
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAnionic CO2 activation in the anionic and di-anionic state of aza-naphthalene†
Chang Jun Park,
One Heo ,
Hyeon Seok Lee,
Kyung Suh Lee and
Sang Hak Lee*
,
Hyeon Seok Lee,
Kyung Suh Lee and
Sang Hak Lee*
Department of Chemistry, Pusan National University, Busan 46241, Korea. E-mail: shlee77@pusan.ac.kr
First published on 29th July 2021
Abstract
Nitrogen-containing polycyclic aromatic hydrocarbon (PAH) is the single basic moiety in N-doped graphene, the only metal-free catalyst reported to date to successfully produce the oxygen reduction reaction. N-doped graphene is quite promising as a material to increase the efficiency of oxygen reduction. In addition, it is known that when carbon dioxide is added to aza-benzene, there will be an associative chemical reaction upon electron attachment between the anionic nitrogen atoms in the aza-benzene and the carbon atom in the carbon dioxide; however, it has previously been reported that when there are more nitrogen atoms in the small aza-benzene moiety, the associative reaction does not always occur. In this study, we carried out a theoretical simulation to determine whether more electrons increase the CO2 reductive reactivity of the aza-naphthalene as a model system of a nitrogen-containing polycyclic aromatic hydrocarbon. We found that even though an associative chemical reaction between nitrogen atoms in the N-PAH and carbon atoms in carbon dioxide did not occur in anionic complexes of aza-naphthalene and carbon dioxide, chemical reactions did occur in all the nitrogen atoms of these complexes when we added an extra excess electron. Therefore, we conclude that the efficiency of CO2 reduction will be increased in nitrogen atoms when more electrons are added to increase their anionic properties.
Introduction
Graphene is the most fertile scientific subject of research in the materials sciences and research to date has shown various applications because of graphene's high thermal conductivity, surface area, and charge carrier mobility.1–3 Due to its electrical properties, research has shown the possibility of using it for single-molecule electronics4 and as a catalyst for water splitting.5Researchers have recently found that nitrogen-doped graphene shows interesting optical and chemical properties. When adding nitrogen atoms to graphene molecules, the bandgap of graphene can be manipulated so that nitrogen-doping graphene changes its optical and chemical properties.6–8 For example, nitrogen-containing graphene has been reported to emit blue luminescence.6 Furthermore, since nitrogen atoms can have a partial charge, they are likely to react to other chemicals. Nitrogen-doped graphene has been reported to have high electrocatalytic activity in the oxygen reduction reaction.7,8
More specifically, Kim and coworkers have recently discovered that nitrogen-containing benzene (aza-benzene) molecules make carbon dioxide molecules react with nitrogen atoms of aza-benzene in the anionic state through excess electron delocalization.9,10 Using photoelectron spectroscopy as well as theoretical calculations, they noted a much higher value of vertical detachment energy (VDE) than adiabatic electron affinity (AEA), which indicates that the structure had a significant difference between the neutral state and anionic state. In addition, M. A. Johnson and coworkers proved this chemical reaction using IR vibrational spectroscopy.11 Both Johnson and Kim concluded that a covalent bond was created between the carbon of carbon dioxide and the nitrogen of aza-benzene. Since the excess electron migrates to the anti-bonding orbital of a CO2 molecule, the bond order of the C–O bond decreased and so this chemical reaction is a reducing reaction of CO2 molecules. This indicates that a reduced CO2 molecule is more likely to react with other chemicals than a neutral CO2 molecule. Thus, this is the first step in converting carbon dioxide molecules to other useful chemicals.12 Similarly, Weber and coworkers found that carbon dioxide molecules reductively interacted with anionic metals and oxygen molecules.13,14 This means that even though the electron affinity of carbon dioxide is slightly negative, it can be further reduced when it creates a molecular complex (or cluster) with other molecules or atoms that have a slightly negative or positive electron affinity.
In various applications, this anionic associative reaction has been used to convert CO2 molecules to other fuel molecules using the anionic form of nitrogen-containing molecules or ionic liquid.15 Research recently has been carried out to convert CO2 molecules using organic catalysts rather than transition-metal-based catalysts.16 The anionic forms of nitrogen-containing organic molecules is a popular current research topic to explore the reducing reaction of CO2 molecules.12,15 X. Y. Luo et al. have shown that when increasing reaction sites of pyridine-containing anionically functionalized ionic liquids, the efficiency of CO2 capture was significantly improved.15 This leads to better efficiency in converting CO2 molecules into useful chemicals. Recently, two-dimensional materials are used to reduce CO2 molecules due to their ultra-large reaction area.17 In particular, a theoretical simulation suggests that nitrogen-containing graphene serves as a catalyst.18,19 Since it has high electrical conductivity, graphene is a good material for electrochemical CO2 reduction. Thus, it is worthwhile to investigate how associative molecular interactions between nitrogen-containing PAH and carbon dioxide contribute to reduce and convert carbon dioxide.
