
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSolvation effect on the ESIPT mechanism of nitrile-substituted ortho-hydroxy-2-phenyl-oxazolines
Hengwei Zhanga,
Wenzhi Lia,
Yuxi Wanga,
Yaping Taob,
Yi Wang a,
Fan Yang*a and
Ziqing Gao*a
a,
Fan Yang*a and
Ziqing Gao*a
aSchool of Biological Engineering, Dalian Polytechnic University, Dalian 116034, P. R. China. E-mail: wangyi@dlpu.edu.cn; gao_zq@dlpu.edu.cn; Fax: +86 0411 86323646; Tel: +86 0411 86323646
bCollege of Physics and Electronic Information, Luoyang Normal University, Luoyang 471022, P. R. China
First published on 26th July 2021
Abstract
In this study, density functional theory (DFT) and time-dependent density functional theory (TD-DFT) were used to unveil the solvation effects on the excited-state intramolecular proton transfer (ESIPT) of nitrile-substituted ortho-hydroxy-2-phenyl-oxazoline (NOHPO) molecules in three different solvents. According to the functional analysis of the reduced density gradient, hydrogen bond in low-polar solvents is stronger compared to that in high-polar solvents, indicating that proton transfer (PT) can be influenced by the polarity of the solvent. Moreover, the geometric parameters and infrared vibration spectrum of NOHPO in different types of solvents in the S0 and S1 state were compared, which confirmed the above results. By analyzing electronic spectra and frontier molecular orbitals, it was found that the spectral properties were affected by different polar solvents. Molecular electrostatic potential surface calculations proved that PT took place between the H2 atom and N3 atom, and the natural population analysis and Hirshfeld charge reveal the charge distribution after photoexcitation. To investigate the ESIPT progress intensively, the potential energy curves of NOHPO in three types of solvents were established. The findings revealed that NOHPO could transform from enol to keto form in the S1 state spontaneously, and ESIPT progress was promoted with the decrease in polarity.
Introduction
Excited-state intramolecular proton transfer (ESIPT), one of the most elementary processes in chemistry and biology, plays a crucial role in photophysics, photochemistry, and photobiology.3–6 Since Weller et al. observed a large Stokes shift in salicylic acid and found the reaction of ESIPT, ESIPT has received considerable attention from experimental and theoretical chemists.The ESIPT progress needs proper distance between the proton donor and proton acceptor to form intramolecular hydrogen bonds, so that the ESIPT reaction may occur after photoexcitation. The hydrogen bond is an important part of ESIPT; consequently, the theory of hydrogen bond enhancement in the excited state has been proposed and has proven numerous mechanisms of ESIPT.7–20 ESIPT is very common in organisms, whether it is a vision circle or the luminescence of green fluorescent protein, they are all relevant to it.21–23 Apart from natural ESIPT progress, an ESIPT unit can be synthesized chemically to mimic the optical performance of green fluorescent protein; we can even get excellent fluorescent materials with high fluorescence efficiency and narrow bandwidth.24 The most important application of ESIPT is the chemical sensor, which is designed by the ESIPT mechanism and is highly selective and highly efficient in detecting inorganic ions, such as Zn2+,25 Cu2+,26 Al3+,27 and As3+.28 Due to the characteristics of ESIPT, some researchers have reported on dual-channel chemical sensors based on ESIPT for detecting Al3+/F−,29 Zn2+/Cd2+,30 Cu2+/Fe3+,31 etc. In addition to detecting inorganic compounds, an ESIPT sensor can also detect organic compounds, and it has been widely used in vivo detection. Compared to other traditional methods (HPLC or MS), fluorometric detection has great selectivity, sensitivity, and in vivo imaging capability. Among them, chemical sensors based on ESIPT have been a widespread concern due to their ultra-fast reaction rate and large Stokes shift, which impede auto-fluorescence in imaging.32,33
Although ESIPT is widely used in chemical sensors, the application of ESIPT also faces numerous challenges. For example, the ESIPT unit may happen concurrently with nonradiative quenching processes, resulting in low emission efficiency and quantum yields. In addition, the oversized ESIPT molecule could influence metabolite traffic within cells in bioimaging.34 Therefore, it is meaningful to design an organic molecule with minimalistic and bright fluorescence for bioimaging and fluorescent sensors. Nitrile-substituted ortho-hydroxy-2-phenyl-oxazolines (NOHPOs) with extremely efficient, low molecular weight, single-benzene fluorophores, have been synthesized in the experiment.35 In the experiment, NOHPOs in different solvents have been studied, but the solvation effect on the ESIPT progress has not been discussed in detail. Therefore, the influence of the different solvent on the ESIPT progress of NOHPO should be investigated to grasp its ESIPT mechanism and develop more efficient ESIPT units.
In the present study, we will theoretically reveal the ESIPT progress of NOHPO in three different solvents: acetonitrile (ACN), dichloromethane (DCM), and cyclohexane (CH). We use DFT and TD-DFT in the S0 and S1 states to optimize all configurations. To compare the hydrogen bond strength in ACN, DCM, and CH by analyzing the infrared vibrational spectra (IR), the functional analysis of reduced density gradient (RDG) isosurfaces and scatter plots was carried out. The frontier molecular orbitals (FMOs), molecular electrostatic potential surface (MEPS), natural population analysis (NPA), and Hirshfeld charge are analyzed to research the charge distribution. More importantly, we established potential-energy curves (PECs) to further illustrate the mechanisms of ESIPT.
