
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceCoumarin–carbazole based functionalized pyrazolines: synthesis, characterization, anticancer investigation and molecular docking†
Mrugesh Patel a,
Nilesh Pandey*c,
Jignesh Timaniyaa,
Paranjay Parikha,
Alex Chauhanb,
Neeraj Jain*b and
Kaushal Patel*a
a,
Nilesh Pandey*c,
Jignesh Timaniyaa,
Paranjay Parikha,
Alex Chauhanb,
Neeraj Jain*b and
Kaushal Patel*a
aDepartment of Advanced Organic Chemistry, P. D. Patel Institute of Applied Sciences, Charotar University of Science and Technology, Gujarat 388421, India. E-mail: kaus_chem@yahoo.com
bDepartment of Biological Sciences, P. D. Patel Institute of Applied Sciences, Charotar University of Science and Technology, Gujarat 388421, India. E-mail: neerajjain.as@charusat.ac.in
cDepartment of Medical Laboratory Technology, Charotar Institute of Paramedical Sciences, Charotar University of Science and Technology, Gujarat 388421, India. E-mail: nileshpandey.cips@charusat.ac.in
First published on 13th August 2021
Abstract
A series of novel pyrazoline scaffolds from coumarin–carbazole chalcones were synthesized. We explored various acetyl, amide, and phenyl substituents at the N-1 position of the pyrazoline core. The synthesized compounds were characterized by FTIR, 1H-NMR, 13C-NMR, DEPT, and mass spectroscopic techniques. The in vitro cytotoxicity study of all the synthesized compounds was evaluated against HeLa, NCI-H520 and NRK-52E cell lines. Compounds 4a and 7b became the most active compounds and exhibited their potential to arrest the cell cycle progression and induce apoptosis in both the cell lines. In addition, molecular docking studies revealed a higher binding affinity of both the molecules with CDK2 protein. Based on the obtained results, a comprehensive analysis is warranted to establish the role of compounds 4a and 7b as promising cancer therapeutic agents.
1. Introduction
Cancer has been one of the major causes of human mortality, and the approximately 10 million deaths in 2020 are predicted to reach up to 16.3 million by 2040.1 Challenges faced concerning cancer treatment include fatal side effects, low selectivity and solubility, adverse drug interactions, and resistance against varied therapeutic agents. That has led to continual research to identify newer compounds and modify current drugs for safe and effective cancer treatment. In this context, natural products have taken a lead which continues to advance until clinical trials wherein most of the new anticancer drugs are structurally modified/optimized natural compounds.2 Generally, the natural products are heterocyclic compounds, i.e., they contain heteroatoms in their skeleton that suit well as anticancer drugs either by affecting tubulin polymerization (vincristine and vinblastine) or inhibiting kinase activity (midostaurin), etc. However, many of these natural compounds also come with the risk mentioned above. Therefore, there is an exigent need to explore modified natural compound mimics as potential anticancer drug candidates.Coumarins are naturally occurring oxygen-containing heterocyclic compounds produced by bacteria, fungi, and numerous plant species like Umbelliferae, Asteraceae, Rutaceae, Leguminosae, etc.3,4 Nearly 1300 coumarin derivatives have been identified as secondary metabolites from the same sources. Coumarin compounds have been shown to exert potential anti-microbial, antiviral, anti-tuberculosis, antioxidant, anticancer effects.5–9 Anticancer properties of coumarin derivative osthole have been potent against hepatocellular carcinoma. The in vivo and in vitro studies showed that osthole induces apoptosis via inhibiting the Akt/NF-κB pathway.10 A coumarin based drug candidate (RKS262) exerted remarkable potency in vivo and in vitro on ovarian cancer cell line OVCAR-3.11 A strategically developed coumarin–monastrol hybrid selectively imparted its anticancer potential over breast cancer cell lines MCF-7 and MDA-MB-231.12 Coumarin derivatives are used to treat prostate cancer, renal cell carcinoma, and leukemia. They have also been reported to inhibit breast and cervical cancer proliferation and malignant melanoma cell lines.13
Like coumarin, carbazole is also a tricyclic nitrogen-containing heterocyclic compound found in various plant species and natural sources. Carbazoles have been shown to possess antibacterial, antifungal, anti-inflammatory, and anticancer activities.14–17 Carbazole derivative Clausenawalline F revealed potent cytotoxicity on lung cancer cell line NCI-H187.18 An arylsulfonyl N-substituted carbazole derivative exhibited potent inhibition over pancreatic cell lines PANC-1 and Capan-2, and preliminary in vivo efficacy studies on mice revealed no side effects.19 Further, carbazole derivatives Ellipticine, alectinib, and midostaurin are used to treat metastatic breast cancer, ALK-positive advanced non-small-cell lung cancer, and acute myeloid leukemia, respectively, as well as reported to show good cytotoxic activity on various cancer cell lines.20
Pyrazoles are another group of heterocyclic compounds from the family of azole group compounds. Azole family members possess compounds that contain a nitrogen atom with at least one other hetero atom as part of a five-membered ring, such as thiazoles, oxazoles, etc. These compounds contain diverse pharmacological and physiological activities.21–24 A reduced form of pyrazole, 2-pyrazoline, has been reported to exert potent antimicrobial,25 anti-inflammatory,26 anti analgesic,26 antioxidant, and anti-cancer27 activities. Several pyrazole-containing drugs have been serving in clinical trials for years, and many are under current research.28 One such pyrazole-containing drug candidate, Crizotinib, demonstrated modest multikinase inhibitory activity in non-small-cell lung carcinoma patients harboring MET exon 14 mutation29 and exhibited antitumor activity in advanced non-small-cell lung cancer patients with ROS1-rearrangement.30 For patients with ALK-positive non-small-cell lung cancer. Similarly, in non-small-cell lung cancer patients with ALK mutations, another pyrazole-containing drug candidate, Lorlatinib, showed antitumor activity in the global phase II trial.31 Additionally, phase I and II study of Zanubrutinib, a pyrazole consisting of drug candidates, revealed anticancer activity in mantle cell lymphoma, a multikinase inhibitor.32 Moreover, pyrazole-containing drug Pazopanib is known to inhibit angiogenesis by blocking tyrosine kinase activity.33 Thus, most coumarin, carbazole, and pyrazole derivatives impart their cytotoxicity via inhibiting kinases that are vital drug target molecules in cancer therapy.
In terms of clinical efficacy, conventional single-target therapy has revealed pharmacokinetic limitations leading to insufficient control on malignancy. Nonetheless, these limitations have opened the gates of research on multi-target hybrid drug therapy to effectively kill cancer cells with minimal side effects. The hybridization of two or more moieties in a single molecule provides a potent hybrid pharmacophore to develop newer anticancer compounds.34
Considering this fact, we merged three potential pharmacophores, coumarin, carbazole, and pyrazole, into one compound to augment the individual molecule's potential for anticancer activity. The merger of two pharmacophores was performed by making chalcone of two moieties. Although there are many ways by which the merger of two pharmacophores is possible but making chalcone of both the moieties is one of the efficient and more applicable methods for the conjunction of coumarin and carbazole by reacting with appropriate hydrazines in ethanol using a catalytic amount of acetic acid.
In the present study, 4a–c, 5a–c, 6a–c, 7a–c, and 8a–c compounds were investigated for cytotoxicity followed by cell cycle and apoptotic analysis in cancer cell lines HeLa and NCI-H520 to assess anticancer activities. Furthermore, in silico molecular docking, it was carried out to predict binding affinity between active compounds and cyclin-dependent kinase 2 (CDK2).
2. Results and discussion
2.1. Chemistry
The target compounds (3a–c) were obtained using the synthetic procedure and the substitution pattern depicted in Scheme 1. The first precursor's acetyl derivatives of coumarins were synthesized according to the reported procedure,35,36 and 9-ethyl-9H-carbazole-3-carbaldehyde was synthesized by initial ethylation of carbazole by the reaction of bromoethane and potassium hydroxide in acetone; the ethylated product was formulated using Vilsmeier–Haack reaction.37 Both the precursor further consumed for the synthesis of chalcone.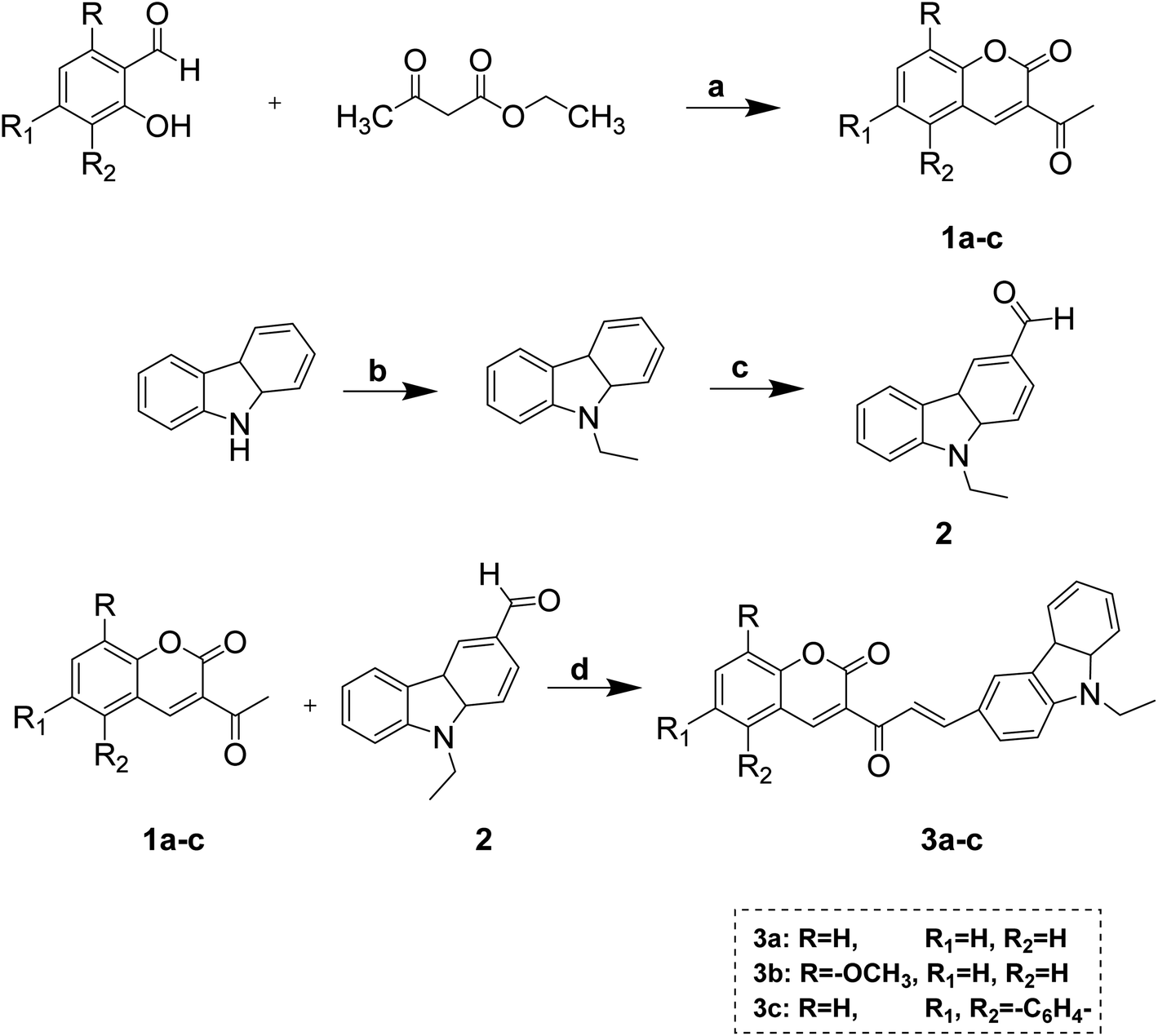 | ||
| Scheme 1 Synthesis of acetyl-coumarin, formyl-N-ethyl carbazole, and coumarin–carbazole chalcone. Reagents and conditions: (a) catalytic amount of piperidine, 70–80 °C, 40 min, (b) C2H5Br/KOH, acetone, r.t., (c) DMF/POCl3, (d) catalytic amount of piperidine, ethanol, 65 °C, 2.5–3 h. | ||
Coumarin–carbazole chalcones (3a–c) were obtained by reacting 3-acetyl coumarins derivatives, 6-formyl carbazole, and piperidine in ethanol. Pyrazoline derivatives (4a–c, 5a–c, 6a–c, 7a–c) were obtained by reacting chalcone (3a–c) with appropriate hydrazines in ethanol using a catalytic amount of acetic acid shown in Scheme 2.
 | ||
| Scheme 2 Synthesis of pyrazoline substituted coumarin–carbazole derivatives. Reagents and conditions: (a) appropriate hydrazine hydrate derivatives, ethanol, a catalytic amount of acetic acid, 65 °C, 1.5–3 h. | ||
The plausible mechanism for pyrazolines synthesis is illustrated in Scheme 3. The mechanism proceeded in two ways: path 1 involved an initial attack of nucleophilic secondary amine-nitrogen of substituted hydrazine on the carbon–carbon double bond of chalcone to form an intermediate underwent cyclization and rearrangement to afford pyrazoline. Path 2 involved an initial attack of nucleophilic terminal nitrogen of substituted hydrazine on the carbonyl carbon to form an intermediate that underwent a second nucleophilic attack of imine nitrogen resulting in cyclization and after rearrangement afforded a pyrazoline ring.
 | ||
| Scheme 3 Plausible reaction mechanism. | ||
However, acetyl derivatives of pyrazolines (8a–c) were obtained by reacting chalcones (3a–c), hydrazine hydrate, acetic acid in ethanol shown in Scheme 4.
 | ||
| Scheme 4 Synthesis of N-acetyl pyrazoline substituted coumarin–carbazole derivatives. Reagents and conditions: (a) hydrazine hydrate, acetic acid, ethanol, 65 °C, 1.5–3 h. | ||
In this study, the Claisen–Schmidt condensation was preferred for the synthesis of chalcones, and these chalcone derivatives were further reacted with hydrazines to obtain the final targeted pyrazolines. The purity of compounds was established by thin-layer chromatography (TLC), and purification was performed using column chromatography.
