains of the micelles are hydrophilic and hold water molecules, which is reflected in the DLS measurements; hence, formulations with a higher content of grafted mPEG (B4, B5, and B6) are larger in size. All these formulations possessed net negative charge on their surfaces (−15 mV to −42 mV), which was due to the OH− groups on the PEG chains of the micelles. The formulations prepared from EC-PEG2 had slightly more negative charge than the formulations prepared from EC-PEG1, which could be explained due to the additional mPEG grafting. The drug-loading efficiency was determined via UV-vis spectrophotometric analysis, and the obtained values are shown in Table 1. The formulations with higher drug-loading efficiencies, namely B3 and B6 (52.73% and 52.51%, respectively), were chosen for further studies.
The functional group interactions relating to the polymers and their formulations in the form of micelles were characterized via FTIR spectroscopy. The FTIR spectrum of EC (Fig. S7A†) showed characteristic peaks at 3485 cm−1, due to O–H stretching, and at 2974 cm−1, 2872 cm−1, and 1376 cm−1, due to C–H stretching and bending. The peak noticed at 1060 cm−1 corresponds to C–O–C groups, while the one at 1120 cm−1 is particular to C–C stretching.31 Bare mPEG showed a broad absorption peak from terminal O–H that appeared at around 3421 cm−1. Characteristic C–H bending bands were observed at 2880 cm−1, 1475 cm−1, 948 cm−1, and 842 cm−1. The peaks that were visible at 1150 cm−1 and 1060 cm−1 were due to the ether linkage of the C–O–C groups (Fig. S7A†).32–34 The spectrum of EC-PEG1, as shown in Fig. S7B,† showed peaks at 3465 cm−1, 2972 cm−1, and 2879 cm−1, which represented both EC and mPEG, and characteristic peaks in the fingerprint region of mPEG at 1112 cm−1, 948 cm−1, and 842 cm−1 are evident32. A peak at 1745 cm−1 was observed due to the formation of ester C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O bonds between CEC and mPEG. Further, similar peaks were observed in the EC-PEG2 spectrum (Fig. S7B†) at 3465 cm−1, 2972 cm−1, 2879 cm−1, 1745 cm−1, 1112 cm−1, 948 cm−1, and 842 cm−1. The FTIR spectrum of DOX (Fig. S7A†) possessed characteristic peaks from N–H stretching at 3527 cm−1, carbonyl CO at 1730 cm−1, aromatic CC stretching at 1620 cm−1, aromatic C–H stretching at 2930 cm−1, and O–H stretching at 3334 cm−1.35,36 The FTIR spectra of the DOX-EC-PEG micelles B3 and B6 are shown in Fig. S7C.† The spectra displayed a broad peak at 3464 cm−1 due to N–H and O–H functional groups being present in the compositions (B3 and B6). C–H stretching peaks present in the EC and mPEG spectra were observed at 2880 cm−1 and 2974 cm−1. A broad peak at 1740 cm−1 was attributed to the characteristic CO groups of DOX and CEC. The sharp peak from C–C stretching at 1120 cm−1, which is characteristic of EC, was also observed; the C–O–C peak at 1060 cm−1 of mPEG merged with this sharp broad peak. The characteristic C–H bending peaks of mPEG at 948 cm−1 and 842 cm−1 were also observed in the B3 and B6 spectra. Observations from the FTIR spectra of B3 and B6 suggested that the prepared micelles were a combination of the copolymer EC-PEG with DOX. Moreover, the peaks of DOX merged with the copolymer EC-PEG due to their interactions and the good distribution in the micelles.
O bonds between CEC and mPEG. Further, similar peaks were observed in the EC-PEG2 spectrum (Fig. S7B†) at 3465 cm−1, 2972 cm−1, 2879 cm−1, 1745 cm−1, 1112 cm−1, 948 cm−1, and 842 cm−1. The FTIR spectrum of DOX (Fig. S7A†) possessed characteristic peaks from N–H stretching at 3527 cm−1, carbonyl CO at 1730 cm−1, aromatic CC stretching at 1620 cm−1, aromatic C–H stretching at 2930 cm−1, and O–H stretching at 3334 cm−1.35,36 The FTIR spectra of the DOX-EC-PEG micelles B3 and B6 are shown in Fig. S7C.† The spectra displayed a broad peak at 3464 cm−1 due to N–H and O–H functional groups being present in the compositions (B3 and B6). C–H stretching peaks present in the EC and mPEG spectra were observed at 2880 cm−1 and 2974 cm−1. A broad peak at 1740 cm−1 was attributed to the characteristic CO groups of DOX and CEC. The sharp peak from C–C stretching at 1120 cm−1, which is characteristic of EC, was also observed; the C–O–C peak at 1060 cm−1 of mPEG merged with this sharp broad peak. The characteristic C–H bending peaks of mPEG at 948 cm−1 and 842 cm−1 were also observed in the B3 and B6 spectra. Observations from the FTIR spectra of B3 and B6 suggested that the prepared micelles were a combination of the copolymer EC-PEG with DOX. Moreover, the peaks of DOX merged with the copolymer EC-PEG due to their interactions and the good distribution in the micelles.