Computational methods
The Gaussian 16 program36 was used to perform all of the theoretical calculations reported in this study. We used DFT and TD-DFT methods with B3LYP-D3(BJ)37–42 functional and TZVP43,44 basis sets to calculate the geometric optimizations in the ground (S0) and first singlet excited state (S1). To ensure the optimized structures at minima without imaginary frequency, we also analyzed vibration frequencies. The polarizable continuum model (PCM) with the integral equation formalism variant (IEF-PCM) was used in our calculations. IEF-PCM was used in three types of solvents (ACN: ε = 35.688, DCM: ε = 8.93, CH: ε = 2.0165). The potential energy curves (PECs) were scanned in the S0 and S1 states to further investigate the ESIPT process, and the N–H bond length (0.05 Å) decreased with a constant step in the S0 state and the O–H bond length (0.05 Å) was decreased with a constant step in the S1 state.The hydrogen bond strength in numerous solvents was compared using the RDG function,45 and the results were calculated and plotted using the Multiwfn software46 and VMD program.47
Result and discussion
Geometric structures, IR vibrational spectra, RDG isosurfaces
The geometric structures of NOHPO molecules were optimized at the B3LYP-D3(BJ)/TZVP level in the S0 and S1 states, as shown in Fig. 1.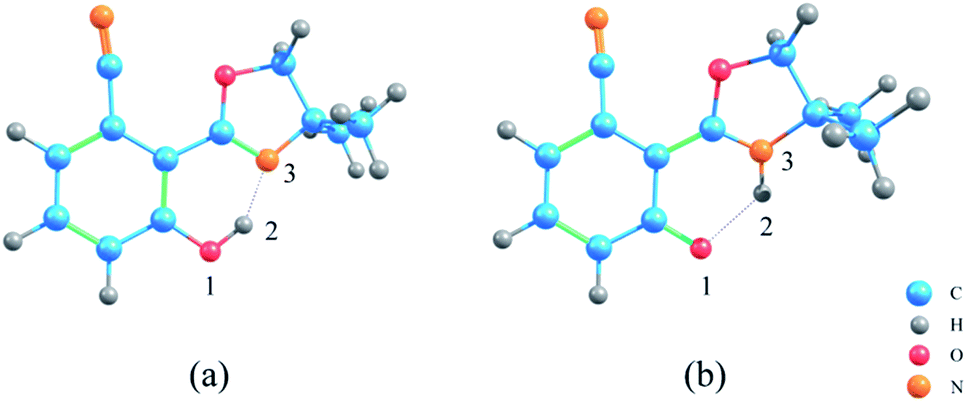 | ||
| Fig. 1 The optimized structures of NOHPO molecules: (a) S0 state and (b) S1 state. | ||
Table 1 shows the primary bond lengths and angles. The hydrogen bond of H2–N3 turns to covalent bond in the S1 state, and its bond length decreases in the trend 0.628 Å (ACN) < 0.631 Å (DCM) < 0.640 Å (CH) in three types of solvents, respectively. Therefore, the hydrogen bond is enhanced in the S1 state. The bond length of O1–H2 in ACN, DCM, and CH are extended from 1.007, 1.006 and 1.003 Å to 1.963, 1.958 and 1.940 Å, respectively. This phenomenon shows that in the S1 state, O1–H2 binding is weakened.
| ACN | DCM | CH | ||||
|---|---|---|---|---|---|---|
| S0 | S1 | S0 | S1 | S0 | S1 | |
| O1–H2 | 1.007 | 1.963 | 1.006 | 1.958 | 1.003 | 1.940 |
| H2–N3 | 1.646 | 1.018 | 1.650 | 1.019 | 1.661 | 1.021 |
| δ(O1–H2–N3) | 147.869 | 125.451 | 147.767 | 125.713 | 147.413 | 126.464 |
The IR vibration frequency analysis is a good way to analyze the strength of the hydrogen bond. In order to test the hydrogen bond strength, H2–N3 is analyzed in three types of solvents. Fig. 2 shows frequencies in ACN, DCM, and CH are 3476 cm−1, 3466 cm−1, 3432 cm−1, respectively, which indicates that the ESIPT reaction occurs more easily in low-polar solvents.
