
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSynergic effects between boron and nitrogen atoms in BN-codoped C59−nBNn fullerenes (n = 1–3) for metal-free reduction of greenhouse N2O gas†
Mehdi D. Esrafili *a,
Adnan Ali Khanbc and
Parisasadat Mousavianad
*a,
Adnan Ali Khanbc and
Parisasadat Mousavianad
aDepartment of Chemistry, Faculty of Basic Sciences, University of Maragheh, P. O. Box 55136-553, Maragheh, Iran. E-mail: esrafili@maragheh.ac.ir
bCentre for Computational Materials Science, University of Malakand, Chakdara, Pakistan
cDepartment of Chemistry, University of Malakand, Chakdara, Pakistan
dDepartment of Chemistry, Azarbaijan Shahid Madani University, Tabriz, Iran
First published on 28th June 2021
Abstract
The geometries, electronic structures, and catalytic properties of BN-codoped fullerenes C59−nBNn (n = 1–3) are studied using first-principles computations. The results showed that BN-codoping can significantly modify the properties of C60 fullerene by breaking local charge neutrality and creating active sites. The codoping of B and N enhances the formation energy of fullerenes, indicating that the synergistic effects of these atoms helps to stabilize the C59−nBNn structures. The stepwise addition of N atoms around the B atom improves catalytic activities of C59−nBNn in N2O reduction. The reduction of N2O over C58BN and C57BN2 begins with its chemisorption on the B–C bond of the fullerene, followed by the concerted interaction of CO with N2O and the release of N2. The resulting OCO intermediate is subsequently transformed into a CO2 molecule, which is weakly adsorbed on the B atom of the fullerene. On the contrary, nitrogen-rich C56BN3 fullerene is found to decompose N2O into N2 and O* species without the requirement for activation energy. The CO molecule then removes the O* species with a low activation barrier. The activation barrier of the N2O reduction on C56BN3 fullerene is just 0.28 eV, which is lower than that of noble metals.
1. Introduction
The increase in atmospheric concentration of greenhouse gases is among the most serious threats to our planet.1,2 These species are able to trap solar energy and emit it within the thermal infrared range. Although greenhouse gases are naturally present in the Earth's atmosphere, human activities like agriculture, fuel combustion and industrial processes have led to a continual build-up of these heat-trapping gases, hence intensifying the greenhouse effect. Nitrous oxide (N2O) is a long-lived greenhouse gas which is recognized as a main contributor to global warming and climate change.3 The other concern about N2O is that it is unstable in the stratosphere and can be easily decomposed into nitrogen oxides (NOx). These NOx are able to interact with the ozone molecule, leading to the ozone layer depletion.3,4 Therefore, considerable efforts have been made to find practical solutions to decrease or remove harmful N2O molecules from the atmosphere.5–8 For instance, many theoretical calculations and experimental studies have focused on the catalytic reduction of N2O by carbon monoxide (CO) as an efficient and possible solution for removing of N2O.6,9–11 Transition-metal (TM) catalysts including cations,12,13 clusters14 and those finely isolated on supports15,16 have been also frequently used as durable and efficient materials to accelerate this process. In addition, some TM-free catalysts like B-,17 Al- (ref. 18) or Si-doped graphene19,20 have also attracted increasing attention to tackle the serious environmental problems caused by N2O emission.In recent decades, carbon-based nanoparticles like fullerenes, nanocones, nanotubes and graphene have drawn a lot of attention in various fields of chemistry and materials science due to their exceptional physicochemical properties, unique specific area and low cost.21–27 Apart from these characteristics, theoretical studies28–31 have proven that the chemical doping with heteroatoms can drastically improve the electronic structure of carbon nanomaterials. For instance, earlier studies32,33 have pointed out that the chemical doping of C60 fullerene with B atoms can substantially enhance its catalytic properties in the oxygen reduction reaction (ORR) due to activation of adsorbed O2 molecules. Recently, Chen and coworkers34 have identified that the C59B fullerene could serve as a superior and robust catalyst for CO oxidation. Since the boron atom has a lower electronegativity than carbon (2.0 vs. 2.5 based on Pauling's scale), the introduction of a boron atom can provide an active catalytic center in C60 to capture O2 molecules. Likewise, N-doping has also been suggested as a useful strategy for tuning the electronic structure and properties of carbon-based nanomaterials.35–38 For example, graphene layers doped with nitrogen atoms are among the most studied systems with novel and superior catalytic activity in different chemical reactions.39–42 In addition to large electronegativity, which causes a positive charge over the nearest carbon atoms, the presence of nitrogen atoms can bring an asymmetric spin density in carbon nanomaterials. Therefore, N-doped carbon nanomaterials have been frequently exploited as metal-free catalysts in various chemical reactions like ORR,43–45 CO oxidation46–48 and NO reduction.49
Some experimental investigations and theoretical calculations have claimed that B and N codoping of carbon-based nanomaterials can offer further intriguing opportunities to realize new metal-free catalysts due to the synergistic effects between B and N atoms.50–53 For instance, Wang et al.54 prepared BN-codoped graphene for the ORR with superior catalytic ability comparable with those of commercial Pt-based catalysts. In addition to such high catalytic performance, a combined experimental and theoretical work has also found the BN codoping can provide 100% selectivity for the ORR process via the four-electron pathway.55 Density functional theory (DFT) computations also confirmed that the active graphitic BN3 sites could improve the O2 (ref. 56 and 57) and N2O adsorptions17 in the BN-codoped graphene. Extensive studies have also proven that the presence of BN moieties in carbon fullerenes can provide active sites for sensing toxic gas molecules.53,58 Furthermore, the difference in electronegativity between B, C and N atoms can polarize the inert carbon-π electron density in these systems, rendering them as excellent substrates for Li-ion batteries59,60 and H2 storage.61 However, little is yet known about the mechanism of N2O reduction and the synergic effects between B and N atoms in the BN-codoped fullerenes.
C60 fullerene, on the other hand, may trap atoms and small molecules to form stable endohedral complexes with tremendous potential uses as superconductors, drug delivery systems, and molecular reactors.62–64 For example, noble gases (Ng = He, Ne, Ar, Kr, Xe) can be introduced into the C60 by applying high temperatures and pressure to a mixture of fullerenes and the target Ng, hitting fullerenes with accelerated ions, or other techniques.65 DFT computations have demonstrated that encapsulating a Ng atom in the C60 fullerene can stabilize the system due to the coupling the electronic states of the encapsulated atom with those of fullerene C atoms.66 Bühl et al.67 found that the negative interaction energies between endohedral He to Xe atoms and C60 increase with the Ng size (Xe > Kr > Ar > Ne > He) owing to enhanced dispersion interactions. Sola and coworkers68 examined the activation energies of the [4+2] Diels–Alder cycloaddition processes in free and Ng endohedral Ng@C60 systems (Ng = He–Xe), and found that the Ng atom has only a little effect on the activation energy of this reaction. According to Bil et al.,69 the addition of an endohedral Ng atom could influence the ozone ring-opening process in Ng@C70O3, but has no effect on the relative stability of the C70O3 isomers. Based on these findings, one might ask whether adding an endohedral Ng atom to BN-codoped C60 fullerenes improves the N2O reduction reaction on these systems.
In the present study, by employing first-principles calculations, we focus on the mechanisms of N2O reduction over C59−nBNn fullerenes (n = 1–3). We show that the successive substitution of N atoms around a pre-doped boron atom can substantially enhance the tendency of C60 fullerene to capture N2O molecule. In particular, because of curvature effects, the chemisorption of a N2O molecule is found to be stronger than that of on the BN-codoped graphene. Due to distinct electronic structure and sizable charge-transfer, the nitrogen-rich C56BN3 fullerene exhibits highest activity for the N2O reduction. Our results propose that C60 fullerenes with a small content of B and N atoms provide exceptional catalytic performance for reduction of N2O at low-temperatures.