The thermal properties of the pure polymers (EC, mPEG, and CEC) and formulations with (B3 and B6) and without (B1 and B4) DOX are shown in Table 2 and Fig. S8.† The glass transition temperature (Tg) of pure EC was recorded to be 115 °C with a melting temperature (Tm) of 233 °C. Similarly, CEC showed Tg and Tm values of 116 °C and 233 °C, respectively. Further, the Tg, crystallization temperature (Tc), and Tm values of mPEG were −66 °C, 29 °C, and 56 °C, respectively. The thermogram of B4 showed Tg, Tc, and Tm values of −29 °C, 21 °C, and 53/176 °C, respectively, which indicated the semi-crystalline nature of EC-PEG. Further, the Tg value of EC was greatly changed after grafting mPEG to CEC. These results suggested that the grafting of PEG to CEC was done successfully. The thermal properties of the B6 micelles were recorded, where the Tg, Tc, and Tm values were −26 °C, 24 °C, and 53/175 °C, respectively. The changes in Tg and Tc indicated the loading of DOX into the micelles. According to the reported literature,37 DOX has a Tm value of 218 °C. The Tm value of DOX was greatly changed based on the thermograms of the DOX-loaded micelles B3 and B6, which indicated that the drug was molecularly dispersed and bound to the polymers.14 This also suggests the improved solubility of crystalline DOX in an aqueous medium when it is entrapped in a polymeric system.
Table 2 The thermal properties of pure polymers and micelles with DOX
| Chemical composition name |
Glass transition temperature (Tg) (°C) |
Crystallization temperature (Tc) (°C) |
Melting temperature (Tm) (°C) |
| EC |
115 |
— |
233 |
| mPEG |
−66 |
29 |
56 |
| CEC |
116 |
— |
233 |
| B1 |
−24 |
24 |
53, 176 |
| B4 |
−29 |
21 |
53, 176 |
| B3 |
−29 |
25 |
53, 176 |
| B6 |
−26 |
24 |
53, 175 |
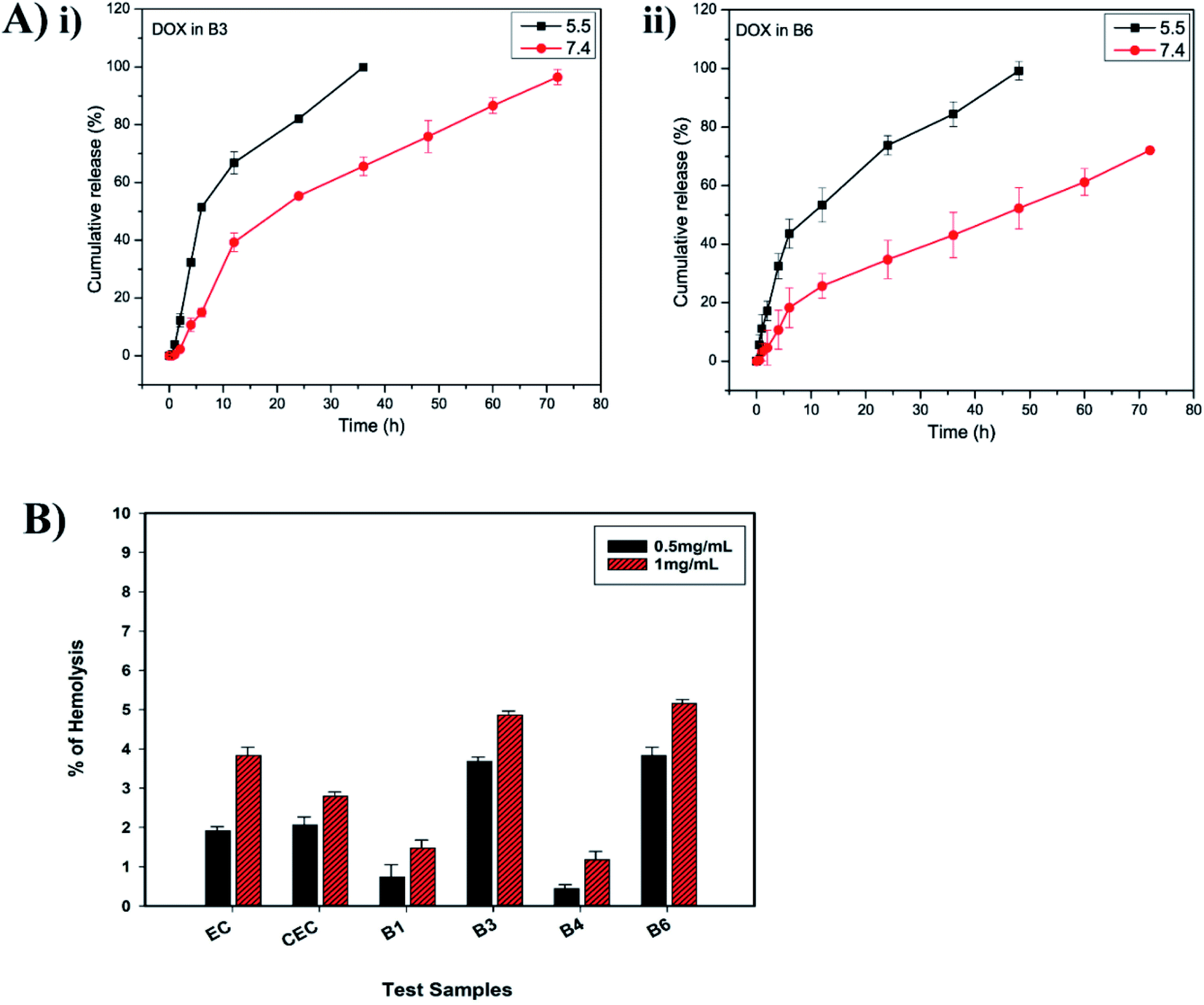
3.3.1 In vitro DOX release. The release of DOX from EC-PEG micelles (B3 and B6) was studied for 72 h via a dialysis method at 37 °C using phosphate buffer at pH 5.5 and 7.4, respectively. The release of DOX was faster at pH 5.5 compared to the release of DOX at pH 7.4 because the ester bonds formed between CEC and mPEG were susceptible to the acidic pH.38,39 Fig. 3A shows the release profiles of DOX at different pH values from B3. At pH 5.5, the B3 micelles recorded 100% release of DOX within 35 h and at pH 7.4, 96% DOX release was recorded within 72 h. The release of DOX from B6 micelles was slow at both pH values compared to that from the B3 micelles. For instance, 99% DOX release was recorded at pH 5.5 within 48 h and 72% DOX release was recorded at pH 7.4 within 72 h. This trend of slow release from B6 micelles compared to from B3 micelles suggested that increased mPEG grafting increased the molecular weight of the copolymer, thus the rate of release was slowed.40 Overall, the sustained release of DOX can be attributed to the encapsulating properties of EC and hydrophobic interactions between DOX and EC.30,41 The pH-dependent release from the micelles can be utilized to deliver DOX at tumor pH, because the tumor extracellular environment is slightly acidic (pH 6.8) and, after endocytosis by cancer cells, the pH of lysosomes/endosomes is also acidic (pH 5.5); there is also minimized release of DOX at normal tissue pH (pH 7.4).42–44
