
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAntioxidant and UV-radiation absorption activity of aaptamine derivatives – potential application for natural organic sunscreens†
Thi Le Anh Nguyen *ab,
Thi Hoai Nam Doanc,
Dinh Hieu Truongab,
Nguyen Thi Ai Nhungd,
Duong Tuan Quange,
Dorra Khirif,
Sonia Taamallif,
Florent Louisf,
Abderrahman El Bakalif and
Duy Quang Dao*a
*ab,
Thi Hoai Nam Doanc,
Dinh Hieu Truongab,
Nguyen Thi Ai Nhungd,
Duong Tuan Quange,
Dorra Khirif,
Sonia Taamallif,
Florent Louisf,
Abderrahman El Bakalif and
Duy Quang Dao*a
aInstitute of Research and Development, Duy Tan University, Da Nang, 550000, Vietnam. E-mail: nguyenthileanh@duytan.edu.vn; daoduyquang@duytan.edu.vn
bFaculty of Natural Sciences, Duy Tan University, Da Nang, 550000, Vietnam
cDepartment of Chemistry, Danang University of Science and Technology, The University of Danang, Da Nang, 550000, Vietnam
dDepartment of Chemistry, University of Sciences, Hue University, Hue, 530000, Vietnam
eDepartment of Chemistry, University of Education, Hue University, Hue, 530000, Vietnam
fUniversité de Lille, CNRS, UMR 8522, PC2A – PhysicoChimie des Processus de Combustion et de l'Atmosphère, 59000 Lille, France
First published on 17th June 2021
Abstract
Antioxidant and UV absorption activities of three aaptamine derivatives including piperidine[3,2-b]demethyl(oxy)aaptamine (C1), 9-amino-2-ethoxy-8-methoxy-3H-benzo[de][1,6]naphthyridine-3-one (C2), and 2-(sec-butyl)-7,8-dimethoxybenzo[de]imidazo[4,5,1-ij][1,6]-naphthyridin-10(9H)-one (C3) were theoretically studied by density functional theory (DFT). Direct antioxidant activities of C1–C3 were firstly evaluated via their intrinsic thermochemical properties and the radical scavenging activity of the potential antioxidants with the HOO˙/HO˙ radicals via four mechanisms, including: hydrogen atom transfer (HAT), single electron transfer (SET), proton loss (PL) and radical adduct formation (RAF). Kinetic calculation reveals that HOO˙ scavenging in water occurs via HAT mechanism with C1 (kapp, 7.13 × 106 M−1 s−1) while RAF is more dominant with C2 (kapp, 1.40 × 105 M−1 s−1) and C3 (kapp, 2.90 × 105 M−1 s−1). Antioxidant activity of aaptamine derivatives can be classified as C1 > C3 > C2. Indirect antioxidant properties based on Cu(I) and Cu(II) ions chelating activity were also investigated in aqueous phase. All three studied compounds show spontaneous and favorable Cu(I) ion chelating activity with ΔG0 being −15.4, −13.7, and −15.7 kcal mol−1, whereas ΔG0 for Cu(II) chelation are −10.4, −10.8, and −2.2 kcal mol−1 for C1, C2 and C3, respectively. In addition, all compounds show UVA and UVB absorption; in which the excitations are determined mostly as π–π* transition. Overall, the results suggest the potential applications of the aaptamines in pharmaceutics and cosmetics, i.e. as a sunscreen and antioxidant ingredient.
1. Introduction
Aaptamines are commonly known natural products, which have been extracted from Aaptos aaptos species in the marine milieu of the Pacific ocean, i.e. Malaysia,1 Vietnam,2 and Indonesia.3 The first aaptamine structures, extracted from a Japanese sponge and characterized by Nakamura et al.,4 are identified as alkaloid-based compounds containing the 1H-benzo[de]-1,6-naphthyridine skeleton. There is a large amount of research which reported different biological activities of aaptamines such as antifungal,5,6 antiviral,7 antimicrobial,8 and anticancer.1,9–13 In particular, antioxidant activity of aaptamines was early predicted and studied. Indeed, aaptamines and isoaaptamine, amongst other marine sponges, were reported for strong antioxidant activity against the DPPH radical.14 In a perspective view, besides the natural and well-disposed origin, the antioxidant properties and other biological activities of the aaptamines can be of human-health-benefit because they help protect the human body from free radicals, fight aging, boost the immune system, and prevent diseases. In the modern concept of antioxidants, it is noteworthy that the antioxidant properties of a compound are based not only on the free radical scavenging processes but also on the repairing activity for biomolecules, such as DNA, lipid or proteins, and on the chelating ability with oxidative transition metal ions, particularly Cu15–19 and Fe ions.20–22 The latter plays an important role in preventing the indirect formation of reactive hydroxyl radical (i.e. HO˙) caused by the Fenton-like reactions between Fe(II) and Cu(I) with hydrogen peroxide (i.e. H2O2).On the other hand, oxidative stress (OS) resulting from long-time ultraviolet radiation (UVR) exposure is identified as one of the main causes for skin aging, DNA skin damage and melanogenesis.23 At the early stage of the sunscreen research started by the 1940s, most of the products targeted minimizing the effect of UVB (280–315 nm) radiation because the direct UVB absorption by DNA generates UV-signature mutations leading to DNA lesions and carcinogenic effect.24 Scientists later discovered that the UVA (315–400 nm) can deeply penetrate into the skin, producing reactive oxygen species (ROS) and reactive nitrogen species (RNS) in human skin that cause DNA as well as other biological molecules to be damaged and so is not less harmful than the UVB to human skin.25,26 One of the common mechanism, for example, is the ROS-mediated cell damage by peroxidation of fatty acids within the phospholipid structure of the membrane. Recently, a broad-spectrum UV filter that covers both UVA and UVB regions is one of the first and foremost criteria for considering an organic sunscreen candidate.27
Moreover, the photo-protective properties of natural products have been earlier reported for natural antioxidants including polyphenols,28 stilbenes,29 hydroxycinamate derivatives.30,31 A large number of studies have shown positive effect of antioxidant in the skin, for both treatment and prevention of inflammation, oxidation, sebaceous glands or melanogenesis.32 Multiple mechanisms of skin photo-protective have been documented.26 In addition, the synthesis of p-hydroxycinnamic diacids such as of ferulic, sinapic, p-coumaric and caffeic diacid results in molecules which show potent antioxidant and UV filter.33 Recently, we reported the antioxidant and photo-protective properties of different natural compounds in which the cycloechinulin and wasabidienone extracted from marine fungus showed the most potential antioxidant and photo-protective properties.34
To the best of our knowledge, although some experimental studies have been performed on the antioxidant properties of aaptamines derivatives, there have not been any computational studies that consider both radical scavenging and photo-protective properties of the aaptamine derivatives. Thus, in this study the antioxidant activities and the UV filter properties of three aaptamine derivatives4 including piperidine[3,2-b]demethyl(oxy)aaptamine (C1), 9-amino-2-ethoxy-8-methoxy-3H-benzo[de][1,6]naphthyridine-3-one (C2), and 2-(sec-butyl)-7,8-dimethoxybenzo[de]imidazo[4,5,1-ij][1,6]-naphthyridin-10(9H)-one (C3) (Fig. 1) were elucidated using density functional theory (DFT). Multi-level thermodynamic and mechanistic approaches were applied to provide better insight into direct antioxidant properties based on the free radical scavenging reactions. Thermodynamic parameters of reaction characteristic of the antioxidant activity of three aaptamines C1–C3 via four common mechanisms: hydrogen transfer (HT), single electron transfer (SET), proton loss (PL), and radical adduct formation (RAF) were focused. Different intrinsic thermochemical parameters including bond dissociation enthalpy (BDE), adiabatic ionization potential (IP) and proton affinity (PA) were calculated in the gas phase (vacuo) and water. The reaction enthalpies (ΔrH0) and standard Gibbs free energies (ΔrG0) of four reactions between the studied compounds and HOO˙/HO˙ radicals were examined and the kinetic calculations were investigated using transition state theory (TST). Remarkably, indirect antioxidant properties based on the Cu(II) ion chelating activity of studied compounds in aqueous phase were also predicted. Finally, the time-dependent density functional theory (TD-DFT) was performed to study the UV absorption activities of the aaptamines. Hopefully, the obtained results contribute to a better understanding of the antioxidant and UVR absorption properties of marine product which allows further practical applications.
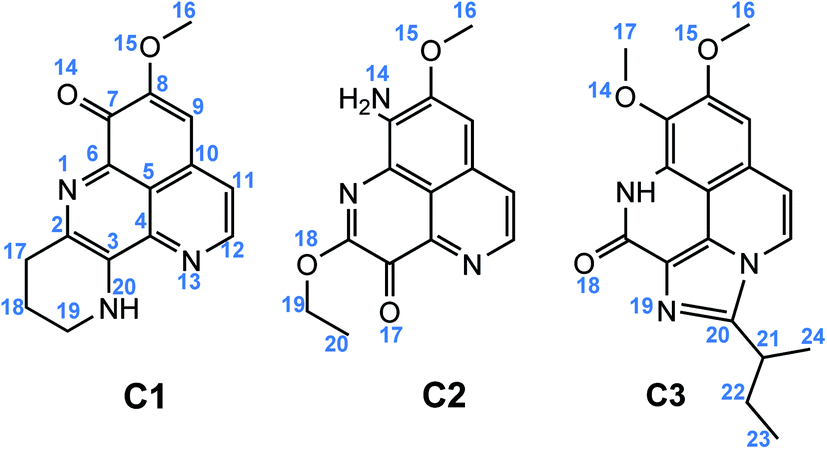 | ||
| Fig. 1 Chemical structure of C1–C3. | ||
2. Computational details
Gaussian 16 revision A.03 package was used to optimize the geometrical and electronic structures of the studied compounds.35 All calculations were performed at the M05-2X/6-311++G(d,p) level of theory.36 The M05-2X is previously reported to have a good benefit for the TS location and kinetics calculation.37,38The four main working mechanisms, including formal hydrogen transfer (FHT), single electron transfer (SET), proton loss (PL) and radical adduct formation (RAF) have been investigated. In the first approach, the intrinsic thermochemical parameters characterizing for three mechanisms FHT, SET, and PL were calculated according to the following reaction equations:
Formal hydrogen transfer (FHT):
| A–H → A˙ + H˙ (BDE); | (R1) |
Single electron transfer (SET):
| A–H → AH+˙ + e− (IP); | (R2) |
Proton transfer (PT):
| A–H → A− + H+ (PA); | (R3) |
Based on the equations (R1)–(R3), intrinsic thermodynamic parameters such as bond dissociation enthalpies (BDE), adiabatic ionization potential (IP), and proton affinities (PA) were calculated in the gas phase and in water as follows:
| BDE (A–H) = H(A˙) + H(H˙) − H(A–H); | (1) |
| IP (A–H) = H(AH˙+) + H(e−) − H(A–H); | (2) |
| PA (A–H) = H(A−) + H(H+) − H(A–H); | (3) |
In order to evaluate the influence of chemical nature of free radicals on the antioxidant potential of the studied compounds vs. representative radicals such as HOO˙, the standard Gibbs free energies of the reaction (ΔrG0) were calculated for HAT, PL, RAF and SET mechanisms according to the (R4)–(R7) reactions ((4)–(7)). The HO˙ radical scavenging reactions were similarly obtained.
| FHT: A–H + HOO˙ → A˙ + HOOH; | (R4) |
| PL: A–H + HOO˙ → A− + HOOH˙+; | (R5) |
| RAF: A–H + HOO˙ → AH–OOH˙; | (R6) |
| SET: A–H + HOO˙ → AH˙+ + HOO−; | (R7) |
Standard Gibbs free energies (ΔrG0) of the reactions were calculated at 298.15 K as follows:
| ΔrG0 (HAT) = [G(A˙) + G(HOOH)] − [G(A–H) + G(HOO˙)]; | (4) |
| ΔrG0 (PL) = [G(A−) + G(HOOH˙+)] − [G(A–H) + G(HOO˙)]; | (5) |
| ΔrG0 (RAF) = G(AH–OOH˙) − G(A–H) − G(HOO˙); | (6) |
| ΔrG0 (SET) = [G(AH˙+) + G(HOO−)] − [G(HOO˙) + G(A–H)]; | (7) |
The kinetics of FHT and RAF reactions in the gas phase and in water studied in this work were based on quantum mechanics-based test for overall free radical scavenging activity (QM-ORSA) protocol37 using the Eyringpy code.41 Details of this calculation can be found in our previous work.34 In brief, reaction rate, k(T), can be calculated using conventional transition state theory (TST) approach.