In a previous study, even though triazine had three nitrogen atoms, two carbon dioxide molecules reacted with only two of the three nitrogen atoms.9 This was attributed to the deficiency of negative charge in the nitrogen atoms. This means that only one excess electron was insufficient to distribute excess charge to all three nitrogen atoms. Thus, we thought that increasing the excess charge in the nitrogen-containing PAH molecules would allow all nitrogen atoms in the nitrogen-containing PAH molecules to react with the carbon dioxide molecules.
In general, dianion molecules are not an electronically stable chemical species and so the excess electron will eject to the vacuum level through autoionization (or auto detachment). Thus, theoretical methods to rigorously study dianionic molecules will require the time-dependent Schrodinger equation; however, Pyykko and coworkers have proved that conventional methods of the time-independent Schrodinger equation also reasonably describe the multiple-charged anionic species.20,21 Furthermore, Wang and coworkers have shown that dianion molecules exist for a fairly long time in the gas phase,22,23 even though they are not electronically stable. Furthermore, through solvation or interaction in a solid-state, they can be electronically stable even in the dianionic state.24
Therefore, the present study's theoretical approach aimed to prove that nitrogen-containing polycyclic aromatic hydrocarbon molecules are more reactive with CO2 molecules in their di-anionic state than in the anionic state (or singly-charged state). Although nitrogen-containing PAH molecules are electronically (energetically) unstable in the dianionic state, they can be in the gas phase and be electronically stable with solvents or in solid form. Thus, in the present study, we theoretically delved into the energetics as well as the reactivity of nitrogen-containing PAH molecules with CO2 molecules in the neutral, anionic, and dianionic states.
Theoretical methods
In order to investigate the reactivity to anionically activate CO2 molecules, we carried out a theoretical calculation to study the reactivity of aza-naphthalene to CO2. DFT calculation have recently been shown feasible to find the energetics and geometry of anionic molecules and their clusters. We employed various functionals, BP86, B3LYP, BPBE, PBEPBE, wb97xd, and M11 to show the feasibility to simulate the molecular geometry and energetics using Gaussian 16.25 All calculations were performed with the basis set of 6-311G**. We obtained the optimized structures, energy and the vibrational frequencies of neutrals, anion, and di-anion of aza-naphthalene and aza-naphthalene–(CO2)n complexes. All three functionals showed very similar results in the calculation as shown in Fig. S7 and Table S1.†Results