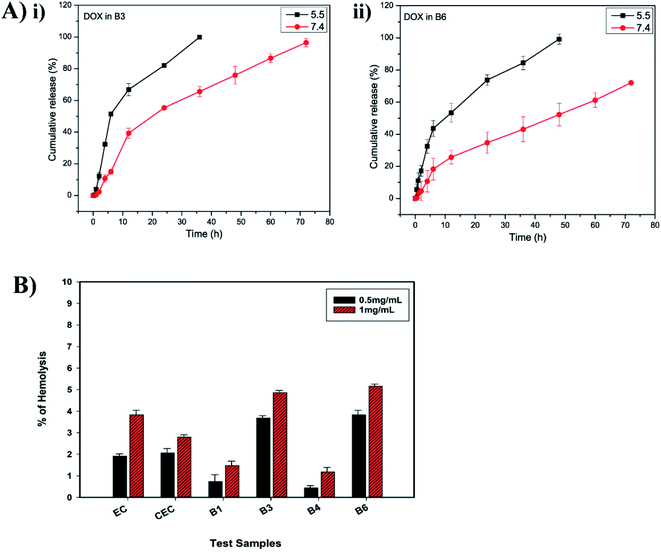 |
| | Fig. 3 (A) (i) DOX release from B3 micelles in PBS at pH 5.5 and 7.4 over 72 h; and (ii) DOX release from B6 micelles in PBS at pH 5.5 and 7.4 over 72 h. (B) Hemolysis percentages upon exposure to test samples: EC, CEC, and B1, B3, B4, and B6 micelles. | |
3.3.2 Hemolysis assays. Hemolysis assays are used to validate the biocompatibility of a biomaterial or polymer. According to ASTM standards (E2524-08), hemolysis for a test sample of above 5% indicates toxicity toward RBCs.45 The test samples EC, CEC, B1, B3, B4, and B6 were assessed for their biocompatibility in human blood at concentrations of 0.5 and 1 mg mL−1. The hemolysis percentages were calculated via eqn (3), considering 100% lysis in DI water and 0% lysis in PBS. As seen in the histogram shown in Fig. 3B, EC displayed less than 5% hemolysis, hence it can be termed a non-hemolytic material, and after modification and the formation of micelles (B1, B3, B4, and B6), the material maintained its hemocompatibility. As observed, CEC had slightly better hemocompatibility than EC, which could be due to the addition of negatively charged carboxylic groups. In the cases of B1 and B4, even at a higher concentration of 1 mg mL−1, the observed hemolysis% values were 1.47 ± 0.2 and 1.17 ± 0.2, respectively. The improved hemocompatibility of the micelles in comparison to EC and CEC can be attributed to the mPEG grafting.46 These results confirm the advantages of the PEGylation of polymers; upon mPEG grafting, the ability of the test materials to induce RBC lysis decreased significantly. In B3 and B6, at 0.5 mg mL−1, the observed hemolysis% values were 3.68 ± 0.1 and 3.82 ± 0.2, respectively, which are within the permissible limit of 5%, whereas at 1 mg mL−1, hemolysis is close to 5%. The toxicity to RBCs in the presence of B3 and B6 samples at higher concentrations was due to DOX. DOX is known to enter RBCs and cause swelling and lysis via building up the osmotic pressure.47 As per the report by Shuai et al.,48 free DOX induces 11% hemolysis at 200 μg mL−1, but when loaded into micelles its toxicity is largely diminished. The reason for the lower hemotoxicity of DOX in EC-PEG micelles in comparison to that reported for free DOX is the entrapment of DOX in the hydrophobic core, thus reducing the amount of DOX available on the surface for interactions with RBCs. As mentioned earlier, the hydrophilic mPEG chains formed a protective cover and, therefore, the RBCs were inhibited from being lysed. In line with these findings, we consider that the developed copolymer EC-PEG is biocompatible and can be used as a potential nanocarrier for DD.