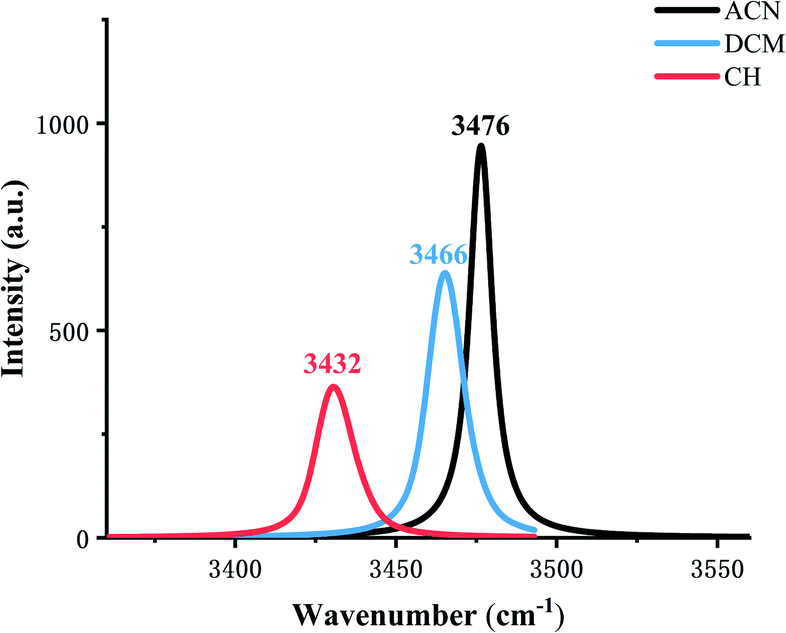 | ||
| Fig. 2 Calculated stretching vibrational frequencies of the H2–N3 bond in the S1 state in three types of solvents. | ||
In addition, the RDG function is used to distinguish different weak interactions mainly to unveil the characters of the hydrogen bonds.1,2 As shown in Fig. 3, the scatter graphs show the weak interactions and the strength of the hydrogen bond in numerous solvents. In Fig. 3(a), blue represents the hydrogen bond, green represents van der Waals interactions and red represents nonbond repulsive interaction. The bluer the isosurfaces, the stronger the hydrogen bond strength is. By comparing the color of RDG isosurfaces in Fig. 3, the hydrogen bond strength in CH is the strongest. Fig. 3(b) shows the hydrogen bond strength in the S1 state in three types of solvents. With the increase in the absolute value of sign((λ2) ρ(r)), the strength of the hydrogen bond is enhanced. As the result, the hydrogen bond strength in three types of solvents orders by CH > DCM > ACN. The above result can also prove that the intensity of the hydrogen bonds is reinforced as the polarity decreasing.
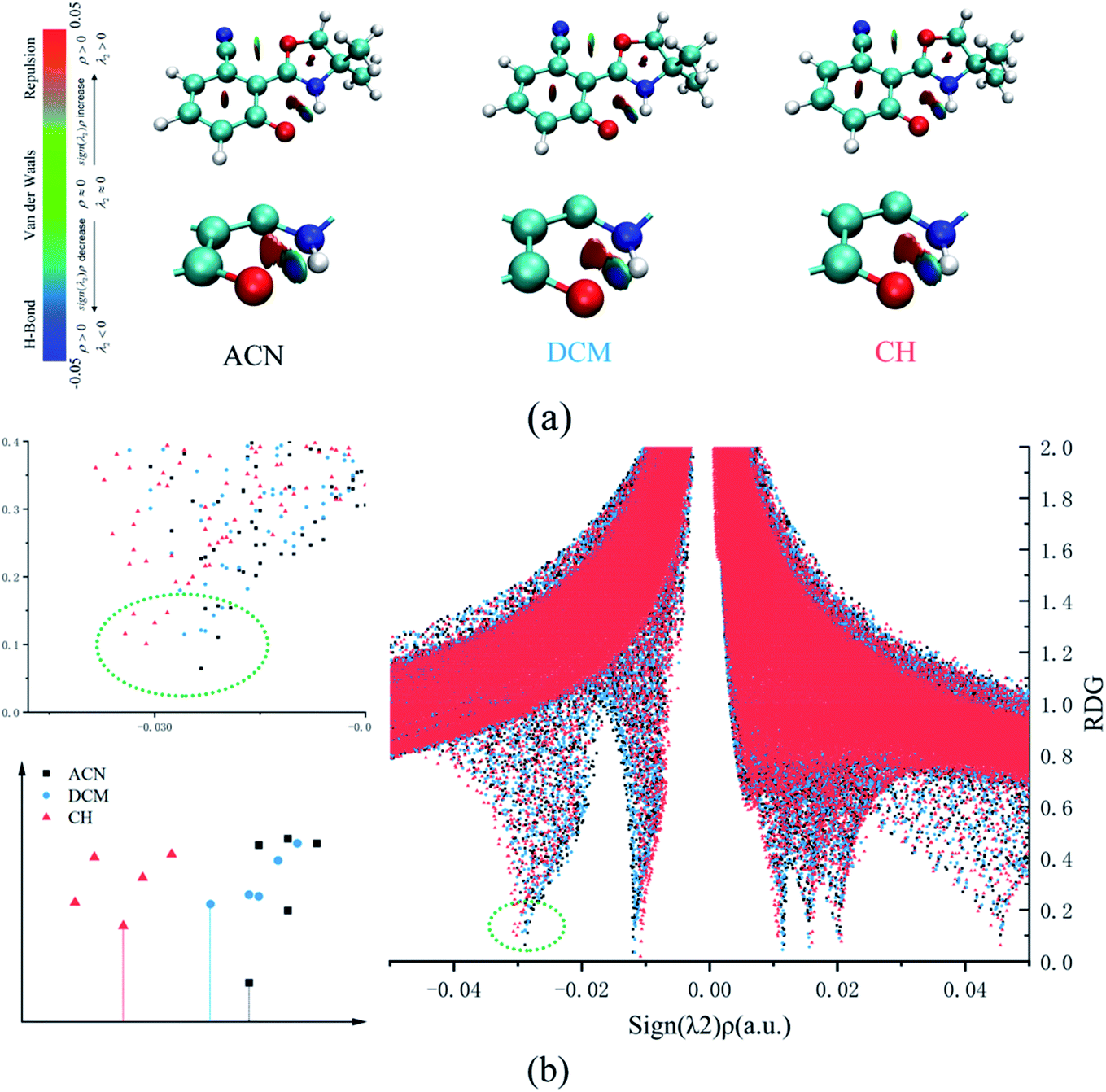 | ||
| Fig. 3 (a) The visual diagram of RDG isosurfaces and the color gradient axis. (b) Scatter graphs of the reduced density gradient (RDG(r)) versus Ω(r) in three types of solvents. | ||
Electronic spectra, FMOs, MEPS, charge distribution
Fig. 4 shows the calculated absorption and fluorescence spectra in three different solvents. The calculated absorption peaks (ACN: 315 nm, DCM: 313 nm, CH: 309 nm) and fluorescence emission peaks (ACN: 468 nm, DCM: 470 nm, CH: 482 nm) are in accord with the experimental data.35 The Stokes shift in three different solvents is 153 nm, 157 nm, and 173 nm, respectively. The calculated