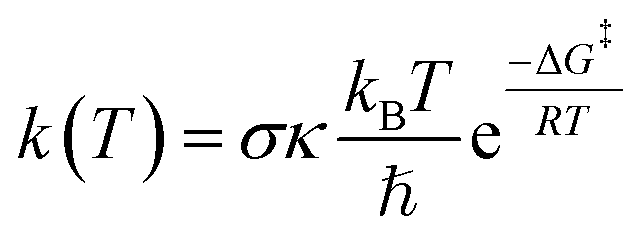 | (8) |
For SET reaction, the Marcus theory44 was applied for the estimation of the electron transfer rate. The energy barrier was obtained as (9).
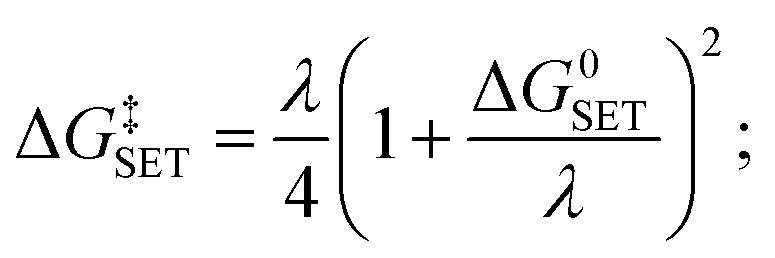 | (9) |
In solvent, diffusion rate kD may be important and greatly contributes to the apparent rate constant kapp. Therefore, the Collins–Kimball theory45 was employed (10).
| kapp = kDk/(kD + k); | (10) |
| kD = 4πRABDABNA; | (11) |
| DAB = kBT/(6πηr); | (12) |
The pKa calculation of three amines was performed following the thermodynamic cycle previously reported.50,51 pKa of the C1–C3 were determined as (13)
pKa = ΔG0deprot,aq/RT![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) ln(10); ln(10);
| (13) |
Cu(I) and Cu(II) ions chelating processes of three antioxidant compounds, now denoted as L, occur via the following reactions:
| L + [Cu·4H2O]+ = [CuL·2H2O]+ + 2H2O; | (R8) |
| L + [Cu·4H2O]2+ = [CuL·2H2O]2+ + 2H2O; | (R9) |
The Gibbs free energies (ΔrG0) of the complex formation were then calculated by (14) and (15) as follows:
| ΔrG0[Cu(I)] = G([CuL·2H2O]+) + 2G(H2O) − G(L) − G([Cu·4H2O]+); | (14) |
| ΔrG0[Cu(II)] = G([CuL·2H2O]2+) + 2G(H2O) − G(L) − G([Cu·4H2O]2+); | (15) |
The standard enthalpies of reaction (ΔH0) were similarly calculated for the complexation reaction (R8) and (R9).
The vertical excitation of C1–C3 in methanol was calculated using TD-DFT approach. A small benchmark of functionals with different exchange correlation XC part, i.e. B3LYP, B98, M06, PBE0, CAMB3LYP, and M05-2X were employed with the same basis set as in the previous part. These functionals are chosen following the recommendation by Jacquemine et al. for low Mean Signed Error (MSE) and Mean Absolute Error (MAE) for singlet excited states.52 Solvent effects were implicitly studied using the Polarization Continuum Model (IEF-PCM).53
3. Results and discussion
3.1. Structure and electronic properties
The optimized geometry and electronic structure of the three studied aaptamine compounds calculated by DFT method at the M05-2X/6-311++G(d,p) level of theory in the gas phase are presented in the Fig. 2. Cartesian coordinates and thermochemical properties of three aaptamines derivatives are resumed in Table S1 of ESI file.†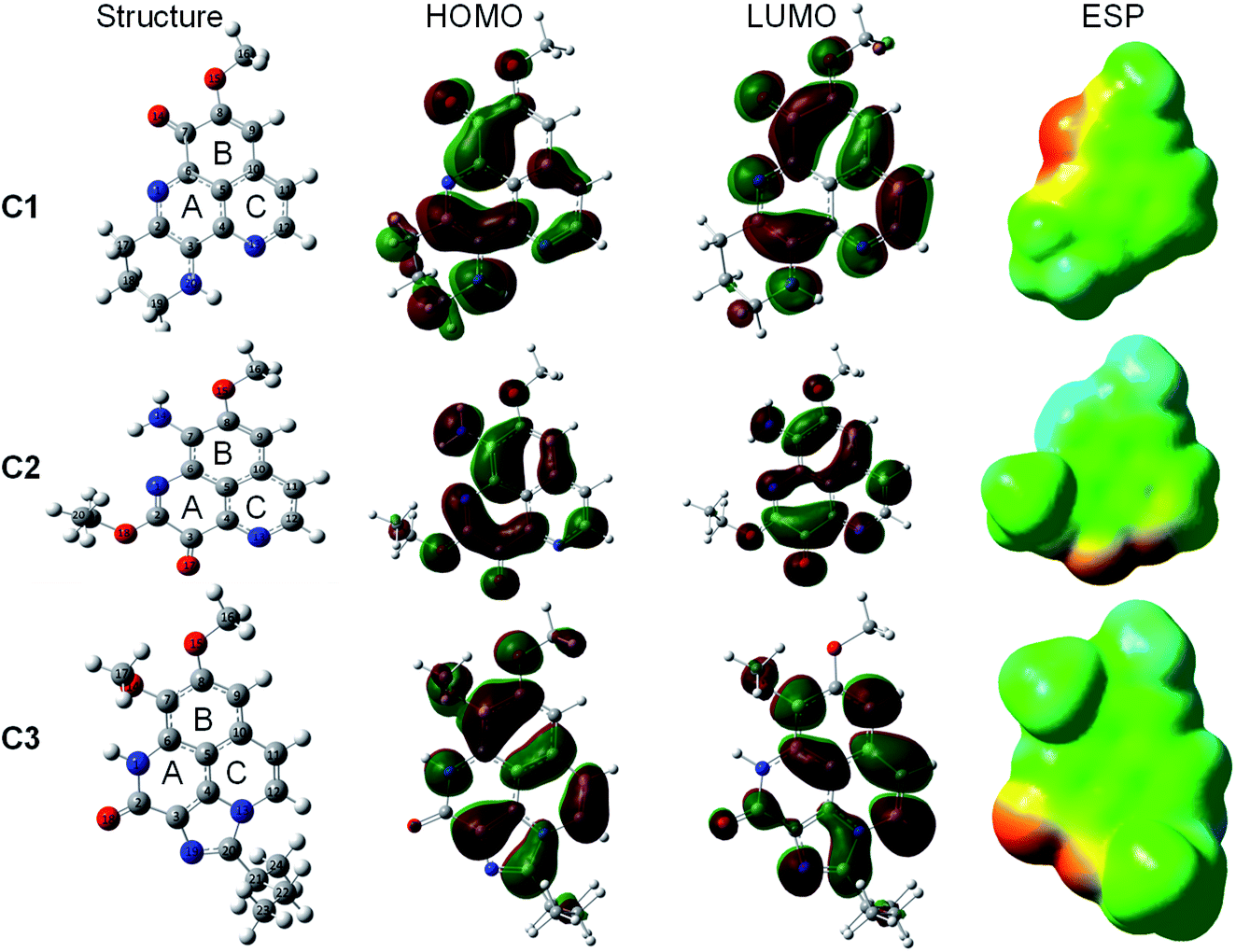 | ||
| Fig. 2 Optimized geometry, HOMO, LUMO, and ESP maps of C1–C3 calculated in the gas phase at the M05-2X/6-311++G(d,p) level of theory (isovalue = 0.02). | ||
Compounds C1–C3 share the benzo[de][1,6]naphthyridine skeleton and similarly have –OCH3 group attached to C8 position. At C7 position, the substituent groups such as ketone (![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O), amine (–NH2) and methoxy (–OCH3) are attached to the B ring, in C1, C2 and C3, respectively. The main difference in the chemical nature of three compounds is the substituent groups found at the A ring, for C1 a N-containing six-membered cycle, for C2 the O and –OC2H5 groups, and for C3 a N-containing five-membered cycle with 2-butyl derivative that shared both A and C ring. Moreover, the electronic distribution on the frontier molecular orbitals such as the highest occupied molecular orbital (HOMO) and the lowest unoccupied molecular orbital (LUMO) shows that all the rings play a key role in electron-donating (HOMO distribution) and electron-accepting (LUMO distribution) reactions with free radicals. For all molecules, the electrostatic potential (ESP) maps display a negative region on the CO groups and the nearby carbons.
O), amine (–NH2) and methoxy (–OCH3) are attached to the B ring, in C1, C2 and C3, respectively. The main difference in the chemical nature of three compounds is the substituent groups found at the A ring, for C1 a N-containing six-membered cycle, for C2 the O and –OC2H5 groups, and for C3 a N-containing five-membered cycle with 2-butyl derivative that shared both A and C ring. Moreover, the electronic distribution on the frontier molecular orbitals such as the highest occupied molecular orbital (HOMO) and the lowest unoccupied molecular orbital (LUMO) shows that all the rings play a key role in electron-donating (HOMO distribution) and electron-accepting (LUMO distribution) reactions with free radicals. For all molecules, the electrostatic potential (ESP) maps display a negative region on the CO groups and the nearby carbons.