3.4 Cell studies
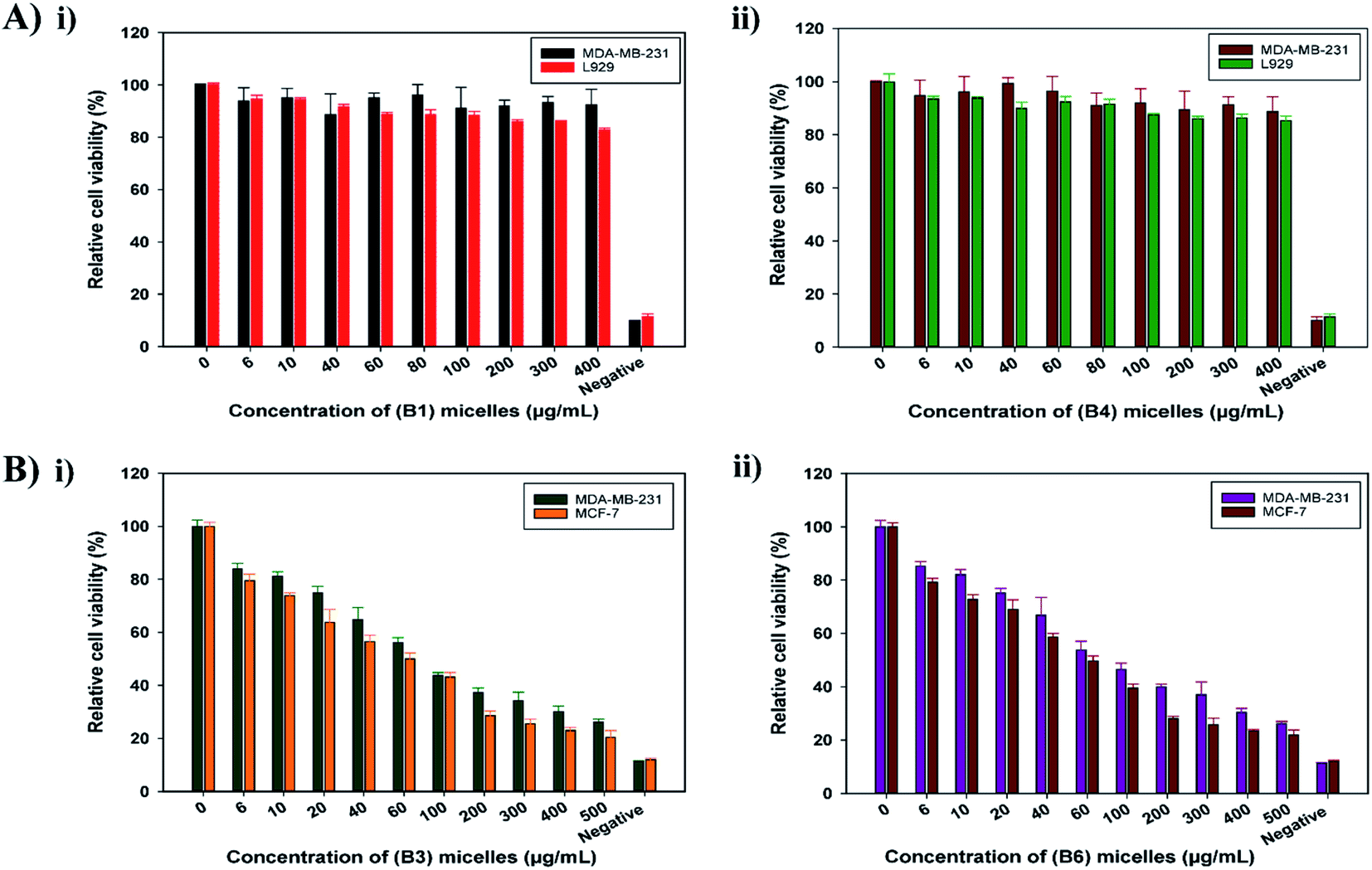
3.4.1 Cytotoxicity assays. The cytotoxic effects of blank and DOX-loaded EC-PEG micelles were determined via MTT assays. The cytotoxic effects of B1 and B4 micelles were investigated toward fibroblast L929 and breast cancer MDA-MB-231 cell lines. DMSO was used to solubilize the formazan crystals formed after the addition of the MTT reagent, which indicated cellular respiratory activity. As shown in Fig. 4, B1 (Fig. 4A(i)) and B4 (Fig. 4A(ii)) micelles exhibited no cellular cytotoxicity for up to 48 h of incubation at varying concentrations. The relative cell viability was above 80% even at the highest micelle concentration of 400 μg mL−1. Thus, this demonstrated the non-cytotoxic nature of the mPEG-grafted EC micelles and established them as a promising drug-delivery vehicle. The cytotoxic effects of B3 and B6 micelles were investigated toward the breast cancer cell lines MDA-MB-231 and MCF-7 for 72 h. From the histogram, as shown in Fig. 4B, the DOX-loaded micelles B3 (Fig. 4B(i)) and B6 (Fig. 4B(ii)) showed a decrease in cell viability as a function of the DOX dose concentration. The concentration of micelles was varied from 0 μg mL−1 to 500 μg mL−1 and the IC50 (the dose inducing 50% cell inhibition) values noted for B3 and B6 toward both cell lines were ∼100 μg mL−1. These results proved that EC-PEG micelles are a biocompatible and non-toxic DDS for delivering DOX at a controlled rate over an extended period of time.
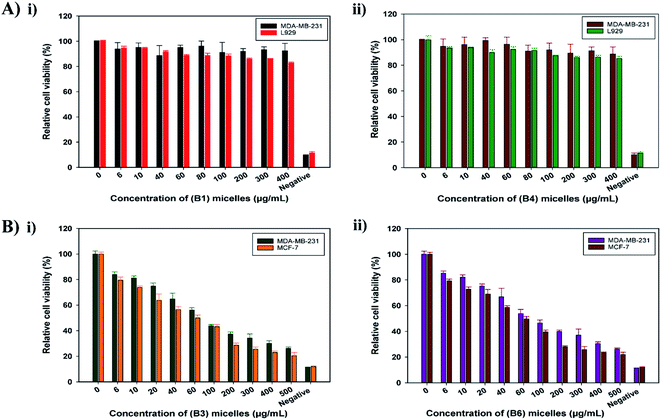 |