2.2. Characterization
All the synthesized compounds were characterized using spectral techniques such as IR, 1H NMR, 13C NMR, DEPT, MS, and elemental analysis. The compounds were then tested for their cytotoxicity over cancer cell lines HeLa and NCI-H520, and normal cell lines NRK-52E. The best performing hybrids were further investigated for cell cycle progress, apoptosis-inducing effects, morphological changes, and molecular docking studies.The spectral analysis data confirmed the synthesized compounds 3a–3c, 4a–4c, 5a–5c, 6a–6c, 7a–7c and 8a–8c. The ring closure reaction of chalcones established the FTIR spectra of final pyrazoline compounds. Infrared spectra revealed two characteristic bands absorption for N–H stretching between 3200–3500 cm−1. Absorption around 1729 cm−1 related to C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretching of δ-lactone of coumarin and absorption between 1600–1680 cm−1 was observed due to stretching of CO of chalcone, amide, and acetyl functional groups of molecules; the band appears for carbonyl of chalcone in this region disappears after the formation of pyrazoline from chalcone which can be seen in pyrazolines which do not possess acetyl and amide functionalities. Stretching bands appear between 1592–1628 cm−1 indicates the conversion of chalcone to pyrazoline as CN bond formation. Chalcone 3a showed bands in infrared region at 3072 cm−1 for aromatic C–H stretching, 2972, 2929 & 2868 cm−1 for aliphatic C–H stretching, 1729 cm−1 for CO stretching of δ-lactone of coumarin, 1677 cm−1 for CO stretching of chalcone, 1569 & 1470 cm−1 for CC stretching. Pyrazoline compound 4a showed bands at 1730 and 1666 cm−1 for CO of coumarin carbonyl and amide carbonyl, respectively. The band at 1605 cm−1 corresponds to –CN confirms the formation of –CN bond and the pyrazine –CN– bonds, while the bands at 1572 and 1467 cm−1 are due to the –CC– bond. The bands at 1133 to 1231 cm−1 were qualified to the (–C–N) vibrations, confirming the pyrazoline ring cyclization in all compounds. A band at 3344 and 3297 cm−1 owing to –NH2 of amide derivative was observed.
O stretching of δ-lactone of coumarin and absorption between 1600–1680 cm−1 was observed due to stretching of CO of chalcone, amide, and acetyl functional groups of molecules; the band appears for carbonyl of chalcone in this region disappears after the formation of pyrazoline from chalcone which can be seen in pyrazolines which do not possess acetyl and amide functionalities. Stretching bands appear between 1592–1628 cm−1 indicates the conversion of chalcone to pyrazoline as CN bond formation. Chalcone 3a showed bands in infrared region at 3072 cm−1 for aromatic C–H stretching, 2972, 2929 & 2868 cm−1 for aliphatic C–H stretching, 1729 cm−1 for CO stretching of δ-lactone of coumarin, 1677 cm−1 for CO stretching of chalcone, 1569 & 1470 cm−1 for CC stretching. Pyrazoline compound 4a showed bands at 1730 and 1666 cm−1 for CO of coumarin carbonyl and amide carbonyl, respectively. The band at 1605 cm−1 corresponds to –CN confirms the formation of –CN bond and the pyrazine –CN– bonds, while the bands at 1572 and 1467 cm−1 are due to the –CC– bond. The bands at 1133 to 1231 cm−1 were qualified to the (–C–N) vibrations, confirming the pyrazoline ring cyclization in all compounds. A band at 3344 and 3297 cm−1 owing to –NH2 of amide derivative was observed.
1H NMR resonance of compounds 3a–c, 4a–c, 5a–c, 6a–c, 7a–c, and 8a–c showed the signal for aliphatic protons of N-ethyl carbazole that appeared between 1.29–1.48 δ ppm for –CH3 as a triplet and 4.31–4.41 δ ppm for –CH2 as a quartet. The –CH2 protons of the pyrazoline ring resonated as a pair of a doublet at 3.38–3.57 (Ha) δ ppm and 3.98–4.25 (Hb) δ ppm. The –CH proton of the pyrazoline ring appeared as a doublet of doublets at 5.50–5.78 (Hx) δ ppm due to vicinal coupling to non-equivalent protons Ha and Hb. In compounds 4a–4c, a broad singlet was observed between 5.40–6.72 δ ppm due to N–H protons of amide-functionality of the pyrazoline ring. Similarly, in compounds 8a–8c, a singlet was observed between 2.34–2.44 δ ppm for –CH3 of acetyl derivatives. All the aromatic protons gave a signal between 6.74–9.49 δ ppm. 1H NMR spectra confirmed the structures of chalcones 3a–3c. In compound 3a, a peak at 1.45 δ ppm appeared as a triplet for –CH3 of N–CH2–CH3, and a peak at 4.37 ppm appeared as a quartet for –CH2– of N–CH2–CH3. The multiplet was observed between 7.26–7.99 δ ppm, a triplet for two aromatic protons observed at 8.12 ppm, a doublet appeared for one proton at 8.38 ppm, and a singlet appeared for one proton at 8.58 ppm for aromatic proton. The 1H NMR spectra of compound 4a showed a pattern of protons of the pyrazoline ring caused by the three hydrogen atoms coupling (Ha, Hb and Hx) 2-pyrazoline ring at 3.50 ppm, dd 4.05 ppm, and dd 5.73 ppm, respectively. The existence of methylene protons (Ha and Hb) as a doublet of doublets postulates the magnetic non-equivalence of these two protons. The alkyl protons (–CH3 and –CH2) appear at the downfield at 1.40 ppm and 4.33 ppm due to carbazole's nitrogen deshielding effect. The two-broad singlet of NH2 group proton for compound 4a was observed at 5.40 ppm. These spectral data unequivocally proved the 2-pyrazoline structure.
13C NMR spectra of all the synthesized pyrazolines showed all carbonyl group signals between 157.63–159.85 δ ppm. The carbonyl of chalcone appears around 186 δ ppm, generally disappears after generation of pyrazoline ring over chalcone functionality. All the aromatic carbons appeared between 108.37–155.88 δ ppm. The 13C NMR spectra of the compounds provided the final carbon skeleton of pyrazolines. The value of the chemical shift of (–CH3) of carbazole carbons appears around 13.79 ppm, while in (–CH2) of carbazole appears around 45.21 ppm. The chemical shifts around 37.52 ppm, 61.03 ppm are due to (–CH2) and (–CHx) of carbons to the pyrazoline; it was observed that CHx signals generally appears around 60 ppm, but in the case of compounds 5a–c, 6a–c, 7a–c due to the presence of pendant phenyl ring that signal shifts towards 65 ppm. The 13C NMR spectra of chalcone 3a give chemical shift of (–CH3) of carbazole carbons appears at 13.85 ppm, while in (–CH2) of carbazole appears at 37.81 ppm. The aromatic ring carbons of compound 3a appear in their desired aromatic region 108–147.56 δ ppm. The peaks at 186.20 ppm and 159.46 ppm are of the CO of chalcone and CO of coumarin carbons. The aromatic ring carbons of pyrazoline derivative 4a appear in their desired aromatic region. The peaks at 158.85 ppm and 155.18 ppm are of the CO of coumarin and CO of amide carbons. Mass spectral analysis of compounds indicated peaks concerning the molecular weight of compounds.