2. Theoretical details
To explore N2O reduction on C59−nBNn fullerenes, we implemented spin-polarized DFT calculations with the Perdew–Burke–Ernzerhof (PBE)70 functional in combination with an all-electron double-ξ numerical basis set involving polarization functions (DNP). The dispersion interactions were corrected with the DFT+D2 scheme of Grimme.71 All the simulations were done in a 25 × 25 × 25 Å cubic box, with a gamma point for the summation in the Brillouin zone. The BN-codoped fullerenes were obtained via replacement of C atom(s) around the B atom of C59B with the N atom(s). The Hirshfeld method was adopted for the population analysis. To search transition states (TS), the complete LST/QST method was employed. All the calculations were done with the DMol3.72,73The relative stability of BN-codoped fullerenes was evaluated by formation energy (Eform), defined as
| Eform = EC59−nBNn − EC60 + (n + 1)μC − (μB + nμN) | (1) |
| Ebind = EC59−nBNn − EB − Edefective | (2) |
The adsorption energies (Ead) of N2O and CO were obtained as the difference between the total energy of the resulting complex (Ecomplex) and sum of the energy of isolated reactant (EX) and fullerene (Efullerene):
| Ead = Ecomplex − (EX + Efullerene) | (3) |
By this equation, a negative Ead value shows that the resulting complex would be stable and its formation is a thermodynamically facile process. To consider the temperature and entropic effects, the adsorption Gibbs free energy changes (ΔGad) were obtained at T = 298.15 K and P = 1 atm, according to the following equation:
| ΔGad = (εcomplex + Gcorr,complex) − [(εX + Gcorr,X) + (εfullerene + Gcorr,X fullerene)] | (4) |
| Gcorr = Hcorr − TStot = (Etot + kBT) − TStot | (5) |
We also investigated the nature of the interactions in N2O and CO adducts of C59−nBNn, which seems to be affected by the composition of fullerene.74,75 The quantum theory of atoms in molecules (QTAIM)76 was used here to investigate the nature of interactions between fullerenes and adsorbed N2O or CO molecules. QTAIM is based on topological analysis of electron density, which determines the nature of a chemical bond by analyzing electron density at a specific point between interacting atoms, known as the bond critical point (BCP).76 Although the gradient of electron density at the BCP is nearly zero, the electron density (ρBCP) and its Laplacian (∇2ρBCP) can provide valuable information about the nature of the interaction. In QTAIM, a covalent interaction is distinguished by a large ρBCP and a negative ∇2ρBCP, whereas a closed-shell interaction is identified by a small ρBCP and a positive ∇2ρBCP. In addition, ∇2ρBCP and total electronic energy density at the BCP (HBCP) may be used to assess the degree of covalency of the interaction between two atoms.77 That is, for a covalent interaction, ∇2ρBCP < 0 and HBCP < 0, for an electrostatic interaction, ∇2ρBCP > 0 and HBCP > 0, and for a partially covalent interaction, ∇2ρBCP > 0 and HBCP < 0. The AIM2000 (ref. 78) was used to perform the QTAIM analyses in this study, using the PBEPBE/6-31++G** wave functions generated by the Gaussian 09 software package.79
3. Results and discussion
3.1. C59−nBNn fullerenes
Fig. 1 depicts the relaxed atomic structures of BN-codoped fullerenes C59−nBNn (n = 1–3), as well as the molecular electrostatic potentials (MEP) and total density of states (TDOS) plots. The equilibrium geometry and electrical structure of C59B heterofullerene and pure C60 are also presented to help understand the effect of B and N doping. A carbon atom of pure C60 is substituted with a boron impurity to produce C59B. The BN-codoped C58BN, C57BN2 and C56BN3 structures were then constructed by successively substituting the carbon atoms around the B atom in C59B with the N atom. Though the close cage structure of C60 is retained in all the systems, the substitution of C atoms with B and N makes some distortions in the vicinity of the dopant atoms. According to Table 1, the formation energy (Eform) of C59B is −5.23 eV, which is in agreement with those reported by other studies.80 This negative Eform value suggests that a B atom may be simply incorporated within the C60 structure and the formation of C59B would be thermodynamically feasible at normal temperatures. The newly formed B–C bonds distances are calculated to be 1.52 and 1.55 Å, which match reasonably with those previous studies.80–82 Table 1 also lists the Eform values of BN-codoped structures. It is seen that as the C atoms around the B in C59B is substituted with the N atoms, the Eform of the fullerene increases to −6.13 (C58BN), −6.66 (C57BN2) and −6.63 eV (C56BN3). This implies that the substitutional nitrogens increase the durability and stabilization of C59B fullerenes, and the BN codoping of C60 is more energetically desirable than the doping of a B atom. The latter can be explained by the donor–acceptor interaction between the vacant orbital of B and lone-pair electrons of the added N atoms. From the Eform values, this donor–acceptor interaction increases as C57BN2 > C56BN3 > C58BN. The average B–N bond distances of C58BN, C57BN2 and C56BN3 are calculated to be 1.50, 1.46 and 1.44 Å, respectively, which are about 6, 3 and 2% longer than the B–C bond of C59B. It is evident from Table 1 that the binding energy (Ebind) of the B atom at the vacancy site also varies with the number of nitrogen dopants around it. Interestingly, the Ebind values of B in the BN-codoped fullerenes are smaller than that of C59B, which can be related to the larger bond formation energy of B–C (≈−4.65 eV) than B–N (≈−4.04 eV).83 Nevertheless, the large negative Ebind values indicate that the B atom in C59−nBNn fullerenes cannot be easily diffused and hence these systems can remain as a stable catalyst even at high temperatures.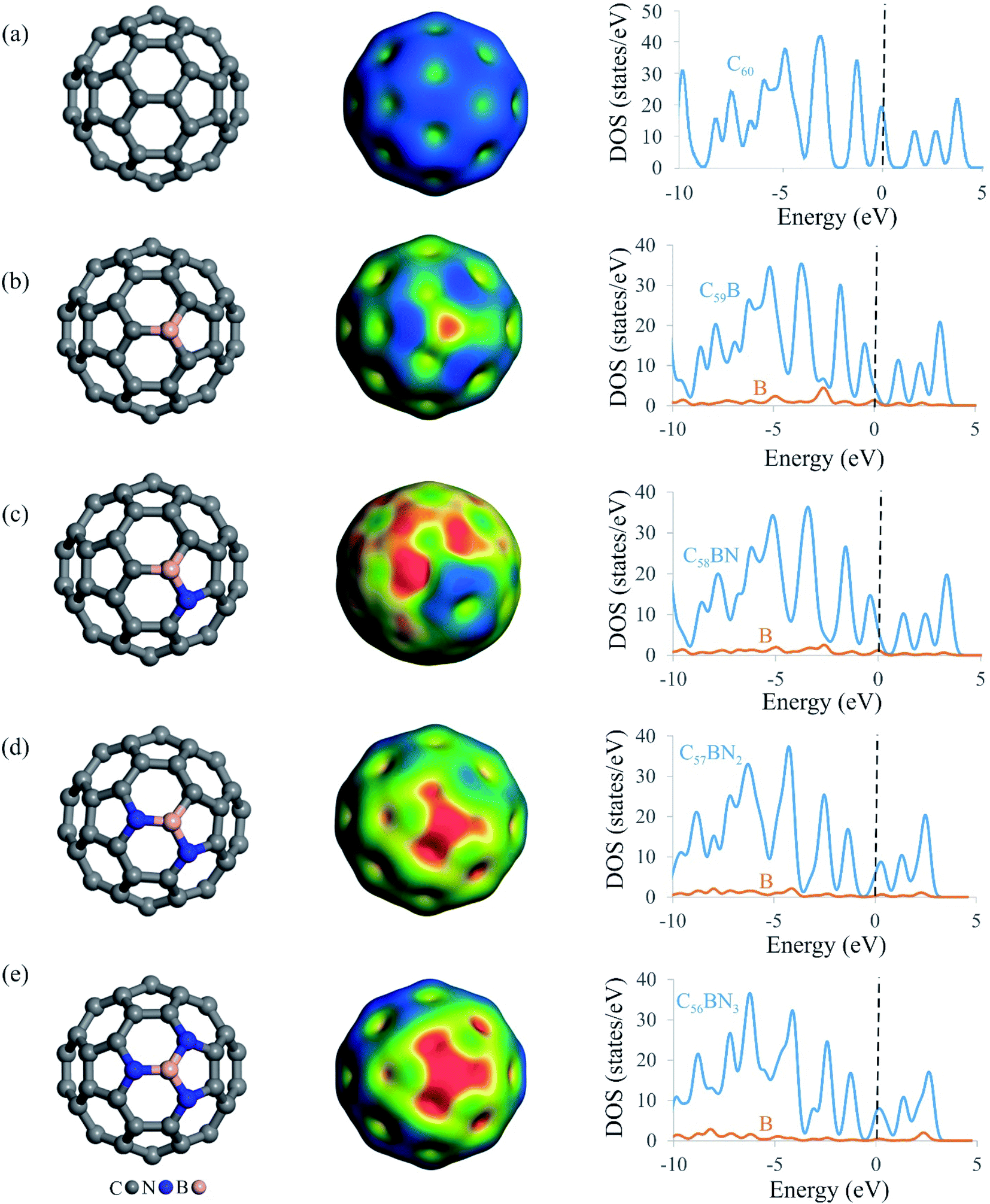 | ||