3.2. Evaluation of antioxidant potential via intrinsic thermochemical parameters
Table 1 presents the intrinsic thermochemical parameters such as BDE, IP and PA which are examined via the FHT (R1), SET (R2) and PL (R3) mechanisms, respectively.| C1 | C2 | C3 | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Pos. | BDE | PA | IP | Pos. | BDE | PA | IP | Pos. | BDE | PA | IP |
| C9 | 111.8 | 371.6 | 168.6(110.0) | C9 | 113.9 | 366.7 | 168.3(109.2) | C9 | 113.7 | 368.9 | 169.3(114.0) |
| C11 | 114.3 | 374.4 | C11 | 113.5 | 376.3 | C11 | 114.9 | 371.3 | |||
| C12 | 106.2 | 383.6 | C12 | 107.3 | 387.6 | C12 | 114.4 | 360.6 | |||
| C16 | 97.1 | 367.1 | C16 | 97.6 | 384.4 | C16 | 96.5 | 384.2 | |||
| C17 | 85.5(85.6) | 366.4 | C19 | 94.7 | 377.8 | C17 | 97.3 | 389.3 | |||
| C18 | 98.9 | 390.7 | C20 | 100.8 | C21 | 84.6(84.9) | 366.3 | ||||
| C19 | 85.7(79.7) | 361.7 | N14 | 92.4(91.6) | 343.5(59.6) | C22 | 95.6 | 396.0 | |||
| C20 | 93.2 | 342.5(60.2) | C23 | 99.8 | 360.7 | ||||||
| C24 | 99.0 | 390.4 | |||||||||
| N1 | 101.8 | 347.2(59.7) | |||||||||
The most favorable H donating positions for C1 and C3 are found at C–H bonds. For example, C1 exhibits the lowest BDE at C17 and C19 positions, being 85.5 and 85.7 kcal mol−1, respectively, whereas C3 has the lowest BDE value at C21 position, i.e. 84.6 kcal mol−1. For C2, the easiest breaking-bond characterized by the lowest BDE value is located at N14 position (92.4 kcal mol−1). It is noteworthy that BDE values of C1 and C3 are much lower than that of the standard antioxidant Trolox in the gas phase (i.e. 91.1 kcal mol−1).54 Consequently, the antioxidant potential of these aaptamines via FHT process can be classified in the following order: C2 < Trolox < C1 < C3. We observed a very slight change of BDE values in water for all compounds C1–C3. This result is as expected, taken into account the neutral nature of the H atom species that is transferred. The only exception is obtained for C1 at C19 position, with a BDE value lowering from 85.7 kcal mol−1 in gas phase to 79.7 kcal mol−1 in water.
The proton donating reaction from the aaptamines to free radical is characterized by the proton affinity (PA, (3)); the lower the PA value, the better the antioxidant potential. As can be seen in Table 1, the lowest PA values are found to be 342.5, 343.5 and 347.2 kcal mol−1 for C1 (at C20 position), C2 (at N14 position) and C3 (at N1 position), respectively. These PA values are quite similar to that of cembrene in the gas phase (i.e. 343.2 kcal mol−1). The proton donating ability of the three compounds in reaction with free radical according to the PL process is in the increasing order: C3 < cembrene ≈ C2 < C1. It is noteworthy that the PL is much preferred in water, for which the PA values are significantly reduced about 60 kcal mol−1. The result is totally in agreement with the nature of the charge specie that is given and with previously reported results.55
Ionization potential (IP), characteristic for SET mechanism, is the minimum energy required to transfer an electron from the studied compound to free radical to form cationic species at ground state. The lower the IP value, the easier the electron transferring ability, thus, the antioxidant activity via SET mechanism will be higher. According to the results presented in Table 1, the adiabatic IP of C1–C3 varied from 168 to 169 kcal mol−1, which is slightly higher than that of Trolox (i.e. 164.6 kcal mol−1) and lower than the one of cembrene (i.e. 171.9 kcal mol−1).54 The antioxidant activities of the studied aaptamines following the SET mechanism increases in the order: Trolox < C2 < C1 < C3 < cembrene. Similar to the proton transfer, the electron transfer is also favorable in water, with IP values reducing from 168–169 kcal mol−1 in gas phase to 109–114 kcal mol−1 in water, while remaining in the same order C2 < C1 < C3.
Overall, three aaptamines C1–C3 are potential antioxidants. In the gas phase, the FHT mechanism can be responsible for the antioxidant properties of the molecules while in water the PL mechanism is particularly favorable.
3.3. HOO˙ and HO˙ free radical scavenging
The antioxidant potential of the studied aaptamines is investigated through the interactions with two representative free radicals, HOO˙ and OH˙, via four distinguished processes: formal hydrogen transfer (FHT), proton loss (PL), radical adduct formation (RAF), and single electron transfer (SET) mechanism ((R4)–(R7)). The Gibbs free energies (ΔrG0) of HAT, PL and RAF reactions between antioxidant and HOO˙ and HO˙ radicals at various positions are shown in Tables 2 and 3, respectively. The reaction via SET mechanism, which is considered position-independent, is separately presented in Table 4.| HAT | PL | RAF | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Pos. | C1 | C2 | C3 | Pos. | C1 | C2 | C3 | Pos. | C1 | C2 | C3 | |
| C9 | 27.3 | 29.5 | 29.2 | C9 | 219.8 | 214.9 | 217.0 | C2 | −1.0 | −9.8(5.2) | — | |
| C11 | 29.8 | 29.0 | 30.5 | C11 | 222.5 | 224.5 | 219.4 | C3 | −7.8 | 10.3 | 7.9 | |
| C12 | 21.7 | 22.8 | 29.9 | C12 | 231.8 | 235.7 | 208.7 | C4 | 14.4 | 2.4 | 2.6 | |
| C16 | 12.7 | 13.1 | 12.0 | C16 | 224.3 | 232.5 | 232.3 | C5 | 19.8 | 19.6 | 11.0 | |
| C17 | 1.0(0.4) | 12.8 | C17 | 214.5 | 237.5 | C6 | −3.6 | 0.1 | 9.6 | |||
| C18 | 14.4 | C18 | 238.9 | C7 | — | −5.6 | −7.4 | |||||
| C19 | 1.2(−5.0) | 10.2(9.1) | C19 | 209.9 | 225.9 | C8 | −12.6(6.5) | −1.5 | 3.7 | |||
| C20 | 8.7 | 16.4 | C20 | 190.6(77.3) | C9 | −6.1 | 7.0 | −0.3 | ||||
| N14 | 7.9(7.1) | N14 | 191.7(77.5) | C10 | 8.3 | 16.5 | 9.8 | |||||
| N1 | 17.4 | N1 | 195.3(76.8) | C11 | −4.0 | −4.3 | −1.4 | |||||
| C21 | 0.2(−0.4) | C21 | 214.4 | C12 | 0.4 | 3.4 | −11.7(3.0) | |||||
| C22 | 11.1 | C22 | 244.1 | |||||||||
| C23 | 15.4 | C23 | 208.9 | |||||||||
| C24 | 14.5 | C24 | 239.0 | |||||||||
| HAT | PL | RAF | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Pos. | C1 | C2 | C3 | Pos. | C1 | C2 | C3 | Pos. | C1 | C2 | C3 |
| C9 | −4.4 | −2.3 | −2.6 | C9 | 437.0 | 432.1 | 434.3 | C2 | −39.3(−23.6) | — | |
| C11 | −1.9 | −2.7 | −1.3 | C11 | 439.8 | 441.7 | 436.6 | C3 | −33.0 | −16.3 | −7.9 |
| C12 | −10.0 | −8.9 | −1.9 | C12 | 449.0 | 453.0 | 425.9 | C4 | −9.8 | −21.7 | −10.4 |
| C16 | −19.1 | −18.6 | −19.7 | C16 | 441.5 | 449.7 | 449.5 | C5 | −4.2 | −3.3 | −2.5 |
| C17 | −30.7(−33.6) | −18.9 | C17 | 431.8 | 454.7 | C6 | −28.1 | −23.2 | −4.5 | ||
| C18 | −17.3 | C18 | 456.1 | C7 | −17.8 | −32.1 | −26.7(−24.6) | ||||
| C19 | −30.5(−38.9) | −21.6(−24.8) | C19 | 427.1 | 443.1 | C8 | −41.6(−21.6) | −27.7 | −16.1 | ||
| C20 | −23.0 | −15.4 | C20 | 407.9(79.7) | C9 | −31.6 | −32.2 | −15 | |||
| N14 | −23.8(−26.8) | N14 | 408.9(80.0) | C10 | −15.0 | −5.7 | −2.1 | ||||
| N1 | −14.4 | N1 | 412.5(79.2) | C11 | −30.7 | −30.7 | −16.6 | ||||
| C21 | −31.6(−34.4) | C21 | 431.7 | C12 | −24.0 | −20.4 | −26.6(−24.2) | ||||
| C22 | −20.6 | C22 | 461.3 | ||||||||
| C23 | −16.4 | C23 | 426.1 | ||||||||
| C24 | −17.3 | C24 | 456.3 | ||||||||
| HOO˙ | HO˙ | |||||
|---|---|---|---|---|---|---|
| C1 | C2 | C3 | C1 | C2 | C3 | |
| Gas | 145.1 | 144.8 | 145.5 | 129.7 | 129.5 | 130.2 |
| Water | 37.4 | 37.2 | 41.1 | 15.3 | 15.2 | 19.0 |
As shown in the Table 2, in the gas phase, the Gibbs free energy values (ΔrG0) for FHT reaction (R4) towards HOO˙ are positive at all H-donating positions, ranging from 0.2 to 30.5 kcal mol−1. This result indicates that the potential for HOO˙ removal via FHT mechanism of all three investigated compounds is not spontaneous and not favorable. Concerning the RAF mechanism (R6), the ΔrG0 found negative values at some specific positions. For example, the C1 compound shows highly negative ΔrG0 of −13.0 and −12.6 kcal mol−1 at C7 and C8 positions, respectively. For C2, HOO˙ RAF process is highly favorable at C2 position with ΔrG0 being −9.8 kcal mol−1. Finally, for C3, the RAF reaction favorably occurs at C2 and C12 positions with ΔrG0 value being −14.2 and −11.7 kcal mol−1, respectively. Thus, the RAF is the only responsible mechanism for the scavenging process towards HOO˙ radical in the gas phase. However, in water, HOO˙ scavenging is only favorable with FHT mechanism but not with RAF. Indeed, spontaneous ΔrG0 are only obtained via FHT mechanism for C1 (at C19 position, −0.5 kcal mol−1) and C3 (at C21 position, −0.4 kcal mol−1). Finally, the proton transferring process (PL, (R5)) has positive ΔrG0 at all positions; thus, the ability to remove HOO˙ free radicals by PL process is neither favored in the gas phase nor in water.
Regarding to the OH˙ scavenging activities (Table 3), the negative ΔrG0 values are obtained at all positions of three studied compounds for both the FHT and RAF processes. This proves that C1–C3 have high scavenging potential towards OH˙ radical via FHT and RAF processes. The most active compound via FHT mechanism is awarded for C1 (C17 position) and C3 (C21 position), with the ΔrG0 of −30.7 and −31.6 kcal mol−1, respectively. The lowest ΔrG0 values for RAF mechanism are obtained for C1 (C8 position) and C2 (C2 position) compounds with the values in the gas phase of −41.6 and −39.3 kcal mol−1, respectively. For the proton transfer process, positive values of ΔrG0 are observed at all the positions for all aaptamines. This result is reasonable because the gas phase is not a favorable medium for the charge transfer process like the proton transfer. In gas phase, the RAF scavenging towards OH˙ is more favorable than the FHT. For example, C1 has the lowest ΔrG0(FHT) being −30.7 kcal mol−1, whereas it has the lowest ΔrG0(RAF) being −41.6 kcal mol−1. In contrast, in water, the FHT is more competitive than the RAF. For example, the lowest ΔrG0(FHT) of C1 is lowered to −38.9 kcal mol−1 at C19, while the lowest ΔrG0(RAF) is obtained for C3 with −24.6 kcal mol−1 at C18 position. In contract to this, the PL process is always not spontaneous and not favorable in any medium.