results show that emission is completely from keto tautomer. Furthermore, with the decrease in the polarity of the solvent, the maximum emission wavelength (from 468 nm to 482 nm) of the fluorescence spectrum exhibits a bathochromic shift and the Stokes shift enlarged. The influence of the solvent environment on the electronic configuration could cause different solvent chromic effects.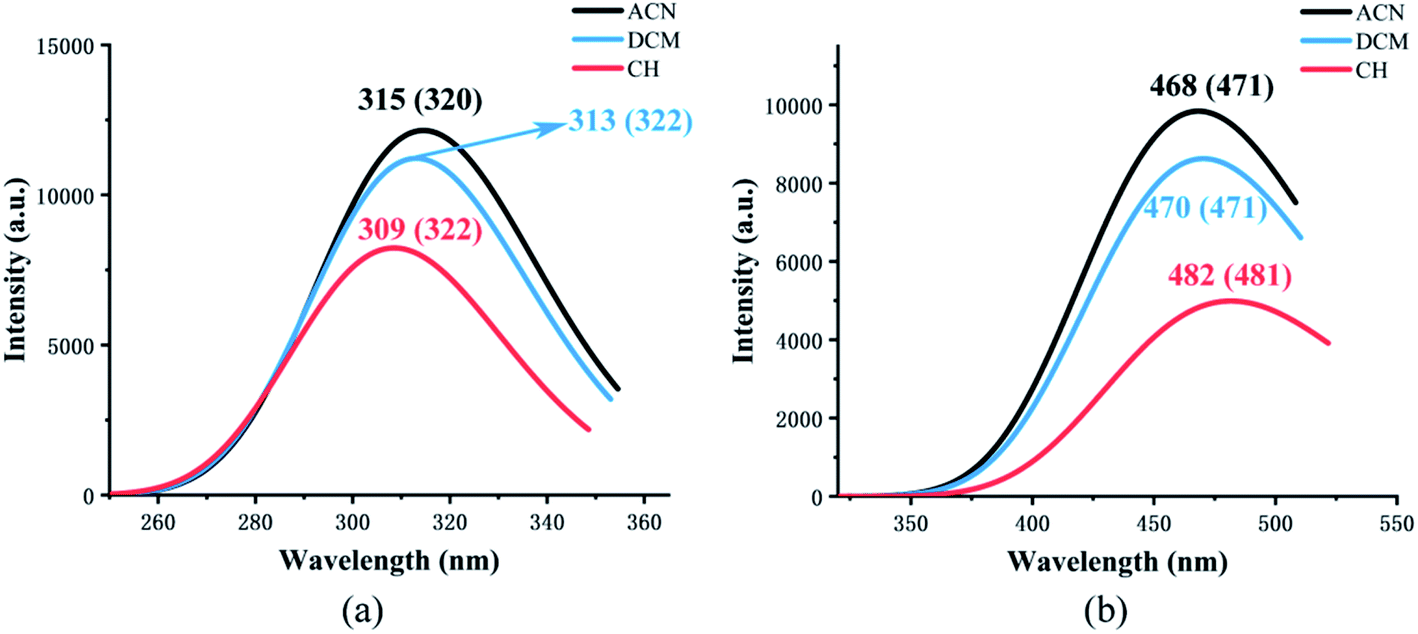 | ||
| Fig. 4 The calculated absorption (a) and fluorescence (b) wavelength of NOHPO in different kinds of solvents. The values in parentheses are experimental data.35 | ||
As is known, charge could be transferred and redistributed after photoexcitation. The molecular charge analysis provides proper evidence for the determination of what drives the proton transfer process. FMOs are studied in order to gain a better understanding of the ESIPT reaction. The results are listed in Table 2. The oscillator strength of NOHPO in the S1 state is 0.3005 (ACN), 0.2777 (DCM), and 0.2036 (CH). Fig. 5 shows the FMOs of NOHPO and the energies in the highest occupied molecular orbital (HOMO) and the lowest unoccupied molecular orbital (LUMO) in ACN, DCM and CH. In the S1 state, the orbital transition only happens from HOMO to LUMO. Hence, only HOMO and LUMO are shown in Fig. 5. The HOMO displays π characteristic and the LUMO displays π* characteristic, so the orbital composition from HOMO to LUMO (96.5% for ACN, 96.1% for DCM, and 94.7% for CH) could be assigned the dominant ππ*-type transition. Hence, the systems from S1 to S0 are more likely to get deactivated by radiation (emitting fluorescence), rather than by non-radiation. It is clear that the transition energies in ACN, DCM and CH are different, which indicates that the polarity of the solvents can affect the ESIPT progress of NOHPO.