2.3. Biology
Cytotoxicity was expressed as the mean IC50 of three independent experiments (Table 1). All the synthesized compounds exhibited cytotoxicity with IC50 values in the range of 9.13–141.8 μM. The majority of synthesized compounds were less active on NRK-52E than the other two cancer cell lines. The chalcone derivatives 3a–3c exhibited low inhibition over all the cell lines as compared to pyrazolines. Two of the all pyrazoline scaffolds, N-amide pyrazoline 4a and m-chloro substituted N-phenyl pyrazoline 7b, exhibited remarkable IC50 at 12.59, 11.36 μM respectively against HeLa and 11.26, 9.13 μM respectively against NCI-H520. Compound 7b also exhibits an IC50 of 24.16 μM over NRK-52E cell lines. Compounds 4b and 7c, which contained a methoxy group on the 8th position of coumarin, showed good activity against HeLa at 29.82, 29.04 μM, respectively, and NCI-H520 22.48, 20.83 μM concentrations, respectively. The rest of the compounds exhibited moderate to low activity on all three cell lines.
| Compounds | IC50, μM | ||
|---|---|---|---|
| HeLa | NCI–H520 | NRK-52E | |
| 3a | 131.6 ± 5.16 | 119.86 ± 3.78 | 94.60 ± 3.97 |
| 3b | 118.7 ± 6.88 | 103.24 ± 4.78 | 115.30 ± 7.23 |
| 3c | 141.8 ± 7.77 | 132.66 ± 4.75 | 82.67 ± 1.87 |
| 4a | 12.59 ± 0.10 | 11.26 ± 0.45 | 28.37 ± 1.29 |
| 4b | 29.82 ± 1.85 | 22.48 ± 2.15 | 40.32 ± 1.92 |
| 4c | 43.48 ± 1.02 | 55.61 ± 1.93 | 59.05 ± 0.91 |
| 5a | 92.50 ± 4.25 | 81.24 ± 1.46 | 102.96 ± 1.65 |
| 5b | 39.03 ± 0.46 | 62.70 ± 1.63 | 70.35 ± 3.68 |
| 5c | 55.00 ± 0.96 | 83.57 ± 1.20 | 121.70 ± 7.03 |
| 6a | 67.03 ± 1.87 | 38.31 ± 0.78 | 83.24 ± 5.14 |
| 6b | 30.80 ± 0.50 | 30.01 ± 0.33 | 51.99 ± 4.27 |
| 6c | 57.51 ± 1.32 | 61.67 ± 1.48 | 79.92 ± 5.10 |
| 7a | 33.44 ± 0.64 | 14.31 ± 0.21 | 40.98 ± 3.80 |
| 7b | 11.36 ± 0.24 | 9.13 ± 0.08 | 24.16 ± 1.73 |
| 7c | 29.04 ± 0.84 | 20.83 ± 0.27 | 57.11 ± 3.59 |
| 8a | 43.18 ± 0.52 | 62.46 ± 1.95 | 116.26 ± 6.95 |
| 8b | 76.78 ± 1.45 | 54.72 ± 0.65 | 97.92 ± 4.99 |
| 8c | 55.85 ± 1.30 | 77.61 ± 0.34 | 81.27 ± 3.77 |
| Cis-platin | 7.75 ± 0.42 | 10.41 ± 1.35 | 12.93 ± 0.40 |
| 5-Fluorouracil | 55.72 ± 2.10 | 8.36 ± 0.45 | 46.68 ± 3.79 |
 | ||
| Fig. 1 Effect of compounds 4a and 7b on the cell cycle of cervical cancer cells HeLa and its graphical presentation. Data are represented mean ± SE. p < 0.05 was considered statistically significant compared to the untreated cells. | ||
 | ||
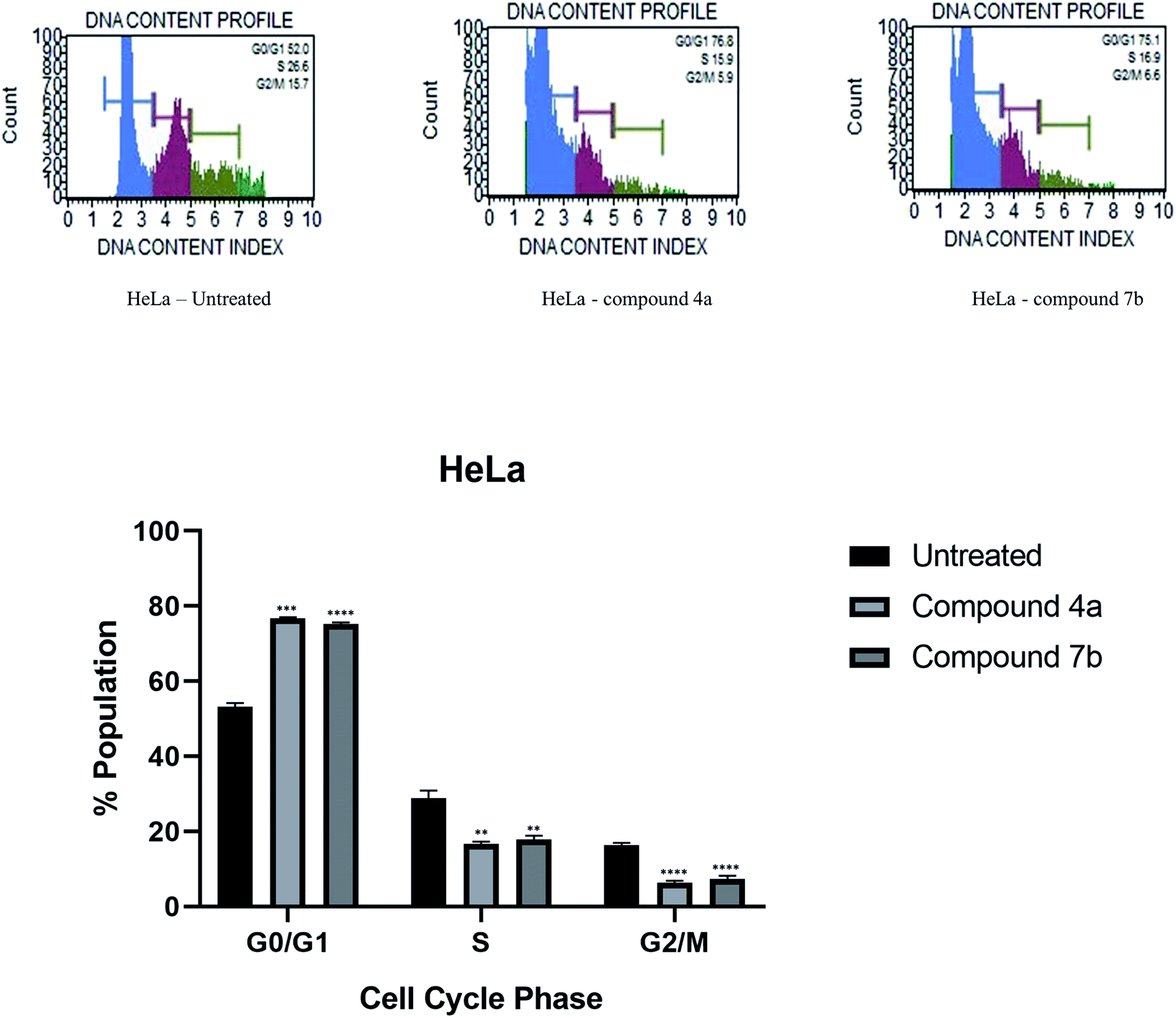
| Fig. 2 Effect of compounds 4a and 7b on the cell cycle of lung cancer cells NCI-H520 and its graphical presentation. Data are represented mean ± SE. p < 0.05 was considered statistically significant compared to the untreated cells. | ||
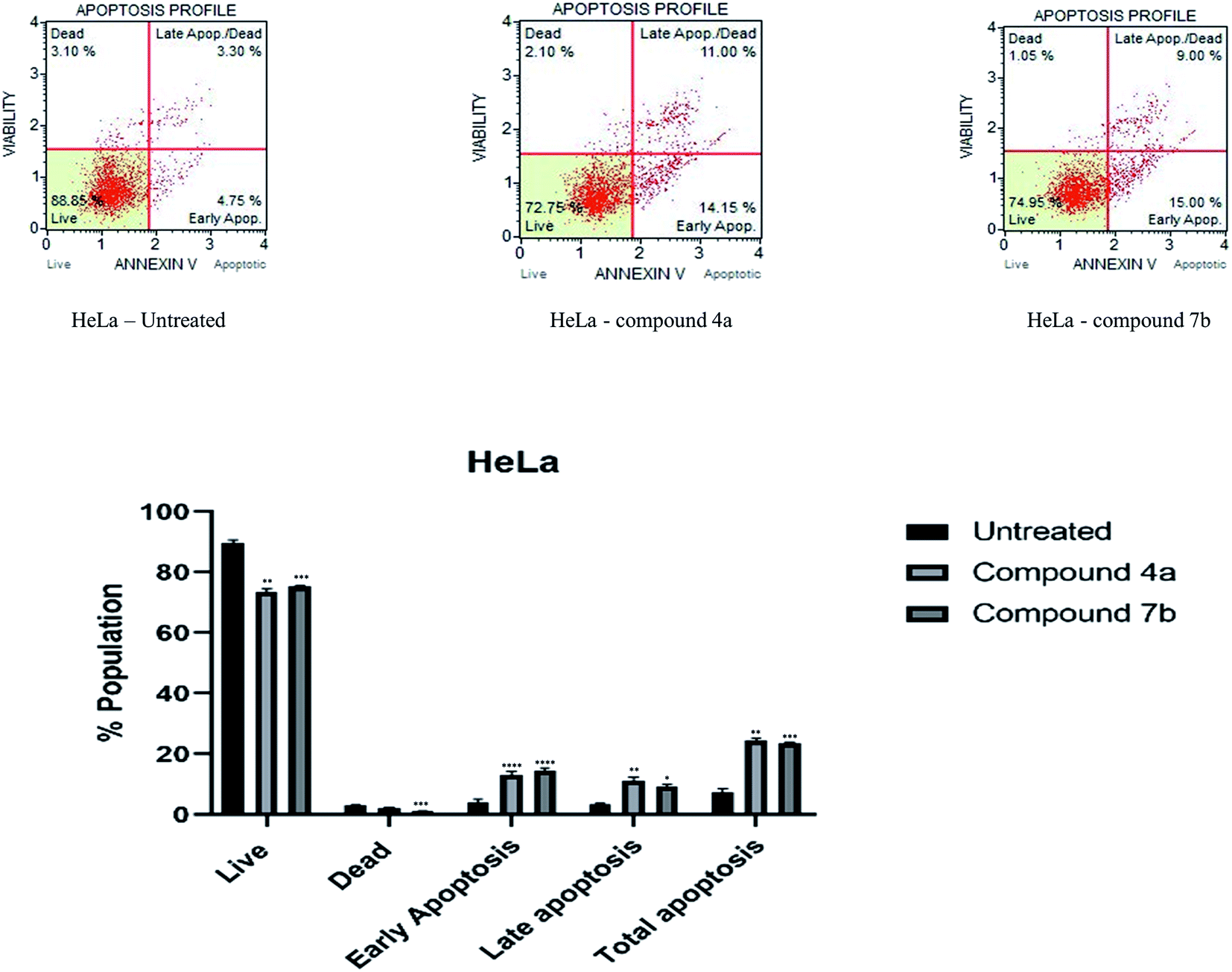 | ||
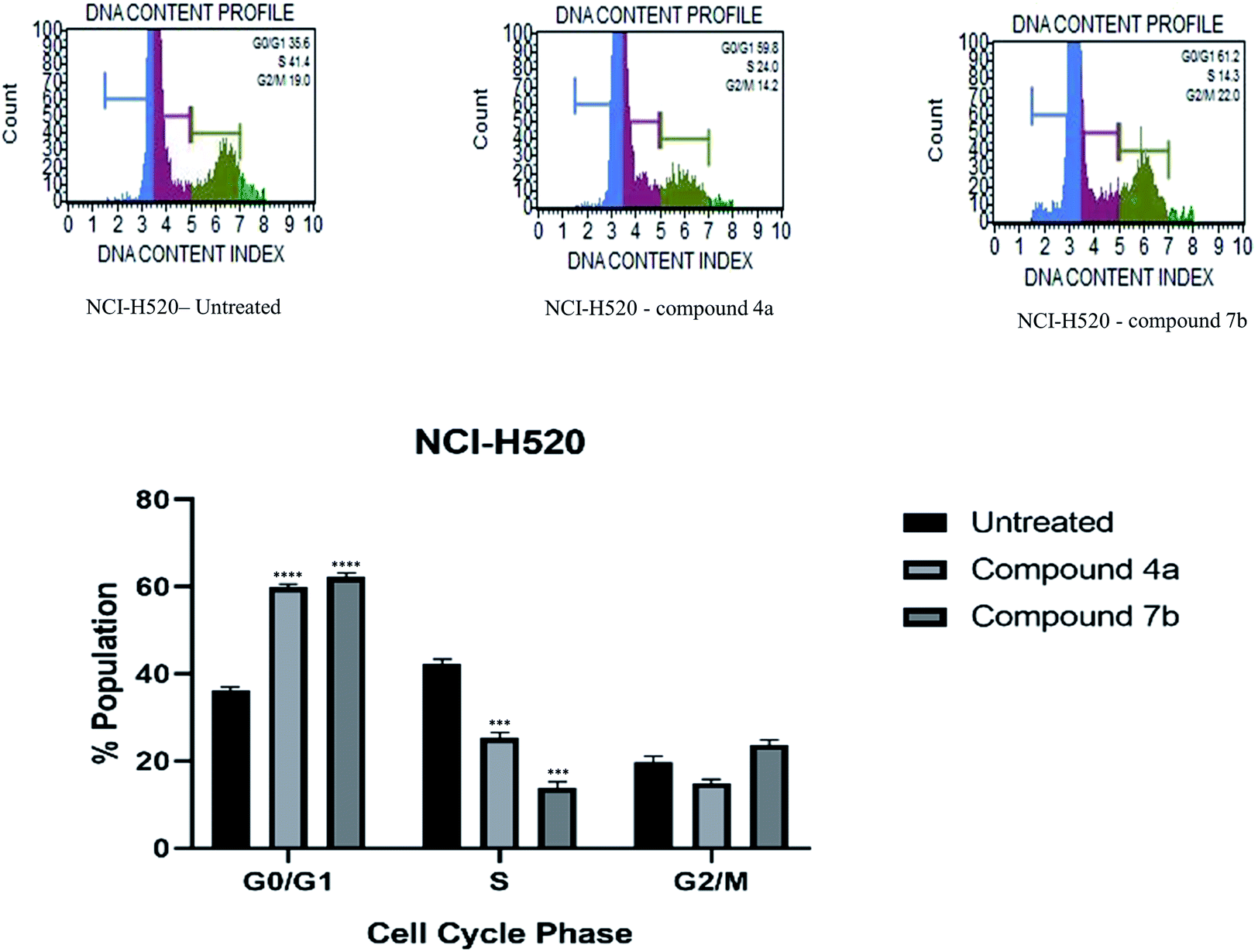
| Fig. 3 Apoptosis of HeLa cells induced by compounds 4a and 7b and its graphical presentation. Data are represented mean ± SE. p < 0.05 is considered statistically significant compared to the untreated cells. | ||
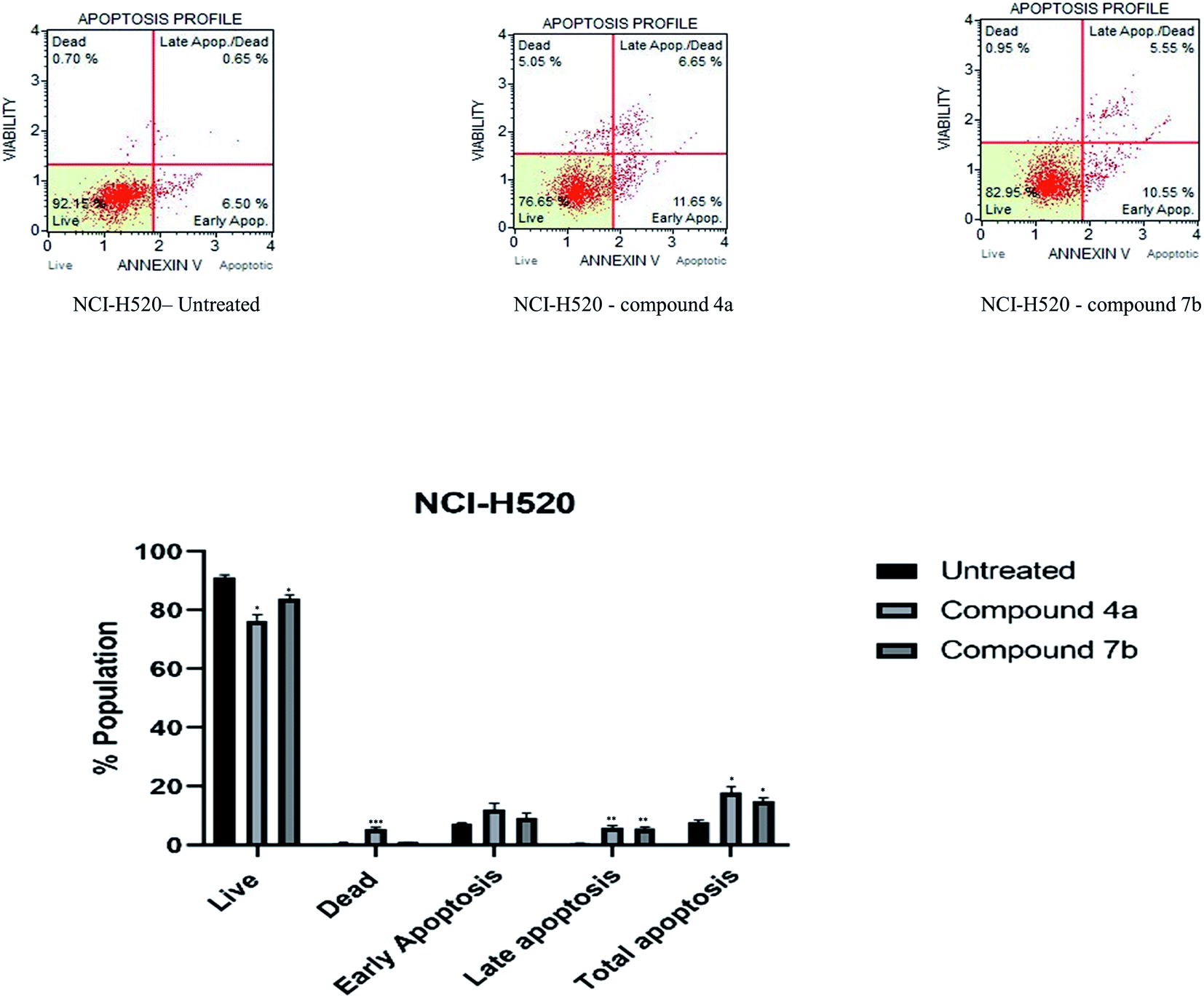 | ||
| Fig. 4 Apoptosis of NCI-H520 cells induced by compounds 4a and 7b and its graphical presentation. Data are represented mean ± SE. p < 0.05 is considered statistically significant compared to the untreated cells. | ||
 | ||
| Fig. 5 Nuclear morphologic changes were observed after 24 h treatment of compounds 4a and 7b on HeLa and NCI-H520 cells. | ||
2.3.5.1. Interaction with CDK2. The docking analysis revealed a higher binding affinity −11.5 kcal mol−1 and −11.9 kcal mol−1 of molecules 4a and 7b, respectively, at the active site of CDK2. The interaction was compared with the roscovitine (PDB ID: 2A4L). Concerning 4a, Ile10 of CDK2 formed a hydrogen bond (2.58 Å) with the amide group. A novel hydrogen bonding was observed where Thr14 of CDK2 was involved in two hydrogen bonds (3.08 Å, 2.38 Å) with the two oxygen present in the coumarin ring. The other bonds showing bond length between 3 and 4 Å in the interaction were (i) a pi-alkyl bond (3.90 Å) between Val18 and the aromatic ring of carbazole and (ii) the carbon–hydrogen bond (3.35 Å) between Gly11 and oxygen of an amide linkage. Several common amino acids such as Leu134, Lys33, Leu83, Phe80, Asp145, and Lys89 also showed interaction with 4a (Fig. 6a); however, the interaction type was not similar to standard roscovitine.41 The analysis of 7b interaction with CDK2 also revealed the formation of two hydrogen bonds (2.65 Å, 2.28 Å). The other interactions with ≈4 Å bond length were: Pi anion (3.96 Å) between Asp86 and substituted phenyl ring of pyrazoline, Pi sigma (3.91) formation of Val18 with the aromatic ring of carbazole, carbon–hydrogen bond (3.21 Å) between Gly13 and oxygen of carbonyl of coumarin, Leu134 forming Pi-alkyl bond (4.01 Å) with pendant methylene of carbazole attached with nitrogen, Lys89 forming pi-alkyl bond (4.04 Å) with chlorine of substituted pyrazoline of phenyl and Met115 forming pi alkyl bond (4.2 Å) with lactone ring of coumarin. Comparing the binding of 7b with PDB ID: 2A4L following interacting amino acids Tyr108, Ala149, Val156, Phe112, Leu137, and Asp111 (Fig. 6b) were detected to be common. An additional hydrogen bond was formed with the Thr14 at the active site signifying its potential inhibitor. The interaction of 4a and 7b with the active site of CDK2 depict its potential as an inhibitor, which needs to be validated through laboratory experimentation.