Moreover, the Gibbs free energies of the SET reactions (R7) are not favorable in our conditions with the ΔrG0 value in the gas phase varying around 145 kcal mol−1 for HOO˙ radical and 130 kcal mol−1 for HO˙ radical (Table 4). Although the ΔrG0 have been significantly decreased in water (15–41 kcal mol−1), the electron transfer is obviously not spontaneous.
Overall, the radical scavenging activities of the studied compounds favorably occur via RAF mechanism in the gas phase while in water it more likely occurs via HAT mechanism. The HO˙ radical scavenging is much more favorable than the reaction with HOO˙. Finally, the antioxidant potential via HAT process of the three studied compounds is classified in the increasing trend: C2 < C3 ≈ C1. For RAF mechanism, C1 and C2 are potent in gas phase while the C3 is more active in water.
3.4. Kinetics
In order to identify the main existing form of C1–C3 (i.e. neutral, protonated or deprotonated forms) at the physiological condition (pH = 7.4), pKa value needs to be determined.56 The pKa of three aaptamines C1–C3 were calculated at the M05-2X/6-311++G(d,p) level of theory. Similar calculation for aniline at the same level of theory was performed for comparison; the obtained value of pKa is 31.1 at 298.15 K for aniline, consistent with the experimental data, i.e. 30.6 or 28.0, reported in DMSO and in water, respectively.57,58 For our aaptamines, the result shows that pKa of C1–C3 are of 22.8, 23.6 and 22.4, respectively. Therefore, the neutral form of C1–C3 in water is considered in the following calculations.The kinetics of the possible reactions was studied with the neutral forms of C1–C3 in gas phase and water, for the OOH˙ scavenging. The Gibbs free energy of activation (ΔG‡) and transition state theory (TST) rate constant (kTST) for all reactions were systematically calculated at the M05-2X/6-311++G(d,p) level of theory. Gibbs free energy of activation and TST rate constants obtained in the gas phase are presented in Table 5. Optimized structures of the transition states (TSs) for FHT and RAF reaction in both phases are presented in the Fig. 3 and 4. Cartesian coordinates and thermochemical properties for TSs of HAT and RAF reactions between three aaptamines derivatives with HOO radical are resumed in Tables S2 and S3, ESI file.†
| Reaction | ΔG‡, 1 M, kcal mol−1 | kTST298.15 K, cm3 per molecule per s |
|---|---|---|
| FHT | ||
| C1@C19H + HOO˙ | 16.8 | 9.10 × 10−19 |
| C2@N14H + HOO˙ | 19.3 | 2.60 × 10−21 |
| C3@C21H + HOO˙ | 19.8 | 2.30 × 10−19 |
| Ascorbic + HOO˙ | 9.2 | 3.10 × 10−15 |
|
||
| RAF | ||
| C1@C8 + HOO˙ | 14.8 | 3.40 × 10−19 |
| C2@C2 + HOO˙ | 14.0 | 3.80 × 10−18 |
| C3@C12 + HOO˙ | 15.6 | 7.60 × 10−21 |
| Ascorbic + HOO˙ | 15.0 | 3.60 × 10−19 |
|
||
| SET | ||
| C1 + HOO˙ | 420.9 | 4.30 × 10−295 |
| C2 + HOO˙ | 378.7 | 3.93 × 10−264 |
| C3 + HOO˙ | 401.3 | 1.05 × 10−280 |
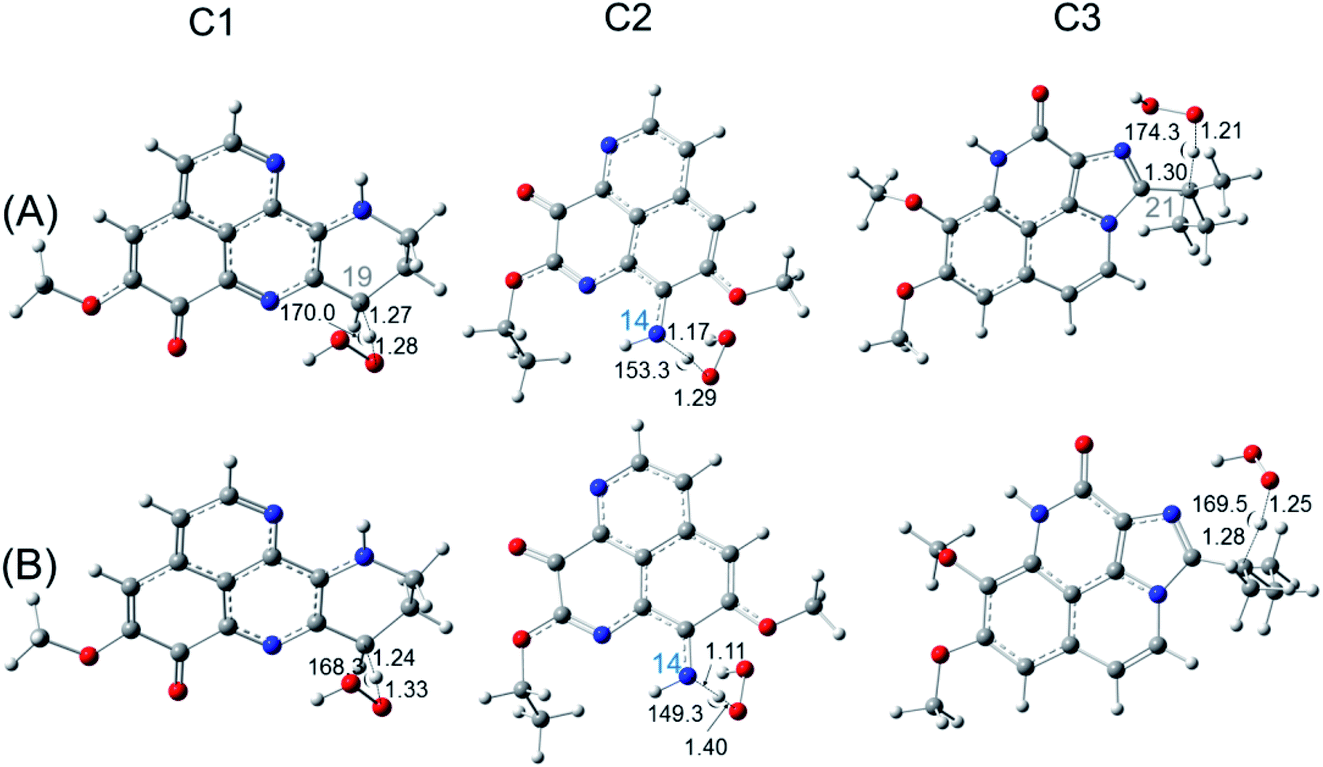 | ||
| Fig. 3 Optimized structures of the transition state for FHT reaction of C1, C2, C3 with HOO˙ radical in the gas phase (A) and in water (B) at the easiest H donating positions. Calculations are performed at the M05-2X/6-311++G(d,p) level of theory. | ||
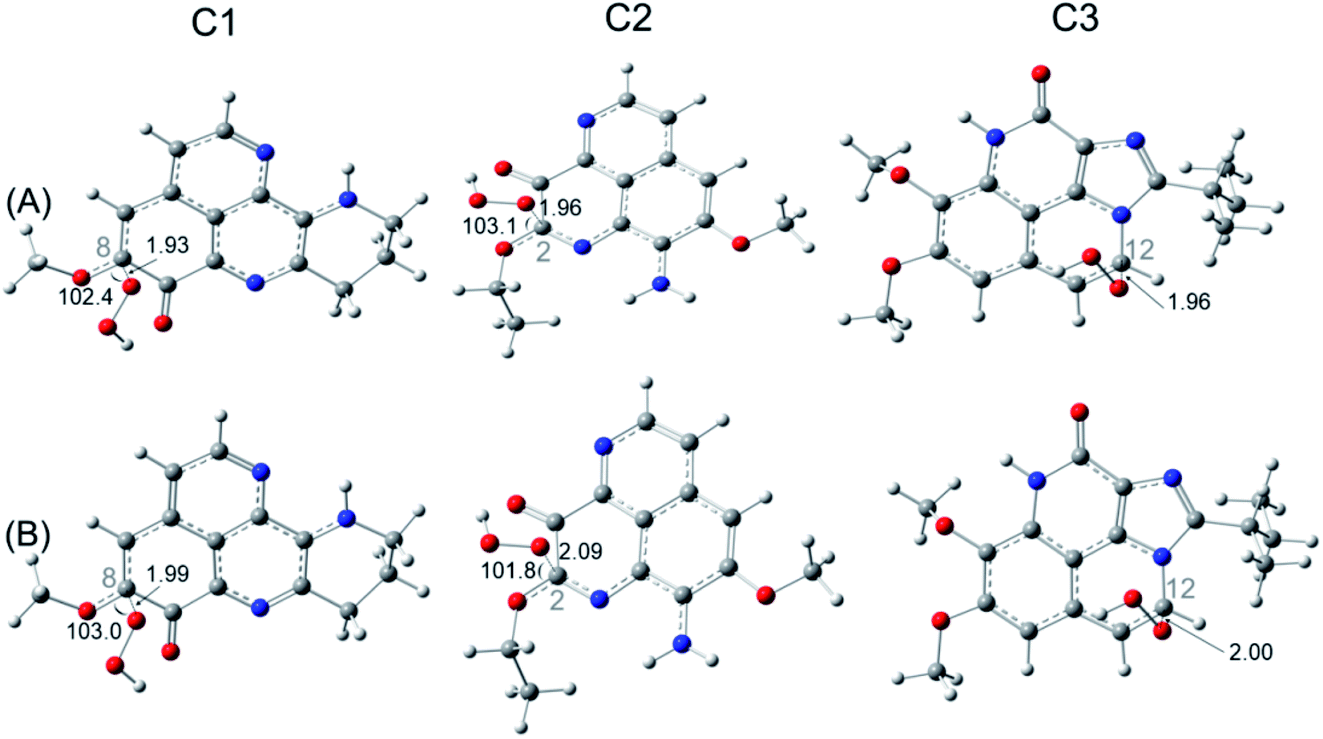 | ||
| Fig. 4 Optimized structures of the transition state for RAF reaction of C1, C2, C3 with HOO˙ radical in the gas phase (A) and in water (B) at the most spontaneous addition positions. Calculations are performed at the M05-2X/6-311++G(d,p) level of theory. | ||
For FHT reaction, the compounds C1 and C3 display the activation Gibbs free energy of 16.8 and 19.8 kcal mol−1, respectively. Similar rate constants are obtained for two compounds C1 and C3 (∼10−19 cm3 per molecule per s), which is much smaller than the one of a standard antioxidant such as ascorbic acid calculated in the same condition (∼10−15 cm3 per molecule per s). For the RAF reaction, the lowest activation Gibbs free energy ΔG‡ (14.0 kcal mol−1) and the highest rate constant (3.80 × 10−18 cm3 per molecule per s) are obtained for C2 at C2 position, indicate the most favorable reaction. At the second place, the C1 is found with similar activation Gibbs free energy as ascorbic acid, values of about 14.8–15.0 kcal mol−1 and rate constants of 3.4–3.6 × 10−19 cm3 per molecule per s. In the contrary, the SET reaction is found with very high activation energy of about 400 kcal mol−1 and near-zero rate constants.
