
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Water-promoted dehydrative coupling of 2-aminopyridines in heptane via a borrowing hydrogen strategy†
Taku Nakayama,
Hidemasa Hikawa *,
Shoko Kikkawa and
Isao Azumaya*
*,
Shoko Kikkawa and
Isao Azumaya*
Faculty of Pharmaceutical Sciences, Toho University, 2-2-1 Miyama, Funabashi, Chiba 274-8510, Japan. E-mail: hidemasa.hikawa@phar.toho-u.ac.jp; isao.azumaya@phar.toho-u.ac.jp
First published on 1st July 2021
Abstract
A synthetic method for dehydrative N-benzylation promoted by water molecules in heptane using a π-benzylpalladium system has been developed. The presence of water significantly accelerates carbon–nitrogen bond formation, which is accomplished in an atom-economical process to afford the corresponding N-monobenzylated products. A crossover experiment afforded H/D scrambled products, which is consistent with a borrowing hydrogen mechanism. Kinetic isotope effect measurements revealed that benzylic carbon–hydrogen bond cleavage was the rate-determining step.
Introduction
The borrowing hydrogen methodology has emerged as a promising greener synthetic strategy for straightforward carbon–nitrogen bond formation utilizing readily available and low-toxicity benzyl alcohols instead of benzyl halides as coupling partners.1 Various catalyst systems using iridium(III),2 ruthenium(II),3 or other metals4 have been developed. Although this strategy affords an attractive atom-economical pathway and is an appealing synthetic shortcut for constructing valuable products, the reactions generally require high temperatures, organic solvents and strong bases. Thus, an efficient system possessing greater catalytic activity that can be performed at lower temperatures in greener solvents without any additives is highly desirable for sustainable carbon–nitrogen bond forming reactions.5 Recently, our group has been exploring a greener borrowing hydrogen methodology involving π-benzylpalladium(II) complexes6 in water.7Recent studies disclosed that hydrophilic interactions play an important role in controlling self-assembly in biological processes.8 In 2008, McLain et al. showed that charge-based interactions play an important role in dipeptide association in aqueous solution.8b In non-polar organic solvents, reverse micelles (RMs), with the polar groups concentrated in the interior of the aggregate, are formed via the self-assembly of surfactant molecules.9 Notably, the presence of water molecules is the driving force for reverse micelle formation in nanosized water pools.9b In 2014, Zhao et al. prepared gold cluster catalysts within interfacially cross-linked reverse micelles via extraction of HAuCl4 in the hydrophilic core for intramolecular alkyne carboxylation.9c
Inspired by these reports, we became interested in designing a new catalytic system via water-assisted self-assembly of polar substrates with a palladium catalyst in non-polar solvents (Scheme 1). We herein present the dehydrative coupling of 2-aminopyridines with benzylic alcohols in heptane using a π-benzylpalladium system, furnishing the corresponding N-benzyl-2-aminopyridines.10 As the reaction begins, in situ generated water accelerates the aggregation of polar substrates with a Pd(0)/TPPMS catalyst in heptane, forming an active π-benzylpalladium catalyst system. The oxidative addition of alcohols to palladium(0) complexes is generally difficult due to the poor performance of the hydroxyl moiety as a leaving group. In the present work, the first example of direct conversion of non-activated benzyl alcohols to the π-benzylPd(II) system has been developed in organic solvents, and applied to the dehydrative cross-coupling reaction. This water-in-oil reaction system is substantially different from our previous method (an oil-in-water type reaction system) concerning the self-assembly between palladium catalysts and polar substrates (Scheme 1A vs. B), and shows a significant advancement over our previous synthetic protocol for more sustainable chemistry.
 | ||
| Scheme 1 Pd-catalyzed dehydrative coupling between aminopyridine 1a and alcohol 2a. | ||
Achieving the direct introduction of various functionalities to aminopyridines, which are found in a wide variety of pharmaceuticals, is of enormous interest.11 Notably, our π-benzylPd(II) system can be applied to a variety of 2-aminopyridine substrates and benzylic alcohols under mild conditions.
Results and discussion
1. Optimization of N-benzylation
We initially examined the Pd-catalyzed reaction between 5-(trifluoromethyl)pyridin-2-amine (1a) and benzyl alcohol (2a) in heptane (Table 1). The reaction was completed in 4 h at 100 °C under air, furnishing the desired product 3a (entry 1). When using 3 equiv. of alcohol 2, the reaction did not proceed after 4 h, but was completed in 16 h (entry 2). Screening of other palladium catalysts found that palladium(II) trifluoroacetate resulted in an almost quantitative yield, whereas palladium(II) dichloride was not applicable (entries 3 and 4). The reaction using tris(dibenzylideneacetone)dipalladium(0) at 120 °C for 16 h gave 3a in 77% yield (entry 5). Although the reaction proceeded to completion using octane instead of heptane (entry 6), the use of other low-polarity organic solvents resulted in lower yields (entries 7–11). Furthermore, little reaction occurred under neat conditions (entry 12). Other water-soluble phosphine ligands such as P(C6H4SO3Na)3 L2 (TPPTS) and (diphenylphosphino)benzoic acids L3–4 were less effective than TPPMS (entries 13–15), and a lower yield was also obtained without a phosphine ligand (entry 16). Since the reaction was completed in 4 h under an Ar atmosphere, oxygen was not essential to the borrowing hydrogen reaction (entry 17).