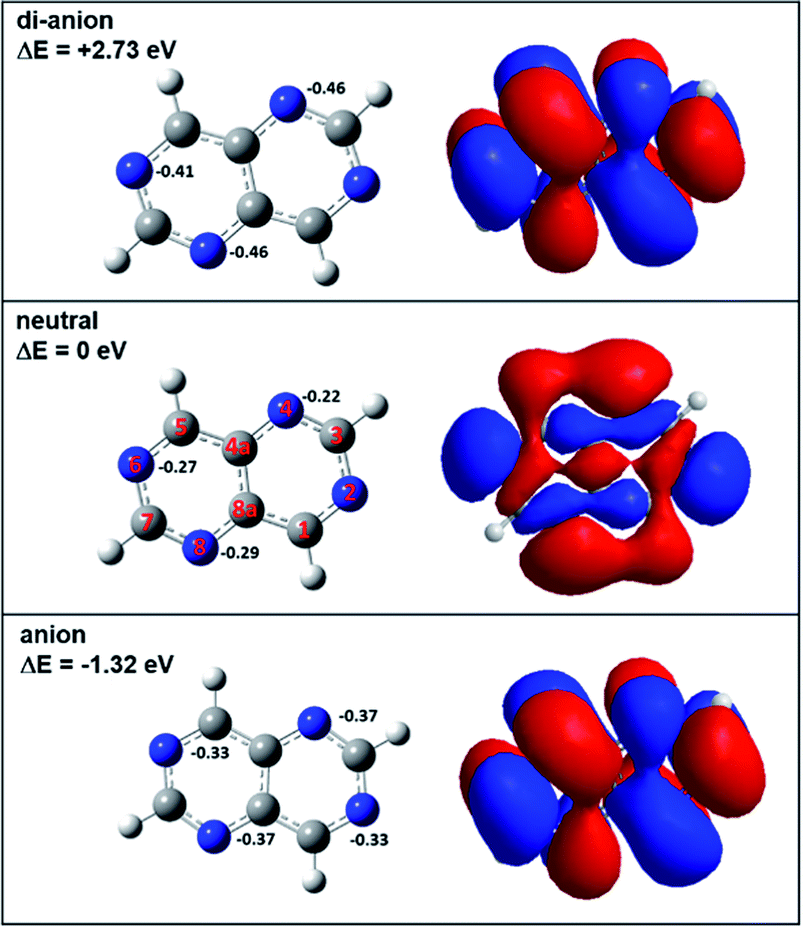
In order to investigate the reactivity of aza-naphthalene in the dianionic state, we first calculated the energetics of tetra-aza-naphthalene (TAN) molecules in neutral, anionic, and di-anionic states. As shown in Fig. 1, the TAN molecule has a positive electron affinity. The adiabatic electron affinity of TAN was 1.32 eV; however, the dianion, as expected, was much less stable than the anion which means that the dianion is electronically unstable. When creating a di-anion from the anion, the di-anionic state is 4.05 eV and 2.73 eV higher than the anion and neutral states, respectively. The molecular orbital of the anionic state has a very stable resonance structure and fewer nodes than in the neutral state. The orbital of the dianionic state is the same as the anionic state which means that the second extra electron to form a dianionic TAN occupies the SOMO (singly occupied molecular orbital) of the anion. In addition, stable geometry was found in the theoretical calculation. This means that the dianionic state of TAN will be a bound state although it may be in a very transient state.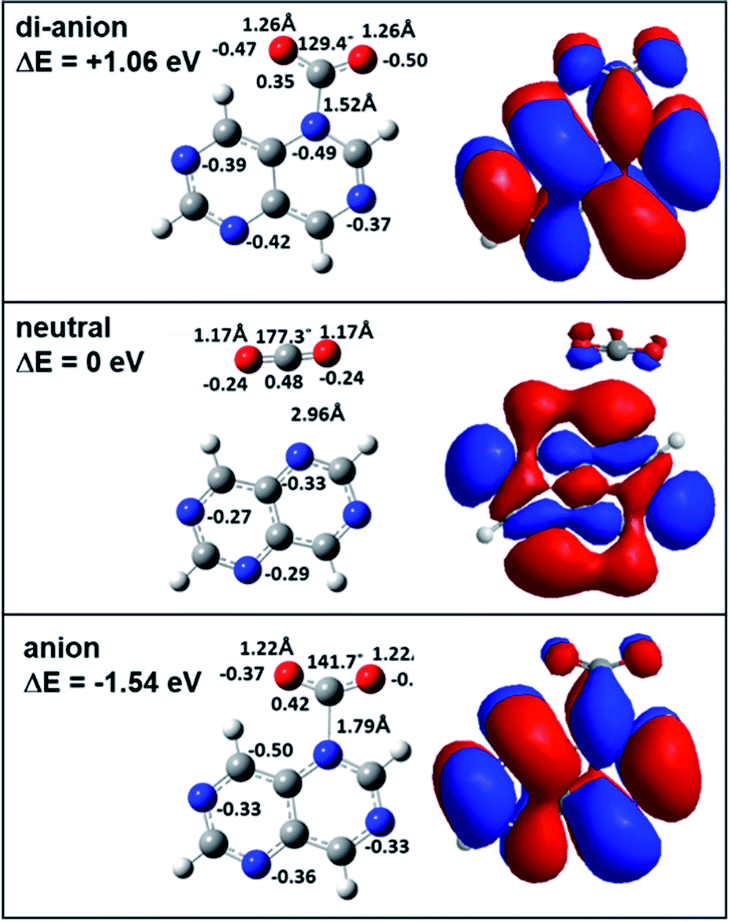 | ||
| Fig. 1 Neutral, anionic, and di-anionic geometries and calculated energies of the 4 (or 8) isomer of TAN1(CO2)1 optimized at the BP86/6-311g** level. Orbitals are highly occupied orbitals: HOMO of neutral and dianion and SOMO of anion. | ||
In the Mulliken charge analysis, we found that the nitrogen charge varied as a function of the position of the nitrogen. Each ionic state shows the same electron charge distribution for the pair of nitrogen atoms in positions 2 and 6 and in positions 4 and 8, as shown in Fig. 1. The symmetry of molecules results in the same charge distribution. As expected, when increasing excess electrons, the negative charge in nitrogen atoms is increasing. Nitrogen atoms in the dianion have much more negative charge than the anion and the neutral. This means that nitrogen atoms in the dianionic state have a higher Fukui value (0.23) than that (0.15) in the anionic state. Therefore, we expect that the dianion will have stronger chemical reactivity with other molecules rather than the anion due to enough negative charge in the nitrogen atoms.