For the reactions in water, kinetics data including the Gibbs free energy of activation ΔG‡, the diffusion rate constants kD, thermal rate constant kT, Eckart-tunneling-corrected rate constants keck, diffusion-corrected rate constants kapp, and the branching ratio Γ (%) for each reaction FHT, RAF and SET are resumed in Table 6.
| Reaction path | ΔG‡, 1 M, kcal mol−1 | kD, M−1 s−1 | kT, M−1 s−1 | keck, M−1 s−1 | kapp, M−1 s−1 | Γ, % |
|---|---|---|---|---|---|---|
| FHT | ||||||
| C1@C19H + HOO˙ | −0.87 | 1.40 × 109 | 7.17 × 106 | 3.22 × 10−15 | 7.13 × 106 | 96 |
| C2@N14H + HOO˙ | 22.2 | 2.60 × 109 | 3.60 × 10−4 | 6.10 × 100 | 6.10 × 100 | 0 |
| C3@C21H + HOO˙ | 5.17 | 1.27 × 109 | 3.84 × 103 | 3.22 × 10−16 | 3.84 × 103 | 1 |
|
||||||
| RAF | ||||||
| C1@C8 + HOO˙ | 7.2 | 2.10 × 109 | 3.00 × 107 | 2.80 × 105 | 2.80 × 105 | 4 |
| C2@C2 + HOO˙ | 5.6 | 2.20 × 109 | 4.20 × 108 | 1.40 × 105 | 1.40 × 105 | 100 |
| C3@C12 + HOO˙ | 7.5 | 2.00 × 109 | 1.90 × 107 | 2.90 × 105 | 2.90 × 105 | 99 |
|
||||||
| SET | ||||||
| C1 + HOO˙ | 47.10 | 8.22 × 109 | 4.56 × 10−21 | — | 4.56 × 10−21 | 0 |
| C2 + HOO˙ | 45.91 | 8.26 × 109 | 3.35 × 10−20 | — | 3.35 × 10−20 | 0 |
| C3 + HOO˙ | 53.19 | 8.47 × 109 | 1.55 × 10−25 | — | 1.55 × 10−25 | 0 |
For FHT reaction at the C19H position of C1, it happens to be a barrierless reaction, with negative Gibbs free energy of activation (−0.87 kcal mol−1). Moreover, the rate constant obtained at this position is of 7.13 × 106 M−1 s−1. On the contrary, the FHT reaction of C3 requires 5.17 kcal mol−1 of Gibbs free energy of activation and appears with a rate constant of only 3.84 × 103 M−1 s−1. Moreover, FHT reaction is not favored for the C2 with very high reaction barrier (22.2 kcal mol−1) and even lower apparent rate constant (6.10 M−1 s−1). For RAF reaction, the reaction barriers (ΔG‡) are found to be 5.6, 7.2 and 7.5 kcal mol−1 for C1, C2, C3, respectively. As can be seen in the Table 6, the diffusion rate constants kD of RAF are of about 109 M−1 s−1, while the thermal rate constants are much smaller (∼105 M−1 s−1). The RAF apparent rate is observed with C2 at the C2 position with kapp = 1.40 × 105 M−1 s−1 while a double kapp values (2.8–2.9 × 105 M−1 s−1) are obtained for C1 and C3. These results show higher reaction rate of C2 and C3 via RAF than via FHT mechanism. Similar as in the gas phase, the SET reaction of all three compounds requires very high activation Gibbs free energy (45–53 kcal mol−1) and occurred at extremely small rate (10−25 to 10−20 M−1 s−1). Overall, we observed a competition of the FHT and RAF reaction for the HOO˙ scavenging depending on the chemical nature of the substituents to the aaptamine. For C1, the FHT reaction is dominant with branching ratio being 96%, while the HOO˙ scavenging via RAF is more favored with C2 and C3 with Γ values being 100% and 99%, respectively.
3.5. Formal H transfer – HAT or PCET?
In order to answer the question how and whether or not the electron and proton are transferred in many important energy conversion processes in chemistry and biology,59 the chemical nature of the FHT process for the aaptamine derivatives with HOO˙ radical is analyzed in this section. Identifying the HAT and PCET mechanisms is a difficult mission because the same reactants and the same products are recognized between these two mechanisms, although the reaction pathways are totally different. Analyses of singly occupied molecular orbital (SOMO) distribution (Fig. 5) and of electronic properties such as NPA charges, atomic spin densities (ASD), natural electron configuration (NEC) calculated at the transition states (TSs) (Table S4, ESI file†) as well as NBO analyses (Table S5, ESI file†) allow addressing this problem.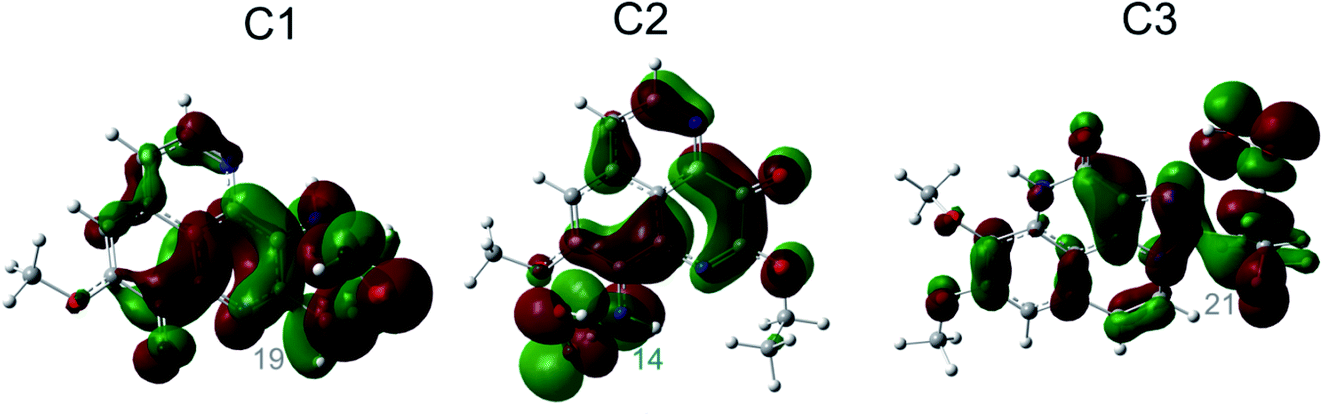 | ||
| Fig. 5 SOMO distributions of the transition states (TSs) for FHT reactions at the easiest H donating positions. | ||
Fig. 5 represents first the SOMO distributions at the TSs for FHT reactions of three aaptamine compounds with HOO˙ radical in the gas phase. It is reported that SOMO of HAT TSs is distributed with significant densities in orbitals along the H-shifting vector between the donor and acceptor, while the one of PCET TSs is orthogonal to the transition vector.60 As can be seen in Fig. 5, the 2p orbitals of the acceptor (i.e. O34 and O44 atom of the HOO˙ radical) and the one of the donor (i.e. C atoms or N14 atom) are interacted and distributed along the H-shifting vector whereas the π-electrons on the HOO˙ radical and the ones on the aromatic rings of aaptamines are tunneled together. This observation consists in the first signal for a H atom transfer (HAT) nature.
To shed more light into changes of electron densities at the transition states of FHT processes, we investigated natural bond orbitals (NBO) analysis (Table S5, ESI file†). As a result, for the FHT reaction between C1 and HOO˙ at the C19H position (C1@C19H + HOO˙), the electron densities are transferred from the first σ-bonding of C19–H bond, σ(1) C19–H, to the first σ-antibonding of O34–O35 bond, σ*(1) O34–O35, with a stabilization energy [E(2)] of 7.7 kcal mol−1. Conversely, the electron densities are also transferred from the lone pairs of electron on O34 atom of HOO˙ radical, LP(1) O34, LP(2) O34 and LP(3) O34, to the σ*(1) C19–H with the E(2) values being 324.3, 27.2 and 255.8 kcal mol−1, respectively. For the reaction C2@N14H + HOO˙, only the electron transfer from σ-antibonding of N14–H bond, σ*(1) N14–H, to the one of O34–O35 bond, σ*(1) O34–O35, is observed with a small E(2) value of 5.5 kcal mol−1. A similar observation is found the reaction C3@C21H + HOO˙. In fact, the electron densities are essentially changed from σ*(1) C21–H to σ*(1) O34–O35 with the stabilization energy E(2) being 1293.7 kcal mol−1. In the reverse trend, the electrons from the lone pair on O44 and O45 atoms, LP(1) O44 and LP(1) O45, of the HOO˙ radical to the first σ-antibonding on C21–C24 bond, σ*(1) C21–C24, with the E(2) values being 5.6 and 77.5 kcal mol−1, respectively.
Furthermore, natural electron configuration (NEC) of the TSs in Table S5† of ESI shows that the mitigated-H species have 1s occupancy 1s0.61–0.67 which likely corresponds to one H atom with configuration 1s1.0. In addition, the ASD values of the H species at the transition state are slightly varying from −0.05014 to −0.01012. This result is in good agreement with the one observed for HAT reaction of α-mangostin with HOO˙ radical.60 Moreover, the NEC of carbon centered-donors in C1 and C3 shows the configurations 2s0.96–1.022p3.20–3.31 likely 2s2p3, while the one of nitrogen-based donor in C2 has the configuration 2s1.392p4.25 likely 2s2p4. In the same time, the H-acceptors (i.e. O34 and O44 atoms) have the electron configurations 2s1.80–1.822p4.49–4.56 which are similar with the 2s22p4. In addition, the spin densities are all distributed at the donors (0.26019–0.46482) and at the acceptors (0.32787–0.42642). These observations support the conclusion that the shifted-H has an atom character rather than a proton one. Thus, the FHT processes of all three aaptamines have the chemical nature of HAT mechanism.