| | Fig. 4 (A) (i) Cytotoxic effects toward MDA-MB-231 and L929 cells treated with B1 micelles for 48 h; and (ii) cytotoxic effects toward MDA-MB-231 and L929 cells treated with B4 micelles for 48 h. (B) (i) Cytotoxic effects toward MDA-MB-231 and MCF-7 cells treated with B3 micelles for 72 h; and (ii) cytotoxic effects toward MDA-MB-231 and MCF-7 cells treated with B3 micelles for 72 h. | |
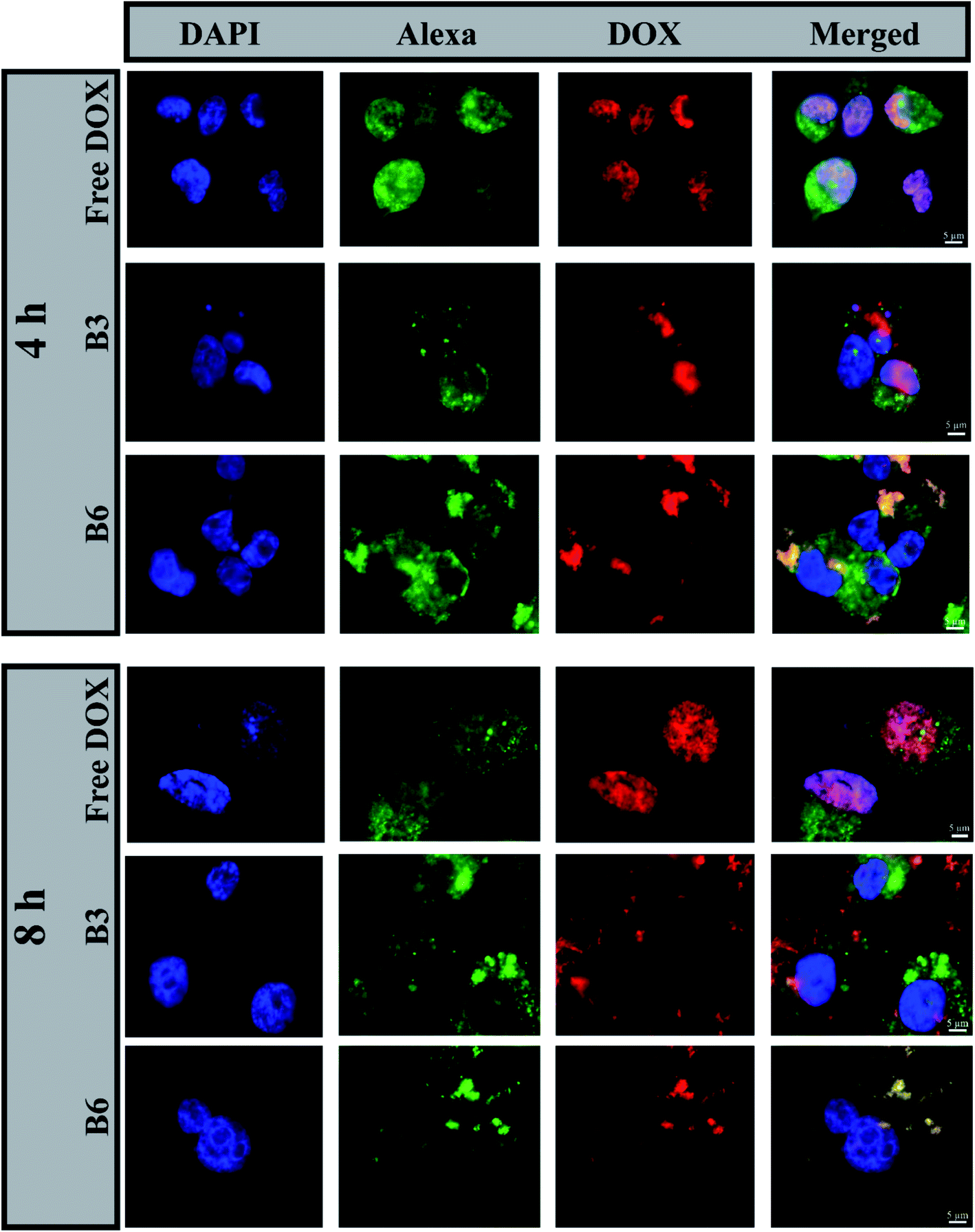
3.4.2 In vitro cellular uptake. The cellular uptake of free DOX and DOX-EC-PEG (B3 and B6) micelles was studied in breast cancer cells MDA-MB-231 via incubating them with the samples in media at 37 °C for 4 h and 8 h, respectively; later, analysis was conducted via epifluorescence microscopy. The nuclei of cells were stained with DAPI, the actin filaments of the cytoskeleton were stained with Alexa Fluor 488 phalloidin, and DOX emitted red fluorescence. In cells treated with free DOX, as shown in Fig. 5, it was observed that DOX was exclusively present in the nuclei. This can be explained based on the fact that DOX can be readily transported into cells via the mechanism of passive diffusion, which is energy independent, where it binds to DNA in the nuclei via intercalation.49,50 In cells treated with B3 and B6 micelles at 100 μg mL−1, it was observed that the red fluorescence of DOX was located in the cytoplasm and nuclear region (Fig. 5, B3 and B6 rows). This observation leads to the inference that the DOX-loaded micelles were taken up by the cells via an endocytosis pathway and were located in the intracellular compartment (endosomes and lysosomes), which is supported by the reported literature.42,51
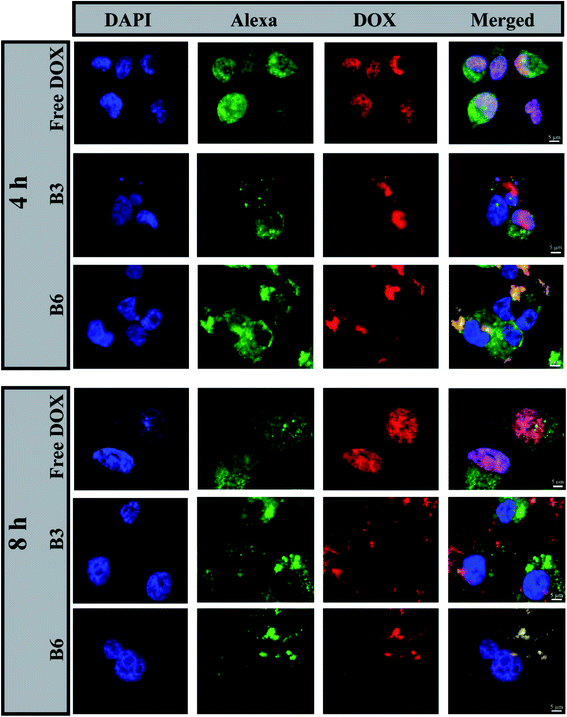 |
| | Fig. 5 Fluorescence images of MDA-MB-231 cells treated with free DOX, B3 micelles, or B6 micelles for 4 h and 8 h. The right panels show merged images of nuclei stained with DAPI (blue), F-actin stained with Alexa Fluor 488 phalloidin (green), and DOX fluorescence (red). The scale bars correspond to 5 μm. | |