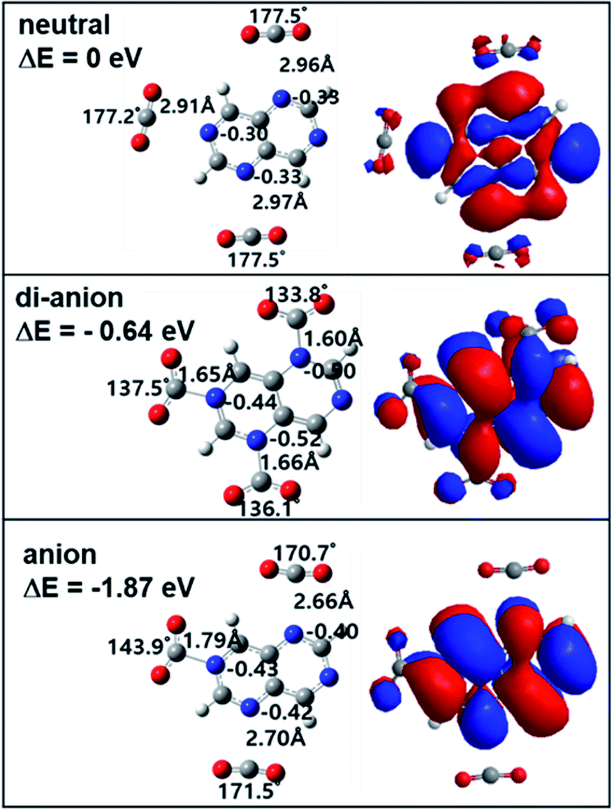
When making the TAN complex with a CO2 molecule, there are two different structures, as shown in Fig. 2 and S1.† Similar to charge distribution, the same two pairs of nitrogen atoms (2 & 6, 4 & 8) share the same chemical structure and so there are two structural isomers depending on where a CO2 molecule binds to one of the two nitrogen atoms in each given pair. In the neutral states, both neutral states show the TAN complexes with CO2 have van der Waals interaction, as shown in Fig. 2 and S1.† This means that the CO2 and nitrogen atom do not change their structures. For the anionic state, the complex shows different features from the neutral state. Similar to previous studies, we also found that the covalent bond between a nitrogen atom of TAN and a carbon atom of CO2 was newly created and the structure of CO2 was changed from a linear to a bent form. The angle of the CO2 molecule bent from 177° (linear) to 142° and 141° (bent), for two isomers. Furthermore, the bond length was elongated as well, from 1.17 Å to 1.22 Å. This indicates that the excess electron migrated to the CO2 moiety and the bond order of CO2 decreased.
 | ||
| Fig. 2 Neutral, anionic, and di-anionic geometries orbitals and calculated energies of TAN optimized at the BP86/6-311g** level. Positive or negative energy values indicate higher or lower than the neutral state, respectively. Orbitals are highly occupied orbitals: HOMO of neutral and dianion and SOMO of anion. | ||
Since the nitrogen atoms of the dianion are more negatively charged than the anion and the neutral, the TAN dianion have much better reactivity as compared with other two. The optimized the geometry of the TAN–CO2 di-anionic complex showed that the bound CO2 moiety shaped a more bent (129° and 131°) form than that of the anionic state and the length of the bond also increased. The Mulliken charge analysis also showed that the CO2 moiety in the complex was more negatively charged. When delocalizing two excess electrons, more charge migrated to the CO2 moiety. This indicated that, in terms of the reduction of CO2 molecules, TAN had greater reactivity in the di-anionic state.
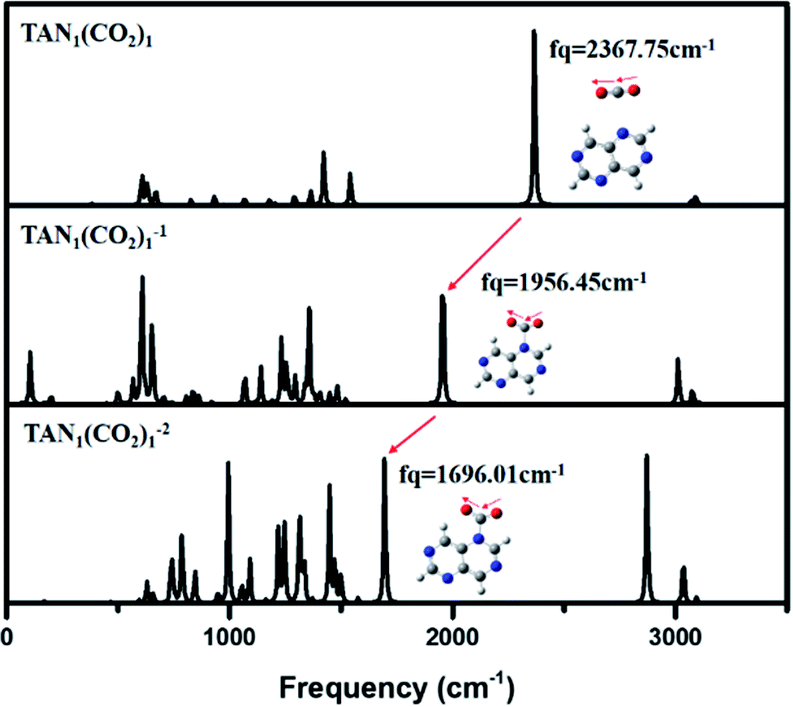 | ||
| Fig. 3 IR spectrum for neutral, anionic, di-anionic of TAN1(CO2)1 when CO2 position is 4 optimized at the BP86/6-311g** level. | ||
Vibrational frequency analysis of the three charge states of the TAN–(CO2)1 complex showed two main changes (Fig. 3). The first was a gradual decrease in frequency of the asymmetric stretching mode of the CO2 moiety in the complex. This means that the bond strength of a CO2 molecule is actually decreased in the anionic and dianionic state due to decreasing the bond order. In the neutral state, the frequency was 2367 cm−1, which was similar to that (2349 cm−1, IR experiment) of the CO2 asymmetric stretching vibrational mode. The second was slight red-shifted and the peak intensity evolves when the complex has more charges. Thus, in terms of molecular properties, CO2 moiety in the complex is the same as a CO2 molecule. At the anionic state, as we found the optimized geometry, the bond order of CO2 was decreased since a covalent bond was created between the carbon of CO2 and a nitrogen atom of the complex. The vibrational frequency of the asymmetric mode of CO2 was decreased to 1956 cm−1. For the dianionic state, the frequency was even more decreased to 1696 cm−1 which is corresponded that the excess charges migrate to CO2 moiety in the dianionic state. This clearly indicates that the CO2 molecule can be more highly reduced at the higher charge state.