| Transition | λ (nm eV−1) | f | OT | CI (%) | ||
|---|---|---|---|---|---|---|
| ACN | Absorption | S0 → S1 | 315/3.93 | 0.3005 | H → L | 96.5% |
| Emission | S1 → S0 | 468/2.65 | 0.2433 | L → H | 96.9% | |
| S2 → S0 | 389/3.18 | 0.0333 | L → H−1 | 95.7% | ||
| DCM | Absorption | S0 → S1 | 313/3.96 | 0.2777 | H → L | 96.1% |
| Emission | S1 → S0 | 470/2.64 | 0.2133 | L → H | 95.6% | |
| S2 → S0 | 394/3.15 | 0.0393 | L → H−1 | 94.4% | ||
| CH | Absorption | S0 → S1 | 309/4.02 | 0.2036 | H → L | 94.7% |
| Emission | S1 → S0 | 482/2.57 | 0.1234 | L → H | 88.4% | |
| L → H−1 | 11.1% | |||||
| S2 → S0 | 408/3.04 | 0.0554 | L → H−1 | 87.2% | ||
| L → H | 11.3% |
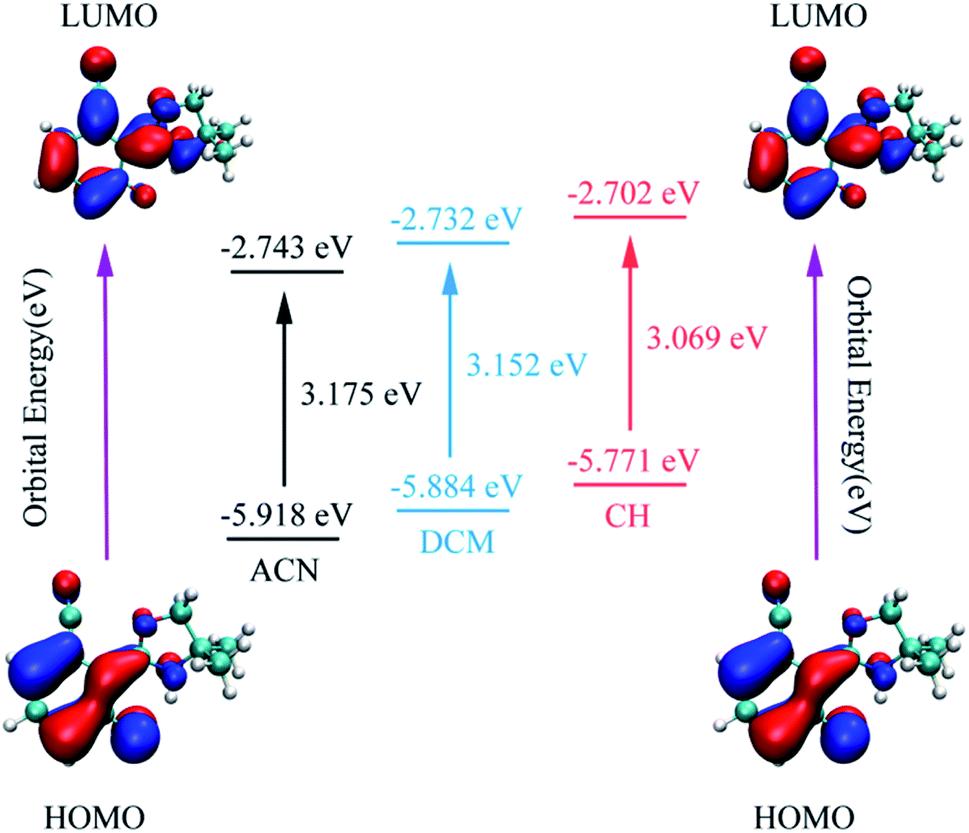 | ||
| Fig. 5 The calculated frontier molecular orbitals (HOMO and LUMO) of NOHPO in the S1 state and its transition energy. | ||
MEPS (molecular electrostatic potential surface) has been widely used to investigate the electrostatic interaction, predicting the reaction sites and properties of the molecule, particularly in the study of intramolecular or intermolecular hydrogen bonding interactions. Fig. 6 shows NOHPO molecules in the S0 state in the ACN solvent. It is clear that there is a drastic polarization distribution of negative and positive electrostatic potentials on the surface. The blue area represents the most negative electrostatic potential, and the red represents the most positive electrostatic potential. The positive electrostatic potential on H2 demonstrates its potential to donate a hydrogen bond, while the negative electrostatic potential on N3 demonstrates its potential to accept a hydrogen bond. This indicates that the proton transfer takes place between the positive electrostatic H2 atom and negative electrostatic N3 atom.
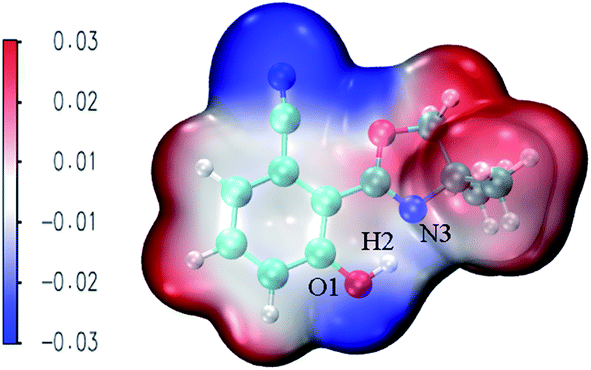 | ||
| Fig. 6 The total electron density isosurfaces mapped with MEPS48 for the NOHPO-enol form in ACN. | ||
Natural population analysis (NPA) and Hirshfeld charge are used to analysis the charge transfer quantificationally, and the results are shown in Table 3. The trend of NPA and Hirshfeld charges are the same. In three types of solvents, the Hirshfeld charges of N3 are −0.143 (ACN), −0.144 (DCM), −0.146 (CH), and O1 are −0.211 (ACN), −0.209 (DCM), −0.200 (CH), respectively, which shows that electronegativity of N3 is enhanced and that of O1 weakened with the polarity of the solvents decreasing. Therefore, as the solvent polarity decreasing, the attraction of N3 to H2 is improved, promoting the progress of ESIPT, which is the same with our above verdict.