3.6. Indirect antioxidant potential via copper ions chelating properties
Besides ferric ion [i.e. Fe(III)], the cupric ion Cu(II) consists in one of the most abundant cations available in most organisms and in human body.18 Although they are themselves not harmful species, their reduction can lead to the formation of very reactive hydroxyl HO˙ radical via the Haber–Weiss cycle.61 Regarding to Cu(II) ion, its complexation with the potential antioxidant compounds could sometime prevent the reduction process that forms Cu(I) ion and HO˙ radical. The fact was that, several potential antioxidants show good copper ions chelating abilities were reported with aminoguanidine,62 pyridoxamine,63 lipoic and dihydrolipoic acids,64 purine,18 citric acid,65 D-penicillamine,66 2-hydroxymelatonin and 4-hydroxymelatonin.16 In this study, the Cu(II) complexes formed by the chelating process of the three aaptamines at different possible chelation sites in water phase are evaluated; the results are presented in Fig. 6. Similar complexes with Cu(I) ion are also presented in Fig. 7. Standard enthalpies (ΔH0) and Gibbs free energies (ΔG0) of the complexation processes ((R8) and (R9)) are determined to evaluate the stability of the obtained complexes. Only bidentate complexes with the hydrated copper ions, i.e. [Cu(I)·4H2O]+ and [Cu(II)·4H2O]2+ are considered because they are more stable than the monodentate ones as showed by several studies in the literature. Cartesian coordinates and thermochemical properties for the complexes between three aaptamines derivatives with Cu(II) and Cu(I) ions are presented in the Tables S6 and S7, ESI.†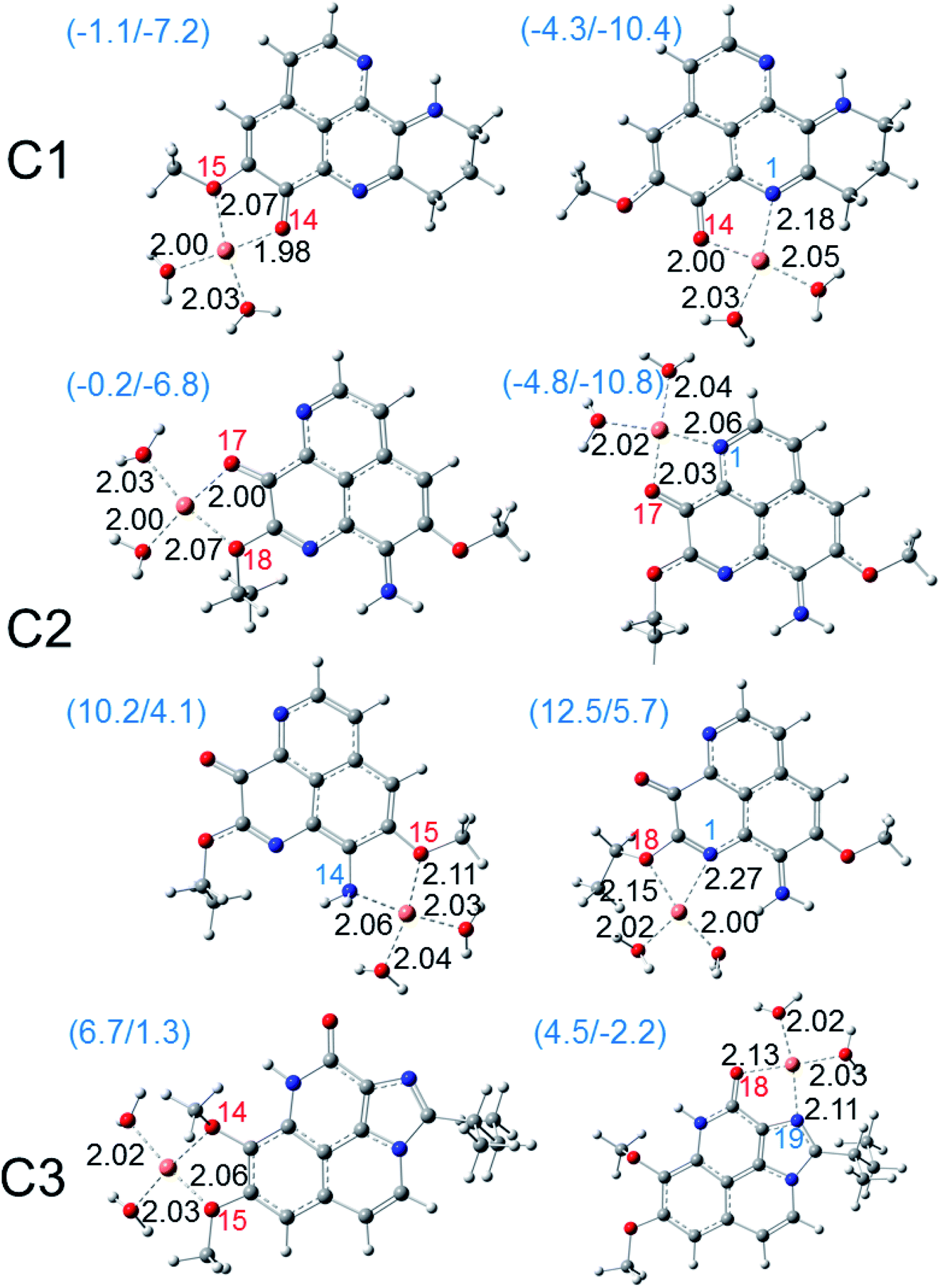 | ||
| Fig. 6 Optimized structures of the bidentate Cu(II) complexes of three aaptamines C1–C3 in water phase calculated at the M05-2X functional with the basis set LanL2DZ for Cu and the 6-311++G(d,p) for other elements. The standard enthalpies (ΔH0) and Gibbs free energies (ΔG0) of the complexation reactions in kcal mol−1 are given in the parentheses. Coordinated oxygen and nitrogen atoms are numbered in red and blue, respectively. Bond distances (in angstroms) are noted in black. | ||
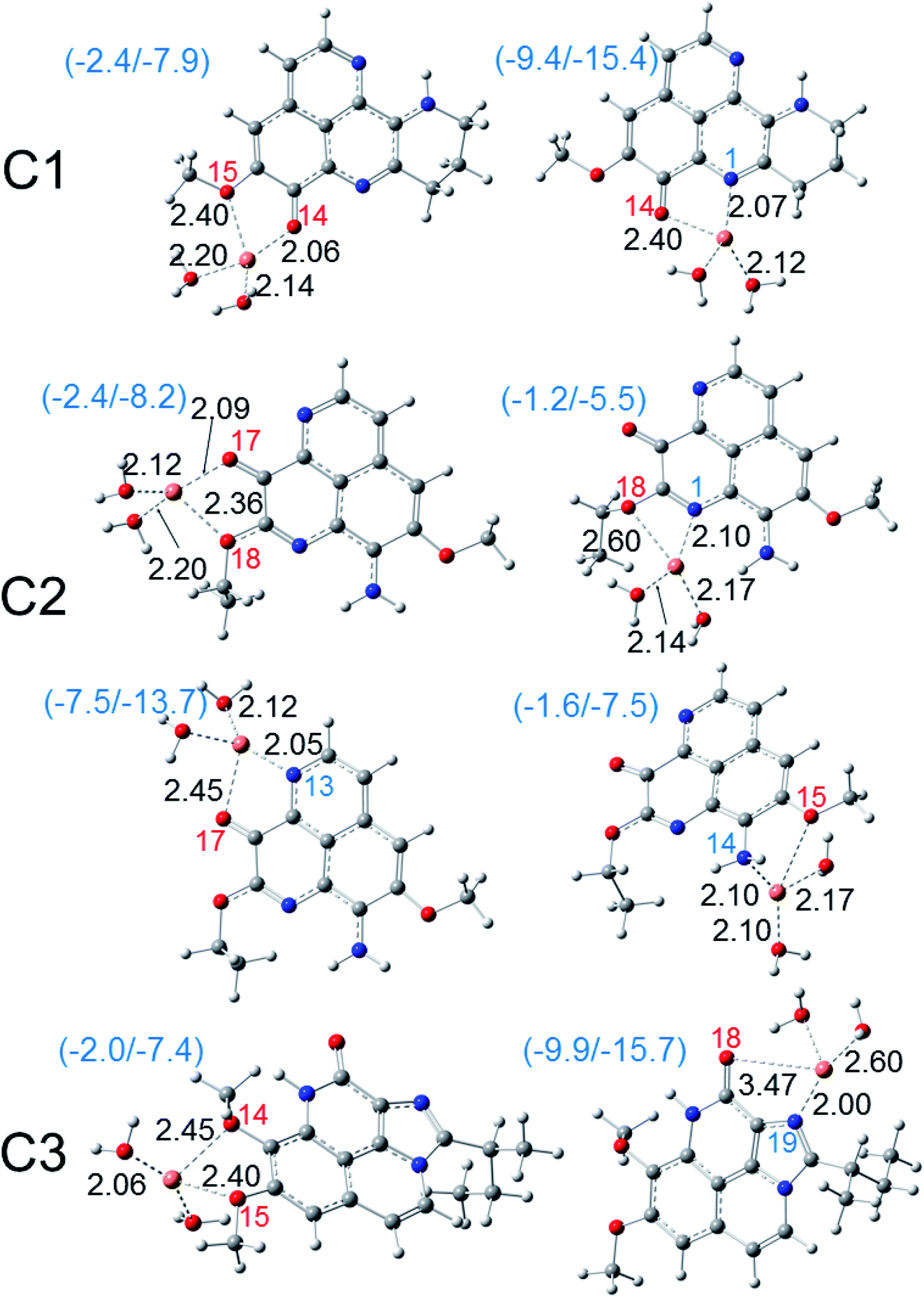 | ||
| Fig. 7 Optimized structures of the bidentate copper(I) complexes of three aaptamines C1–C3 in water phase calculated at the M05-2X functional with the basis set LanL2DZ for Cu and the 6-311++G(d,p) for other elements. The standard enthalpies (ΔH0) and Gibbs free energies (ΔG0) of the complexation reactions in kcal mol−1 are given in the parentheses. Coordinated oxygen and nitrogen atoms are numbered in red and blue, respectively. Bond distances (in angstroms) are noted in black. | ||
As can be seen in Fig. 6, all three aaptamine compounds show spontaneous and favorable Cu(II) ion chelating activities with negative ΔH0 and ΔG0 values. Indeed, the most favorable chelating site for C1 is found at O14/N1 with ΔH0/ΔG0 being −4.3/−10.4 kcal mol−1. Similarly, the most favorable site for C2 is at O17/N1 with ΔH0/ΔG0 being −4.8/−10.8 kcal mol−1, and for C3 being at O18/N19 (ΔH0/ΔG0 being 4.5/−2.2 kcal mol−1). Thus, the Cu(II) ion chelating activities of three studied compounds can be ranged in the increasing trend: C3 < C1 < C2. The Cu(II) chelation of the studied aaptamines are better than the one of aminoguanidine (ΔG0, 13.8 kcal mol−1),62 2-hydroxymelatonin and 4-hydroxymelatonin (ΔG0, −5.59 and 0.63 kcal mol−1, respectively)16 and D-penicillamine (ΔG0, 0.3 kcal mol−1).66
Furthermore, it is noteworthy that the Cu(I) complexation is more favorable than the one of Cu(II) ion for all three compounds at the same chelation site. For example, the ΔH0 and ΔG0 values of the complexation process at the O14/N1 site of C1 are −9.4 and −15.4 kcal mol−1, respectively. For C2 at the O17/N13 site, the formed Cu(I) complex is also stable with ΔH0/ΔG0 being −7.5/−13.7 kcal mol−1, while the ΔH0/ΔG0 values for C3 at the O18/N19 chelating site are equal to −9.9/−15.7 kcal mol−1. It is noted that the Cu(I) chelating activities are in the increasing order: C2 < C1 < C3, which is in the reverse order in comparison with the Cu(II) ones.
Thus, all three aaptamine derivatives present the spontaneous and favorable formation of the complexes with both Cu(II) and Cu(I) hydrated ions in water phase. This reduces the availability of these ions to participate in the Haber–Weiss cycle in forming the hydroxyl radical.