This showed that the complex in the di-anionic state had greater reactivity, in terms of CO2 reduction, than that in the neutral or anionic states, yet in terms of energetics, the dianion was much more unstable than the anion or even the neutral state. This means that, even though the dianion of the TAN molecule exhibited high reactivity, it appeared to only transiently exist. Thus, we were curious whether the energetics of dianion might be lower than that of at least the neutral state as the number of CO2 molecules increases.
For TAN1·(CO2)2, the CO2 binding position is different for each of four isomers, as shown in Fig. S2.† In the anionic state, all isomers in the complex showed a similar electron binding energy: two CO2 molecules bound to the 2/4 (1.78 eV), 2/6 (1.87 eV), 4/6 (1.79 eV), and 4/8 (1.65 eV) nitrogen positions; however, in the di-anionic states, only the 2/6-TAN1·(CO2)2 isomer showed a comparably high adiabatic electron binding energy (0.54 eV); the other isomers had a binding energy value close to 0 eV, as shown in Fig. S2.†
For optimized geometry of the complex in the neutral state, the two CO2 molecules are bound to two of the nitrogen atoms in the TAN molecule through a van der Waals interaction and thus the structure of the two CO2 molecules is little changed, as in the TAN1·(CO2)1 complex. All four isomers show a very similar structural property, namely that the bound CO2 molecules maintain their linear structure.
For the anion state, the reactivities of the TAN molecule differed with the binding position of CO2 molecules. This complex also had four isomers, as shown in Fig. S2.† These four isomers can be categorized according to the CO2 binding position on the nitrogen atoms of the TAN molecule. For the two isomers with the 2/6 and 4/8 CO2 binding sites, two CO2 molecules bound to opposite nitrogen atoms in the TAN; for the two isomers with the 2/4 and 4/6 binding sites, two CO2 molecules bound to neighboring nitrogen atoms in the TAN. As we expected, the two CO2 molecules that bound to the nitrogen atoms in the 2/6 and 4/8 isomers covalently interacted with only one CO2 molecule in the anionic state. This covalent bond required enough negative charge in the nearby nitrogen atom of TAN and enough positive charge in the carbon atom of the CO2. Thus, in the case of the 2/6 and 4/8 complexes, neighboring nitrogen atoms likely had too much negative charge, so they were repulsed, thus delocalizing the excess charge. This explains why the anionic complex created only one covalent bond. As for the 2/6 and 4/8 complexes, only the 4/8 complex created two covalent bonds between CO2 molecules and nitrogen atoms of TAN; this is because the distance between the two nitrogen atoms was enough to overcome the repulsion caused by the excess charge. By contrast, in the case of the 2/6 complex, even though the two nitrogen atoms were on opposite sides, they were sufficiently close to have enough negative charge. This suggests that in the anionic state, when there are two CO2 molecules, TAN will have a selective reactivity to reduce CO2 molecules.
As for the dianionic state, interestingly, all four isomers showed both CO2 molecules reduced: both CO2 molecules covalently interacted with nitrogen atoms in TAN, as shown in Fig. S2.† This indicates that when TAN molecules are electronically, in particular anionically, charged, they are likely to have stronger reactivity. Thus, when they are in the di-anionic state, the associative interaction is dominant between TAN and CO2 molecules. In terms of the energetics, only the 2/6 complex is meaningfully more stable than neutral: 0.54 eV more stable than the neutral state. However, the other three isomers are almost in the same energy state as in the neutral state.
Fig. 4 and S3† shows that the structure and energetics of the neutral, anion, and dianion of the TAN1·(CO2)3 complex. This complex has two isomers: the 2,4,6 complex and the 4,6,8 complex describing CO2 binding positions. In the singly charged anion, both isomers have similar electron binding energy: 1.93 eV for the 2,4,6 complex and 1.87 eV for the 4,6,8 complex. As shown in Fig. 5, two of the three CO2 molecules covalently interact with TAN in the 2,4,6 complex, which is very similar to the 2,6 complex of the TAN1·(CO2)2. Like the 2,6 complex, the two CO2 molecules that are the farthest apart in the TAN molecule are the two that are able to create covalent bonds while at the same time, the nitrogen atom to which the CO2 bound had insufficient negative charge, thus allowing a van der Waals interaction between each of the two CO2 molecules and the TAN. The 4,6,8-TAN1·(CO2)3 complex is also similar to the 4/8 TAN1·(CO2)2 complex. CO2 molecules that bound to the nitrogen atoms in positions 4 and 8 failed to covalently interact with the TAN, but instead, the only CO2 molecule to covalently interact was the one that bound to the nitrogen atom in position 6. The failure to covalently bind was probably due to excess charge deficiency as well as charge repulsion inside of the TAN molecule.