4. Conclusions
In this study, we synthesized a new amphiphilic copolymer via grafting PEG to EC. The developed copolymer was self-assembled into micelles in an aqueous system via a dialysis method, and the micelle properties were confirmed via DLS, TEM, and fluorescence spectroscopy studies; the CMCs of the copolymers EC-PEG1 and EC-PEG2 were recorded to be 0.03 mg mL−1 and 0.00193 g mL−1, respectively. The sizes of the DOX-encapsulated EC-PEG nano-micelles were larger than the size of the blank micelles, which was confirmed via DLS. The DOX-loaded micelles demonstrated faster release at an acidic pH of 5.5, whereas slow DOX release was observed at physiological pH of 7.4. Hemolysis assay and MTT assay studies confirmed that the micelles were biocompatible and non-toxic toward normal cells. Further, the DOX-loaded micelles showed toxicity toward the cancerous cells MDA-MB-231 and MCF-7, and fluorescence microscopy images showed the internalization of the DOX-loaded micelles in MDA-MB-231 cells. According to the above observations, we concluded that the developed micelles can be used as a potential DDS for cancer therapy. In the future scope of this work, the micelles could be tailored with various targeting moieties to achieve site-specific targeted delivery.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
The authors are grateful to the funding agencies CSIR (MLP034726, CSC0302, CSC0134) and DBT-SRF, New Delhi. The authors also are thankful to Dr Neha Tiwari and Mr Prashant Yadav for support in designing the synthesis scheme and for input relating to NMR evaluations.