3.7. UV radiation absorption properties
The vertical excitation of the molecules C1–C3 is studied with TD-DFT using different functionals such as B3LYP, B98, M06, PBE0, CAM-B3LYP and M05-2X as previously recommended for accuracy.52 In order to compare with experimental data reported in MeOH,4 all calculation is performed in MeOH using the implicit model IEF-PCM. The lowest absorption wavelength of each compound calculated by six above-mentioned functionals is presented in the Table S8† of ESI in comparison with the measured data.As can be seen in Table S8,† among the different methods, a fairly consistent result in comparison with the experimental data is obtained with CAM-B3LYP and M05-2X functional. For example, the lowest absorption of C1 calculated in MeOH by M05-2X is found at 423 nm while the experimental value was reported at 398 nm in the same solvent (25 nm deviation). Similar deviation of about 30 nm was obtained for C2. However, a much higher deviation is observed with C3, for which a shorter wavelength of 292 nm is absorbed vs. 350 nm in measurement. The other methods, which comprise a portion of 20–30% HF in exchange correlation, give much less-comparative absorption. For example, the B3LYP functional (20% HF) provides much higher absorption wavelengths, i.e. 471, 482 and 332 nm for C1–C3, respectively. As discussed in the literature, the main drawback of TD-DFT consists of the underestimation of the vertical excited energy, for which a significant error up to 0.4 eV can be found.67 However, for comparative purpose, the TD-DFT results in general can still provide a benefit when it treats with the same kind of molecules.
The vertical excited energy of some lowest excitations calculated by TD/M05-2X/6-311++G(d,p) as well as the oscillator strength and the nature of the corresponding electronic transition are presented in Table 7. Vertical excitations of C1, C2, and C3 are found at 2.93, 2.87 and 4.24 eV, respectively. In all cases, we observed mainly the contribution of the HOMO to LUMO transition for the lowest lying excitation of C1–C3. This contribution varies from 96% for C1, 97% for C2 and 86% for C3. As predicted in the electronic properties part, the HOMO and LUMO of three compounds involved mostly the rings of aaptamine derivatives, then all the lowest absorption is mainly characterized as π–π* transition.
| Cp | ES | E/eV | A/nm | f | Transition | % |
|---|---|---|---|---|---|---|
| C1 | S1 | 2.93 | 423 | 0.3553 | H → L | 96 |
| S3 | 3.60 | 345 | 0.1614 | H−1 → L | 91 | |
| S7 | 4.84 | 257 | 0.4625 | H−2 → L | 47 | |
| H → L+1 | 43 | |||||
| S8 | 5.33 | 233 | 0.1603 | H → L+2 | 92 | |
| 5.72 | 212 | 0.0277 | H−1 → L+1 | 62 | ||
| H → L+3 | 24 | |||||
| C2 | S1 | 2.87 | 435 | 0.2938 | H → L | 97 |
| S3 | 3.78 | 328 | 0.1196 | H−1 → L | 86 | |
| H−3 → L | 6 | |||||
| S6 | 5.01 | 248 | 0.6778 | H → L+1 | 77 | |
| H−1 → L | 8 | |||||
| H−3 → L | 8 | |||||
| S8 | 5.24 | 237 | 0.1734 | H → L+2 | 83 | |
| H−3 → L | 8 | |||||
| C3 | S1 | 4.24 | 292 | 0.1447 | H → L | 86 |
| H−1 → L+1 | 8 | |||||
| S3 | 4.90 | 253 | 0.5688 | H → L+1 | 45 | |
| H → L+2 | 21 | |||||
| H−1 → L | 15 | |||||
| H−1 → L+2 | 9 | |||||
| S4 | 5.22 | 238 | 0.1517 | H−1 → L+2 | 72 | |
| H−1 → L | 13 | |||||
| H → L+2 | 6 | |||||
| H → L+1 | 5 | |||||
| S5 | 5.46 | 227 | 0.9586 | H → L+2 | 55 | |
| H−1 → L | 15 | |||||
| H−1 → L+1 | 8 | |||||
| H−1 → L+2 | 8 | |||||
| H → L+1 | 7 | |||||
| S7 | 5.71 | 217 | 0.2460 | H-1 → L+2 | 49 | |
| H-2 → L | 18 | |||||
| H → L+2 | 15 | |||||
| H → L+6 | 5 |
Finally, the Fig. 8 displays the energy gap and energy level of the frontier molecular orbitals that participated to the lowest electronic transition of the C1–C3. One can easily notice that: (i) there is a much lower gap (4.9 eV) for C1 and C2 in comparison with 6.5 eV gap in case of C3, and (ii) possessing the same energy level for HOMO, the energy level of LUMO of C1 and C2 is much lower than that of C3. All results indicate that the C1 and C2 can be more easily excited than C3. This agrees with the TD-DFT results, that the C1 and C2 absorbed the deep blue and UVA radiation (423/345 and 435/328 nm) while the C3 is effective UVB (292 nm) absorption agent. The results suggest the use of aaptamines C1–C3 as photo-protective agents.
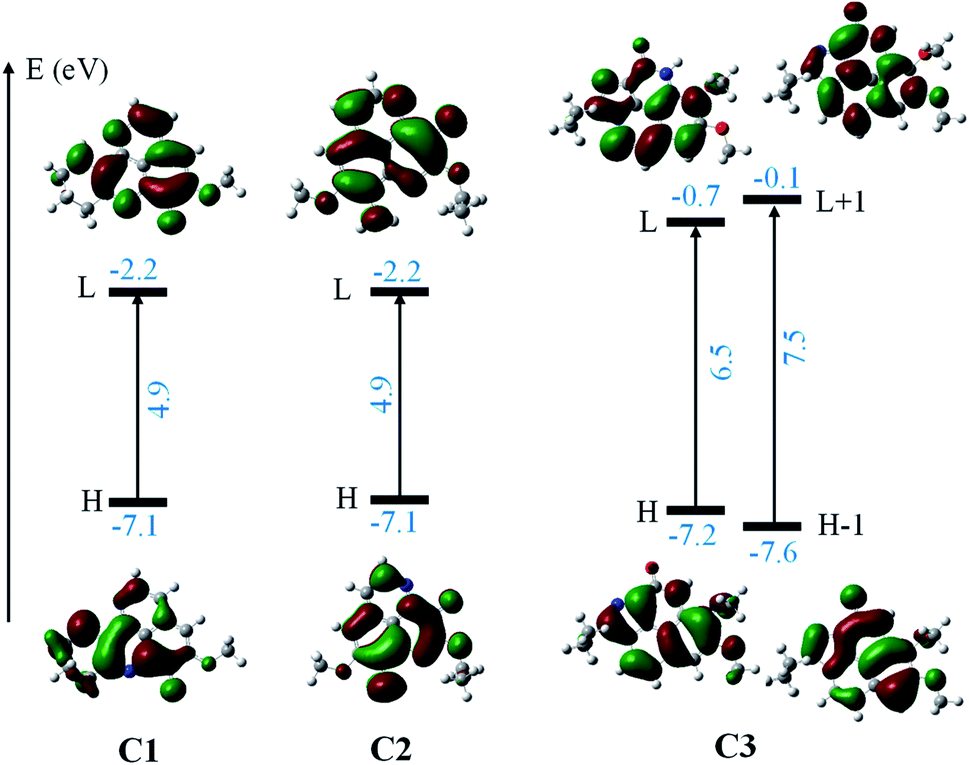 | ||
| Fig. 8 The energy gap and energy levels of the frontier molecular orbitals which participated to the lowest electronic transition of C1–C3. The result is obtained at the TD-DFT/M05-2X/6-311++G(d,p) level in MeOH (IEF-PCM). | ||
4. Conclusions
The antioxidant activities of three aaptamines (C1, C2 and C3) extracted from sponges was investigated in the gas phase and water using DFT method according to four mechanisms: HAT, SET, PL and RAF. First, all of these investigated compounds exhibited the best antioxidant activity via RAF mechanism, for which antioxidant potential is classified in the increasing order C2 < C1 < C3 for HOO˙ and C3 < C2 < C1 for HO˙ free radical scavenging activity. Furthermore, HAT mechanism is elucidated as the second competitive mechanism, in particularly for HO˙ quenching and in water. Thermodynamically, the antioxidant activities via four studied processes are in the decreasing order as follows: RAF > HAT > SET > PL (gas) and HAT > RAF > SET > PL (water). Kinetic calculation shows that the HAT mechanism is the most favorable path for HOO˙ scavenging in water with C1 while the RAF is more competitive with C2 and C3. Second, all three aaptamine derivatives exhibit spontaneous and favorable complexation with both the Cu(I) and Cu(II) hydrated ions in water. The Cu(II) ion chelating activity is classified in the increasing order: C3 < C1 < C2, whereas the Cu(I) complexation is in the reverse order: C2 < C1 < C3. Third, all compounds are effective in the UVA and UVB absorption. Within the range of our study, the M05-2X level provides the best performance for calculation of vertical excited energy using TD-DFT. These results promote aaptamine derivatives as natural antioxidant and anti-UV agents for the use in human healthcare such as in pharmaceutics and cosmetics.Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
This research is funded by Vietnam National Foundation for Science and Technology Development (NAFOSTED) under grant number 103.01-2019.380. The authors are grateful for the help in kinetic calculation from Dr Thi Chinh Ngo (Duy Tan University). The authors are also grateful to the Gridchem (http://www.seagrid.org) for providing computer resources using the Extreme Science and Engineering Discovery Environment (XSEDE) supported by the USA National Science Foundation grant number ACI-10535. Dorra Khiri, Sonia Taamalli, Florent Louis, and Abderrahman El Bakali appreciated the support from the LABEX CaPPA (Chemical and Physical Properties of the Atmosphere), which is funded by the French National Research Agency (ANR) through the PIA (Programme d’Investissement d’Avenir) under contract ANR-11-LABX-0005-01 and also the Regional Council “Hauts de France” and the “European Funds for Regional Economic Development”.References
- K. Shaari, K. C. Ling, Z. Mat Rashid, T. P. Jean, F. Abas, S. M. Raof, Z. Zainal, N. H. Lajis, H. Mohamad and A. M. Ali, Mar. Drugs, 2009, 7, 1–8 CrossRef CAS PubMed.
- L. K. Shubina, A. I. Kalinovsky, S. N. Fedorov, O. S. Radchenko, V. A. Denisenko, P. S. Dmitrenok, S. A. Dyshlovoy, V. B. Krasokhin and V. A. Stonik, Nat. Prod. Commun., 2009, 4, 1085–1088 CrossRef CAS PubMed.
- C.-D. Pham, R. Hartmann, W. E. G. Müller, N. de Voogd, D. Lai and P. Proksch, J. Nat. Prod., 2013, 76, 103–106 CrossRef CAS PubMed.
- H. Nakamura, J. Kobayashi, Y. Ohizumi and Y. Hirata, Tetrahedron Lett., 1982, 23, 5555–5558 CrossRef CAS.
- H.-B. Yu, F. Yang, F. Sun, J. Li, W.-H. Jiao, J.-H. Gan, W.-Z. Hu and H.-W. Lin, Mar. Drugs, 2014, 12(12), 6003–6013 CrossRef PubMed.
- L. K. Shubina, A. I. Kalinovsky, S. N. Fedorov, O. S. Radchenko, V. A. Denisenko, P. S. Dmitrenok, S. A. Dyshlovoy, V. B. Krasokhin and V. A. Stonik, Nat. Prod. Commun., 2009, 4(8), 1085–1088 CrossRef CAS.