 | ||
| Fig. 4 Neutral, anionic, and di-anionic geometries and calculated energies of 4,6,8 isomer of TAN1(CO2)3 optimized at the BP86/6-311g** level. Orbitals are highly occupied orbitals: HOMO of neutral and dianion and SOMO of anion. | ||
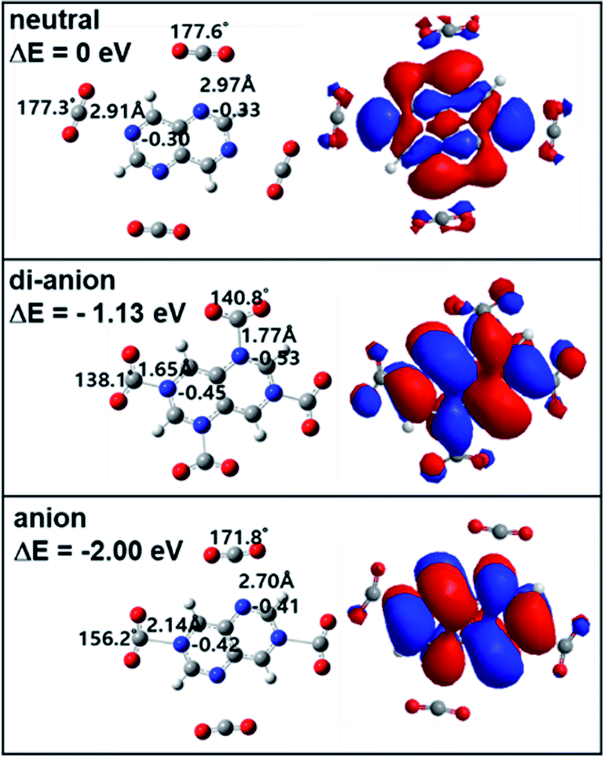 | ||
| Fig. 5 Neutral (top), anionic (bottom), and di-anionic (middle) geometries and calculated energies of TAN1(CO2)4 optimized at the BP86/6-311g** level. Orbitals are highly occupied orbitals: HOMO of neutral and dianion and SOMO of anion. | ||
Interestingly, in the case of dianion, both isomers showed that all three CO2 molecules covalently bound to the TAN molecule. This indicates that, when a TAN molecule has one more excess electron charge, it has a greater effective reactivity to activate CO2 molecules. In terms of the energetics, the 2,4,6 complex (0.92 eV respect to the neutral state) is slightly more stable than that of the 4,6,8 complex.
For TAN1·(CO2)4, in the anionic state, only two CO2 molecules were reduced in the 2,6 positions of nitrogen atoms and the other two CO2 molecules had van der Waals interaction with the 4,8 position of nitrogen atoms as shown in Fig. 5. On the other hand, as we expected, all four CO2 molecules were reduced in the dianionic state. In terms of geometric change, CO2 molecules in the 2,6 positions were slightly different from CO2 molecules in the 4,8 positions: the bent angle of CO2 and the bond length between the carbon of CO2 and the nitrogen of TAN. The reason is, we think, simply due to the asymmetric electron charge distribution in the TAN molecule.
Through vibrational spectrum analysis, we found that, in the neutral state, the CO2 asymmetric stretching mode of all four CO2 molecules was decoupled from the TAN molecule and so the vibrational frequency was almost the same: 2367 cm−1 for CO2 in the 4,8 positions and 2368 cm−1 for CO2 in the 2,6 positions. In the anionic state, there was structural similarity and the vibration of each pair of CO2 molecules was coupled as shown in Fig. 6; hence, one pair of CO2 molecules created covalent bonds, but the other pair did not. Thus, the asymmetric stretching mode was split into two peaks in the spectrum: 2180 cm−1 for CO2 molecules in the 4,8 positions and 2344 cm−1 for CO2 molecules in the 2,6 positions. Furthermore, the peak intensity of the covalently interacting CO2 molecules exceeded that of the vibrations of the other two CO2 molecules. In the dianionic state, all four CO2 molecules created covalent bonds with the TAN molecule and so the frequency and the intensity of the CO2 asymmetric stretching vibration were similar in all four molecules, as shown in Fig. 6.