 | ||
| Fig. 6 (a) In silico docking model of CDK2 protein with the ligand, 4a shows 2D and 3D images generated with Biovia Discovery Studio Visualizer. (b) In silico docking model of CDK2 protein with the ligand, 7b shows 2D and 3D images generated by Biovia Discovery Studio Visualizer. | ||
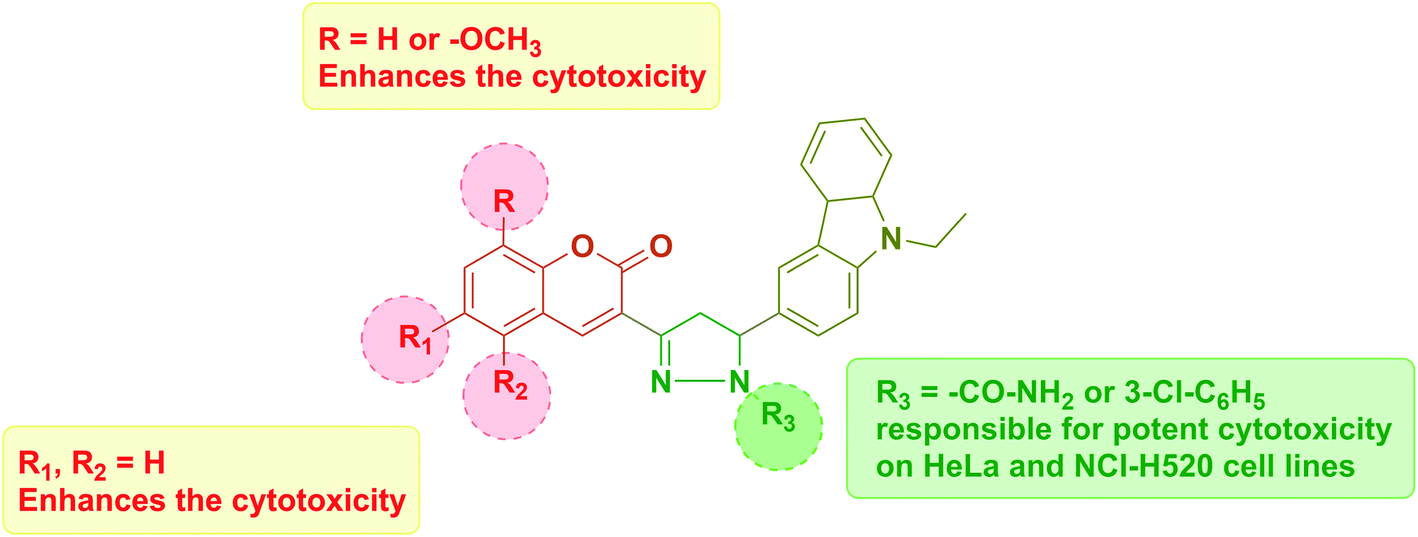 | ||
| Fig. 7 Structural activity relationship (SAR). | ||
3. Conclusion
In summary, this investigation represents an efficient mild conditioned route for the synthesis of novel coumarin–carbazole containing pyrazoline derivatives. This final hybrid structure includes many pharmacophores in single molecules that may result in varied structural diversity. Compounds 4a and 7b possessing N-amide pyrazoline and m-chloro substituted N-phenyl pyrazoline were most potent against cancer cell lines HeLa and NCI-H520, whereas moderately active on normal rat epithelial NRK-52E cell line. The excellent cytotoxic potency of both the compounds was also investigated and found to potentially arrest the cell cycle phase in the G0/G1 phase and induce apoptosis in both the cell lines. The molecular docking study of both the potent candidates on CDK2 rationalized their inhibition with high docking score. We expect these active functions to endow molecules to contribute to the broader range of applications. This study provides valuable information for structurally designing and developing potent coumarin hybrids as promising cancer therapeutic agents.4. Materials and methods
4.1. Chemistry
Reagents and solvents were obtained from commercial sources and used without further purification. All melting points were determined by using Precision Digital Melting/Boiling Point Apparatus by VEEGO (Model: Vmp-D/Ds). Thin-layer chromatography (TLC, aluminium plates coated with silica gel 60 F254, 0.25 mm thickness, Merck) was used for monitoring the progress of all reactions, purity, and homogeneity of the synthesized compounds. FT-IR spectra were recorded using potassium bromide disc on Nicolet 6700 FT-IR spectrophotometer, and only the characteristic peaks are reported. Elemental analysis was carried out on Perkin-Elmer 2400 Series II CHNS/O Analyzer. In addition, 1H-NMR and 13C-NMR spectra were recorded using DMSO-d6 and CDCl3 as a solvent on a Bruker Avance spectrometer at a 400 MHz frequency 100 MHz, respectively, using TMS as an internal standard. The splitting pattern of NMR signals is as singlet (s), doublet (d), doublet of doublets (dd), doublet of doublets of doublets (ddd), triplet (t), quartet (q), multiplet (m). The coupling constant (J) is given in Hertz (Hz). Mass spectra were documented on Shimadzu QP2010 Spectrometer.4.1.1.1. General synthetic procedure for compounds 3a–c. A mixture of 6-formyl, 9-ethyl carbazole (2) (3 mmol), various 3-acetyl coumarin (1a–c) (3 mmol), and piperidine (0.3 mmol) in 30 mL of ethanol was stirred at 70 °C for 45–60 min. Then, the mixture was filtered, and the precipitated chalcone product was washed with cold ethanol.
4.1.1.2. (E)-3-(3-(9-Ethyl-9H-carbazol-3-yl)acryloyl)-2H-chromen-2-one (3a). Yield: 85%; mp 194–196 °C (lit42); yellow solid; IR spectrum, νmax, cm−1: 3072 (aromatic C–H stretching), 2972, 2929 and 2868 (aliphatic C–H stretching), 1729 (C
O stretching of δ-lactone of coumarin), 1677 (CO stretching of chalcone), 1569 & 1470 (CC); 1H NMR (CDCl3) δ, ppm (J, Hz): 1.45 (3H, t, J = 7.2 Hz, CH3 of carbazole), 4.37 (2H, q, J = 7.2 Hz, CH2 of carbazole), 7.29 (1H, t, J = 7.2 Hz, Ar-H), 7.33 (1H, t, J = 7.6 Hz, Ar-H), 7.40 (3H, t, J = 7.2 Hz, Ar-H), 7.49 (1H, t, J = 7.2 Hz, Ar-H), 7.61–7.66 (2H, m, Ar-H), 7.83 (1H, dd, J = 1.6 Hz and 7.2 Hz, Ar-H), 7.96–7.99 (1H, m, Ar-H), 8.12 (2H, t, J = 8.0 Hz, Ar-H), 8.38 (1H, d, J = 1.2 Hz, Ar-H), 8.58 (1H, s, Ar-H); 13C NMR (CDCl3): δ (ppm) 186.20(CO of chalcone), 159.46(CO of coumarin), 155.17(C), 147.56(Ar-CH), 147.05(AR-CH), 141.76(C), 140.49(C), 133.92(CH), 129.91(CH), 126.88(CH), 126.35(CH), 125.96(C), 125.85(C), 124.88(CH), 123.50(C), 122.93(C), 122.43(CH), 120.87(CH), 120.75(CH), 119.83(CH), 118.72(C), 116.66(CH), 108.91(CH), 37.81(CH2 of carbazole), 13.85(CH3 of carbazole). Anal. calcd for C26H19NO3: elemental analysis: C, 79.37; H, 4.87; N, 3.56%. Found: C, 79.39; H, 4.86; N, 3.52%.
4.1.1.3. (E)-3-(3-(9-Ethyl-9H-carbazol-3-yl)acryloyl)-8-methoxy-2H-chromen-2-one (3b). Yield: 82%; mp 245–246 °C (lit42); yellow solid; IR spectrum, νmax, cm−1: 3056 (aromatic C–H stretching), 2971, 2929 & 2838 (aliphatic C–H stretching), 1729 (C
O stretching of δ-lactone of coumarin), 1651 (CO stretching of chalcone), 1573 & 1471 (CC); 1H NMR (CDCl3) δ, ppm (J, Hz): 1.46 (3H, t, J = 7.2 Hz, CH3 of carbazole), 4.38 (2H, q, J = 7.2 Hz, CH2 of carbazole), 4.0 (3H, s, –OCH3 of coumarin), 7.17 (1H, dd, J = 7.2 Hz and 2.0 Hz, Ar-H), 7.23–7.30 (3H, m, Ar-H), 7.42 (2H, dd, J = 8.0 Hz and 2.8 Hz), 7.50 (1H, t, J = 7.2 Hz, Ar-H), 7.84 (1H, dd, J = 7.2 Hz and 1.6 Hz, Ar-H), 7.98 (1H, d, J = 15.6 Hz, Ar-H), 8.13 (2H, t, J = 7.2 Hz, Ar-H), 8.39 (1H, d, J = 0.8 Hz, Ar-H), 8.57 (1H, s, Ar-H); 13C NMR (CDCl3): δ (ppm) 186.30(CO of chalcone), 158.94(CO of coumarin), 147.79(CH), 147.09(CH), 147.07(C), 144.90(C), 141.76(C), 140.49(C), 126.90(CH), 126.33(CH), 126.04(C), 125.98(C), 124.71(CH), 123.49(C), 122.95(C), 122.50(CH), 121.13(CH), 120.91(CH), 120.77(CH), 119.82(CH), 119.36(C), 115.56(CH), 108.90(CH), 56.40(–OCH3 of coumarin), 37.82(CH2 of carbazole), 13.85(CH3 of carbazole). Anal. calcd for C27H21NO4: elemental analysis: C, 76.58; H, 5.00; N, 3.31%. Found: C, 76.58; H, 4.98; N, 3.31%.
4.1.1.4. (E)-2-(3-(9-Ethyl-9H-carbazol-3-yl)acryloyl)-3H-benzo[f]chromen-3-one (3c). Yield: 87%; mp 240–243 °C (lit42); orange solid; IR spectrum, νmax, cm−1: 3052 (aromatic C–H stretching), 2970, 2929, 2890 (aliphatic C–H stretching), 1723 (C
O stretching of δ-lactone of coumarin), 1654 (CO stretching of chalcone), 1572 & 1468 (CC); 1H NMR (CDCl3) δ, ppm (J, Hz): 1.45 (3H, t, J = 7.2 Hz, CH3 of carbazole), 4.36 (2H, q, J = 7.2 Hz, CH2 of carbazole), 7.28 (1H, t, J = 7.2 Hz, Ar-H), 7.40–7.43 (2H, m, Ar-H), 7.48–7.51 (2H, m, Ar-H), 7.60 (1H, t, J = 7.2 Hz, Ar-H), 7.74 (1H, ddd, J = 8.0 Hz, 6.4 Hz, 1.2 Hz, Ar-H), 7.86 (1H, dd, J = 7.2 Hz and 1.6 Hz, Ar-H), 7.91 (1H, d, J = 8.0 Hz, Ar-H), 8.07 (1H, d, J = 9.2 Hz, Ar-H), 8.14 (3H, m, Ar-H), 8.40 (2H, d, J = 8.0 Hz, Ar-H), 9.40 (1H, s, Ar-H); 13C NMR (CDCl3): δ (ppm) 186.13 (CO of chalcone), 159.66(CO of coumarin), 155.88(C), 146.94(CH), 143.57(CH), 141.72(C), 140.47(C), 135.78(CH), 130.27(C), 129.84(C), 129.17(CH), 129.07(CH), 126.90(CH), 126.54(CH), 126.32(CH), 126.10(C), 123.86(C), 123.49(C), 122.95(C), 122.46(CH), 121.90(CH), 120.97(CH), 120.77(CH), 119.81(CH), 116.56(CH), 113.26(C), 108.89(CH), 37.80(CH2 of carbazole), 13.85(CH3 of carbazole). Anal. calcd for C30H21NO3: elemental analysis: C, 81.25; H, 4.77; N, 3.16%. Found: C, 81.22; H, 4.76; N, 3.15%.*One carbon appears equivalent in 3a, 3b, and 3c compounds.
4.1.1.5. General synthetic procedure for compounds 4a–c, 5a–c, 6a–c, 7a–c. A mixture of various chalcone (3a–c) (3 mmol), various hydrazines such as semicarbazide, phenylhydrazine, 4-methyl phenylhydrazine and 3-chloro phenylhydrazine (3 mmol), and acetic acid (0.3 mmol) in 30 mL of ethanol was stirred at 65 °C for 90–180 min. It was then poured in water (75 mL), and the crude solid obtained was extracted with chloroform (3 × 50 mL). The organic layer was washed with 10% sodium bicarbonate solution (50 mL), water (50 mL), and dried over anhydrous sodium sulfate. Distillation of chloroform in vacuum gave gummy material subjected to column chromatography using petroleum ether (60–80 °C)
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :CHCl3 (5:5) as an eluent to afford final products 4a–c, 5a–c, 6a–c, 7a–c.
:CHCl3 (5:5) as an eluent to afford final products 4a–c, 5a–c, 6a–c, 7a–c.