| Fig. 1 Local atomic structures (left) molecular electrostatic potential isosurfaces (MEP, middle) and TDOS plots (right) of (a) C60, (b) C59B, (c) C58BN, (d) C57BN2 and (e) C56BN3. The dashed line in TDOS plots shows the Fermi level. The color range in the MEP maps is from blue (more negative = −0.001 a.u.) to red (more positive = 0.078 a.u.). | ||
| Fullerene | Eform | Ebind | qB |
|---|---|---|---|
| C59B | −5.23 | −8.74 | 0.09 |
| C58BN | −6.13 | −7.92 | 0.10 |
| C57BN2 | −6.66 | −8.55 | 0.16 |
| C56BN3 | −6.63 | −8.17 | 0.21 |
Since N2O molecule is a polar molecule, it is expected that its adsorption and subsequent catalytic reduction are directly related to charge distribution of the catalyst surface. Hence, we provided the MEP isosurfaces of C59B and C59−nBNn fullerenes in Fig. 1. For the all systems studied, there is a positive electrostatic potential region developed on the B atom, indicating the possibility of this site to interact with nucleophilic species. As the electronegativity of N is greater than that of B, hence, C56BN3 indicates the largest electrostatic potential among the C59−nBNn systems. From Fig. 1, one can see that there is a quite small potential on the B atom of C58BN which is because of the charge transfer from the lone-pair of N atom into unfilled orbital of B. This builds up a sizable electron density in the region between the B and N atoms as evidenced from the corresponding MEP isosurface. However in the case of C57BN2 and C56BN3, the addition of second and third N atoms withdraws the electron density around the B atom and hence induces a large positive electrostatic potential over the B atom. These results are fully consistent with the Hirshfeld analysis results in Table 1, in which the substitution of first N atom has a negligible influence on the charge of the B atom. However, the incorporation of second and third N atoms induces a larger positive charge over the B atom, a phenomenon which is beneficial for the N2O adsorption. Accompanied with these charge redistributions, the electronic structure of C59−nBNn systems is also regulated as the number of involved nitrogen atoms grows. From the TDOS maps in Fig. 1, it is seen that the successive addition of N atoms tends to shift the band gap of C59B above the Fermi level. Compared to others, C56BN3 exhibits the smallest band gap due to increase in the donor states around the Fermi level. The latter phenomenon may modify greatly the surface reactivity of the BN-codoped fullerenes and facilities the charge-transfer between these systems and N2O molecule.
The dynamic stability of C59−nBNn fullerenes is also studied by the first-principle molecular dynamic simulations in a period of 2000 fs (2 ps) and at the temperature of 600 K (Fig. S2 of ESI†). It is seen that the cage structure of BN-codoped fullerenes are almost preserved after the simulations due to strong chemical bonding between the B, C and N atoms. In particular, the lack of any geometry buckling around the B atom suggests that these systems are dynamically stable and therefore can be utilized as a durable and stable catalyst at high temperatures.
3.2. N2O adsorbed configurations
After screening out the surface reactivity and electronic structure of C59−nBNn fullerenes, we probe the adsorption of N2O molecule on these systems. Fig. 2 compares the geometry of N2O molecule adsorbed on the C59−nBNn fullerenes, and Table 2 summarizes the adsorption energies (Ead), electron-transfer values (QCT) and Gibbs free energy changes (ΔGad) due to the adsorption of N2O. According to earlier studies,12,84,85 N2O can be adsorbed on catalyst surfaces via three different configurations, namely, linear and [2+2]- or [3+2]-cycloaddition. Our results indicate that the linear configuration, in which N2O interacts with the B atom of C59−nBNn fullerenes via its N- or O-end is metastable, and it is readily converted into a stable chemisorbed configuration by overcoming a small activation barrier. In the case of C59B, C58BN and C57BN2, in which there is at least one carbon atom around the B, the O and N atoms of N2O bind with the B–C bond through a [3+2]-cycloaddition. From Table 2, the adsorption energies (Ead) of N2O are calculated to be −0.54, −0.62 and −0.70 eV on C59B, C58BN and C57BN2, respectively, indicating that the tendency of fullerenes to adsorb N2O molecule is improved with the successive incorporation of N atoms. The B–O (N–C) bond distances between N2O and C59−nBNn are 1.52 (1.54), 1.49 (1.53) and 1.46 (1.53) Å for n = 0, 1 and 2, respectively. This reveals that the interaction between the fullerene and N2O is enhanced by increasing the N dopants around the B atom. Moreover, the Hirshfeld analysis discloses that the adsorbed N2O extracts about 0.20 electrons from