|
||||
|---|---|---|---|---|
| Entry | Catalyst | Ligand | Solvent | NMR yield (%) |
| a Optimizations were performed with 1 mmol of 1a and 5 mmol of 2a in the presence of 5 mol% Pd catalyst and 10 mol% TPPMS (10 mol%) in solvent (4 mL) at 100 °C for 4 h under air.b 3 equiv. of alcohol 2a.c For 16 h.d Conducted at 120 °C for 16 h.e 2.5 mol%.f Methylcyclohexane.g Under Ar. | ||||
| 1 | Pd(OAc)2 | L1 | Heptane | Quant |
| 2 | Pd(OAc)2 | L1 | Heptane | 0b (quant)c |
| 3 | Pd(TFA)2 | L1 | Heptane | 97 |
| 4 | PdCl2 | L1 | Heptane | 0 |
| 5d | Pd2(dba)3·CHCl3e | L1 | Heptane | 77 |
| 6 | Pd(OAc)2 | L1 | Octane | Quant |
| 7 | Pd(OAc)2 | L1 | Hexane | 2 |
| 8 | Pd(OAc)2 | L1 | MCHf | 38 |
| 9 | Pd(OAc)2 | L1 | Toluene | 4 |
| 10 | Pd(OAc)2 | L1 | (CHCl2)2 | 0 |
| 11 | Pd(OAc)2 | L1 | 1,4-Dioxane | 20 |
| 12 | Pd(OAc)2 | L1 | None | 8 |
| 13 | Pd(OAc)2 | L2 | Heptane | 5 |
| 14 | Pd(OAc)2 | L3 | Heptane | 0 |
| 15 | Pd(OAc)2 | L4 | Heptane | 6 |
| 16 | Pd(OAc)2 | None | Heptane | 30 |
| 17g | Pd(OAc)2 | L1 | Heptane | 99 |
 |
||||

2. Reaction scope
Next, we explored the scope of 2-aminopyridines 1 and benzylic alcohols 2 capable of undergoing a borrowing hydrogen reaction (Scheme 2). A variety of 2-aminopyridines 1 were well tolerated, giving the desired products 3b–e in 75–96% yields (Scheme 2A). The reaction of simple 2-aminopyridine, bearing no substituent at the 5-position, also proceeded smoothly (3f, 90%). Advantageously, a substrate containing a sensitive reducible nitro group could be utilized, furnishing the corresponding desired product 3g, albeit in low yield. The reaction of 6-aminonicotinic acid gave an extremely poor result (only 7% isolated yield of N-benzylated product 3h). To our delight, the utilization of benzyl alcohol (2a) directly as a coupling partner could be applied to 3-aminopyridine, 2-aminoquinoline and nicotinamide (niacinamide) as amine substrates, furnishing the corresponding pharmaceutically active N-benzylated motifs in moderate to excellent yields (3i 88%; 3j, 82%; 3k, 66%).12 Unfortunately, no reactions occurred when using 4-aminopyridine, 2-aminopyrimidine and 2-(methylamino)pyridine since the amino groups had poor nucleophilicity. Benzylic alcohols with electron-donating groups were effective coupling partners (3j–s, 65–93%) (Scheme 2B). A sterically hindered 2-methoxybenzyl alcohol (2s) was well tolerated to obtain the N-benzylated product 3s in 84% yield. Furthermore, the reactions of electron-deficient fluorobenzyl alcohols afforded the desired products 3t–v in 56–88% yields, although in octane, higher temperatures were required to achieve better yields. In contrast, no reaction proceeded when using 4-nitrobenzyl alcohol and 2-phenylethyl alcohol, likely because the corresponding π-benzyl Pd(II) cation species was not generated. | ||
| Scheme 2 Substrate scope of N-benzylation. Yield of isolated products. a3 equiv. of alcohol. bNMR yield. c10 mol% Pd(OAc)2, 20 mol% TPPMS, 150 °C, 24 h in octane. | ||
3. Effect of water
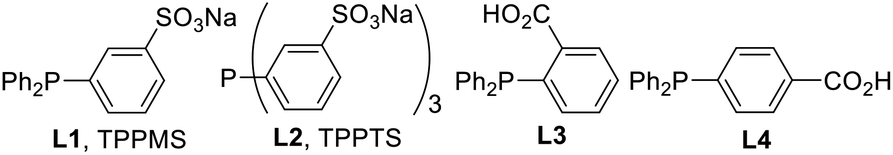
To measure the full reaction profile, dehydrative coupling between 2-aminopyridine 1a and alcohol 2a at 100 °C in heptane was monitored by 1H NMR spectroscopy. The formation of N-benzylated product 3a along with benzaldehyde (4a) was observed (Fig. 1A). No imine intermediate was detected, suggesting its rapid hydrogenation to the corresponding desired product 3a. The consumption of alcohol 2a followed pseudo-first-order kinetics including the presence of two mechanistic periods (0–2 h: k1 = 0.01 h−1; 2–4 h: k2 = 0.35 h−1) (Fig. 1B). Based on these results, we hypothesized that in situ generated water molecules might promote the dehydrogenation of 2a. Therefore, we investigated the detailed reaction mechanism, especially the role of water molecules in heptane.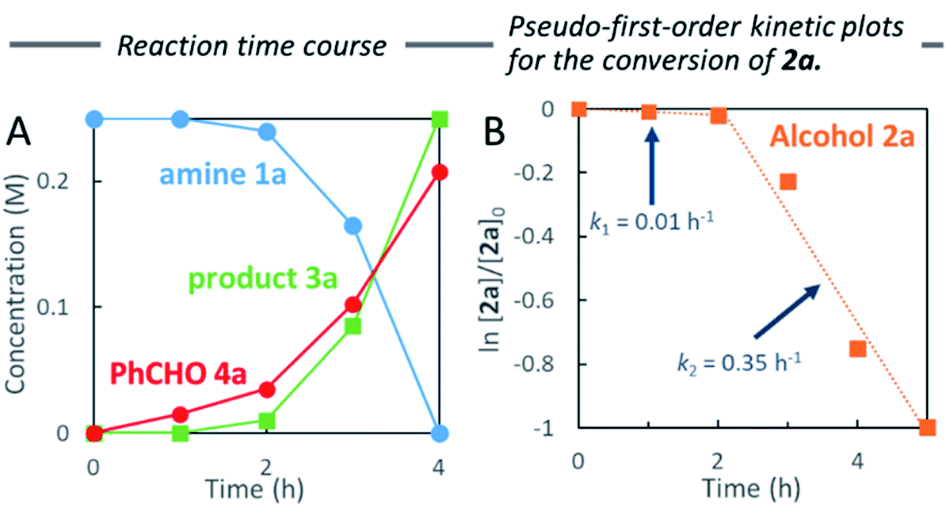 | ||