4.1.1.6. 5-(9-Ethyl-9H-carbazol-3-yl)-3-(2-oxo-2H-chromen-3-yl)-4,5-dihydro-1H-pyrazole-1-carboxamide (4a). Yield: 71%; mp 152–154 °C; yellow solid; IR spectrum, νmax, cm−1: 3448 & 3277 (amine stretching), 3048 (aromatic C–H stretching), 2973, 2929 (aliphatic C–H stretching), 1730 (C
O stretching of δ-lactone of coumarin), 1666 (CO stretching of amide), 1605 (CN), 1572 & 1467 (CC), 1231 & 1133 (C–N); 1H NMR (CDCl3) δ, ppm (J, Hz): 1.39 (3H, t, J = 7.2 Hz, –CH3 of carbazole), 3.50 (1H, dd, J = 13.6 Hz and 5.2 Hz, Ha of pyrazoline), 4.05 (1H, dd, J = 12.0 and 6.4 Hz, Hb of pyrazoline), 4.33 (2H, q, J = 7.2 Hz, –CH2 of carbazole), 5.40 (2H, broad singlet, –NH2 of pyrazoline ring), 5.72 (1H, dd, J = 8.0 Hz and 5.4 Hz, Hx of pyrazoline), 7.18 (1H, t, J = 7.2 Hz, Ar-H), 7.25–7.38 (5H, m, Ar-H), 7.43 (1H, td, J = 7.6 Hz and 1.2 Hz, Ar-H), 7.54–7.59 (2H, m, Ar-H), 7.96 (1H, s, Ar-H), 8.05 (1H, d, J = 7.6 Hz, Ar-H), 8.32 (1H, s, Ar-H); 13C NMR (CDCl3): δ (ppm) 158.85(CO of coumarin), 155.18(CO of amide), 153.89(C), 148.30(C), 140.50(CH), 140.27(C), 139.45(C), 132.93(C), 132.56(CH), 128.63(CH), 125.70(CH), 124.82(CH), 123.33(CH), 121.12(C), 122.78(C), 120.55(CH), 119.70(C), 118.85(C), 118.73(CH), 117.49(CH), 116.52(CH), 108.81(CH), 108.43(CH), 61.03(CHx of pyrazoline), 45.21(CH2 of pyrazoline), 37.52(CH2 of carbazole), 13.79(CH3 of carbazole). Anal. calcd for C27H22N4O3: elemental analysis: C, 71.99; H, 4.92; N, 12.44%. Found: C, 71.97; H, 4.93; N, 12.47%. MS m/z: 473.2 (M + 23), LCMS: 451.3 (M + 1).
4.1.1.7. 5-(9-Ethyl-9H-carbazol-3-yl)-3-(8-methoxy-2-oxo-2H-chromen-3-yl)-4,5-dihydro-1H-pyrazole-1-carboxamide (4b). Yield: 75%; mp 169–172 °C; light yellow solid; IR spectrum, νmax, cm−1: 3490 & 3379 (amine stretching), 3051 (aromatic C–H stretching), 2971 & 2931 (aliphatic C–H stretching), 1709 (C
O stretching of δ-lactone of coumarin), 1687 (CO stretching of amide), 1603 (CN), 1576 & 1480 (CC), 1135 & 1230 (C–N); 1H NMR (DMSO-d6) δ, ppm: 1.35 (3H, t, J = 7.2 Hz, CH3 of carbazole), 3.38 (1H, d, J = 6.0 Hz, Ha protons of pyrazoline), 3.94 (3H, s, –OCH3 of coumarin), 4.02 (1H, d, J = 9.6 Hz, Hb protons of pyrazoline), 4.40 (2H, q, J = 6.8 Hz, CH2 of carbazole) 5.61 (1H, dd, J = 8.0 Hz and 5.6 Hz, Hx protons of pyrazoline), 6.46 (2H, broad singlet, –NH2 of pyrazoline ring), 7.16 (1H, t, J = 7.2 Hz, Ar-H), 7.25 (2H, d, J = 6.0 Hz, Ar-H), 7.27–7.51 (3H, m, Ar-H), 7.94 (1H, s, Ar-H), 8.06 (1H, d, J = 7.6 Hz, Ar-H), 8.16 (2H, s, Ar-H), 8.65 (1H, s, Ar-H); 13C NMR (DMSO-d6): δ (ppm) 157.93(CO of coumarin), 154.75(CO of amide), 146.67(C), 146.30(C), 142.71(C), 140.88(CH), 139.74(C), 138.71(C), 133.88(C), 125.51(CH), 124.59(CH), 123.29(CH), 122.08(C), 122.00(C), 120.05(CH), 119.92(CH), 119.56(C), 119.36(C), 118.49(CH), 117.12(CH), 114.41(CH), 108.81(CH), 108.68(CH), 60.37(CHx of pyrazoline), 55.92(–OCH3 of coumarin), 44.71(CH2 of pyrazoline), 36.98(CH2 of carbazole), 13.84(CH3 of carbazole). Anal. calcd For C28H24N4O4: elemental analysis: C, 69.99; H, 5.03; N, 11.66%. Found: C, 69.94; H, 5.01; N, 11.69%. MS m/z: 503.2 (M + 23).
4.1.1.8. 5-(9-Ethyl-9H-carbazol-3-yl)-3-(3-oxo-3H-benzo[f]chromen-2-yl)-4,5-dihydro-1H-pyrazole-1-carboxamide (4c). Yield: 74%; mp 241–244 °C; dark yellow solid; IR spectrum, νmax, cm−1: 3483 & 3275 (amine stretching), 3050 (aromatic C–H stretching), 2972 & 2929 (aliphatic C–H stretching), 1726 (C
O stretching of δ-lactone of coumarin), 1680 (CO stretching of amide), 1628 (CN), 1567 & 1451 (CC), 1231& 1130 (C–N); 1H NMR (DMSO-d6) δ, ppm (J, Hz): 1.34 (3H, t, J = 7.2 Hz, CH3 of carbazole), 3.46 (1H, dd, J = 16.8 Hz and 5.6 Hz Ha protons of pyrazoline), 4.02 (1H, dd, J = 12.4 Hz and 6.0 Hz, Hb protons of pyrazoline), 4.40 (2H, q, J = 7.2 Hz, –CH2 of carbazole), 5.64 (1H, dd, J = 12.0 Hz and 5.6 Hz, Hx protons of pyrazoline), 6.72 (2H, broad singlet, –NH2 of pyrazoline ring), 7.16 (1H, t, J = 7.6 Hz, Ar-H), 7.34–7.50 (4H, m, Ar-H), 7.51–7.64 (2H, m, Ar-H), 7.78–8.07 (5H, m, Ar-H), 8.75 (1H, d, J = 8.2 Hz, Ar-H), 9.49 (1H, s, Ar-H); 13C NMR (DMSO-d6): δ (ppm) 158.46(CO of coumarin), 154.93(CO of amide), 153.33(C), 146.64(C), 139.74(C), 138.71(C), 136.19(CH), 134.10(C), 133.69(CH), 129.96(C), 128.99(C), 128.76(CH), 128.19(CH), 126.08(CH), 125.51(CH), 123.36(CH), 122.53(CH), 122.08(C), 122.02(C), 120.06(CH), 118.49(CH), 118.40(C), 117.17(CH), 116.20(CH), 113.26(C), 108.82(CH), 108.69(CH), 60.54(CHx of pyrazoline), 44.85(CH2 of pyrazoline), 36.98(CH2 of carbazole), 13.56(CH3 of carbazole). Anal. calcd for C31H24N4O3: elemental analysis: C, 74.39; H, 4.83; N, 11.19%. Found: C, 74.42; H, 4.81; N, 11.20%. MS m/z: 523.2 (M + 23).
4.1.1.9. 3-(5-(9-Ethyl-9H-carbazol-3-yl)-1-phenyl-4,5-dihydro-1H-pyrazol-3-yl)-2H-chromen-2-one (5a). Yield: 65%; mp 232–235 °C; orange solid; IR spectrum, νmax, cm−1: 3043 (aromatic C–H stretching), 2977 & 2930 (aliphatic C–H stretching), 1729 (C
O stretching of δ-lactone of coumarin), 1598 (CN), 1563, 1528 & 1494 (CC), 1231 & 1133 (C–N); 1H NMR (CDCl3) δ, ppm (J, Hz): 1.41 (3H, t, J = 7.2 Hz, CH3 of carbazole), 3.47 (1H, dd, J = 12.4 Hz and 7.6 Hz, Ha protons of pyrazoline), 4.18 (1H, dd, J = 12.8 and 5.6 Hz, Hb protons of pyrazoline), 4.34 (2H, q, J = 7.2 Hz, CH2 of carbazole), 5.52 (1H, dd, J = 7.2 Hz and 5.2 Hz, Hx protons of pyrazoline), 6.78–6.80 (1H, m, Ar-H), 7.16–7.43 (10H, m, Ar-H), 7.46 (1H, dd, J = 8.0 Hz and 1.2 Hz, Ar-H), 7.50 (1H, td, J = 8.0 Hz and 1.2 Hz, Ar-H), 7.59 (1H, dd, J = 7.6 Hz and 1.2 Hz, Ar-H), 8.01 (1H, s, Ar-H), 8.06 (1H, d, J = 7.6 Hz, Ar-H), 8.43 (1H, s, Ar-H); 13C NMR (CDCl3): δ (ppm) 159.68(CO of coumarin), 153.56(C), 144.34(C), 143.05(C), 140.32(C), 139.47(C), 137.48(CH), 132.80(C), 131.40(CH), 128.95(CH), 128.16(CH), 127.70(CH), 126.57(CH), 126.11(CH), 125.55(CH), 124.68(CH), 123.35(C), 121.06(CH), 120.55(CH), 119.76(C), 119.59(CH), 118.86(C), 117.73(C), 116.42(CH), 113.82(C), 109.16(CH), 108.53(CH), 108.37(CH), 65.60(CHx of pyrazoline), 45.86(CH2 of pyrazoline), 37.62(CH2 of carbazole), 13.84(CH3 of carbazole). Anal. calcd for C32H25N3O2: elemental analysis: C, 79.48; H, 5.21; N, 8.69%. Found: C, 79.51; H, 5.22; N, 8.68%. MS m/z: 506.2 (M + 23).
4.1.1.10. 3-(5-(9-Ethyl-9H-carbazol-3-yl)-1-phenyl-4,5-dihydro-1H-pyrazol-3-yl)-8-methoxy-2H-chromen-2-one (5b). Yield: 64%; mp 136–138 °C; dark-orange solid; IR spectrum, νmax, cm−1: 3046 (aromatic C–H stretching), 2971 & 2931 (aliphatic C–H stretching), 1727 (C
O stretching of δ-lactone of coumarin), 1598 (CN), 1571 & 1495 (CC), 1131 & 1231 (C–N); 1H NMR (CDCl3) δ, ppm (J, Hz): 1.41 (3H, t, J = 7.2 Hz, CH3 of carbazole), 3.46 (1H, dd, J = 14.8 Hz and 7.6 Hz, Ha protons of pyrazoline), 3.95 (3H, s, –OCH3), 4.18 (1H, dd, J = 12.4 and 6.0 Hz Hb protons of pyrazoline), 4.33 (2H, q, J = 7.2 Hz, CH2 of carbazole), 5.51 (1H, dd, J = 7.6 Hz and 4.8 Hz, Hx protons of pyrazoline), 6.79 (1H, t, J = 2.8 Hz, Ar-H), 7.03–7.46 (12H, m, Ar-H), 8.00 (1H, s, Ar-H), 8.05 (1H, d, J = 8.4 Hz, Ar-H), 8.41 (1H, s, Ar-H); 13C NMR (DMSO-d6): δ (ppm) 159.14(CO of coumarin), 146.98(C), 144.37(C), 143.14(C), 140.33(CH), 139.47(C), 137.66(CH), 132.81(C), 129.04(CH), 128.95(CH), 127.69(C), 125.84(CH), 125.56(CH), 124.50(CH), 124.35(C), 123.43(CH), 122.65(C), 121.28(C), 120.56(CH), 120.24(C), 119.75(CH), 119.71(C), 118.85(CH), 117.74(CH), 113.83(CH), 113.41(CH), 109.15(CH), 108.53(CH), 65.64(CHx of pyrazoline), 56.28(–OCH3), 45.89(CH2 of pyrazoline), 37.69(CH2 of carbazole), 13.84(CH3 of carbazole). Anal. calcd for chemical formula: C33H27N3O3: elemental analysis: C, 77.17; H, 5.30; N, 8.18%. Found: C, 77.20; H, 5.27; N, 8.16%. MS m/z: 536.2 (M + 23).
4.1.1.11. 2-(5-(9-Ethyl-9H-carbazol-3-yl)-1-phenyl-4,5-dihydro-1H-pyrazol-3-yl)-3H-benzo[f] chromen-3-one (5c). Yield: 68%; mp 243–245 °C; Crimson-red solid; IR spectrum, νmax, cm−1: 3051 (aromatic C–H stretching), 2978 & 2930 (aliphatic C–H stretching), 1726 (C
O stretching of δ-lactone of coumarin), 1595 (CN), 1561 & 1467 (CC), 1136 & 1228 (C–N); 1H NMR (DMSO-d6) δ, ppm (J, Hz): 1.29 (3H, t, J = 7.2 Hz, CH3 of carbazole), 3.44 (1H, d, J = 6.0 Hz, Ha protons of pyrazoline), 4.14 (1H, dd, J = 12.8 Hz and 5.2 Hz, Hb protons of pyrazoline), 4.41 (2H, q, J = 7.2 Hz, CH2 of carbazole), 5.72 (1H, dd, J = 6.4 Hz and 6.0 Hz, Hx protons of pyrazoline), 6.74 (1H, t, J = 6.8 Hz, Ar-H), 7.15–7.67 (11H, m, Ar-H), 7.75 (1H, t, J = 7.6 Hz, Ar-H) 8.07 (1H, dd, J = 7.2 Hz, Ar-H), 7.13 (2H, t, J = 7.6 Hz, Ar-H), 8.19 (1H, d, J = 9.2 Hz, Ar-H), 9.10 (1H, s, Ar-H); 13C NMR (CDCl3): δ (ppm) 159.83(CO of coumarin), 153.32(C), 144.31(C), 143.43(C), 140.30(C), 139.46(C), 133.22(CH), 132.83(CH), 130.46(C), 129.19(C), 129.06(CH), 129.01(CH), 128.29(CH), 126.21(CH), 125.86(CH), 123.42(CH), 123.33(C), 122.62(C), 121.95(CH), 120.58(CH), 119.84(C), 119.78(CH), 118.86(CH), 117.75(CH), 116.63(CH), 113.92(C), 113.82(CH), 109.19(CH), 108.54(CH), 65.61(CHx of pyrazoline), 45.92(CH2 of pyrazoline), 37.62(CH2 of carbazole), 13.88(CH3 of carbazole). Anal. calcd for C36H27N3O2: elemental analysis: C, 81.03; H, 5.10; N, 7.87%. Found: C, 81.06; H, 5.12; N, 7.89%. MS m/z: 556.2 (M + 23).