the fullerene, which occupy its empty 3π* orbital, leading to bending of the N–N–O angle. The latter is verified by the PDOS plots in Fig. 2, in which the successive addition of N atoms downshifts the 3π* orbital of N2O. From the Hirshfeld population analysis, the oxygen atom of N2O is negatively charged by 0.13, 0.15 and 0.16|e| on C59B, C58BN and C57BN2, respectively. Besides, the N–O bond length in the free N2O molecule is 1.19 Å, which is stretched to 1.37, 1.40 and 1.42 Å on C59B, C58BN and C57BN2, respectively. These results show that increasing N impurities in C59−nBNn results in the further activation of N2O. From Table 2, it is seen that the Gibbs free energy changes (ΔGad) due to the adsorption of N2O on C59B, C58BN and C57BN2 fullerenes are negative, suggesting the thermodynamic feasibility of this process. Nevertheless, the differences between the Ead and ΔGad values indicate that the temperature and entropy effects are essential factors, owning to their 0.31–0.52 eV contribution to the adsorption energy.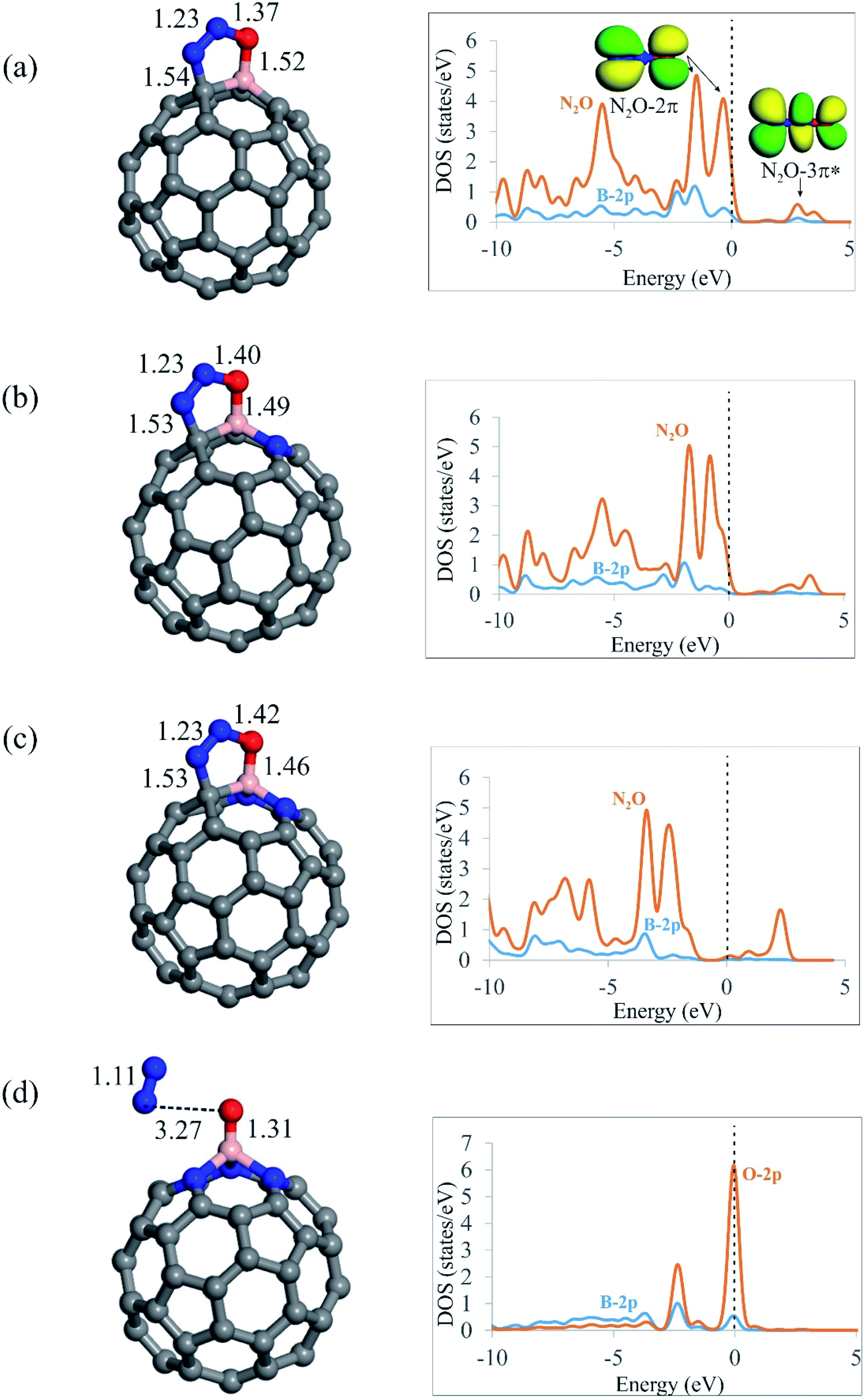 | ||
| Fig. 2 The optimized local atomic structures, key bond lengths (in Å), and PDOS plots of N2O adsorbed onto (a) C59B, (b) C58BN, (c) C57BN2 and (d) C56BN3 fullerenes. In the PDOS plots, the Fermi level is indicated by the dashed line. | ||
| Fullerene | N2O | CO | ||||
|---|---|---|---|---|---|---|
| Ead | QCT | ΔGad | Ead | QCT | ΔGad | |
| a The values within the parenthesis refers to the adsorption of N2O and CO molecules onto endohedral Kr@C59−nBNn systems. | ||||||
| C59B | −0.54 (−0.53) | 0.21 (0.21) | −0.08 (−0.07) | −0.92 (−0.92) | 0.24 (0.24) | −0.38 (−0.37) |
| C58BN | −0.62 (−0.60) | 0.23 (0.23) | −0.10 (−0.09) | −0.35 (−0.34) | 0.21 (0.20) | 0.14 (0.15) |
| C57BN2 | −0.70 (−0.68) | 0.23 (0.22) | −0.22 (−0.20) | −0.35 (−0.34) | 0.22 (0.21) | 0.13 (0.14) |
| C56BN3 | −1.95 (−1.93) | 0.41 (0.40) | −1.64 (−1.62) | −0.25 (−0.24) | 0.20 (0.19) | 0.23 (0.23) |
On the other hand, it is found that the [3+2]-cycloaddition configuration is energetically unfavorable for N2O on the nitrogen-rich C56BN3 fullerene, since it is spontaneously dissociated into N2 molecule and an O atom (O*) attached to the B atom (Fig. 2d). It is necessary to state that a similar behavior has been also reported for other surfaces like Si-doped19 or BN-codoped graphene.17 As discussed above, the successive introduction of N impurities can largely reinforce the electron-donating property of B-doped fullerene. Hence, one can expect that electron-rich C56BN3 pumps a sizeable electron density to 3π* orbital of N2O, leading to break of its N–O bond. The adsorption energy of N2O on C56BN3 is −1.95 eV, which indicates that the dissociation of this molecule is energetically favorable. Also, this implies that the O* atom is tightly attached to B atom. The Hirshfeld analysis discloses that the O atom adsorbed on the B atom is negatively charged by about 0.40|e|, which suggests its efficient electron donating property. Like other BN-codoped fullerenes as discussed above, the negative ΔGad value for the N2O adsorption on C56BN3 indicates that it is a thermodynamically facile process at ambient temperature mainly due to the increase of entropy. Considering the PDOS plot in Fig. 2d, it is observed that the O-2p states are favorably coupled with the B-2p states at the Fermi level. This observation agrees well with the calculated Ead and ΔGad values, implying that the O* is strongly adsorbed on the B atom.
As noted before, one of the key questions that has inspired this study is the effect of encapsulating a Ng atom on the catalytic activities of the C59−nBNn fullerenes. To achieve this goal, a Kr atom was put in the center of the fullerenes, and the N2O adsorption was then studied over the resulting systems (Fig. S3†). The B–O bond lengths between encapsulated fullerenes and N2O molecule range from 1.31 to 1.52 Å, which is nearly equal to those reported for empty C59−nBNn systems. The Kr atom is stably inserted within the C59−nBNn fullerenes in all studied systems due to its negative adsorption energy (≈−0.54 eV). Meanwhile, following encapsulation, the Kr atom obtains a positive charge of 0.25|e|, suggesting that the Kr atom redistributes the electron density within the fullerene. As a result, the N2O adsorption energies on Kr@C59−nBNn fullerenes are slightly lower than those on empty C59−nBNn fullerenes. It is also worth noting that the Kr encapsulation has no effect on the order of the N2O adsorption energies on C59−nBNn fullerenes. These findings support previous findings68 that adding a noble gas into C60 has little effect on its surface reactivity and hence catalytic properties.
The calculated ρBCP, ∇2ρBCP and HBCP values associated with the B–O BCPs of N2O adsorbed complexes are shown in Table 3. It is seen that the ρBCP value at the B–O BCPs increases as the number of the N atoms increases, which is consistent with the adsorption energies discussed above. However, the ∇2ρBCP values at these BCPs are all positive, but the HBCP values are all negative, suggesting that the B–O bonds in these systems are partly covalent. Importantly, the associated HBCP values vary as C56BN3 > C57BN2 > C58BN > C59B, showing that the degree of covalency in the B–O bonds increases with the number of doped N atoms.