| Fig. 1 Monitoring the reaction. | ||
Thus, we conducted the reaction while adding different amounts of water (0.5, 1 and 2 mmol) (Scheme 3). As expected, the initial reaction rate in heptane dramatically increased upon addition of 0.5 mmol water compared to no water. The addition of 1 mmol water resulted in almost the same outcome (60%). In contrast, no benzylation was observed in the presence of excess water (2 mmol). When changing the additional water ratio, the reaction was accelerated in the following order: 0.5 mmol ≈ 1 mmol > no addition of water (in situ generated 0.35 mmol of water molecules after 3 h) ≫ 2 mmol, suggesting that traces of water present in situ (≤1 mmol) promoted aggregation between the polar substrates and the Pd catalyst in heptane, forming the active π-benzylpalladium(II) catalyst system. Since the formation of reverse micelles via aggregation of polar substrates did not occur by adding excess water, the water-in-oil type reaction employing the π-benzylpalladium species did not take place. Indeed, the addition of water (1 mmol) was not effective without stirring or addition of n-Bu4NBr.
 | ||
| Scheme 3 N-Benzylation in the presence of different amounts of water. | ||
Water-promoted dehydrogenation of 2a in heptane exhibited pseudo-first-order plots with a higher primary kinetic solvent isotope effect (KSIE = 4.6). This value was much larger than the KSIE value of 1.4 measured in water reactions (Fig. 2A and B),7b suggesting that hydrogen bonds play a significant role in the dehydrative coupling reaction in heptane.
 | ||
| Fig. 2 Comparison of pseudo-first-order kinetic plots for reaction of 2a in heptane (A) with addition of H2O vs. D2O, and (B) in H2O vs. D2O. | ||
4. Deuterium labeling experiment
We performed a crossover experiment by subjecting an equimolar amount of deuterated benzyl alcohol 2a-d2 (5 mmol) and 3-methylbenzyl alcohol 2r (5 mmol) to the standard reaction conditions (Scheme 4). If the nucleophilic substitution of alcohol 2r (dehydrative Tsuji–Trost type reaction) proceeds, deuterium labeled product 3r-d would not be obtained. In contrast, a borrowing hydrogen reaction would afford the H/D scrambled N-benzylated product 3r-d. As expected, a mixture of crossover product 3r-d and non-crossover product 3r was obtained in 18% yield. The results obtained from a 13C NMR analysis showed that the carbon of the methylene group exhibited a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1:1 triplet for 3r-d. Furthermore, the ratio of the integrated values in the 1H NMR spectrum showed 18% incorporation of deuterium at the benzylic position of 3r-d (see ESI†). This experimental evidence clearly indicates a borrowing hydrogen reaction and rules out an alternative Pd-catalyzed SN2-type substitution reaction.
:1:1 triplet for 3r-d. Furthermore, the ratio of the integrated values in the 1H NMR spectrum showed 18% incorporation of deuterium at the benzylic position of 3r-d (see ESI†). This experimental evidence clearly indicates a borrowing hydrogen reaction and rules out an alternative Pd-catalyzed SN2-type substitution reaction.