References
- D. Hwang, J. D. Ramsey and A. V. Kabanov, Adv. Drug Delivery Rev., 2020, 156, 80–118 CrossRef CAS PubMed
 .
. - J. Gong, M. Chen, Y. Zheng, S. Wang and Y. Wang, J. Controlled Release, 2012, 159, 312–323 CrossRef CAS PubMed .
- Z. Ahmad, A. Shah, M. Siddiq and H.-B. Kraatz, RSC Adv., 2014, 4, 17028–17038 RSC .
- H. Cabral and K. Kataoka, J. Controlled Release, 2014, 190, 465–476 CrossRef CAS PubMed .
- J. Fang, H. Nakamura and H. Maeda, Adv. Drug Deliv. Rev., 2011, 63, 136–151 CrossRef CAS PubMed .
- S. Wilhelm, A. J. Tavares, Q. Dai, S. Ohta, J. Audet, H. F. Dvorak and W. C. W. Chan, Nat. Rev. Mater., 2016, 1, 1–12 Search PubMed .
- S. Biswas, P. Kumari, P. M. Lakhani and B. Ghosh, Eur. J. Pharm. Sci., 2016, 83, 184–202 CrossRef CAS PubMed .
- S. Sabra, M. Abdelmoneem, M. Abdelwakil, M. T. Mabrouk, D. Anwar, R. Mohamed, S. Khattab, A. Bekhit, K. Elkhodairy and M. Freag, Curr. Pharmaceut. Des., 2017, 23, 5213–5229 CAS .
- X. Su, Z. Yang, K. B. Tan, J. Chen, J. Huang and Q. Li, Carbohydr. Polym., 2020, 241, 116259 CrossRef CAS PubMed .
- S. Muschert, F. Siepmann, B. Leclercq, B. Carlin and J. Siepmann, J. Controlled Release, 2009, 135, 71–79 CrossRef CAS PubMed .
- B. Balzus, M. Colombo, F. F. Sahle, G. Zoubari, S. Staufenbiel and R. Bodmeier, Int. J. Pharm., 2016, 513, 247–254 CrossRef CAS PubMed .
- S. Leitner, S. Grijalvo, C. Solans, R. Eritja, M. J. García-Celma and G. Calderó, Carbohydr. Polym., 2020, 229, 115451 CrossRef CAS PubMed .
- W. Yuan, J. Yuan, F. Zhang and X. Xie, Biomacromolecules, 2007, 8, 1101–1108 CrossRef CAS PubMed .
- R. Gref, A. Domb, P. Quellec, T. Blunk, R. H. Müller, J. M. Verbavatz and R. Langer, Adv. Drug Deliv. Rev., 2012, 64, 316–326 CrossRef .
- E. Lasseuguette, Cellulose, 2008, 15, 571–580 CrossRef CAS .
- T. Kaldéus, M. Nordenström, A. Carlmark, L. Wågberg and E. Malmström, Carbohydr. Polym., 2018, 181, 871–878 CrossRef PubMed .
- D. Shen and Y. Huang, Polymer, 2004, 45, 7091–7097 CrossRef CAS .
- Q. Zhao, B. Han, Z. Wang, C. Gao, C. Peng and J. Shen, Nanomedicine, 2007, 3, 63–74 CrossRef CAS PubMed .
- J. Araki, M. Wada and S. Kuga, Langmuir, 2001, 17, 21–27 CrossRef CAS .
- J. Chen, X. Qiu, J. Ouyang, J. Kong, W. Zhong and M. M. Q. Xing, Biomacromolecules, 2011, 12, 3601–3611 CrossRef CAS PubMed .
- M. Hirota, N. Tamura, T. Saito and A. Isogai, Carbohydr. Polym., 2009, 78, 330–335 CrossRef CAS .
- A. E. J. De Nooy, A. C. Besemer and H. Van Bekkum, Recl. Trav. Chim. Pays-Bas, 1994, 113, 165–166 CrossRef CAS .