| NPA charge | Hirshfeld charge | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| O1 | H2 | N3 | O1 | H2 | N3 | |||||||
| S0 | S1 | S0 | S1 | S0 | S1 | S0 | S1 | S0 | S1 | S0 | S1 | |
| ACN | −0.653 | −0.732 | 0.503 | 0.463 | −0.528 | −0.518 | −0.211 | −0.389 | 0.102 | 0.122 | −0.143 | −0.048 |
| DCM | −0.651 | −0.727 | 0.504 | 0.463 | −0.528 | −0.521 | −0.209 | −0.385 | 0.102 | 0.121 | −0.144 | −0.050 |
| CH | −0.641 | −0.710 | 0.504 | 0.463 | −0.530 | −0.530 | −0.200 | −0.369 | 0.104 | 0.119 | −0.146 | −0.057 |
Potential-energy curves
The PECs of NOHPO in the S0 and S1 states are established to unveil the effects of different solvents on the progress of ESIPT. PECs are shown in Fig. 7. The S0 state has a lower relative energy than that in the S1 state, indicating that NOHPO is unstable in the S1 state. The reaction potential energy of ESIPT in the S1 state is 17.74 kcal mol−1 in ACN, 17.86 kcal mol−1 in DCM, and 18.44 kcal mol−1 in CH, as shown in Fig. 7. With photoexcitation, H2 can transfer to N3 easily, so that the NOHPO molecule can transform from enol tautomer to keto tautomer. This also indicates that low-polar solvents facilitate the reaction of proton transfer. Proton transfer reaction barriers in the S0 state are 4.34 kcal mol−1 (ACN), 4.61 kcal mol−1 (DCM), and 5.48 kcal mol−1 (CH), respectively. NOHPO molecules can be transformed from enol tautomer to keto tautomer due to the low reaction barrier of proton transfer. However, the unstable keto tautomer, whose energy is high, would return to a stable enol tautomer.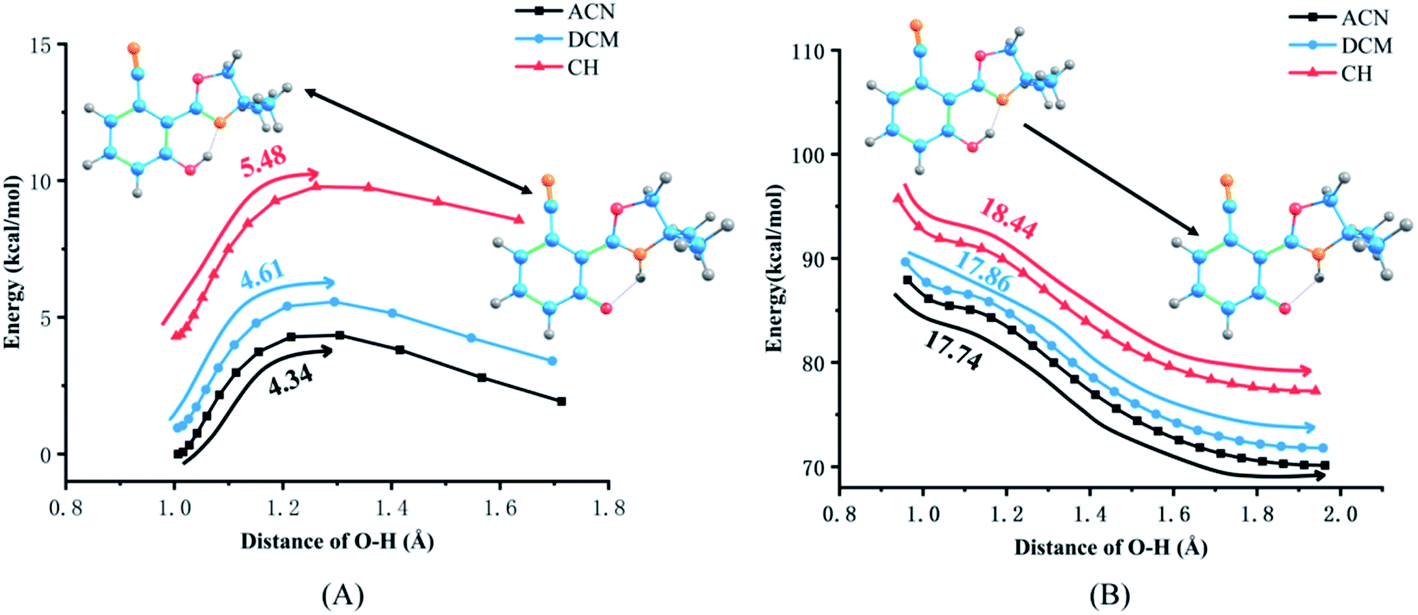 | ||
| Fig. 7 Potential energy curves of the S0 (A) and S1 (B) states of NOHPO as a function of the O1–H2 bond length in ACN, DCM, CH. | ||
In summary, the progress of ESIPT can generalize that NOHPO molecules can be excited to the S1 state upon photoexcitation. In the S1 state, all NOHPO molecules transform to tautomeric structures via ESIPT, which when back to the S0 state via radiation transition finally turn back to the normal structure spontaneously.