4.1.1.12. 3-(5-(9-Ethyl-9H-carbazol-3-yl)-1-(p-tolyl)-4,5-dihydro-1H-pyrazol-3-yl)-2H-chromen-2-one (6a). Yield: 66%; mp 221–223 °C; orange solid; IR spectrum, νmax, cm−1: 3033 (aromatic C–H stretching), 2975 & 2915 (aliphatic C–H stretching) 1725 (C
O stretching of δ-lactone of coumarin), 1604 (CN), 1562 & 1465 (CC), 1134 & 1230 (C–N); 1H NMR (CDCl3) δ, ppm (J, Hz): 1.44 (3H, t, J = 7.2 Hz, CH3 of carbazole), 2.24 (3H, s, –CH3), 3.48 (1H, dd, J = 10.4 Hz and J = 8.0 Hz, Ha protons of pyrazoline), 4.19 (1H, dd, J = 12.8 and 5.2 Hz, Hb protons of pyrazoline), 4.36 (2H, q, J = 7.2 Hz, CH2 of carbazole), 5.51 (1H, dd, J = 8.0 Hz and 4.8 Hz, Hx protons of pyrazoline), 7.0 (2H, d, J = 8.4 Hz, Ar-H), 7.10 (2H, d, J = 8.4 Hz, Ar-H), 7.23 (1H, t, J = 7.2 Hz, Ar-H), 7.28–7.39 (2H, m, Ar-H), 7.46–7.54 (2H, m, Ar-H), 7.40 (3H, t, J = 6.8, Ar-H), 7.60 (1H, d, J = 7.6 Hz, Ar-H), 8.04 (1H, s, Ar-H), 8.08 (1H, d, J = 7.6 Hz, Ar-H), 8.43 (1H, s, Ar-H); 13C NMR-APT (CDCl3): δ (ppm) 159.74(CO of coumarin), 153.46(C), 142.48(C), 142.14(C), 140.28(C), 139.42(C), 137.15(CH), 132.89(C), 131.29(CH), 129.50(CH), 129.09(C), 128.11(CH), 125.82(CH), 124.67(CH), 123.46(CH), 123.29(C), 122.63(C), 121.10(C), 120.57(CH), 119.63(C), 118.83(CH), 117.79(CH), 116.40(CH), 113.87(CH), 109.13(CH), 108.52(CH), 65.83(CHx of pyrazoline), 45.80(CH2 of pyrazoline), 37.61(CH2 of carbazole), 20.55(CH3 of pendant phenyl ring), 13.84(CH3 of carbazole). Anal. calcd for C33H27N3O2: elemental analysis: C, 79.66; H, 5.47; N, 8.44%. Found: C, 79.72; H, 5.50; N, 8.42%. MS m/z: 520.1 (M + 23).
4.1.1.13. 3-(5-(9-Ethyl-9H-carbazol-3-yl)-1-(p-tolyl)-4,5-dihydro-1H-pyrazol-3-yl)-8-methoxy-2H-chromen-2-one (6b). Yield: 56%; mp 124–126 °C; red solid; IR spectrum, νmax, cm−1: 3040 (aromatic C–H stretching), 2971 & 2930 (aliphatic C–H stretching) 1726 (C
O stretching of δ-lactone of coumarin), 1606 (CN), 1572 & 1479 (CC), 1232 (C–N); 1H NMR (CDCl3) δ, ppm (J, Hz): δ (ppm) 1.43 (3H, t, J = 7.2 Hz, CH3 of carbazole), 2.24 (3H, s, –CH3), 3.47 (1H, dd, J = 10.4 Hz and 8.0 Hz, Ha protons of pyrazoline), 3.98 (3H, s, –OCH3), 4.20 (1H, dd, J = 12.4 Hz and 5.6 Hz, Hb protons of pyrazoline), 4.36 (2H, q, J = 7.2 Hz, –CH2 of carbazole), 5.50 (1H, dd, J = 8.0 Hz and 4.8 Hz, Hx protons of pyrazoline), 6.99 (2H, d, J = 8.4 Hz, Ar-H), 7.05–7.52 (10H, m, Ar-H), 8.03 (1H, s, Ar-H), 8.08 (1H, d, J = 7.6 Hz, Ar-H), 8.41 (1H, s, Ar-H); 13C NMR (CDCl3): δ (ppm) 159.14(CO of coumarin), 146.98(C), 144.37(C), 143.14(C), 140.33(C), 139.47(C), 137.66(CH), 132.81(C), 129.04(CH), 128.95(CH), 127.69(C), 125.84(CH), 125.56(CH), 124.50(CH), 124.35(C), 123.43(CH), 122.65(C), 121.28(C), 120.56(CH), 120.24(C), 119.75(CH), 119.71(C), 118.85(CH), 117.74(CH), 113.83(CH), 113.41(CH), 109.15(CH), 108.53(CH), 65.64(CHx of pyrazoline), 56.28(–OCH3 of coumarin ring), 45.89(CH2 of pyrazoline), 37.69(CH2 of carbazole), 20.55(CH3 of Pendant phenyl ring), 13.84(CH3 of carbazole). Anal. calcd for C34H29N3O3: elemental analysis: C, 77.40; H, 5.54; N, 7.96%. Found: C, 77.38; H, 5.47; N, 7.96%. MS m/z: 550.2 (M + 23).
4.1.1.14. 2-(5-(9-Ethyl-9H-carbazol-3-yl)-1-(p-tolyl)-4,5-dihydro-1H-pyrazol-3-yl)-3H-benzo[f] chromen-3-one (6c). Yield: 65%; mp 229–232 °C; Crimson red solid; IR spectrum, νmax, cm−1: 3049 (aromatic C–H stretching), 2977 & 2910 (aliphatic C–H stretching) 1727 (C
O stretching of δ-lactone of coumarin), 1608 (CN) 1560 & 1466 (CC), 1227 & 1136 (C–N); 1H NMR (CDCl3) δ, ppm (J, Hz): 1.44 (3H, t, J = 7.2 Hz, CH3 of carbazole), 2.26 (3H, s, –CH3), 3.54 (1H, dd, J = 10.8 Hz and 7.6 Hz, Ha protons of pyrazoline), 4.25 (1H, dd, J = 8.8 and 5.6 Hz, Hb protons of pyrazoline), 4.36 (2H, q, J = 7.2 Hz, –CH2 of carbazole), 5.54 (1H, dd, J = 7.6 Hz and 4.8 Hz, Hx protons of pyrazoline), 7.03 (2H, d, J = 8.4 Hz, Ar-H), 7.16 (2H, d, J = 8.4 Hz, Ar-H), 7.22 (1H, d, J = 7.6 Hz, Ar-H), 7.37–7.49 (5H, m, Ar-H), 7.61 (1H, t, J = 8.0 Hz, Ar-H), 7.76 (1H, t, J = 7.6 Hz, Ar-H), 7.94 (2H, t, J = 9.6 Hz, Ar-H), 8.07–8.10 (2H, m, Ar-H), 8.46 (1H, d, J = 8.4 Hz, Ar-H), 9.21 (1H, s, Ar-H); 13C NMR (CDCl3): δ (ppm) 159.85(CO of coumarin), 153.20(C), 142.89(C), 142.18(C), 140.28(C), 139.43(C), 132.95(C), 132.84(CH), 132.65(CH), 130.45(C), 129.53(CH), 129.17(C), 129.11(C), 129.03(CH), 128.23(CH), 126.16(CH), 125.82(CH), 123.48(CH), 123.31(C), 122.64(C), 121.96(CH), 120.58(CH), 119.93(C), 119.27(C), 118.83(CH), 117.80(CH), 116.62(CH), 113.92(CH), 109.14(CH), 108.52(CH), 65.89(CHx of pyrazoline), 45.86(CH2 of pyrazoline), 37.61(CH2 of carbazole), 20.59(CH3 of Pendant phenyl ring), 13.87(CH3 of carbazole). Anal. calcd for C37H29N3O2: elemental analysis: C, 81.15; H, 5.34; N, 7.67%. Found: C, 81.18; H, 5.31; N, 7.66%. MS m/z: 570.2 (M + 23).
4.1.1.15. 3-(1-(3-Chlorophenyl)-5-(9-ethyl-9H-carbazol-3-yl)-4,5-dihydro-1H-pyrazol-3-yl)-2H-chromen-2-one (7a). Yield: 70%; mp 189–191 °C; light orange solid; IR spectrum, νmax, cm−1: 3046 (aromatic C–H stretching), 2975 & 2929 (aliphatic C–H stretching), 1727 (C
O stretching of δ-lactone of coumarin), 1594 (CN), 1562 & 1481 (CC); 1H NMR (CDCl3) δ, ppm (J, Hz): 1.46 (3H, t, J = 7.2 Hz, CH3 of carbazole), 3.54 (1H, dd, J = 11.2 Hz and 7.6 Hz, Ha protons of pyrazoline), 4.22 (1H, dd, J = 12.4 Hz and 6.0 Hz, Hb protons of pyrazoline), 4.38 (2H, q, J = 7.6 Hz, CH2 of carbazole), 5.53 (1H, dd, J = 7.6 Hz and 5.4 Hz, Hx protons of pyrazoline), 6.79 (1H, d, J = 7.6 Hz, Ar-H), 6.89 (1H, d, J = 7.6 Hz and 1.2 Hz, Ar-H), 7.06 (1H, t, J = 8.0 Hz, Ar-H), 7.25 (1H, d, J = 7.6 Hz, Ar-H), 7.30–7.57 (8H, m, Ar-H), 7.65 (1H, d, J = 7.6 Hz, Ar-H), 8.02 (1H, s, Ar-H), 8.11 (1H, d, J = 7.6 Hz, Ar-H), 8.50 (1H, s, Ar-H); 13C NMR (CDCl3): δ (ppm) 159.64(CO of coumarin), 153.66(C), 145.26(C), 144.14(C), 140.34(C), 139.55(C), 138.20(CH), 134.82(C), 132.18(C), 131.73(CH), 129.98(CH), 128.37(CH), 125.98(CH), 124.80(CH), 123.42(C), 123.27(CH), 122.58(C), 120.68(C), 120.60(C), 120.59(CH), 119.47(CH), 118.96(CH), 117.64(CH), 116.47(CH), 113.94(CH), 111.54(CH), 109.29(CH), 108.62(CH), 65.28(CHx of pyrazoline), 46.04(CH2 of pyrazoline), 37.66(CH2 of carbazole), 13.89(CH3 of carbazole). Anal. calcd for C32H24ClN3O2: elemental analysis: C, 74.20; H, 4.67; N, 8.11%. Found: C, 74.19; H, 4.70; N, 8.13%. MS m/z: 540.2 (M + 23).
4.1.1.16. 3-(1-(3-Chlorophenyl)-5-(9-ethyl-9H-carbazol-3-yl)-4,5-dihydro-1H-pyrazol-3-yl)-8-methoxy-2H-chromen-2-one (7b). Yield: 68%; mp 130–132 °C; orange solid; IR spectrum, νmax, cm−1: 3049 & 2930 (aromatic C–H), 1727 (C
O, δ-lactone), 1592 (CN), 1484 (CC), 1274 & 1271(C–N); 1H NMR (CDCl3) δ, ppm (J, Hz): 1.48 (3H, t, J = 7.6 Hz, –CH3 of carbazole), 3.51 (1H, dd, J = 11.6 Hz and 7.6 Hz, Ha protons of pyrazoline), 3.98 (3H, t, J = 11.2 Hz, –OCH3), 4.22 (1H, dd, J = 12.4 Hz and 6.0 Hz, Hb protons of pyrazoline), 4.35 (2H, m, CH2 of carbazole), 5.50 (1H, dd, J = 7.2 Hz and 5.6 Hz, Hx protons of pyrazoline), 6.75 (1H, d, J = 8.0 Hz, Ar-H), 6.85 (1H, d, J = 8.0 Hz, Ar-H), 7.05 (1H, t, J = 8.0 Hz, Ar-H), 7.09 (1H, d, J = 8.0 Hz, Ar-H), 7.20–7.24 (3H, m, Ar-H), 7.36–7.40 (3H, m, Ar-H), 7.41 (1H, d, J = 8.4 Hz, Ar-H), 7.46 (1H, d, J = 7.2 Hz, Ar-H), 7.99 (1H, s, Ar-H), 8.08 (1H, d, J = 8.8 Hz, Ar-H), 8.48 (1H, s, Ar-H); 13C NMR (CDCl3): δ (ppm) 159.08(C), 146.93(C), 145.24(C), 144.19(C), 143.24(C), 140.30(C), 139.51(C), 138.37(CH), 134.77(C), 132.14(C), 129.94(CH), 125.92(CH), 124.61(CH), 123.36(C), 123.25(CH), 122.55(C), 120.86(C), 120.57(CH), 120.07(C), 119.80(CH), 119.41(CH), 118.90(CH), 117.59(CH), 113.89(CH), 113.54(CH), 111.48(CH), 109.24(CH), 108.57(CH), 65.28(CHx of pyrazoline), 56.24(–OCH3 of coumarin), 46.03(CH2 of pyrazoline), 37.62(CH2 of carbazole), 13.87(CH3 of carbazole). Anal. calcd for C33H26ClN3O3: elemental analysis: C, 72.32; H, 4.78; N, 7.67%. Found: C, 72.18; H, 4.81; N, 7.65%. MS m/z: 570.2 (M + 23).