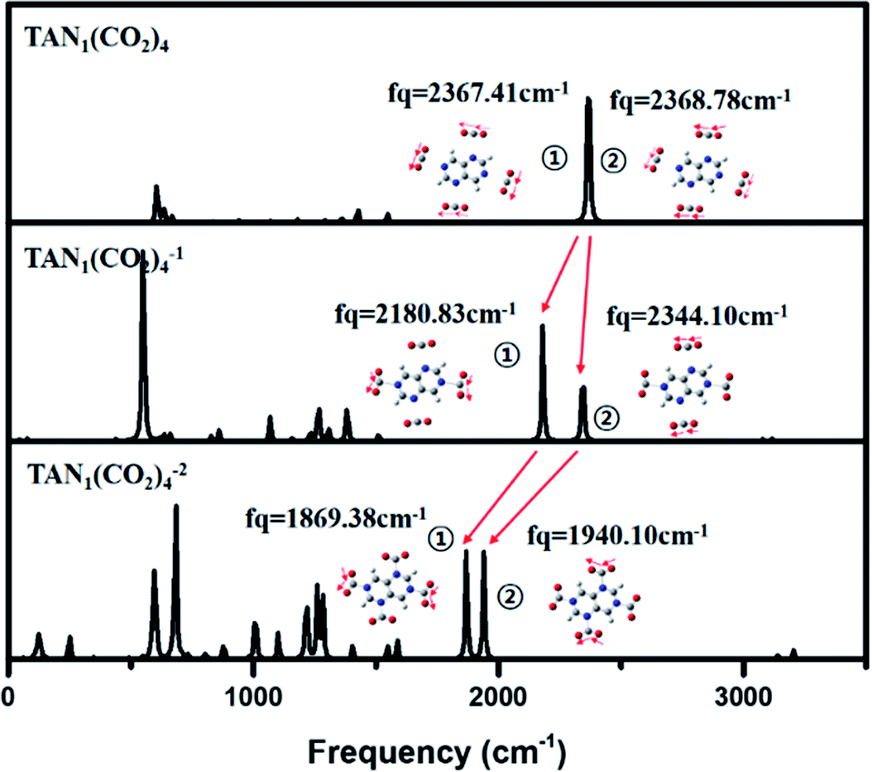 | ||
| Fig. 6 IR spectrum for neutral, anionic, di-anionic of TAN1(CO2)4 when CO2 position is 2, 4, 6, 8 optimized at the BP86/6-311g** level. | ||
Discussion
Accordingly, tetra-aza-naphthalene has the capability to covalently interact with CO2 molecules in the anionic and dianionic states. This indicates that excess electrons are needed to create a chemical reaction between nitrogen-containing PAH and CO2 molecules. Interestingly, when tetra-aza-naphthalene bound to more than three CO2 molecules, anionic tetra-aza-naphthalene did not have a covalent interaction with all three of CO2 molecules. Nevertheless, when adding one more electron to the complex, creating a dianion, all three or four CO2 molecules in the complex covalently bonded to the nitrogen atoms in aza-naphthalene. A similar tendency was found with other aza-naphthalenes, as shown in the ESI.† In addition, all complexes more strongly interacted with CO2 molecules in the dianionic state than in the anionic state. This indicates that nitrogen atoms need to be more negatively charged to create a covalent reaction. Therefore, we conclude that the molecule in the dianionic state was more reactive to CO2 molecules than in the anionic state.As for tetra-aza-naphthalene molecules, the charge density in the dianionic state is about 1.7 × 1019 electrons m−2, which is about 1.0 A cm−2. Considering that the charge density of the graphene nanoribbon is about 0.1 to 1 × 108 A cm−2, it is reasonable to think that the charge density of a nitrogen-containing PAH molecule in the dianionic state is sufficient to create a chemical reaction.26 This means that, in order to capture or reduce CO2 molecules using the nitrogen-containing graphene, if an electric current higher than 1.0 A cm−2 is applied, the chemical reaction to reduce CO2 molecules will occur more efficiently. Thus, our perspective is that the nitrogen-containing PAH molecule or graphene, when in the negatively charged state, can be a good catalytic material to convert CO2 molecules into useful chemicals.
Conclusions
Reducing CO2 molecules is the first step in converting the molecule into useful chemicals. Many nitrogen-containing molecules have been used to capture CO2 molecules. To capture CO2 molecules through a reductive interaction, we found that the charge state of nitrogen-containing molecules was an important factor in increasing the reactivity. In this theoretical study, we investigated tetra-aza-naphthalene, which can serve as a building block for nitrogen-doped graphene and found that it has a higher reactivity to capture CO2 molecules in the dianionic state than in the anionic state. When we approximated the charge density in the molecule, it was about 1.7 × 1019 electrons m−2, which is a comparable charge density in the range of the regular current of graphene or nano-sized graphene (nano-ribbon). Therefore, our perspective is that it is of fundamental importance to make the electric current higher than 1.0 A cm−2 to convert CO2 molecules when nitrogen-containing PAH or graphene is used.Author contributions
C. J. P., O. H., H. S. L, K. S. L. and S. H. L. designed and performed the research. C. J. P. and S. H. L. wrote the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by a 2 Year Research Grant of Pusan National University.Notes and references
- K. S. Novoselov, A. K. Geim, S. V. Morozov, D. Jiang, M. I. Katsnelson, I. V. Grigorieva, S. V. Dubonos and A. A. Firsov, Nature, 2005, 438, 197–200 CrossRef CAS PubMed.
- S. V. Morozov, K. S. Novoselov, M. I. Katsnelson, F. Schedin, D. C. Elias, J. A. Jaszczak and A. K. Geim, Phys. Rev. Lett., 2008, 100, 016602 CrossRef CAS PubMed.