 | ||
| Scheme 4 Deuterium labeling experiment. | ||
5. Mechanistic studies
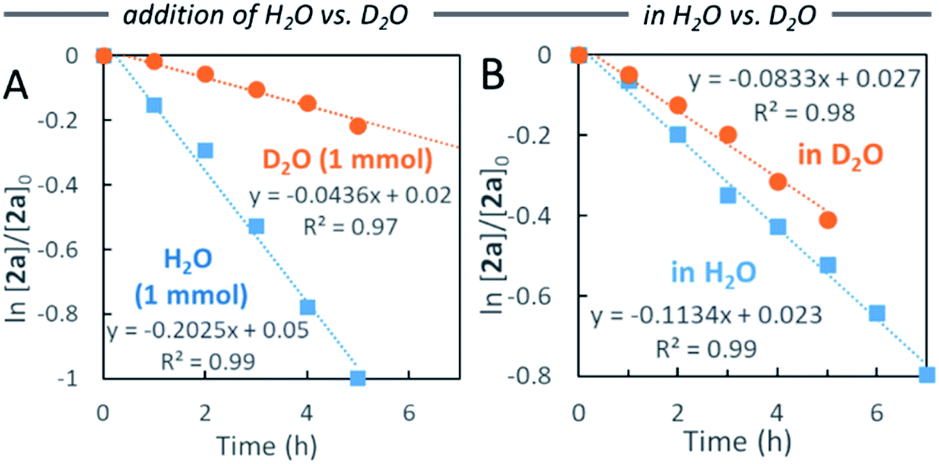
First, we examined the Pd-catalyzed disproportionation of alcohol 2a (Scheme 5A). When the alcohol 2a was converted to aldehyde 4a, the generation of toluene (5a) was clearly observed by 1H NMR spectroscopy. This result suggested that β-hydride elimination of benzylPd(II)-alkoxide complex B led to the aldehyde 4a, which then reacted with benzylPd(II) hydride species C via reductive elimination to form toluene 5a with regenerated Pd(0)Ln.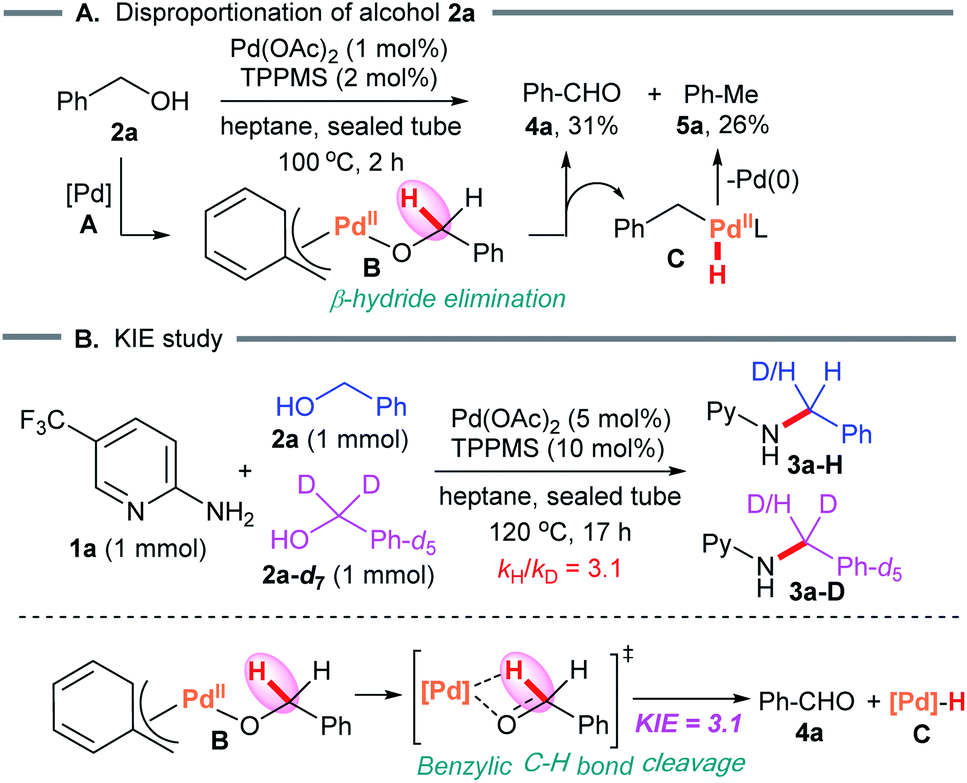 | ||
| Scheme 5 Mechanistic studies. | ||
We next studied the kinetic isotope effect in an intermolecular competition experiment using benzyl alcohol and its deuterium-labeled analog. As expected, a kinetic isotope effect (KIE) of 3.1 was observed for the reaction with substrate 1a (Scheme 5B). A KIE of this magnitude is in agreement with the benzylic C–H bond cleavage featured in the turnover limiting step.
6. Mechanistic considerations
Considering the results above, a proposed catalytic cycle for the borrowing hydrogen reaction in heptane is illustrated in Scheme 6.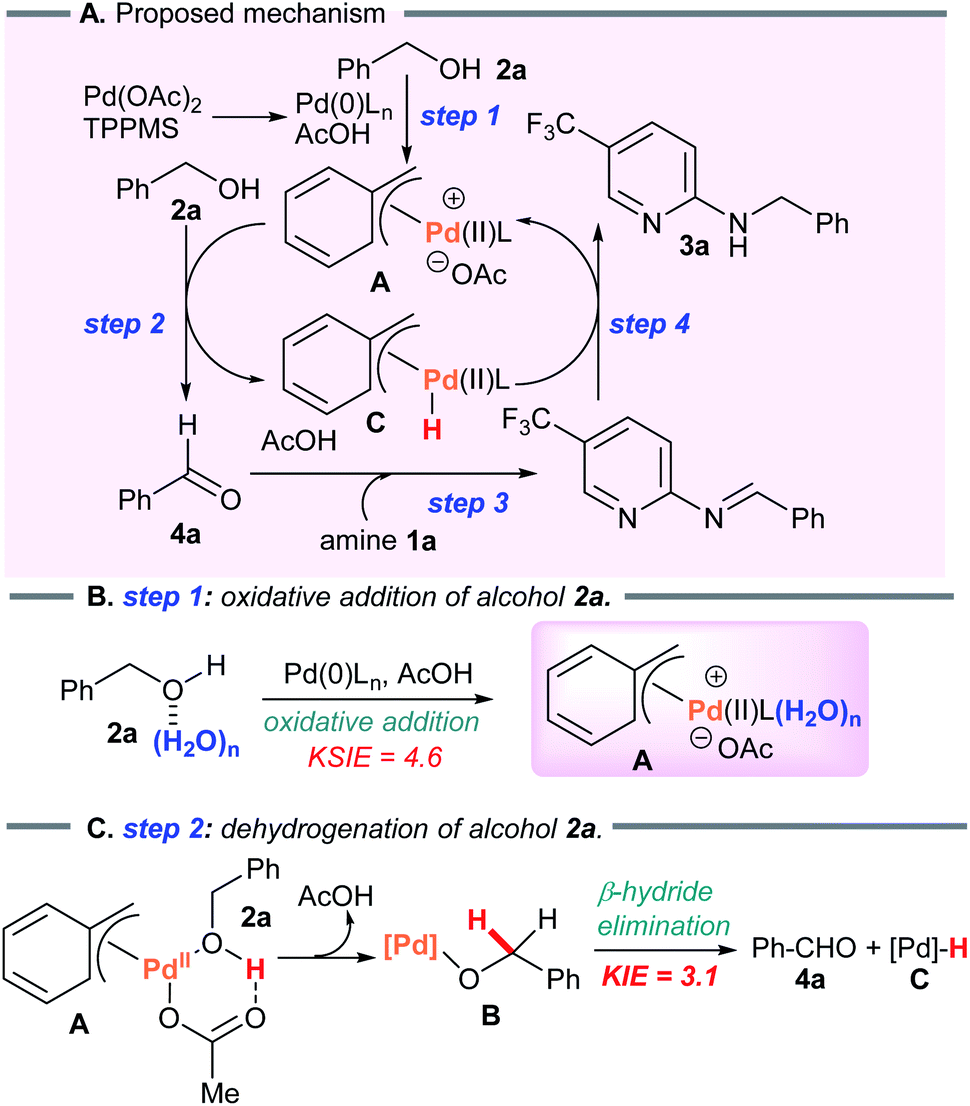 | ||
| Scheme 6 Proposed catalytic cycle. | ||
To verify the formation of the π-benzylpalladium catalyst system, we examined the Tsuji–Trost type reaction of aniline (1w) in heptane (Scheme 7A). An electron-sufficient amine substrate should react with the π-benzylPd(II) species by nucleophilic substitution. As expected, the reactivity of 1w was significantly different from that of 2-aminopyridine 1a, furnishing not only N-benzylaniline (3w) but also dibenzylated product 6w (3w, 20%; 6w, 34%). Furthermore, when using 3w as a starting material, the corresponding dibenzylated product 6w was formed via the condensation of an alcohol with a benzylic C–H bond (Scheme 7B). These results provide convincing evidence supporting the formation of the π-benzylPd(II) species. This catalyst system enables the dehydrative N-benzylation C–H bond activation cascade reaction of electron-rich analog 3w. The η1-σ-benzyl nucleophile attacks an electrophilic η3-π-benzyl moiety in