| Fullerene | N2O | CO | ||||
|---|---|---|---|---|---|---|
| ρBCP | ∇2ρBCP | HBCP | ρBCP | ∇2ρBCP | HBCP | |
| C59B | −0.54 (−0.53) | 0.21 (0.21) | −0.08 (−0.07) | −0.92 (−0.92) | 0.24 (0.24) | −0.38 (−0.37) |
| C58BN | −0.62 (−0.60) | 0.23 (0.23) | −0.10 (−0.09) | −0.35 (−0.34) | 0.21 (0.20) | 0.14 (0.15) |
| C57BN2 | −0.70 (−0.68) | 0.23 (0.22) | −0.22 (−0.20) | −0.35 (−0.34) | 0.22 (0.21) | 0.13 (0.14) |
| C56BN3 | −1.95 (−1.93) | 0.41 (0.40) | −1.64 (−1.62) | −0.25 (−0.24) | 0.20 (0.19) | 0.23 (0.23) |
3.3. CO adsorbed configurations
From Fig. 3, it is seen that CO molecule prefers to interact with the B atom of C59−nBNn fullerenes via its C atom. The Ead of CO on C59B is −0.92 eV, which agrees well with those reported by other DFT studies.58 Meanwhile, this quite large negative value indicates the chemisorption nature of the adsorption. On the contrary, our results in Table 2 show that tendency of C59−nBNn fullerenes to capture CO molecule decreases with the addition of N atoms around the B atom. In particular, the Ead value of CO on nitrogen rich C56BN3 structure is −0.25 eV, which demonstrates the physisorption of this molecule. Besides, the binding distances between the fullerenes and C atom of CO are 1.55, 1.63, 1.64 and 1.67 Å for C59B, C58BN, C57BN2 and C56BN3, respectively, indicating that the CO interaction with the B atom is weakened with the increasing the content of N dopants. Note that the order of Ead values of CO on C59−nBNn is not correlated with the positive charges on the B atom or MEP analysis results. This may suggest that the electrostatic interaction between the B atom and CO has a negligible contribution in the resulting complexes. Moreover, the values of ΔGad in Table 2 indicate that unlike C59B, the CO adsorption would not be thermodynamically possible at room temperature. According to Table 2, the CO adsorption is associated with a charge-transfer from CO molecule into fullerene with the values in the range of 0.20–0.24|e|. Considering the PDOS plots in Fig. 3, one can see that there is a noticeable interaction between the CO-5σ and the vacant B-2p states below the Fermi level especially for the C59B system. Also, a weak orbital interaction occurs between CO-2π* and B atom, which accounts for the lengthening of C–O bond in these systems.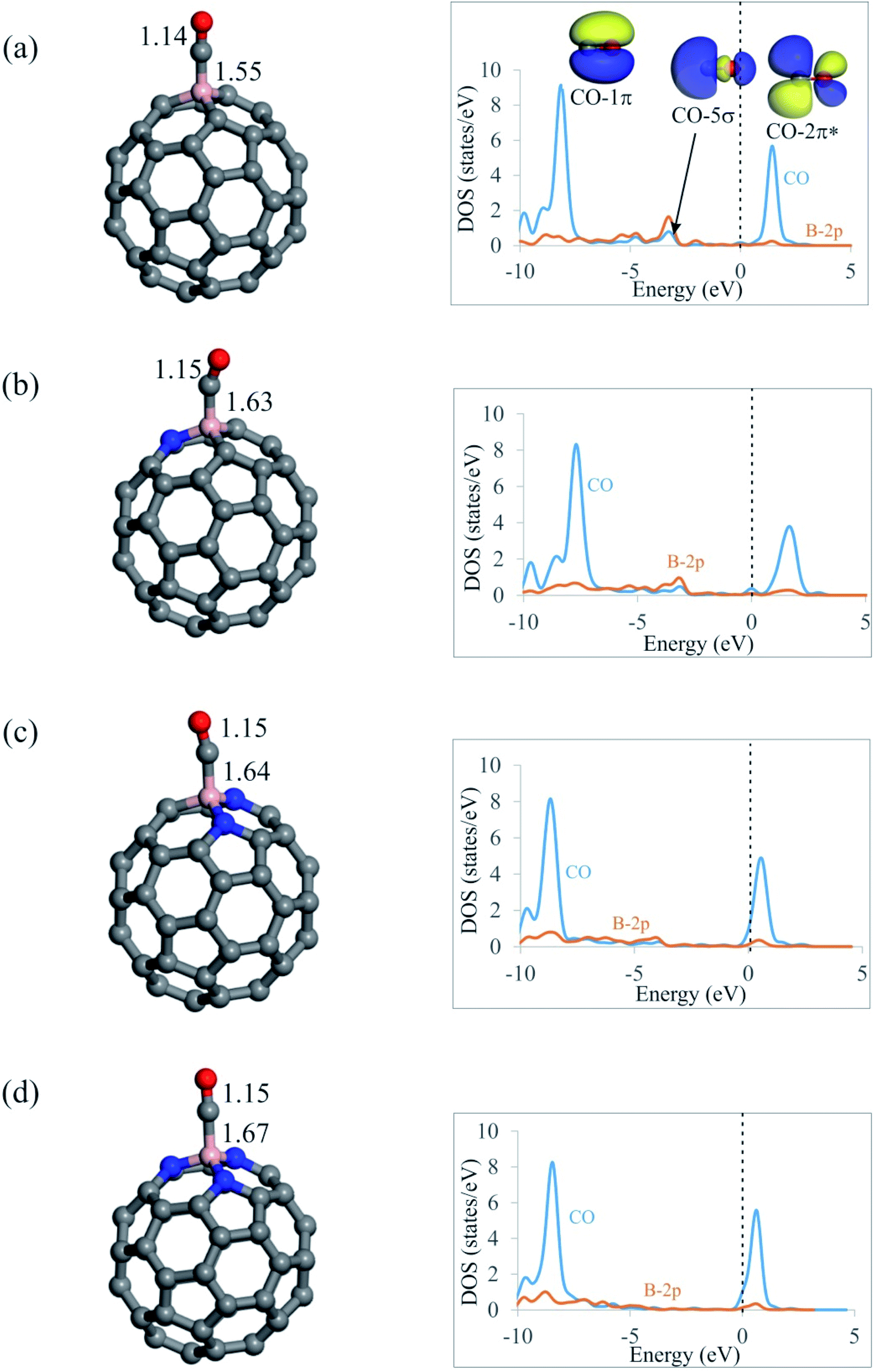 | ||
| Fig. 3 The optimized local atomic structures, binding distances (in Å), and PDOS plots of CO adsorbed onto (a) C59B, (b) C58BN, (c) C57BN2 and (d) C56BN3 fullerenes. In the PDOS plots, the Fermi level is indicated by the dashed line. | ||
Table 2 shows that adding a Kr atom to the C59−nBNn fullerenes has little or no influence on the Ead, QCT and ΔGad values of CO molecule. That is, with the exception of Kr@C59B, CO adsorption is still weak and belongs to physisorption. Furthermore, the calculated ρBCP values show that, among the C59−nBNn fullerenes studied, C56BN3 forms the weakest complex with the CO molecule (Table 3). Also, the positive ∇2ρBCP and negative HBCP values at the B–C BCPs indicate that these bonds are partly covalent. The HBCP values in Table 3 show that, with the exception of C59B complexes, the B–N bonds in the N2O adsorbed complexes are more covalent than the B–C bonds in the CO adsorbed systems.
The above findings clearly demonstrate that increasing the N coordination to the B atom of C59B would be beneficial for N2O capture, which is the starting point for the N2O reduction process. The surrounding B–N bonds, in particular, make C56BN3 an effective catalyst for the spontaneous decomposition of N2O. More importantly, BN-codoped fullerenes have a lower capacity to trap CO molecules than C59B, suggesting that they should be extremely resistant to CO poisoning. As a result, BN-codoped fullerenes appear to have excellent catalytic activity in the reduction of N2O. The N2O reduction on the C59−nBNn systems is compared in detail in the next section.