Conclusion
To sum up, the solvation effect and the ESIPT mechanism in three different solvents are illustrated by employing DFT and TD-DFT methods with the IEFPCM solvation model. Related structure parameters and PECs imply that the hydrogen bond is greatly strengthened in the S1 state, so that H2 can transfer to N3 easily, and the hydrogen bond in H2–N3 transforms into a covalent bond. The results of electronic spectra show that the maximum emission wavelength of the fluorescence spectrum has a bathochromic shift as the polarity decreases, so the range of emission wavelength can be regulated by solvents. In addition, the IR vibration spectrum, RDG analysis, charge distribution and PECs imply that ESIPT are favorable as the polarity decreases. Thus, the ESIPT of NOHPO in the different solvents is in the order of CH > DCM > ACN. The solvation effect is proved, and the relevance between the hydrogen bond and ESIPT in numerous solvents are also investigated.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the Open Project of SKLMRD (the open fund of the State Key Laboratory of Molecular Reaction Dynamics in DICP, CAS).References
- J. Zhao, H. Dong, H. Yang and Y. Zheng, Org. Chem. Front., 2018, 5, 2710–2718 RSC.
- X. Luo, Y. Yang and Y. Li, J. Mol. Liq., 2020, 319, 114145 CrossRef CAS.
- W.-M. Kwok, C. Ma and D. L. Phillips, J. Am. Chem. Soc., 2008, 130, 5131–5139 CrossRef CAS PubMed.
- T. Schultz, E. Samoylova, W. Radloff, I. V. Hertel, A. L. Sobolewski and W. Domcke, Science, 2004, 306, 1765–1768 CrossRef CAS PubMed.
- M. Rini, Science, 2003, 301, 349–352 CrossRef CAS PubMed.
- D. Sicinska, D. G. Truhlar and P. Paneth, J. Am. Chem. Soc., 2001, 123, 7683–7686 CrossRef CAS PubMed.
- Y. Qi, Z. Tang, H. Zhan, Y. Wang, Y. Zhao, X. Fei, J. Tian, L. Yu and J. Liu, Spectrochim. Acta Mol. Biomol. Spectrosc., 2020, 224, 117359 CrossRef CAS PubMed.
- Z. Tang, Y. Qi, Y. Wang, P. Zhou, J. Tian and X. Fei, J. Phys. Chem. B, 2018, 122, 3988–3995 CrossRef CAS PubMed.
- W. Xu, S. Liu, H. Sun, X. Zhao, Q. Zhao, S. Sun, S. Cheng, T. Ma, L. Zhou and W. Huang, J. Mater. Chem., 2011, 21, 7572 RSC.
- G.-J. Zhao and K.-L. Han, Acc. Chem. Res., 2012, 45, 404–413 CrossRef CAS PubMed.
- G.-J. Zhao, R.-K. Chen, M.-T. Sun, J.-Y. Liu, G.-Y. Li, Y.-L. Gao, K.-L. Han, X.-C. Yang and L. Sun, Chem. –Eur. J., 2008, 14, 6935–6947 CrossRef CAS PubMed.
- G.-J. Zhao, J.-Y. Liu, L.-C. Zhou and K.-L. Han, J. Phys. Chem. B, 2007, 111, 8940–8945 CrossRef CAS PubMed.
- Z. Tang, Y. Wang, D. Bao, M. Lv, Y. Yang, J. Tian and L. Dong, J. Phys. Chem. A, 2017, 121, 8807–8814 CrossRef CAS PubMed.
- Z. Tang, Y. Yang, Y. Yang, Y. Wang, J. Tian and X. Fei, J. Lumin., 2018, 194, 785–790 CrossRef CAS.
- P. Zhou and K. Han, Acc. Chem. Res., 2018, 51, 1681–1690 CrossRef CAS PubMed.
- X. Li, Y. Guo and D. Yang, Theor. Chem. Acc., 2020, 139, 181 Search PubMed.
- Y. Yang, W. Shi, Y. Chen, F. Ma and Y. Li, J. Lumin., 2021, 229, 117698 CrossRef CAS.
- L. Jia and Y. Liu, Spectrochim. Acta Mol. Biomol. Spectrosc., 2020, 242, 118719 CrossRef CAS PubMed.
- A. Shahraki, A. Ebrahimi, S. Rezazadeh and R. Behazin, Mol. Syst. Des. Eng., 2021, 6, 66–79 RSC.
- G. Zhao, Y. Yang, C. Zhang, Y. Song and Y. Li, J. Lumin., 2021, 230, 117741 CrossRef CAS.
- C.-C. Hsieh, P.-T. Chou, C.-W. Shih, W.-T. Chuang, M.-W. Chung, J. Lee and T. Joo, J. Am. Chem. Soc., 2011, 133, 2932–2943 CrossRef CAS PubMed.
- T. Chatterjee, M. Mandal, A. Das, K. Bhattacharyya, A. Datta and P. K. Mandal, J. Phys. Chem. B, 2016, 120, 3503–3510 CrossRef CAS PubMed.