the bis-benzylPd(II) complex D, generating a new benzylic C–C bond. This proposed mechanism is consistent with the nucleophilic reactivity of bis-π-allylpalladium complexes reported by Yamamoto et al.19 Indeed, Pd-catalyzed benzylic C–H benzylation of electron-sufficient N-benzylpyridine substrate 3a did not occur (Scheme 7C). Furthermore, the treatment of 3a with D2O (1 equiv.) as the D-atom source in heptane showed no deuterium incorporation at the benzylic position of 3a (The reaction in D2O as a solvent also gave the same result, see ESI†). These results clearly demonstrated that Pd-catalyzed benzylic C–H bond cleavage of 3a did not occur, and therefore rule out D/H exchange between N-benzylated product 3a-d2 and 3r in our crossover experiment (see Scheme 4).
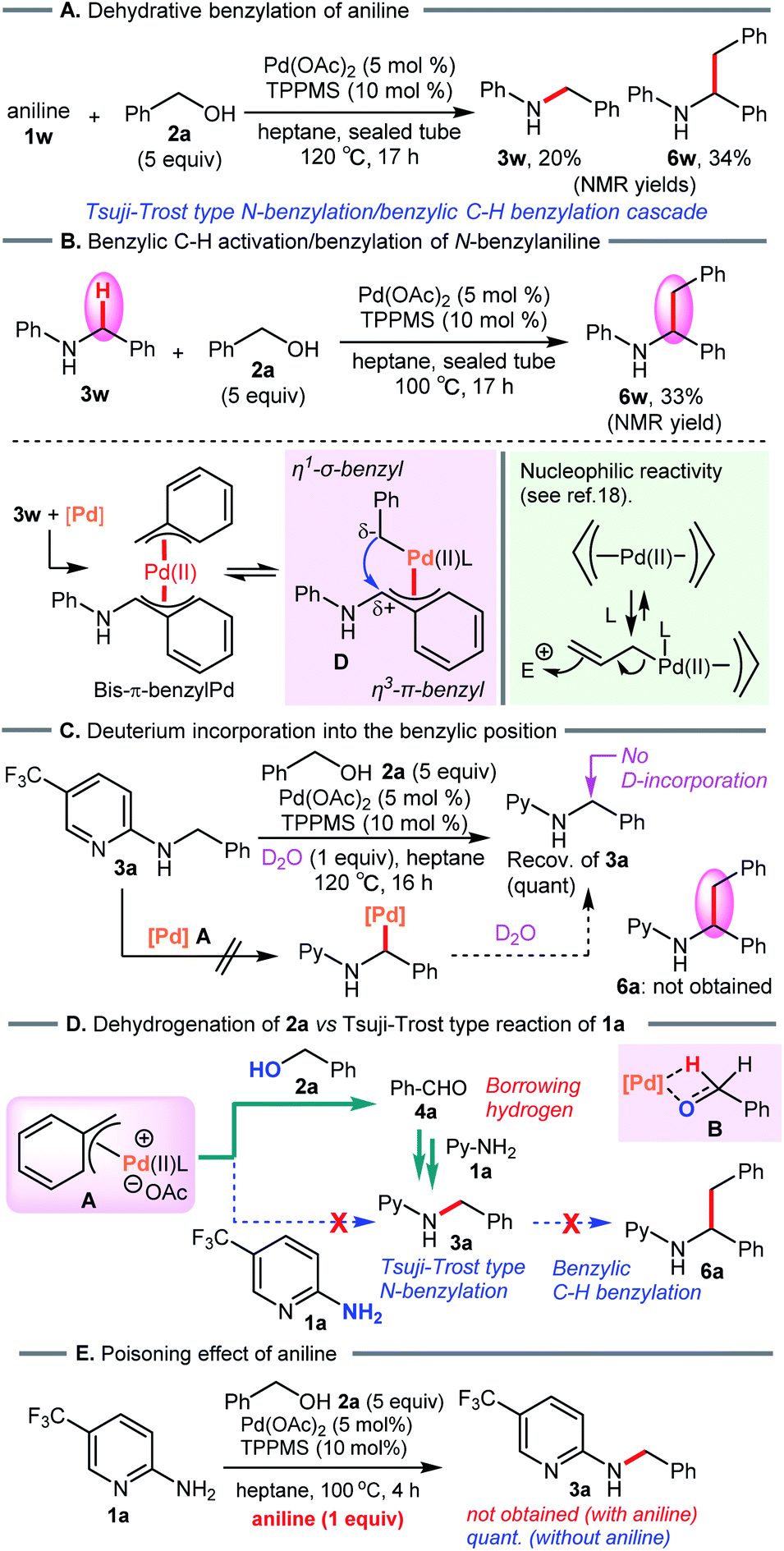 | ||
| Scheme 7 (A) and (B) Pd-catalyzed benzylation of aniline. (C) Deuterium labeling study. (D) Proposed reaction pathway. (E) N-Benzylation of 2-aminopyridine 1a in the presence of aniline. | ||
On the basis of these results, dehydrogenation of benzyl alcohol (2a) by π-benzylPd(II) complex A proceeds faster than nucleophilic substitution of species A, and leaves the weak 2-aminopyridine nucleophile 1a intact (Scheme 7D). Subsequently, reductive amination of 1a with the resulting aldehyde 4a leads to 3a via the borrowing hydrogen pathway. This is consistent with the result of the crossover experiment (see Scheme 4). When adding aniline (1w), N-benzylated product 3a was not obtained due to the poisoning effect of aniline (Scheme 7E).
Having established a successful dehydrative N-benzylation strategy, we compared the catalytic activity of our π-benzylpalladium catalyst system with other efficient systems (Scheme 8). While the Pd(0)/TPPMS-catalyzed reaction in heptane proceeded to completion within 4 h (see Table 1), the previous borrowing hydrogen protocols10c,20 resulted in no reaction. Furthermore, the Pd-catalyzed reaction in water (our previous work)7b was not effective (only 40% yield), clearly demonstrating the superiority of the present catalytic strategy using heptane.
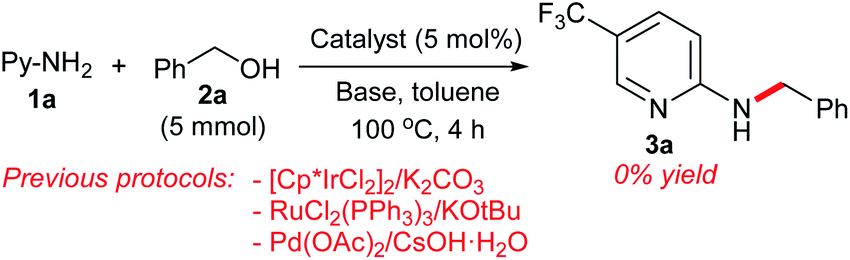 | ||
| Scheme 8 Comparison of the catalytic activity. | ||