3.4. N2O reduction
Now we explore the possible reduction mechanisms for reduction of N2O on BN-codoped fullerenes (C58BN, C57BN2 and C56BN3). Based on the earlier investigations,86 depending on the order of adsorption energies of N2O and CO on the catalyst, N2O reduction can proceed via two main pathways, i.e., the Eley–Rideal (E–R) and Langmuir–Hinshelwood (L–H). In the E–R mechanism, which is also known as the stepwise pathway, a N2O molecule is initially adsorbed and decomposed on the catalyst and then a CO molecule interacts with the remaining O* species to generate CO2 molecule. In contrast, the L–H or concerted mechanism involves the simultaneous binding of N2O and CO molecules to the catalyst. Both these mechanisms are investigated here to realize the most energetically favorable pathway of N2O reduction. The relaxed atomic structures and energy profiles of these competing pathways are given in Fig. 4–6. | ||
| Fig. 4 The potential energy diagram and relevant bond distances (in Å) for direct decomposition of N2O over (a) C58BN and (b) C57BN2 fullerenes. | ||
 | ||
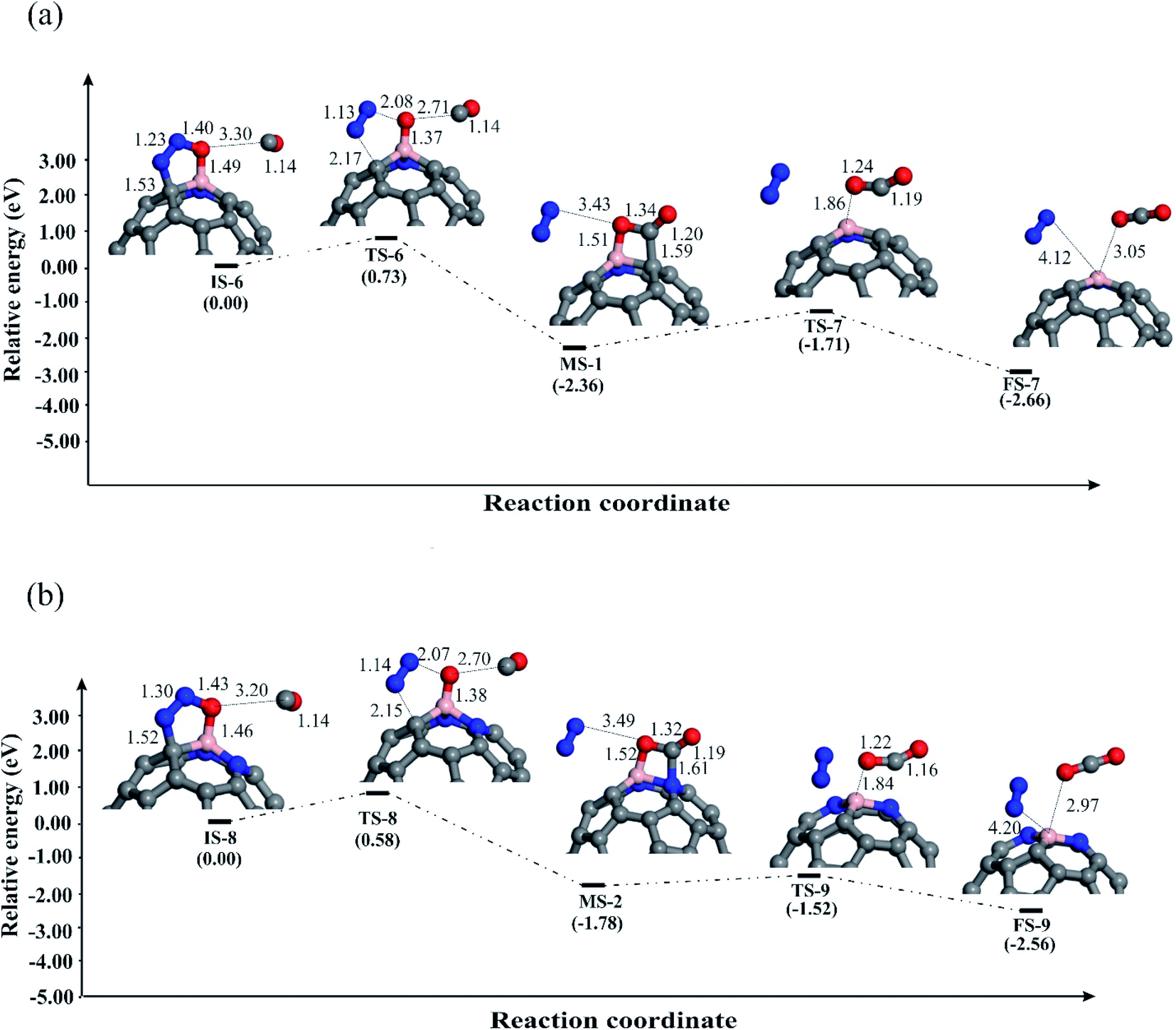
| Fig. 5 The potential energy diagram and relevant bond distances (in Å) for removing O* atom by CO over (a) C58BN, (b) C57BN2 and (c) C56BN3 fullerenes. | ||
 | ||
| Fig. 6 The potential energy diagram and relevant bond distances (in Å) for reduction of N2O by CO molecule through the L–H mechanism over (a) C58BN and (b) C57BN2 fullerenes. | ||
Let us first discuss the E–R mechanism for the N2O reduction reaction on the BN-codoped fullerenes. As discussed above, N2O is adsorbed on the C58BN and C57BN2 fullerenes via a [3+2]-cycloaddition, while it is spontaneously decomposed on C56BN3. This suggests that the first step of the N2O reduction is already completed on the latter system and hence we only focus on the N2O decomposition mechanism on C58BN and C57BN2. Fig. 4a and b indicate the reaction pathways for the N2O → N2 + O* elementary reaction on C58BN and C57BN2, respectively. To start the reaction, N–O bond of the adsorbed N2O is elongated and the negative charge on the O* atom is continuously increased. Passing over the transition states TS-1 and TS-2 with an activation barrier (Ebar) of 0.88 and 0.75 eV, respectively, N2O is decomposed on the C58BN and C57BN2. At the final state, the O atom is attached on the B atom, while N2 molecule is separated from the fullerene with a binding distance of ≈3 Å. An interesting finding here is that the Ebar for the decomposition of N2O varies with the positive charge on the B atom. That is, the Ebar values for the decomposition of N2O on C57BN2 fullerenes is less than the corresponding value obtained for the C58BN, confirming that the catalytic performance of the fullerene is improved with the addition of N atoms around the B. Besides, these Ebar values are less than the reported value for the noble-metal based Ag7Au6 catalyst (1.1 eV).87 It should point out that unlike C57BN2, the O* moiety resulting from the N2O decomposition is strongly chemisorbed on the B–C bond of C58BN through the formation of an epoxide-like structure. Note that a similar species has been also found on the C59B structure,34 suggesting that C58BN is readily oxidized during the N2O reduction process.
To complete the catalytic cycle, the remaining O* should be removed by CO molecule. To examine the energy profile, we added the CO molecule on the O* to form the corresponding initial state (Fig. 5). Next, CO approaches the O* and by passing the corresponding transition state, the CO2 molecule is achieved. Like C59B,34 the Ebar value for the removing O* on C58BN is relatively high (1.73 eV), owning to the strong adsorption of this species on the B–C bond. On the contrary, the energy barrier for the CO + O* → CO2 on C57BN2 and C56BN3 fullerenes are 0.52 and 0.28 eV, respectively, which are small enough to be easily overcome at normal temperatures. The CO2 is finally separated from the fullerenes, and this restores the catalyst to begin a new N2O reduction process.
We further explored the N2O reduction process through the L–H mechanism, in which N2O and CO molecules are first coadsorbed on fullerenes. Fig. 6 depicts the obtained geometric structures for involved species in this mechanism. The shortest binding distance between N2O and CO in the initial state (IS-6 and IS-8) is about 3 Å, hinting at the weak nature of the interaction between them. Then, CO moves towards the N2O until a stable OCO intermediate is obtained on the fullerene. Though this reaction step is exothermic on both BN-codoped structures, the energy barriers and the reaction energies are not similar. An interesting finding is that the energy barrier to obtain the OCO species decreases as the nitrogen content around the B atom increases. Due to large activation of N2O on the C57BN2, a relatively smaller Ebar is obtained (0.36 eV), which is comparable to the mentioned value for the Fe-doped graphene (0.83 eV).86 Also, these Ebar values are lower than that of over C59B (1.95 eV),88 suggesting that the BN codoping is able to substantially improve the catalytic properties of the C60 fullerene. In the next step, the OCO is converted to CO2 molecule as shown in Fig. 6. This elementary reaction is also exothermic but requests an activation barrier of 0.73 (C58BN) and 0.58 eV (C57BN2), which seems to be easily provided at room temperature. The resulting CO2 molecule has a small Ead value, so it can be simply separated from the active site of the fullerenes.