- M. Han, B. Ozyilmaz, Y. Zhang, P. Jarillo-Herero and P. Kim, Phys. Status Solidi B, 2007, 244, 4134–4137 CrossRef CAS.
- K. I. Bolotin, K. J. Sikes, Z. Jiang, M. Klima, G. Fudenberg, J. Hone, P. Kim and H. L. Stormer, Solid State Commun., 2008, 146, 351–355 CrossRef CAS.
- T. F. Yeh, J. Cihlar, C. Y. Chang, C. Cheng and H. S. Teng, Mater. Today, 2013, 16, 78–84 CrossRef CAS.
- B. X. Zhang, H. Gao and X. L. Li, New J. Chem., 2014, 38, 4615–4621 RSC.
- B. Zheng, J. Wang, F. B. Wang and X. H. Xia, Electrochem. Commun., 2013, 28, 24–26 CrossRef CAS.
- G. Lemes, D. Sebastian, E. Pastor and M. J. Lazard, J. Power Sources, 2019, 438, 227036 CrossRef CAS.
- S. H. Lee, N. Kim, D. G. Ha and S. K. Kim, J. Am. Chem. Soc., 2008, 130, 16241–16244 CrossRef CAS PubMed.
- S. Y. Han, I. Chu, J. H. Kim, J. K. Song and S. K. Kim, J. Chem. Phys., 2000, 113, 596–601 CrossRef CAS.
- M. Z. Kamrath, R. A. Relph and M. A. Johnson, J. Am. Chem. Soc., 2010, 132, 15508–15511 CrossRef CAS PubMed.
- E. E. Barton, D. M. Rampulla and A. B. Bocarsly, J. Am. Chem. Soc., 2008, 130, 6342–6344 CrossRef CAS PubMed.
- A. D. Boese, H. Schneider, A. N. Gloss and J. M. Weber, J. Chem. Phys., 2005, 122, 154301 CrossRef PubMed.
- H. Schneider, A. D. Boese and J. M. Weber, J. Chem. Phys., 2005, 123, 074316 CrossRef PubMed.
- X. Y. Luo, Y. Guo, F. Ding, H. Q. Zhao, G. K. Cui, H. R. Li and C. M. Wang, Angew. Chem., Int. Ed., 2014, 53, 7053–7057 CrossRef CAS PubMed.
- B. Kumar, M. Asadi, D. Pisasale, S. Sinha-Ray, B. A. Rosen, R. Haasch, J. Abiade, A. L. Yarin and A. Salehi-Khojin, Nat. Commun., 2013, 4, 2819 CrossRef.
- Y. Q. Sun, C. Li, Y. X. Xu, H. Bai, Z. Y. Yao and G. Q. Shi, Chem. Commun., 2010, 46, 4740–4742 RSC.
- G. L. Chai and Z. X. Guo, Chem. Sci., 2016, 7, 1268–1275 RSC.
- X. R. Zhu and Y. F. Li, WIRES Comput. Mol. Sci., 2019, 9, e1416 CAS.
- P. Pyykko and Y. F. Zhao, J. Phys. Chem., 1990, 94, 7753–7759 CrossRef CAS.
- P. Pyykko and Y. F. Zhao, Mol. Phys., 1990, 70, 701–714 CrossRef CAS.
- X. Li, A. E. Kuznetsov, H. F. Zhang, A. I. Boldyrev and L. S. Wang, Science, 2001, 291, 859–861 CrossRef CAS PubMed.
- M. O. Winghart, J. P. Yang, M. Vonderach, A. N. Unterreiner, D. L. Huang, L. S. Wang, S. Kruppa, C. Riehn and M. M. Kappes, J. Chem. Phys., 2016, 144, 054305 CrossRef.
- A. I. Boldyrev and L. S. Wang, Chem. Rev., 2005, 105, 3716–3757 CrossRef CAS PubMed.
- G. W. T. M. J. Frisch, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, X. Li, M. Caricato, A. V. Marenich, J. Bloino, B. G. Janesko, R. Gomperts, B. Mennucci, H. P. Hratchian, J. V. Ortiz, A. F. Izmaylov, J. L. Sonnenberg, D. Williams-Young, F. Ding, F. Lipparini, F. Egidi, J. Goings, B. Peng, A. Petrone, T. Henderson, D. Ranasinghe, V. G. Zakrzewski, J. Gao, N. Rega, G. Zheng, W. Liang, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell, J. A. Montgomery Jr, J. E. Peralta, F. Ogliaro, M. J. Bearpark, J. J. Heyd, E. N. Brothers, K. N. Kudin, V. N. Staroverov, T. A. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. P. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, J. M. Millam, M. Klene, C. Adamo, R. Cammi, J. W. Ochterski, R. L. Martin, K. Morokuma, O. Farkas, J. B. Foresman, and D. J. Fox, Gaussian 16, Revision C.01, Gaussian, Inc., Wallingford CT, 2016 Search PubMed.
- R. Murali, Y. X. Yang, K. Brenner, T. Beck and J. D. Meindl, Appl. Phys. Lett., 2009, 94, 243114 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1ra04202e |
| This journal is © The Royal Society of Chemistry 2021 |