To highlight the synthetic utility, we performed the N-benzylation on a gram scale (see ESI†). N-Benzylated product 3a (1.56 g) was facilely isolated in 88% yield from pyridine substrate 1a with alcohol 2a. The developed simple operations also avoided the use of column chromatography for a gram scale reaction.
Conclusions
In summary, we report an efficient palladium-catalyzed dehydrative N-benzylation in heptane via the borrowing hydrogen methodology. The strategy provides an efficient method for the facile synthesis of benzylaminopyridines, which are found in a wide variety of pharmaceuticals. Notably, the use of a Pd(0)/TPPMS catalyst in a non-polar heptane solvent with a trace amount of water is critical for our catalytic system. Water molecules assist in the aggregation of polar substrates followed by the formation of an active π-benzylpalladium catalyst system in heptane, which significantly boosts the borrowing hydrogen reaction. We expect that this study will aid in the design of new catalytic systems using non-polar solvents, and will serve as an entry point for reaction discovery.Experimental
General procedure
To a sealed tube were added 2-aminopyridines 1 (1 mmol), Pd catalyst (0.05 mmol), phosphine ligand (0.1 mmol), alcohols 2 (5 mmol) and heptane (4 mL). The resulting reaction mixture was heated at 120 °C for 17 h under air. After completion of the reaction, the mixture was cooled to room temperature. The reaction mixture was diluted with water, and then extracted with EtOAc. The combined organic phases were washed with brine, dried over MgSO4, and filtered and concentrated to dryness under reduced pressure. The resulting residue was purified by flash column chromatography on silica gel (eluent: n-hexane/EtOAc) to obtain desired products 3.Conflicts of interest
There are no conflicts to declare.Acknowledgements
AcknowledgementsThis work was supported by JSPS KAKENHI grant number 19K07003.Notes and references
- For a recent review see: (a) T. Irrgang and R. Kempe, Chem. Rev., 2019, 119, 2524–2549 CrossRef CAS PubMed; (b) A. Corma, J. Navas and M. J. Sabater, Chem. Rev., 2018, 118, 1410–1459 CrossRef CAS PubMed.