4. Conclusions
We used dispersion-corrected DFT calculations to explore the catalytic and electronic structure properties of BN-codoped C60 fullerenes. The results showed that by successively doping N atoms around the B in C59B, the tendency of the fullerene to interact with N2O molecule increases. The B atom in C56BN3 acts as an efficient catalytic site for the spontaneous decomposition of N2O because the surrounding B–N bonds hinder the formation of secondary C–N bond between the fullerene and N2O. Furthermore, the fact that BN-codoped fullerenes have a low tendency for capturing CO molecule implies that they are tolerant to CO poisoning. The adsorption energies of N2O and CO are not influenced by the introduction of a Kr atom inside the C59−nBNn fullerenes. The activation barriers for N2O reduction via the L–H mechanism are lower than those for the E–R mechanism over C57BN2 and C56BN3, indicating that this reaction would occur via the former pathway. Even though that N2O reduction is exothermic over the C59−nBNn fullerenes, the obtained activation barriers vary with number of nitrogen atoms in these systems. Nitrogen dopants surrounding the B atom, for example, inhibit the poisoning of C57BN2 and C56BN3 fullerenes by the O* moiety. The nitrogen-rich C56BN3 fullerene has the highest catalytic activity for N2O reduction due to its unique electronic structure and charge-transfer. We hope that this new understanding will inspire the development of highly efficient metal-free catalysts based on carbon fullerenes, as well as provide a theoretical basis for future studies into the removal of hazardous N2O and CO pollutants from the environment.Conflicts of interest
There are no conflicts to declare.References
- S. A. Montzka, E. J. Dlugokencky and J. H. Butler, Nature, 2011, 476, 43–50 CrossRef CAS PubMed.
- A. Tatar, A. Barati-Harooni, A. Najafi-Marghmaleki, A. Mohebbi, M. M. Ghiasi, A. H. Mohammadi and A. Hajinezhad, Int. J. Greenhouse Gas Control, 2016, 49, 47–54 CrossRef.
- D. J. Wuebbles, Science, 2009, 326, 56–57 CrossRef CAS PubMed.
- R. Portmann, J. Daniel and A. Ravishankara, Philos. Trans. R. Soc., B, 2012, 367, 1256–1264 CrossRef CAS PubMed.
- J.-D. Lee, W.-P. Fang, C.-S. Li and C.-H. Cheng, J. Chem. Soc., Dalton Trans., 1991, 1923–1927 RSC.
- J. A. Ryder, A. K. Chakraborty and A. T. Bell, J. Phys. Chem. B, 2002, 106, 7059–7064 CrossRef CAS.
- P. Pannopard, P. Khongpracha, C. Warakulwit, S. Namuangruk, M. Probst and J. Limtrakul, ChemPhysChem, 2012, 13, 583–587 CrossRef CAS PubMed.
- J. s. A. Luque-Urrutia and A. Poater, Inorg. Chem., 2017, 56, 14383–14387 CrossRef CAS PubMed.
- A. Heyden, B. Peters, A. T. Bell and F. J. Keil, J. Phys. Chem. B, 2005, 109, 1857–1873 CrossRef CAS PubMed.
- A. Heyden, N. Hansen, A. T. Bell and F. J. Keil, J. Phys. Chem. B, 2006, 110, 17096–17114 CrossRef CAS PubMed.
- E. Pachatouridou, E. Papista, E. F. Iliopoulou, A. Delimitis, G. Goula, I. V. Yentekakis, G. E. Marnellos and M. Konsolakis, J. Environ. Chem. Eng., 2015, 3, 815–821 CrossRef CAS.
- A. L. Yakovlev, G. M. Zhidomirov and R. A. van Santen, Catal. Lett., 2001, 75, 45–48 CrossRef CAS.
- V. Blagojevic, G. Orlova and D. K. Bohme, J. Am. Chem. Soc., 2005, 127, 3545–3555 CrossRef CAS PubMed.
- S. M. Hamilton, W. S. Hopkins, D. J. Harding, T. R. Walsh, P. Gruene, M. Haertelt, A. Fielicke, G. Meijer and S. R. Mackenzie, J. Am. Chem. Soc., 2010, 132, 1448–1449 CrossRef CAS PubMed.
- G. Walther, D. J. Mowbray, T. Jiang, G. Jones, S. Jensen, U. J. Quaade and S. Horch, J. Catal., 2008, 260, 86–92 CrossRef CAS.
- S. Pornsatitworakul, S. Phikulthai, S. Namuangruk and B. Boekfa, Int. Conference on IEEE, 2015, pp. 225–229 Search PubMed.
- M. D. Esrafili, Chem. Phys. Lett., 2018, 705, 44–49 CrossRef CAS.
- M. D. Esrafili, F. Mohammadian-Sabet and P. Nematollahi, RSC Adv., 2016, 6, 64832–64840 RSC.
- J.-X. Zhao, Y. Chen and H.-G. Fu, Theor. Chem. Acc., 2012, 131, 1242 Search PubMed.
- A. Junkaew, S. Namuangruk, P. Maitarad and M. Ehara, RSC Adv., 2018, 8, 22322–22330 RSC.
- N. Martin, M. Altable, S. Filippone and A. Martin-Domenech, Synlett, 2007, 2007, 3077–3095 CrossRef.
- B. C. Thompson and J. M. Fréchet, Angew. Chem., Int. Ed., 2008, 47, 58–77 CrossRef CAS PubMed.
- A. K. Geim, Science, 2009, 324, 1530–1534 CrossRef CAS PubMed.
- B. F. Machado and P. Serp, Catal. Sci. Technol., 2012, 2, 54–75 RSC.
- P. Lazar, F. e. Karlický, P. Jurečka, M. s. Kocman, E. Otyepková, K. r. Šafářová and M. Otyepka, J. Am. Chem. Soc., 2013, 135, 6372–6377 CrossRef CAS PubMed.
- Y. García-Rodeja, M. Solà and I. Fernández, J. Org. Chem., 2016, 82, 754–758 CrossRef PubMed.
- K. Kakaei, M. D. Esrafili and A. Ehsani, in Interface Science and Technology, ed. K. Kakaei, M. D. Esrafili and A. Ehsani, Elsevier, 2019, vol. 27, pp. 339–386 Search PubMed.
- T. Wehling, K. Novoselov, S. Morozov, E. Vdovin, M. Katsnelson, A. Geim and A. Lichtenstein, Nano Lett., 2008, 8, 173–177 CrossRef CAS PubMed.
- X. Wang, X. Li, L. Zhang, Y. Yoon, P. K. Weber, H. Wang, J. Guo and H. Dai, Science, 2009, 324, 768–771 CrossRef CAS PubMed.
- B. Guo, L. Fang, B. Zhang and J. R. Gong, Insci. J., 2011, 80–89 CrossRef CAS.
- M. D. Esrafili and P. Mousavian, Appl. Surf. Sci., 2018, 455, 808–814 CrossRef CAS.
- Q. Z. Li, J. J. Zheng, J. S. Dang and X. Zhao, ChemPhysChem, 2015, 16, 390–395 CrossRef CAS PubMed.
- M. D. Esrafili and S. Heidari, Comput. Theor. Chem., 2019, 11, 50–57 CrossRef.
- K.-Y. Chen, S.-Y. Wu and H.-T. Chen, ACS Omega, 2020, 5, 28870–28876 CrossRef CAS PubMed.
- F. Gao, G. L. Zhao, S. Yang and J. J. Spivey, J. Am. Chem. Soc., 2013, 135, 3315–3318 CrossRef CAS PubMed.
- H. Wang, M. Xie, L. Thia, A. Fisher and X. Wang, J. Phys. Chem. Lett., 2014, 5, 119–125 CrossRef CAS PubMed.
- D. C. Camacho-Mojica, E. Muñoz-Sandoval and F. López-Urías, Carbon, 2017, 116, 381–390 CrossRef.
- Z. Xie, M. Chen, S. G. Peera, C. Liu, H. Yang, X. Qi, U. P. Kumar and T. Liang, ACS Omega, 2020, 5, 5142–5149 CrossRef CAS PubMed.
- K. Gong, F. Du, Z. Xia, M. Durstock and L. Dai, Science, 2009, 323, 760–764 CrossRef CAS PubMed.
- Y. Feng, F. Li, Z. Hu, X. Luo, L. Zhang, X.-F. Zhou, H.-T. Wang, J.-J. Xu and E. Wang, Phys. Rev. B: Condens. Matter Mater. Phys., 2012, 85, 155454 CrossRef.