- B. Ding, Y. qing Ye, J. Cheng, K. Wang, J. Luo and B. Jiang, Carbohydr. Res., 2008, 343, 3112–3116 CrossRef CAS PubMed .
- A. Isogai, T. Saito and H. Fukuzumi, Nanoscale, 2011, 3, 71–85 RSC .
- T. Heinze, T. Liebert and A. Koschella, Esterification of polysaccharides, Springer Science & Business Media, 2006 Search PubMed .
- D. Shen, H. Yu and Y. Huang, J. Polym. Sci., Part A: Polym. Chem., 2005, 43, 4099–4108 CrossRef CAS .
- Y. Li, R. Liu, W. Liu, H. Kang, M. Wu and Y. Huang, J. Polym. Sci., Part A: Polym. Chem., 2008, 46, 6907–6915 CrossRef CAS .
- J. Zhu, X.-T. Dong, X.-L. Wang and Y.-Z. Wang, Carbohydr. Polym., 2010, 80, 350–359 CrossRef CAS .
- Y. Dong and K. J. Edgar, Polym. Chem., 2015, 6, 3816–3827 RSC .
- S. Lv, Z. Tang, M. Li, J. Lin, W. Song, H. Liu, Y. Huang, Y. Zhang and X. Chen, Biomaterials, 2014, 35, 6118–6129 CrossRef CAS PubMed .
- V. Suthar, A. Pratap and H. Raval, Bull. Mater. Sci., 2000, 23, 215–219 CrossRef CAS .
- Y. Li, R. Liu and Y. Huang, J. Appl. Polym. Sci., 2008, 110, 1797–1803 CrossRef CAS .
- P. Shoaeifar, M. Abbasian and A. A. Entezami, J. Polym. Res., 2007, 14, 45–52 CrossRef CAS .
- H. Asadi, K. Rostamizadeh, D. Salari and M. Hamidi, J. Microencapsulation, 2011, 28, 406–416 CrossRef CAS PubMed .
- D. Depan, J. Shah and R. D. K. Misra, Mater. Sci. Eng., C, 2011, 31, 1305–1312 CrossRef CAS .
- A. Neacşu, Thermochim. Acta, 2018, 661, 51–58 CrossRef .
- D. Missirlis, R. Kawamura, N. Tirelli and J. A. Hubbell, Eur. J. Pharm. Sci., 2006, 29, 120–129 CrossRef CAS PubMed .
- K. Rostamizadeh, M. Manafi, H. Nosrati, H. K. Manjili and H. Danafar, New J. Chem., 2018, 42, 5937–5945 RSC .
- T. D. Carrillo-Castillo, J. S. Castro-Carmona, A. Luna-Velasco and E. A. Zaragoza-Contreras, e-Polym., 2020, 20, 624–635 CAS .
- Y.-Z. Du, Q. Weng, H. Yuan and F.-Q. Hu, ACS Nano, 2010, 4, 6894–6902 CrossRef CAS PubMed .
- K. Wasilewska and K. Winnicka, Materials, 2019, 12, 3386 CrossRef CAS PubMed .
- S. Lv, M. Li, Z. Tang, W. Song, H. Sun, H. Liu and X. Chen, Acta Biomater., 2013, 9, 9330–9342 CrossRef CAS PubMed .
- Z. Wang, X. Deng, J. Ding, W. Zhou, X. Zheng and G. Tang, Int. J. Pharm., 2018, 535, 253–260 CrossRef CAS PubMed .
- C. R. Justus, L. Dong and L. V. Yang, Front. Physiol., 2013, 4, 354 Search PubMed .
- J. Choi, V. Reipa, V. M. Hitchins, P. L. Goering and R. A. Malinauskas, Toxicol. Sci., 2011, 123, 133–143 CrossRef CAS PubMed .
- S. V. Lale, A. Kumar, F. Naz, A. C. Bharti and V. Koul, Polym. Chem., 2015, 6, 2115–2132 RSC .
- D. Lu, J. Liang, Y. Fan, Z. Gu and X. Zhang, Adv. Eng. Mater., 2010, 12, B496–B503 CrossRef .
- X. Shuai, H. Ai, N. Nasongkla, S. Kim and J. Gao, J. Controlled Release, 2004, 98, 415–426 CrossRef CAS PubMed .
- Y. Cao, Y. Gu, H. Ma, J. Bai, L. Liu, P. Zhao and H. He, Int. J. Biol. Macromol., 2010, 46, 245–249 CrossRef CAS PubMed .
- H. Wang, Y. Zhao, Y. Wu, Y.-l. Hu, K. Nan, G. Nie and H. Chen, Biomaterials, 2011, 32, 8281–8290 CrossRef CAS PubMed .
- H. C. Arora, M. P. Jensen, Y. Yuan, A. Wu, S. Vogt, T. Paunesku and G. E. Woloschak, Cancer Res., 2012, 72, 769–778 CrossRef CAS PubMed .
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1ra04242d |
|
| This journal is © The Royal Society of Chemistry 2021 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence ab,
Naresh Killi
ab,
Naresh Killi