- Recent examples, see: (a) M. Onoda and K.-i. Fujita, Org. Lett., 2020, 22, 7295–7299 CrossRef CAS PubMed; (b) F. Kallmeier, R. Fertig, T. Irrgang and R. Kempe, Angew. Chem., Int. Ed., 2020, 59, 11789–11793 CrossRef CAS PubMed; (c) W. Yao, Z.-C. Duan, Y. Zhang, X. Sang, X.-F. Xia and D. Wang, Adv. Synth. Catal., 2019, 361, 5695–5703 CrossRef CAS; (d) C. M. Wong, R. T. McBurney, S. C. Binding, M. B. Peterson, V. R. Goncales, J. J. Gooding and B. A. Messerle, Green Chem., 2017, 19, 3142–3151 RSC.
- For recent examples, see: (a) N. Biswas, R. Sharma and D. Srimani, Adv. Synth. Catal., 2020, 362, 2902–2910 CrossRef CAS; (b) M. A. Shaikh, S. G. Agalave, A. S. Ubale and B. Gnanaprakasam, J. Org. Chem., 2020, 85, 2277–2290 CrossRef CAS PubMed; (c) Y. Pan, Z. Luo, X. Xu, H. Zhao, J. Han, L. Xu, Q. Fan and J. Xiao, Adv. Synth. Catal., 2019, 361, 3800–3806 CrossRef CAS; (d) S. Zhang, J. J. Ibrahim and Y. Yang, Org. Chem. Front., 2019, 6, 2726–2731 RSC.
- Recent examples, see: (a) N. Hofmann, L. Homberg and K. C. Hultzsch, Org. Lett., 2020, 22, 7964–7970 CrossRef CAS PubMed; (b) A. Kaithal, P. van Bonn, M. Hoelscher and W. Leitner, Angew. Chem., Int. Ed., 2020, 59, 215–220 CrossRef CAS PubMed; (c) Y. Qiu, Y. Zhang, L. Jin, L. Pan, G. Du, D. Ye and D. Wang, Org. Chem. Front., 2019, 6, 3420–3427 RSC; (d) V. Goyal, J. Gahtori, A. Narani, P. Gupta, A. Bordoloi and K. Natte, J. Org. Chem., 2019, 84, 15389–15398 CrossRef CAS PubMed; (e) Z. Xu, D.-S. Wang, X. Yu, Y. Yang and D. Wang, Adv. Synth. Catal., 2017, 359, 3332–3340 CrossRef CAS.
- (a) C. Risi, M. Calamante, E. Cini, V. Faltoni, E. Petricci, F. Rosati and M. Taddei, Green Chem., 2020, 22, 327–331 RSC; (b) M. Huang, Y. Li, J. Liu, X.-B. Lan, Y. Liu, C. Zhao and Z. Ke, Green Chem., 2019, 21, 219–224 RSC; (c) N. H. Luo, Y. H. Zhong, H. L. Wen and R. Luo, ACS Omega, 2020, 5, 27723–27732 CrossRef CAS PubMed; (d) Y. M. A. Yamada, H. Ohta, Y. Yuyama and Y. Uozumi, Synthesis, 2013, 2093–2100 CrossRef CAS; (e) R. Kawahara, K.-I. Fujita and R. Yamaguchi, Adv. Synth. Catal., 2011, 353, 1161–1168 CrossRef CAS.
- (a) J. Le Bras and J. Muzart, Eur. J. Org. Chem., 2016, 2565–2593 CrossRef CAS; (b) H. Hikawa, R. Ichinose, S. Kikkawa and I. Azumaya, Green Chem., 2018, 20, 1297–1305 RSC.
- (a) H. Hikawa, R. Tan, A. Tazawa, S. Kikkawa and I. Azumaya, Eur. J. Org. Chem., 2020, 539–547 CrossRef CAS; (b) H. Hikawa, H. Imamura, S. Kikkawa and I. Azumaya, Green Chem., 2018, 20, 3044–3049 RSC; (c) H. Hikawa, T. Koike, K. Izumi, S. Kikkawa and I. Azumaya, Adv. Synth. Catal., 2016, 358, 784–791 CrossRef CAS.
- (a) N. H. Rhys and A. K. Soper, J. Phys. Chem. B, 2015, 119, 15644–15651 CrossRef CAS PubMed; (b) S. E. McLain, A. K. Soper, I. Daidone, J. C. Smith and A. Watts, Angew. Chem., Int. Ed., 2008, 47, 9059–9062 CrossRef CAS PubMed.
- (a) L.-C. Lee, X. Xing and Y. Zhao, ACS Appl. Mater. Interfaces, 2017, 9, 38436–38444 CrossRef CAS PubMed; (b) A. Khoshnood and A. Firoozabadi, Langmuir, 2015, 31, 5982–5991 CrossRef CAS PubMed; (c) L.-C. Lee and Y. Zhao, J. Am. Chem. Soc., 2014, 136, 5579–5582 CrossRef CAS PubMed.