- H. Miao, S. Li, Z. Wang, S. Sun, M. Kuang, Z. Liu and J. Yuan, Int. J. Hydrogen Energy, 2017, 42, 28298–28308 CrossRef CAS.
- R. Shi, J. Zhao, S. Liu, W. Sun, H. Li, P. Hao, Z. Li and J. Ren, Carbon, 2018, 130, 185–195 CrossRef CAS.
- L. Yu, X. Pan, X. Cao, P. Hu and X. Bao, J. Catal., 2011, 282, 183–190 CrossRef CAS.
- P. Zhang, J. S. Lian and Q. Jiang, Phys. Chem. Chem. Phys., 2012, 14, 11715–11723 RSC.
- F. Studt, Catal. Lett., 2013, 143, 58–60 CrossRef CAS.
- Y. Tang, W. Chen, Z. Shen, S. Chang, M. Zhao and X. Dai, Carbon, 2017, 111, 448–458 CrossRef CAS.
- M. D. Esrafili and S. Asadollahi, Appl. Surf. Sci., 2018, 463, 526–534 CrossRef.
- X. Liu, Y. Sui, T. Duan, C. Meng and Y. Han, Catal. Sci. Technol., 2015, 5, 1658–1667 RSC.
- X. Zhang, Z. Lu, Y. Tang, Z. Fu, D. Ma and Z. Yang, Phys. Chem. Chem. Phys., 2014, 16, 20561–20569 RSC.
- L. Ferrighi, M. I. Trioni and C. Di Valentin, J. Phys. Chem. C, 2015, 119, 6056–6064 CrossRef CAS.
- L. Qin, L. Wang, X. Yang, R. Ding, Z. Zheng, X. Chen and B. Lv, J. Catal., 2018, 359, 242–250 CrossRef CAS.
- S. Wang, J. Feng, Q. Meng, B. Cao and G. Tian, J. Solid State Electrochem., 2019, 23, 2377–2390 CrossRef CAS.
- M. D. Esrafili and H. Janebi, Mol. Phys., 2020, 118, e1631495 CrossRef.
- S. Wang, L. Zhang, Z. Xia, A. Roy, D. W. Chang, J. B. Baek and L. Dai, Angew. Chem., Int. Ed., 2012, 51, 4209–4212 CrossRef CAS PubMed.
- Y. Zheng, Y. Jiao, L. Ge, M. Jaroniec and S. Z. Qiao, Angew. Chem., 2013, 125, 3192–3198 CrossRef.
- J.-i. Ozaki, T. Anahara, N. Kimura and A. Oya, Carbon, 2006, 44, 3358–3361 CrossRef CAS.
- J.-i. Ozaki, N. Kimura, T. Anahara and A. Oya, Carbon, 2007, 45, 1847–1853 CrossRef CAS.
- B. Gao and G. Chen, RSC Adv., 2019, 9, 21626–21636 RSC.
- Y.-X. Yu, Phys. Chem. Chem. Phys., 2013, 15, 16819–16827 RSC.
- H. Jiang, T. Zhao, L. Shi, P. Tan and L. An, J. Phys. Chem. C, 2016, 120, 6612–6618 CrossRef CAS.
- S. Nachimuthu, P.-J. Lai and J.-C. Jiang, Carbon, 2014, 73, 132–140 CrossRef CAS.
- S. Guha and K. Nakamoto, Coord. Chem. Rev., 2005, 249, 1111–1132 CrossRef CAS.
- A. Takeda, Y. Yokoyama, S. Ito, T. Miyazaki, H. Shimotani, K. Yakigaya, T. Kakiuchi, H. Sawa, H. Takagi and K. Kitazawa, Chem. Commun., 2006, 912–914 RSC.
- R. B. Ross, C. M. Cardona, D. M. Guldi, S. G. Sankaranarayanan, M. O. Reese, N. Kopidakis, J. Peet, B. Walker, G. C. Bazan and E. Van Keuren, Nat. Mater., 2009, 8, 208–212 CrossRef CAS PubMed.
- A. A. Popov, S. Yang and L. Dunsch, Chem. Rev., 2013, 113, 5989–6113 CrossRef CAS PubMed.
- F. Cimpoesu, S. Ito, H. Shimotani, H. Takagi and N. Dragoe, Phys. Chem. Chem. Phys., 2011, 13, 9609–9615 RSC.
- M. Bühl, S. Patchkovskii and W. Thiel, Chem. Phys. Lett., 1997, 275, 14–18 CrossRef.
- S. Osuna, M. Swart and M. Sola, Phys. Chem. Chem. Phys., 2011, 13, 3585–3603 RSC.
- A. Bil and C. A. Morrison, J. Phys. Chem. A, 2012, 116, 3413–3419 CrossRef CAS PubMed.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865–3868 CrossRef CAS PubMed.
- S. Grimme, J. Comput. Chem., 2006, 27, 1787–1799 CrossRef CAS PubMed.
- B. Delley, J. Chem. Phys., 1990, 92, 508–517 CrossRef CAS.
- B. Delley, J. Chem. Phys., 2000, 113, 7756–7764 CrossRef CAS.
- C. O. Ulloa, M. Ponce-Vargas and A. Munoz-Castro, J. Phys. Chem. C, 2018, 122, 25110–25117 CrossRef CAS.
- J. C. Gonzalez, S. Mondal, F. Ocayo, R. Guajardo-Maturana and A. Muñoz-Castro, Int. J. Quantum Chem., 2020, 120, e26080 Search PubMed.
- R. Bader, Atoms in Molecules: A Quantum Theory, Clarendon Press, Oxford, 1990 Search PubMed.
- I. Rozas, I. Alkorta and J. Elguero, J. Am. Chem. Soc., 2000, 122, 11154–11161 CrossRef CAS.
- F. Biegler-König and J. Schönbohm, J. Comput. Chem., 2002, 23, 1489–1494 CrossRef PubMed.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, X. Li, M. Caricato, A. Marenich, J. Bloino, B. G. Janesko, R. Gomperts, B. Mennucci, H. P. Hratchian, J. V. Ortiz, A. F. Izmaylov, J. L. Sonnenberg, D. Williams-Young, F. Ding, F. Lipparini, F. Egidi, J. Goings, B. Peng, A. Petrone, T. Henderson, D. Ranasinghe, V. G. Zakrzewski, J. Gao, N. Rega, G. Zheng, W. Liang, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell, J. A. Montgomery Jr, J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, T. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, J. M. Millam, M. Klene, C. Adamo, R. Cammi, J. W. Ochterski, R. L. Martin, K. Morokuma, O. Farkas, J. B. Foresman, and D. J. Fox, Gaussian 09, Revision A.02, Gaussian, Inc., Wallingford CT, 2016 Search PubMed.
- Y. Wang, M. Jiao, W. Song and Z. Wu, Carbon, 2017, 114, 393–401 CrossRef CAS.
- M. D. Esrafili and N. Mohammadirad, Mol. Phys., 2017, 115, 1633–1641 CrossRef CAS.
- C. Ramachandran, J. Phys. Chem. A, 2017, 121, 1708–1714 CrossRef.
- R. Sanderson, Chemical Bonds and Bonds Energy, Elsevier, 2012 Search PubMed.
- A. L. Yakovlev, G. M. Zhidomirov and R. A. van Santen, J. Phys. Chem. B, 2001, 105, 12297–12302 CrossRef CAS.
- P. Nematollahi and M. D. Esrafili, RSC Adv., 2016, 6, 59091–59099 RSC.
- S. Wannakao, T. Nongnual, P. Khongpracha, T. Maihom and J. Limtrakul, J. Phys. Chem. C, 2012, 116, 16992–16998 CrossRef CAS.
- S. T. Oyama, Mechanisms in Homogeneous and Heterogeneous Epoxidation Catalysis, Elsevier, 2011 Search PubMed.
- M. D. Esrafili and S. Heydari, ChemistrySelect, 2019, 4, 2267–2274 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1ra04046d |
| This journal is © The Royal Society of Chemistry 2021 |