- J. J. Bowling, H. K. Pennaka, K. Ivey, S. Wahyuono, M. Kelly, R. F. Schinazi, F. A. Valeriote, D. E. Graves and M. T. Hamann, Chem. Biol. Drug Des., 2008, 71, 205–215 CrossRef CAS PubMed.
- G. Rajivgandhi, S. N. kumar, G. Ramachandran and N. Manoharan, Biocatal. Agric. Biotechnol., 2019, 17, 628–637 CrossRef.
- H.-B. Yu, F. Yang, F. Sun, G.-Y. Ma, J.-H. Gan, W.-Z. Hu, B.-N. Han, W.-H. Jiao and H.-W. Lin, J. Nat. Prod., 2014, 77, 2124–2129 CrossRef CAS PubMed.
- S. A. Dyshlovoy, S. N. Fedorov, L. K. Shubina, A. S. Kuzmich, C. Bokemeyer, G. Keller-Von Amsberg and F. Honecker, BioMed Res. Int., 2014, 2014, 469309 Search PubMed.
- S. A. Dyshlovoy, S. Venz, L. K. Shubina, S. N. Fedorov, R. Walther, C. Jacobsen, V. A. Stonik, C. Bokemeyer, S. Balabanov and F. Honecker, J. Proteomics, 2014, 96, 223–239 CrossRef CAS PubMed.
- T. Hamada, Y. Matsumoto, C.-S. Phan, T. Kamada, S. Onitsuka, H. Okamura, T. Iwagawa, N. Arima, F. Tani and C. S. Vairappan, Nat. Prod. Commun., 2019, 14, 1–3 CrossRef.
- C.-D. Pham, R. Hartmann, W. E. G. Müller, N. de Voogd, D. Lai and P. Proksch, J. Nat. Prod., 2013, 76, 103–106 CrossRef CAS.
- S. Takamatsu, T. W. Hodges, I. Rajbhandari, W. H. Gerwick, M. T. Hamann and D. G. Nagle, J. Nat. Prod., 2003, 66, 605–608 CrossRef CAS PubMed.
- P. De Luna, E. A. C. Bushnell and J. W. Gauld, J. Phys. Chem. A, 2013, 117, 4057–4065 CrossRef CAS.
- A. Pérez-González, A. Galano, J. R. Alvarez-Idaboy, D. X. Tan and R. J. Reiter, Biochim. Biophys. Acta, Gen. Subj., 2017, 1861, 2206–2217 CrossRef PubMed.
- R. Castañeda-Arriaga, J. R. Alvarez-Idaboy and N. Mora-Diez, RSC Adv., 2016, 6, 107924–107932 RSC.
- R. Castañeda-Arriaga, A. Pérez-González, J. R. Alvarez-Idaboy and A. Galano, Int. J. Quantum Chem., 2018, 118, e25527 CrossRef.
- T. H. D. Thao, V. T. N. Dung and D. Q. Dao, Vietnam J. Chem., 2019, 57, 696–701 CrossRef CAS.
- G. Mazzone, J. Phys. Chem. A, 2019, 123, 9560–9566 CrossRef CAS PubMed.
- A. Galano, G. Mazzone, R. Alvarez-Diduk, T. Marino, J. R. Alvarez-Idaboy and N. Russo, Annu. Rev. Food Sci. Technol., 2016, 7, 335–352 CrossRef CAS PubMed.
- D. H. Truong, N. T. A. Nhung and D. Q. Dao, Comput. Theor. Chem., 2020, 1185, 112905 CrossRef CAS.
- J. Krutmann, A. Bouloc, G. Sore, B. A. Bernard and T. Passeron, J. Dermatol. Sci., 2017, 85, 152–161 CrossRef PubMed.
- J. Cadet, T. Douki, J.-L. Ravanat and P. Di Mascio, Photochem. Photobiol. Sci., 2009, 8, 903–911 CrossRef CAS PubMed.
- A. Fourtanier, D. Moyal and S. Seite, Photochem. Photobiol. Sci., 2012, 11, 81–89 CrossRef CAS PubMed.
- S. Dunaway, R. Odin, L. Zhou, L. Ji, Y. Zhang and A. L. Kadekaro, Front. Pharmacol., 2018, 9, 392 CrossRef.
- M. S. Latha, J. Martis, V. Shobha, R. Sham Shinde, S. Bangera, B. Krishnankutty, S. Bellary, S. Varughese, P. Rao and B. R. Naveen Kumar, J Clin Aesthet Dermatol., 2013, 6, 16–26 CAS.
- R. Stevanato, M. Bertelle and S. Fabris, Regul. Toxicol. Pharmacol., 2014, 69, 71–77 CrossRef CAS PubMed.
- J. V Freitas, F. S. G. Praça, M. V. L. B. Bentley and L. R. Gaspar, Int. J. Pharm., 2015, 484, 131–137 CrossRef PubMed.
- D. D. Peres, F. D. Sarruf, C. A. de Oliveira, M. V. R. Velasco and A. R. Baby, J. Photochem. Photobiol., B, 2018, 185, 46–49 CrossRef CAS PubMed.
- S. Scalia and M. Mezzena, Photochem. Photobiol., 2010, 86, 273–278 CrossRef CAS PubMed.
- H. Masaki, J. Dermatol. Sci., 2010, 58, 85–90 CrossRef CAS PubMed.
- B. Rioux, C. Peyrot, M. M. Mention, F. Brunissen and F. Allais, Antioxidants, 2020, 9, 331–345 CrossRef CAS PubMed.
- D. Q. Dao, T. T. T. Phan, T. L. A. Nguyen, P. T. H. Trinh, T. T. Van Tran, J. S. Lee, H. J. Shin and B.-K. Choi, J. Chem. Inf. Model., 2020, 60, 1329–1351 CrossRef CAS PubMed.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, X. Li, M. Caricato, A. V. Marenich, J. Bloino, B. G. Janesko, R. Gomperts, B. Mennucci, H. P. Hratchian, J. V. Ortiz, A. F. Izmaylov, J. L. Sonnenberg, D. Williams-Young, F. Ding, F. Lipparini, F. Egidi, J. Goings, B. Peng, A. Petrone, T. Henderson, D. Ranasinghe, V. G. Zakrzewski, J. Gao, N. Rega, G. Zheng, W. Liang, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell, J. A. Montgomery Jr, J. E. Peralta, F. Ogliaro, M. J. Bearpark, J. J. Heyd, E. N. Brothers, K. N. Kudin, V. N. Staroverov, T. A. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. P. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, J. M. Millam, M. Klene, C. Adamo, R. Cammi, J. W. Ochterski, R. L. Martin, K. Morokuma, O. Farkas, J. B. Foresman and D. J. Fox, Gaussian 16 software, 2016 Search PubMed.
- Y. Zhao, N. E. Schultz and D. G. Truhlar, J. Chem. Theory Comput., 2006, 2, 364–382 CrossRef PubMed.
- A. Galano and J. R. Alvarez-Idaboy, J. Comput. Chem., 2013, 34, 2430–2445 CrossRef CAS PubMed.
- A. Galano and J. R. Alvarez-Idaboy, J. Comput. Chem., 2014, 35, 2019–2026 CrossRef CAS PubMed.
- J. E. Bartmess, J. Phys. Chem., 1994, 98, 6420–6424 CrossRef CAS.
- Z. Markovic, J. Tošovic, D. Milenkovic and S. Markovic, Comput. Theor. Chem., 2016, 1077, 11–17 CrossRef CAS.
- E. Dzib, J. L. Cabellos, F. Ortíz-Chi, S. Pan, A. Galano and G. Merino, Int. J. Quantum Chem., 2019, 119, e25686 CrossRef.
- Y. Okuno, Chem. - Eur. J., 1997, 3, 212–218 CrossRef CAS PubMed.
- S. W. Benson, The foundations of chemical kinetics, Krieger Pub Co., Malabar, Florida, 1982 Search PubMed.
- R. A. Marcus, Annu. Rev. Phys. Chem., 1964, 15, 155 CrossRef CAS.
- F. C. Collins and G. E. Kimball, J. Colloid Sci., 1949, 4, 425–437 CrossRef CAS.
- M. V. Smoluchowski, Z. Phys. Chem., 1918, 92, 129 Search PubMed.
- D. G. Truhlar, J. Chem. Educ., 1985, 62, 104 CrossRef CAS.
- A. Einstein, Ann. Phys., 1905, 322, 549–560 CrossRef.
- G. G. Stokes, Mathematical and Physical Papers, Cambridge University Press, 1905 Search PubMed.
- Y. H. Jang, L. C. Sowers, T. Çaǧin and W. A. Goddard, J. Phys. Chem. A, 2001, 105, 274–280 CrossRef CAS.
- G. C. Shields, Computational Approaches for the Prediction of pKa Values, CRC Press, 2013 Search PubMed.
- D. Jacquemin, B. Mennucci and C. Adamo, Phys. Chem. Chem. Phys., 2011, 13, 16987–16998 RSC.
- J. Tomasi, B. Mennucci and E. Cancès, J. Mol. Struct., 1999, 464, 211–226 CrossRef CAS.
- D. Farmanzadeh and M. Najafi, J. Serb. Chem. Soc., 2016, 81, 277–290 CrossRef CAS.
- T. C. Ngo, T. H. Nguyen and D. Q. Dao, J. Chem. Inf. Model., 2019, 59, 766–776 CrossRef CAS PubMed.
- L. Muñoz-Rugeles, A. Galano and J. R. Alvarez-Idaboy, Phys. Chem. Chem. Phys., 2017, 19, 15296–15309 RSC.
- F. G. Bordwell, D. Algrim and N. R. Vanier, J. Org. Chem., 1977, 42, 1817–1819 CrossRef CAS.
- F. G. Bordwell and D. J. Algrim, J. Am. Chem. Soc., 1988, 110, 2964–2968 CrossRef CAS.
- D. R. Weinberg, C. J. Gagliardi, J. F. Hull, C. F. Murphy, C. A. Kent, B. C. Westlake, A. Paul, D. H. Ess, D. G. McCafferty and T. J. Meyer, Chem. Rev., 2012, 112, 4016–4093 CrossRef CAS PubMed.
- A. Martínez, A. Galano and R. Vargas, J. Phys. Chem. B, 2011, 115, 12591–12598 CrossRef PubMed.
- F. Haber and J. Weiss, Naturwissenschaften, 1932, 20, 948–950 CrossRef CAS.
- G. García-Díez, R. Ramis and N. Mora-Diez, ACS Omega, 2020, 5, 14502–14512 CrossRef PubMed.
- J. Ortega-Castro, M. Adrover, J. Frau, J. Donoso and F. Muñoz, Chem. Phys. Lett., 2009, 475, 277–284 CrossRef CAS.
- R. Monreal-Corona, J. Biddlecombe, A. Ippolito and N. Mora-Diez, Antioxidants, 2020, 9, 674 CrossRef CAS PubMed.
- A. Martínez, R. Vargas and A. Galano, Comput. Theor. Chem., 2018, 1133, 47–50 CrossRef.
- A. Martínez, R. Vargas and A. Galano, Int. J. Quantum Chem., 2018, 118, e25457 CrossRef.
- A. Dreuw and M. Head-Gordon, J. Am. Chem. Soc., 2004, 126, 4007–4016 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1ra04146k |
| This journal is © The Royal Society of Chemistry 2021 |