4.1.1.17. 2-(1-(3-Chlorophenyl)-5-(9-ethyl-9H-carbazol-3-yl)-4,5-dihydro-1H-pyrazol-3-yl)-3H-benzo[f]chromen-3-one (7c). Yield: 61%; mp 234–236 °C; orange solid; IR spectrum, νmax, cm−1: 3051 (aromatic C–H stretching), 2976 & 2930 (aliphatic C–H stretching) 1727 (C
O stretching of δ-lactone of coumarin), 1592 (CN), 1561 & 1483 (CC), 1132 & 1228 (C–N); 1H NMR (CDCl3) δ, ppm (J, Hz): 1.44 (3H, s, CH3 of carbazole), 3.57 (1H, d, J = 13.2 Hz, Ha protons of pyrazoline), 4.31 (3H, m, Hb proton of pyrazoline and CH2 of carbazole), 5.55 (1H, s, Hx proton of pyrazoline), 6.80 (1H, s, Ar-H), 6.94 (1H, s, Ar-H), 7.07 (1H, s, Ar-H), 7.23 (1H, s, Ar-H), 7.40–7.47 (6H, m, Ar-H), 7.63 (1H, s, Ar-H), 7.79 (1H, s, Ar-H), 7.93–8.10 (4H, m, Ar-H), 8.49 (1H, d, J = 6.8 Hz, Ar-H), 9.25 (1H, s, Ar-H); 13C NMR (CDCl3): δ (ppm) 159.75(CO of coumarin), 153.51(C), 145.22(C), 144.55(C), 140.31(C), 139.52(C), 134.81(C), 133.85(CH), 133.17(CH), 132.20(C), 130.49(C), 130.45(C), 129.98(CH), 129.19(C), 129.07(CH), 128.43(CH), 126.30(CH), 125.94(CH), 123.40(C), 123.25(CH), 122.56(C), 122.00(CH), 120.58(CH), 119.46(CH), 118.92(CH), 117.61(CH), 116.60(CH), 113.89(CH), 113.82(C), 111.58(CH), 109.27(CH), 108.58(CH), 65.28(CHx of pyrazoline), 46.06(CH2 of pyrazoline), 37.64(CH2 of carbazole), 13.88(CH3 of carbazole). Anal. calcd for C36H26ClN3O2: elemental analysis: C, 76.12; H, 4.61; N, 7.40%. Found: C, 76.19; H, 4.60; N, 7.42%. MS m/z: 590.2 (M + 23).
4.1.1.18. General synthetic procedure for compounds 8a–c. A mixture of various coumarin–carbazole chalcone (3a–c) (3 mmol), hydrazine hydrate (6 mmol), and acetic acid (6 mmol) in 30 mL of ethanol was stirred at 65 °C for 90–180 min. It was then poured in water (75 mL), and the crude solid obtained was extracted with chloroform (3 × 50 mL). The organic layer was washed with 10% sodium bicarbonate solution (50 mL), water (50 mL), and dried over anhydrous sodium sulfate. Distillation of chloroform in vacuum gave gummy material subjected to column chromatography using petroleum ether (60–80 °C)
:CHCl3 (5:5) as an eluent to afford product 8a–c.
4.1.1.19. 3-(1-Acetyl-5-(9-ethyl-9H-carbazol-3-yl)-4,5-dihydro-1H-pyrazol-3-yl)-2H-chromen-2-one (8a). Yield: 70%; mp 200–202 °C; greenish yellow solid; IR spectrum, νmax, cm−1: 3049 (aromatic C–H stretching), 2976 & 2930 (aliphatic C–H stretching) 1722 (C
O stretching of δ-lactone of coumarin), 1659 (CO stretching of acetyl), 1606 (CN), 1568&1486 (CC), 1231 & 1146 (C–N); 1H NMR (DMSO-d6) δ, ppm (J, Hz): 1.29 (3H, t, J = 7.2 Hz, CH3 of carbazole), 2.34 (3H, s, –CH3 of acetyl), 3.37 (1H, m, Ha protons of pyrazoline), 3.98 (1H, dd, J = 12.0 Hz and J = 6.8 Hz, Hb protons of pyrazoline), 4.41 (2H, q, J = 7.6 Hz, –CH2 of carbazole), 5.70 (1H, dd, J = 7.2 Hz and 4.8 Hz, Hx protons of pyrazoline), 7.17 (1H, t, J = 7.2 Hz, Ar-H), 7.30 (1H, dd, J = 6.8 Hz and 1.6 Hz, Ar-H), 7.38–7.45 (3H, m, Ar-H), 7.57 (2H, t, J = 8.0 Hz, Ar-H), 7.67 (1H, t, J = 8.0 Hz, Ar-H), 7.87 (1H, dd, J = 6.4 Hz and J = 1.2 Hz, Ar-H), 7.98 (1H, d, J = 1.2 Hz, Ar-H), 8.14 (1H, d, J = 7.6 Hz, Ar-H), 8.61 (1H, s, Ar-H); 13C NMR (DMSO-d6): δ (ppm) 167.67(CO of acetyl), 157.94(CO of coumarin), 153.43(C), 150.76(C), 141.78(CH), 139.81(C), 138.78(C), 132.92(CH), 129.31(CH), 126.13(C), 125.77(CH), 124.87(CH), 123.49(CH), 122.01(C), 121.93(C), 120.39(CH), 119.24(C), 118.72(C), 118.63(CH), 117.48(CH), 116.02(CH), 109.25(CH), 109.08(CH), 59.81(CHx of pyrazoline), 44.31(CH2 of pyrazoline), 36.93(CH2 of carbazole), 21.81(CH3 of pendant acetyl group), 13.66(CH3 of carbazole). Anal. calcd for C28H23N3O3: elemental analysis: C, 74.82; H, 5.16; N, 9.35%. Found: C, 74.87; H, 5.12; N, 9.36%. MS m/z: 472.2 (M + 23).
4.1.1.20. 3-(1-Acetyl-5-(9-ethyl-9H-carbazol-3-yl)-4,5-dihydro-1H-pyrazol-3-yl)-8-methoxy-2H-chromen-2-one (8b). Yield: 68%; mp 246–247 °C; yellow solid; IR spectrum, νmax, cm−1: 3048 (aromatic C–H stretching), 2964 & 2928 (aliphatic C–H stretching), 1724 (C
O stretching of δ-lactone of coumarin), 1660 (CO stretching of acetyl), 1604 (CN), 1573 & 1482 (CC), 1143 & 1231 (C–N); 1H NMR (DMSO-d6) δ, ppm (J, Hz): 1.39 (3H, t, J = 7.2 Hz, CH3 of carbazole), 2.44 (3H, s, –CH3 of acetyl), 3.54 (1H, dd, J = 14.4 Hz and J = 4.4 Hz, Ha protons of pyrazoline), 3.97 (3H, s, –OCH3), 4.04 (1H, dd, J = 12.0 Hz and 7.2 Hz, Hb protons of pyrazoline), 4.31 (2H, q, J = 7.2 Hz, –CH2 of carbazole), 5.78 (1H, dd, J = 7.6 Hz and 4.4 Hz, Hx protons of pyrazoline), 7.12 (1H, d, J = 8.0 Hz, Ar-H), 7.15–7.45 (7H, m, Ar-H), 7.93 (1H, s, Ar-H), 8.05 (1H, d, J = 7.6 Hz, Ar-H), 8.44 (1H, s, Ar-H); 13C NMR (DMSO-d6): δ (ppm) 169.05(CO of acetyl), 158.75(CO of coumarin), 150.93(C), 147.20(C), 143.91(C), 141.07(CH), 140.41(C), 139.58(C), 132.39(C), 125.79(CH), 124.87(CH), 123.65(CH), 123.22(C), 122.88(C), 120.66(CH), 120.28(C), 120.19(CH), 119.62(C), 118.83(CH), 117.71(CH), 114.63(CH), 108.84(CH), 108.52(CH), 60.99(CHx of pyrazoline), 56.42(–OCH3 of coumarin), 44.89(CH2 of pyrazoline), 37.66(CH2 of carbazole), 22.20(CH3 of pendant acetyl group), 13.90(CH3 of carbazole). Anal. calcd for C29H25N3O4: elemental analysis: C, 72.64; H, 5.26; N, 8.76%. Found: C, 72.58; H, 5.31; N, 8.74%. MS m/z: 502.2 (M + 23).
4.1.1.21. 2-(1-Acetyl-5-(9-ethyl-9H-carbazol-3-yl)-4,5-dihydro-1H-pyrazol-3-yl)-3H-benzo[f]chromen-3-one (8c). Yield: 61%; mp 158–160 °C; dark yellow solid; IR spectrum, νmax, cm−1: 3051 (aromatic C–H stretching), 2974 & 2932 (aliphatic C–H stretching), 1731 (C
O stretching of δ-lactone of coumarin), 1660 (CO stretching of acetyl), 1600 (CN), 1568 & 1489 (CC), 1144 & 1228 (C–N); 1H NMR (DMSO-d6) δ, ppm (J, Hz): 1.29 (3H, t, J = 7.2 Hz, –CH3 of carbazole), 2.41 (3H, s, –CH3 of acetyl), 3.50 (1H, dd, J = 13.6 Hz and 4.8 Hz, Ha protons of pyrazoline), 4.03 (1H, dd, J = 12.0 Hz and 6.4 Hz, Hb protons of pyrazoline), 4.41 (2H, q, J = 7.2 Hz, CH2 of carbazole), 5.73 (1H, dd, J = 7.2 Hz and 4.8 Hz, Hx protons of pyrazoline), 7.17 (1H, t, J = 7.2 Hz, Ar-H), 7.34 (1H, dd, J = 6.8 Hz and J = 2.0 Hz, Ar-H), 7.43 (1H, t, J = 7.6 Hz, Ar-H), 7.58–7.62 (3H, m, Ar-H), 7.64 (1H, t, J = 7.2 Hz, Ar-H), 7.76 (1H, t, J = 8.0 Hz, Ar-H), 8.02 (1H, d, J = 1.2 Hz, Ar-H), 8.05 (1H, d, J = 7.6 Hz, Ar-H), 8.15 (1H, d, J = 7.6 Hz, Ar-H), 8.23 (1H, d, J = 9.2 Hz, Ar-H), 8.67 (1H, d, J = 8.8 Hz, Ar-H), 9.18 (1H, s, Ar-H); 13C NMR (DMSO-d6): δ (ppm) 167.82(CO of acetyl), 157.63(CO of coumarin), 153.49(C), 150.84(C), 139.79(C), 138.79(C), 137.60(CH), 134.35(CH), 133.04(C), 129.93(C), 128.93(CH), 128.81(C), 128.55(CH), 126.32(CH), 125.74(CH), 123.54(CH), 122.55(CH), 122.03(C), 121.95(C), 120.39(CH), 118.63(CH), 118.24(C), 117.54(CH), 116.42(CH), 112.88(C), 109.24(CH), 109.07(CH), 59.82(CHx of pyrazoline), 44.24(CH2 of pyrazoline), 36.93(CH2 of carbazole), 21.90(CH3 of pendant acetyl group),13.65(CH3 of carbazole). Anal. calcd for C32H25N3O3: elemental analysis: C, 76.94; H, 5.04; N, 8.41%. Found: C, 76.98; H, 5.01;; N, 8.38%. MS m/z: 522.2 (M + 23).