- (a) S. P. Shan and A. M. Seayad, ACS Catal., 2013, 3, 2536–2540 CrossRef; (b) X. Yu, L. Jiang, Q. Li, Y. Xie and Q. Xu, Palladium-Catalyzed N-Alkylation of Amides and Amines with Alcohols Employing the Aerobic Relay Race Methodology, Chin. J. Chem., 2012, 30, 2322–2332 CrossRef CAS; (c) A. Martínez-Asencio, M. Yus and D. J. Ramón, Synthesis, 2011, 3730–3740 Search PubMed.
- (a) S. Hiranaka, Y. Tega, K. Higuchi, T. Kurosawa, Y. Deguchi, M. Arata, A. Ito, M. Yoshida, Y. Nagaoka and T. Sumiyoshi, ACS Med. Chem. Lett., 2018, 9, 884–888 CrossRef CAS PubMed; (b) M. Ohmi, Y. Shishido, T. Inoue, K. Ando, A. Fujiuchi, A. Yamada, S. Watanabe and K. Kawamura, Bioorg. Med. Chem. Lett., 2014, 24, 5364–5368 CrossRef CAS PubMed.
- (a) S. J. Taylor, F. Soleymanzadeh, A. B. Eldrup, N. A. Farrow, I. Muegge, A. Kukulka, A. K. Kabcenell and S. De Lombaert, Bioorg. Med. Chem. Lett., 2009, 19, 5864–5868 CrossRef CAS PubMed; (b) S. R. Inglis, R. K. Jones, G. W. Booker and S. M. Pyke, Bioorg. Med. Chem. Lett., 2006, 16, 387–390 CrossRef CAS PubMed.
- (a) C. Amatore, L. El Kaim, L. Grimaud, A. Jutand, A. Meignie and G. Romanov, Eur. J. Org. Chem., 2014, 4709–4713 CrossRef CAS; (b) B. P. Fors, P. Krattiger, E. Strieter and S. L. Buchwald, Org. Lett., 2008, 10, 3505–3508 CrossRef CAS PubMed; (c) F. Ozawa, A. Kubo and T. Hayashi, Chem. Lett., 1992, 2177–2180 CrossRef CAS.
- J. Le Bras and J. Muzart, Eur. J. Org. Chem., 2017, 3528–3548 CrossRef CAS.
- (a) X. Ma, J. Yu, C. Han, Q. Zhou, M. Ren, L. Li and L. Tang, Adv. Synth. Catal., 2019, 361, 1023–1027 CrossRef CAS; (b) X. Ma, J. Yu, Q. Zhou, R. Yan, L. Zheng and L. Wang, J. Org. Chem., 2019, 84, 7468–7473 CrossRef CAS PubMed; (c) S. Sawadjoon, P. J. R. Sjoeberg, A. Orthaber, O. Matsson and J. S. M. Samec, Chem.–Eur. J., 2014, 20, 1520–1524 CrossRef CAS PubMed; (d) Y. Gumrukcu, B. de Bruin and J. N. H. Reek, ChemSusChem, 2014, 7, 890–896 CrossRef CAS PubMed; (e) H. Kinoshita, H. Shinokubo and K. Oshima, Org. Lett., 2004, 6, 4085–4088 CrossRef CAS PubMed.
- (a) L.-H. Lu, Z. Wang, W. Xia, P. Cheng, B. Zhang, Z. Cao and W.-M. He, Chin. Chem. Lett., 2019, 30, 1237–1240 CrossRef CAS; (b) L.-H. Lu, S.-J. Zhou, M. Sun, J.-L. Chen, W. Xia, X. Yu, X. Xu and W.-M. He, ACS Sustainable Chem. Eng., 2019, 7, 1574–1579 CrossRef CAS; (c) C. Wu, L.-H. Lu, A.-Z. Peng, G.-K. Jia, C. Peng, Z. Cao, Z. Tang, W.-M. He and X. Xu, Green Chem., 2018, 20, 3683–3688 RSC.
- (a) T. Privalov, C. Linde, K. Zetterberg and C. Moberg, Organometallics, 2005, 24, 885–893 CrossRef CAS; (b) B. A. Steinhoff, I. A. Guzei and S. S. Stahl, J. Am. Chem. Soc., 2004, 126, 11268–11278 CrossRef CAS PubMed; (c) J. A. Mueller, C. P. Goller and M. S. Sigman, J. Am. Chem. Soc., 2004, 126, 9724–9734 CrossRef CAS PubMed; (d) J. A. Mueller, D. R. Jensen and M. S. Sigman, J. Am. Chem. Soc., 2002, 124, 8202–8203 CrossRef CAS PubMed.
- (a) L.-M. Wang, K. Kobayashi, M. Arisawa, S. Saito and H. Naka, Org. Lett., 2019, 21, 341–344 CrossRef CAS PubMed; (b) L.-M. Wang, K. Jenkinson, A. E. Wheatley, K. Kuwata, S. Saito and H. Naka, ACS Sustainable Chem. Eng., 2018, 6, 15419–15424 CrossRef CAS.
- (a) H. Nakamura, H. Iwama and Y. Yamamoto, Chem. Commun., 1996, 1459–1460 RSC; (b) H. Nakamura and Y. Yamamoto, J. Am. Chem. Soc., 1996, 118, 6641–6647 CrossRef CAS.
- (a) K. Fujita, Z. Li, N. Ozeki and R. Yamaguchi, Tetrahedron Lett., 2003, 44, 2687–2690 CrossRef CAS; (b) S. Manojveer, S. Salahi, O. F. Wendt and M. T. Johnson, J. Org. Chem., 2018, 83, 10864–10870 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Copies of 1H and 13C NMR spectra for all compounds. See DOI: 10.1039/d1ra04118e |
| This journal is © The Royal Society of Chemistry 2021 |