- G. Cui, Z. Lan and W. Thiel, J. Am. Chem. Soc., 2012, 134, 1662–1672 CrossRef CAS PubMed.
- Y.-H. Hsu, Y.-A. Chen, H.-W. Tseng, Z. Zhang, J.-Y. Shen, W.-T. Chuang, T.-C. Lin, C.-S. Lee, W.-Y. Hung, B.-C. Hong, S.-H. Liu and P.-T. Chou, J. Am. Chem. Soc., 2014, 136, 11805–11812 CrossRef CAS PubMed.
- S. Janakipriya, S. Tamilmani and S. Thennarasu, RSC Adv., 2016, 6, 71496–71500 RSC.
- S. Liu, M. Qin, Q. Lu, L. Lin, C.-K. Wang, J. Fan and Y. Song, Spectrochim. Acta Mol. Biomol. Spectrosc., 2021, 254, 119685 CrossRef CAS PubMed.
- O. Alici and D. Aydin, J. Photochem. Photobiol. Chem., 2021, 404, 112876 CrossRef CAS.
- Y. Cheng, S. Wang, J. Zhang, J. Cao and Y. Qu, J. Mol. Struct., 2020, 1221, 128824 CrossRef CAS.
- G. Kumar, I. Singh, R. Goel, K. Paul and V. Luxami, Spectrochim. Acta Mol. Biomol. Spectrosc., 2021, 247, 119112 CrossRef CAS PubMed.
- S. Paul and P. Banerjee, Sens. Actuators, B, 2021, 329, 129172 CrossRef CAS.
- R. Kaushik, R. Sakla, N. Kumar, A. Ghosh, V. D. Ghule and D. A. Jose, Sens. Actuators, B, 2021, 328, 129026 CrossRef CAS.
- K. Anusuyadevi, S. P. Wu and S. Velmathi, J. Photochem. Photobiol. Chem., 2020, 403, 112875 CrossRef CAS.
- P. Zhang, Y. Xiao, Q. Zhang, Z. Zhang, H. Yu and C. Ding, New J. Chem., 2019, 43, 7620–7627 RSC.
- S. Benson, A. Fernandez, N. D. Barth, F. de Moliner, M. H. Horrocks, C. S. Herrington, J. L. Abad, A. Delgado, L. Kelly, Z. Chang, Y. Feng, M. Nishiura, Y. Hori, K. Kikuchi and M. Vendrell, Angew. Chem., Int. Ed., 2019, 58, 6911–6915 CrossRef CAS PubMed.
- D. Gobel, D. Duvinage, T. Stauch and B. J. Nachtsheim, J. Mater. Chem. C, 2020, 14 Search PubMed.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, X. Li, M. Caricato, A. V. Marenich, J. Bloino, B. G. Janesko, R. Gomperts, B. Mennucci, H. P. Hratchian, J. V. Ortiz, A. F. Izmaylov, J. L. Sonnenberg, D. Williams-Young, F. Ding, F. Lipparini, F. Egidi, J. Goings, B. Peng, A. Petrone, T. Henderson, D. Ranasinghe, V. G. Zakrzewski, J. Gao, N. Rega, G. Zheng, W. Liang, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell, J. A. Montgomery Jr., J. E. Peralta, F. Ogliaro, M. J. Bearpark, J. J. Heyd, E. N. Brothers, K. N. Kudin, V. N. Staroverov, T. A. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. P. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, J. M. Millam, M. Klene, C. Adamo, R. Cammi, J. W. Ochterski, R. L. Martin, K. Morokuma, O. Farkas, J. B. Foresman and D. J. Fox, Gaussian 16, Revision C.01, Wallingford CT, 2016 Search PubMed.
- C. Lee, W. Yang and R. G. Parr, Phys. Rev. B: Condens. Matter Mater. Phys., 1988, 37, 785–789 CrossRef CAS PubMed.
- B. Miehlich, A. Savin, H. Stoll and H. Preuss, Chem. Phys. Lett., 1989, 157, 200–206 CrossRef CAS.
- E. Cancès, B. Mennucci and J. Tomasi, J. Chem. Phys., 1997, 107, 3032–3041 CrossRef.
- S. Miertuš, E. Scrocco and J. Tomasi, Chem. Phys., 1981, 55, 117–129 CrossRef.
- B. Mennucci, E. Cancès and J. Tomasi, J. Phys. Chem. B, 1997, 101, 10506–10517 CrossRef CAS.
- R. Cammi and J. Tomasi, J. Comput. Chem., 1995, 16, 1449–1458 CrossRef CAS.
- A. Schäfer, C. Huber and R. Ahlrichs, J. Chem. Phys., 1994, 100, 5829–5835 CrossRef.
- A. Schäfer, H. Horn and R. Ahlrichs, J. Chem. Phys., 1992, 97, 2571–2577 CrossRef.
- E. R. Johnson, S. Keinan, P. Mori-Sánchez, J. Contreras-García, A. J. Cohen and W. Yang, J. Am. Chem. Soc., 2010, 132, 6498–6506 CrossRef CAS PubMed.
- T. Lu and F. Chen, J. Comput. Chem., 2012, 33, 580–592 CrossRef CAS PubMed.
- W. Humphrey, A. Dalke and K. Schulten, J. Mol. Graph., 1996, 14, 33–38 CrossRef CAS PubMed.
- T. Lu and F. Chen, J. Mol. Graph. Model., 2012, 38, 314–323 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2021 |