4.2. Cell culture
The cervical cancer cell line HeLa, lung cancer cell line NCI-H520 and normal rat kidney epithelial cell line NRK-52E were procured from the National Centre for Cell Science, Pune, India. All the cell lines were grown on 25 cm2 vented culture flasks (Corning, USA). HeLa and NRK-52E cell lines grown using Dulbecco's Modified Eagle Medium (DMEM) (Thermofisher, USA) and NCI-H520 cell line grown using Roswell Park Memorial Institute (RPMI) (Thermofisher, USA) respectively, supplemented with 10% heat-inactivated fetal bovine serum (Thermofisher, USA), and 100 U mL−1 penicillin, 100 μg mL−1 streptomycin and 0.25 μg mL−1 amphotericin B (Thermofisher, USA), in a 37 °C incubator with 5% CO2 and 95% humidity.4.3. Cytotoxicity assay
Cytotoxicity of synthesized compounds was tested utilizing MTT [3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide] assay. The cells were briefly seeded in triplicates into a 96-wells tissue culture plate at a density of 104 cells per well and incubated for 24 h at 37 °C with 5% CO2. The wells were then treated with the compounds tested at concentrations ranging from 5 to 300 μM and incubated further at 37 °C for 24 h. After the incubation period, the compounds were aspirated, and the wells were washed with phosphate-buffered saline (PBS). Afterward, 300 μL of media and 25 μL of MTT solution (5 mg mL−1 in PBS) were added to each well, followed by 3 h of incubation. Later, MTT was removed, and the formed crystal formazan was dissolved in 100 μL dimethyl sulfoxide (DMSO). The MTT assay absorbance was measured at 570 nm on an ELISA plate reader (Molecular Devices, USA). The relationship between surviving cells and drug concentration was plotted to get each cell line's survival curve after treatment with the specified compound. The 50% inhibitory concentration (IC50), the concentration required to cause toxic effects in 50% of intact cells, was estimated using nonlinear regression analysis using Graph pad prism (version 7.0) software.4.4. Cell cycle analysis
The effect of compounds 4a and 7b on cell cycle progression was analyzed using the propidium iodide staining. Firstly, HeLa and NCI-H520 cells were seeded into the 6-well plate at the density of 1 × 105 cells and incubated for 24 h at 37 °C with 5% CO2. The cells were then treated with the IC50 concentrations of compounds 4a and 7b. After 24 h incubation, the cells were fixed with ice-cold 70% ethanol at −20 °C for 16 h. After fixation, cells were washed with Dulbecco's Phosphate-Buffered Saline (DPBS) (GIBCO®, Thermo Scientific) and allowed to incubate in DPBS containing Triton X-100 (0.2%) at 4 °C for 45 min. The cells were then treated with RNase A (100 μg mL−1) and incubated at 37 °C for 1 h. Finally, cells were stained with Propidium Iodide (20 μg mL−1) for 30 min and analyzed on the Muse™ Cell Analyzer (Luminex, USA).4.5 Apoptosis analysis
Apoptosis assay of potent compounds 4a and 7b was carried out using Muse annexin V & dead cell kit (MCH100105) on Muse cell analyzer. It analyzes and gives statistics about non-apoptotic cells, early apoptotic cells, late apoptotic cells, dead cells, and nuclear debris. During the investigation of the impact of compounds 4a and 7b on cell lines, cultured cells were treated with an appropriate concentration of compounds for appropriate incubation time to induce apoptosis. They then treated cells were trypsinized and washed with cold PBS. Next, cells were resuspended in 1% BSA and 1% FBS. After that 100 μL of the cell, the suspension was taken in a vial and mixed with 100 μL of Muse Annexin V & Dead cell reagent. The mixture was incubated for 20 minutes at room temperature and processed for apoptosis analysis on Muse Cell Analyzer.4.6. Detection of morphological changes
HeLa and NCI-H520 cells were seeded in a 6-well cell culture plate and incubated for 24 h at 37 °C with 5% CO2 supply. The cells were treated at IC50 concentrations of compound 4a (μM) and 7b (μM) and allowed to incubate for 24 h. After the incubation, cells were washed and fixed with 4% p-formaldehyde for 15 min and permeabilized with 0.1% Triton X-100. The cells were stained using DAPI (1 mg mL−1) for 10 min and observed under a fluorescence microscope (brand) with the DAPI excitation filter. The images were processed and analyzed by ZEN 3.2 (blue edition).4.7. Molecular docking studies
The structure of ligands was prepared in ChemDraw Ultra v19.0 and assigned appropriate 2D orientation. The drawn structures were analyzed for connection error in bond order. The molecules' energy minimization was carried out by Chem3D ultra using MMFF94 forcefield and 0.1 RMS gradient. Molecular docking analysis of compound 4a and 7b showing the highest in vitro cytotoxicity was performed to determine the binding mechanism of these compounds with the target protein CDK2. The target protein CDK2 (PDB ID: 2A4L) was extracted from Protein Data Bank (PDB). Initially, the PDB files of selected proteins were processed by deleting ligands and non-proteins using Discover Studio visualizer v20.1.0.19295. Further, the protein structures were prepared by removing water molecules and then the addition of Kollman charges and polar hydrogen atoms. Next, the ligands were processed by merging Gastiger charges to their structures. Finally, both ligand and protein structures were converted to pdbqt file using AutoDock Tools 1.5.6. AutodockVina v1.1.2 carried out docking calculations.43 The dimension of the grid used for both the proteins was kept 40 × 40 × 40 Å and separated by 0.375 Å grid-point spacing. The X, Y, and Z coordinates (grid centers) in the grid were specified, which differed for both the receptors (Table 2). The exhaustiveness for the docking run was set to the default value of eight. The ligand's binding energy was calculated using the default Lamarckian genetic algorithm of the AutoDock Vina software package. After completing the docking run, the protein–ligand complexes were analyzed using Biovia Discovery Studio Visualizer v20.1.0.19295 and Pymol molecular graphics software. Biovia Discovery Studio Visualizer v20.1.0.19295 generated the representative 2D and 3D structures showing protein–ligand structures.| Protein | Centre X (Å) | Centre Y (Å) | Centre Z (Å) |
|---|---|---|---|
| 2A4L | 100.865 | 101.747 | 79.893 |
| 6QGG | −15.226 | 14.319 | −9.635 |
4.8. Statistical analysis
The data are shown as mean ± SE of triplicate independent analysis. The IC50 values were determined by nonlinear regression analysis. One-way ANOVA calculated the statistical difference between means with Tukey's multiple comparisons test. All the statistical tests were performed using GraphPad Prism version 8.0.0 for Windows, GraphPad Software, San Diego, California USA, and considered significant when the p-value was less than 0.05.Data availability
All data generated or analyzed during this study are included in this published article (and its ESI†).Author contributions
Mr Mrugesh Patel and Dr Kaushal Patel conceived and designed the experiments based on the core chemistry, performed the experiments, analyzed the data, performed the computation work, prepared figures and/or tables, authored and reviewed drafts of the manuscript, and approved the final draft.Mr Mrugesh Patel, Dr Nilesh Pandey, and Dr Neeraj Jain conceived and designed the experiments based on the core biology, validation of results, prepared figures and/or tables, authored and reviewed manuscript drafts, and approved the final draft.
Dr Alex Chauhan, Mr Paranjay Parikh, and Mr Jignesh Timaniya provided the platform to analyze the data, authored and reviewed drafts of the manuscript, and approved the final draft.
Conflicts of interest
There are no conflicts of interest.Acknowledgements
The authors express their sincere thanks to the Department of Advanced Organic Chemistry, P. D. Patel Institute of Applied Sciences, Charotar University of Science and Technology (CHARUSAT) for providing research facilities and Dr Franky Shah, Senior Scientific Officer, and Dr Jigna Joshi, Gujarat Cancer and Research Institute, Ahmedabad, India for extending the Cell Analyzer facility.References
- H. Sung, J. Ferlay, R. L. Siegel, M. Laversanne, I. Soerjomataram, A. Jemal and F. Bray, Ca-Cancer J. Clin., 2021, 71, 209–249 CrossRef PubMed.
- D. J. Newman and G. M. Cragg, J. Nat. Prod., 2016, 79, 629–661 CrossRef CAS PubMed.
- L. Wang, H. L. Wu, X. L. Yin, Y. Hu, H. W. Gu and R. Q. Yu, Spectrochim. Acta, Part A, 2017, 170, 104–110 CrossRef CAS PubMed.
- S. Vilar, E. Quezada, L. Santana, E. Uriarte, M. Yanez, N. Fraiz, C. Alcaide, E. Cano and F. Orallo, Bioorg. Med. Chem. Lett., 2006, 16, 257–261 CrossRef CAS PubMed.
- K. Venkata Sairam, B. M. Gurupadayya, B. I. Vishwanathan, R. S. Chandan and D. K. Nagesha, RSC Adv., 2016, 6, 98816–98828 RSC.
- D. A. Ostrov, J. A. Hernández Prada, P. E. Corsino, K. A. Finton, N. Le and T. C. Rowe, Antimicrob. Agents Chemother., 2007, 51, 3688–3698 CrossRef CAS PubMed.
- Y. Zhang, B. Zou, Z. Chen, Y. Pan, H. Wang, H. Liang and X. Yi, Bioorg. Med. Chem. Lett., 2011, 21, 6811–6815 CrossRef CAS PubMed.
- M. Curini, F. Epifano, F. Maltese, M. C. Marcotullio, S. P. Gonzales and J. C. Rodriguez, Aust. J. Chem., 2003, 56, 59–60 CrossRef CAS.
- A. Manvar, A. Bavishi, A. Radadiya, J. Patel, V. Vora, N. Dodia, K. Rawal and A. Shah, Bioorg. Med. Chem. Lett., 2011, 21, 4728–4731 CrossRef CAS PubMed.
- L. Zhang, G. Jiang, F. Yao, Y. He, G. Liang, Y. Zhang, B. Hu, Y. Wu, Y. Li and H. Liu, PLoS One, 2012, 7, e37865 CrossRef CAS PubMed.
- R. K. Singh, T. S. Lange, K. K. Kim and L. Brard, Invest. New Drugs, 2011, 29, 63–72 CrossRef CAS PubMed.
- K. V. Sashidhara, S. R. Avula, K. Sharma, G. R. Palnati and S. R. Bathula, Eur. J. Med. Chem., 2013, 60, 120–127 CrossRef CAS PubMed.
- E. K. Akkol, Y. Genç, B. Karpuz, E. Sobarzo-Sánchez and R. Capasso, Cancers, 2020, 12, 1–25 Search PubMed.
- S. Butler, R. Wang, S. L. Wunder, H. Y. Cheng and C. S. Randall, Biophys. Chem., 2006, 119, 307–315 CrossRef CAS PubMed.
- A. Zall, D. Kieser, N. Höttecke, E. C. Naumann, B. Thomaszewski, K. Schneider, D. T. Steinbacher, R. Schubenel, S. Masur, K. Baumann and B. Schmidt, Bioorg. Med. Chem., 2011, 19, 4903–4909 CrossRef CAS PubMed.
- C. Ito, S. Katsuno, M. Itoigawa, N. Ruangrungsi, T. Mukainaka, M. Okuda, Y. Kitagawa, H. Tokuda, H. Nishino and H. Furukawa, J. Nat. Prod., 2000, 63, 125–128 CrossRef CAS PubMed.
- H. J. Knölker and K. R. Reddy, Chem. Rev., 2002, 102, 4303–4427 CrossRef PubMed.
- W. Maneerat, T. Ritthiwigrom, S. Cheenpracha, T. Promgool, K. Yossathera, S. Deachathai, W. Phakhodee and S. Laphookhieo, J. Nat. Prod., 2012, 75, 741–746 CrossRef CAS PubMed.
- X. You, D. Zhu, W. Lu, Y. Sun, S. Qiao, B. Luo, Y. Du, R. Pi, Y. Hu, P. Huang and S. Wen, RSC Adv., 2018, 8, 17183–17190 RSC.
- S. Issa, A. Prandina, N. Bedel, P. Rongved, S. Yous, M. Le Borgne and Z. Bouaziz, J. Enzyme Inhib. Med. Chem., 2019, 34, 1321–1346 CrossRef CAS PubMed.
- H. Z. Zhang, Z. L. Zhao and C. H. Zhou, Eur. J. Med. Chem., 2018, 144, 444–492 CrossRef CAS PubMed.
- A. Ayati, S. Emami, A. Asadipour, A. Shafiee and A. Foroumadi, Eur. J. Med. Chem., 2015, 97, 699–718 CrossRef CAS PubMed.
- D. Havrylyuk, O. Roman and R. Lesyk, Eur. J. Med. Chem., 2016, 113, 145–166 CrossRef CAS PubMed.
- P. H. Parikh, J. B. Timaniya, M. J. Patel and K. P. Patel, Med. Chem. Res., 2020, 29, 538–548 CrossRef CAS.
- I. M. Khan, M. Islam, S. Shakya, K. Alam, N. Alam and M. Shahid, Bioorg. Chem., 2020, 99, 103779 CrossRef CAS PubMed.
- A. R. Zabiulla Gulnaz, Y. H. E. Mohammed and S. A. Khanum, Bioorg. Chem., 2019, 92, 103220 CrossRef PubMed.
- D. C. Liu, M. J. Gao, Q. Huo, T. Ma, Y. Wang and C. Z. Wu, J. Enzyme Inhib. Med. Chem., 2019, 34, 829–837 CrossRef CAS PubMed.
- K. Karrouchi, S. Radi, Y. Ramli, J. Taoufik, Y. N. Mabkhot, F. A. Al-aizari and M. Ansar, Molecules, 2018, 23, 134 CrossRef PubMed.
- A. Drilon, J. W. Clark, J. Weiss, S.-H. I. Ou, D. R. Camidge, B. J. Solomon, G. A. Otterson, L. C. Villaruz, G. J. Riely, R. S. Heist, M. M. Awad, G. I. Shapiro, M. Satouchi, T. Hida, H. Hayashi, D. A. Murphy, S. C. Wang, S. Li, T. Usari, K. D. Wilner and P. K. Paik, Nat. Med., 2020, 26, 47–51 CrossRef CAS PubMed.
- A. T. Shaw, G. J. Riely, Y.-J. Bang, D.-W. Kim, D. R. Camidge, B. J. Solomon, M. Varella-Garcia, A. J. Iafrate, G. I. Shapiro, T. Usari, S. C. Wang, K. D. Wilner, J. W. Clark and S.-H. I. Ou, Ann. Oncol., 2019, 30, 1121–1126 CrossRef CAS PubMed.
- B. J. Solomon, B. Besse, T. M. Bauer, E. Felip, R. A. Soo, D. R. Camidge, R. Chiari, A. Bearz, C.-C. Lin, S. M. Gadgeel, G. J. Riely, E. H. Tan, T. Seto, L. P. James, J. S. Clancy, A. Abbattista, J.-F. Martini, J. Chen, G. Peltz, H. Thurm, S.-H. I. Ou and A. T. Shaw, Lancet Oncol., 2018, 19, 1654–1667 CrossRef CAS PubMed.
- C. S. Tam, S. Opat, D. Simpson, G. Cull, J. Munoz, T. J. Phillips, W. S. Kim, S. Rule, S. K. Atwal, R. Wei, W. Novotny, J. Huang, M. Wang and J. Trotman, Blood Adv., 2021, 5, 2577–2585 CrossRef PubMed.
- G. Ranieri, M. Mammì, E. Donato Di Paola, E. Russo, L. Gallelli, R. Citraro, C. D. Gadaleta, I. Marech, M. Ammendola and G. De Sarro, Crit. Rev. Oncol. Hematol., 2014, 89, 322–329 CrossRef PubMed.
- K. Nepali, S. Sharma, M. Sharma, P. M. S. Bedi and K. L. Dhar, Eur. J. Med. Chem., 2014, 77, 422–487 CrossRef CAS PubMed.
- V. G. Bhila, C. V. Patel, N. H. Patel and D. I. Brahmbhatt, Med. Chem. Res., 2013, 22, 4338–4346 CrossRef CAS.
- M. Patel and K. Patel, Heterocycl. Commun., 2019, 25, 146–151 CAS.
- Z. Li and Y. Xing, Res. Chem. Intermed., 2012, 38, 2223–2228 CrossRef CAS.
- J. Kapuscinski, Biotech. Histochem., 1995, 70, 220–233 CrossRef CAS PubMed.
- D. Donjerkovic and D. W. Scott, Cell Res., 2000, 10, 1–16 CrossRef CAS PubMed.
- S. Cory and J. M. Adams, Nat. Rev. Cancer, 2002, 2, 647–656 CrossRef CAS PubMed.
- W. F. De Azevedo, S. Leclerc, L. Meijer, L. Havlicek, M. Strnad and S. H. Kim, Eur. J. Biochem., 1997, 243, 518–526 CrossRef CAS PubMed.
- Y.-F. Sun and Y.-P. Cui, Dyes Pigm., 2008, 78, 65–76 CrossRef CAS.
- O. Trott and A. J. Olson, J. Comput. Chem., 2009, 31, 455–461 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1ra03970a |
| This journal is © The Royal Society of Chemistry 2021 |