
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceCovalent immobilization of gold nanoparticles on a plastic substrate and subsequent immobilization of biomolecules†
Mimari Matsumotoa,
Kazuki Kanekoa,
Manami Haraa,
Masaki Matsui a,
Kenta Moritaa and
Tatsuo Maruyama*ab
a,
Kenta Moritaa and
Tatsuo Maruyama*ab
aDepartment of Chemical Science and Engineering, Graduate School of Engineering, Kobe University, 1-1 Rokkodai, Nada-ku, Kobe 657-8501, Japan. E-mail: tmarutcm@crystal.kobe-u.ac.jp
bResearch Center for Membrane and Film Technology, Kobe University, 1-1 Rokkodai, Nada, Kobe 657-8501, Japan
First published on 2nd July 2021
Abstract
We propose a novel approach to stably immobilize gold nanoparticles (AuNPs) on a plastic substrate and demonstrate that the modified substrate is also capable of immobilizing biomolecules. To immobilize citrate-capped AuNPs, an acrylic substrate was simply dip-coated in a functional polymer solution to decorate the outermost surface with amino groups. Electrostatic interactions between AuNPs and the amino groups immobilized the AuNPs with a high density. The AuNP-modified acrylic substrate was transparent with a red tint. A heat treatment promoted the formation of amide bonds between carboxy groups on the AuNPs and amino groups on the substrate surface. These covalent bonds stabilized the immobilized AuNPs and the resulting substrate was resistant to washing with acid and thiol-containing solutions. The surface density of AuNPs was controlled by the surface density of amino groups on the substrate surface, which was in turn controlled by the dip-coating in the functional polymer solution. We attempted to immobilize functional biomolecules on the AuNPs-functionalized plastic surface by two different approaches. An enzyme (horseradish peroxidase) was successfully immobilized on the AuNPs through amide formation and 5′-thiolated DNA was also immobilized on the AuNPs through S–Au interactions. These chemistries allow for simultaneous immobilization of two different kinds of biomolecules on a plastic substrate without loss of their functional properties.
Introduction
Since the end of the last century, many studies have reported a variety of physical and chemical properties of metal nanoparticles that are promising for potential applications in optical, electronic, chemical, and biomedical fields. For example, gold nanoparticles (AuNPs) have strong absorption in the visible light region, which arises from surface plasmon resonance (SPR).1 The SPR phenomenon of AuNPs has been widely used in a number of analytical, optical, and biochemical studies. Although there have been many comprehensive studies of colloidal AuNPs in solution, there have been a limited number of works examining the great potential of two dimensional (2D) assemblies of monodisperse AuNPs on solid substrates. Integration of the functional properties of metal nanoparticles on a solid surface would enforce electronic and optical devices.2–4 Several groups showed promise for applications in analytical and electronic devices: electrochemical impedance spectroscopy,5 sensitive electrochemical detection,6 surface-enhanced Raman scattering,7,8 biosensing,9–12 memory devices,13,14 cell scaffolds,15 asymmetric functionalization of AuNPs,16,17 catalysts,18 and conductive surfaces.19 These studies are distinct from those of simple deposition of AuNPs on a surface.20–22 The chemistry of AuNPs immobilization is classified as an electrostatic interaction,11,23–27 S–Au bonds,11,23,27–31 other covalent bonds,13,32 or hydrophobic interactions.19 In many reports, the surfaces of glass and Si substrates are functionalized with various silane coupling agents to create interactions between AuNPs and the functionalized surfaces.33 The well-established synthesis of AuNPs uses tetrachloroauric acid and citric acid to produce a colloidal solution of citrate-capped AuNPs. Because of the negative surface charge of the AuNPs, electrostatic interactions are most frequently used to immobilize AuNPs on the functionalized surfaces of solid substrates.26Despite many studies of immobilization on glass and Si substrates, there have been few reports describing AuNPs immobilization on polymeric (plastic) substrates.19,34,35 Plastic substrates are typically transparent, disposable, lightweight, and easy to mold. These characteristics are beneficial for applications of 2D arrays of immobilized AuNPs in biosensing, electrical devices, cell culturing, and medical diagnostics. We have previously reported an approach based on functionalization with amino and carboxy groups at the outermost surface of a plastic substrate by dip-coating with a functional polymer.36–38 This method allowed control of the surface density of these functional groups on the substrate and was applicable to a variety of plastic substrates. In the present study, we aimed to establish a simple approach for stably immobilizing AuNPs on a surface of a transparent plastic substrate at a controlled density. A plastic substrate was functionalized with amino groups on its surface by the dip-coating approach and AuNPs were immobilized on the surface at a high density. Investigations with different washing solutions indicated that the electrostatic interactions alone were not sufficient to achieve stable immobilization; however, the formation of a covalent bonds between carboxy groups of AuNPs and amino groups of the substrate surface stabilized the AuNPs on the surface. Therefore, we succeeded in covalent immobilization of biomolecules on the AuNPs-immobilized plastic substrate based on two different chemistries.
Experimental
Materials
Synthesis of monomers and the functional polymer (termed PMBA, Fig. 1), synthesis of AuNPs, quantification of amino groups on a surface and surface morphology observation are described in the ESI.†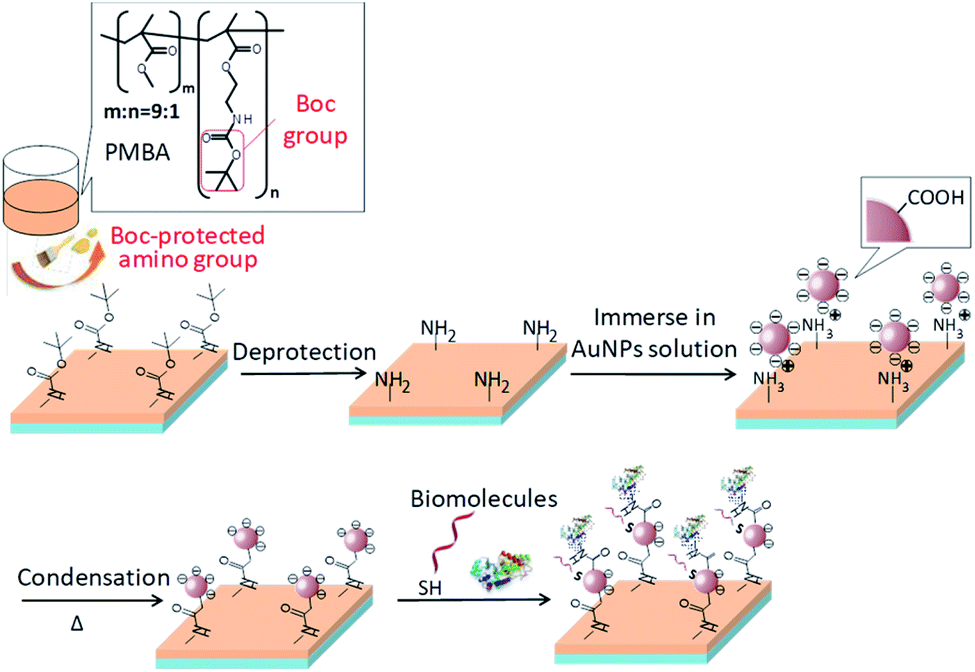 | ||
| Fig. 1 Schematic illustration of the processes of AuNPs immobilization on a plastic substrate and subsequent biomolecule immobilization. | ||
Dip-coating of a plastic substrate with a functional polymer (PMBA) to display amino groups on its surface36,38
An acrylic substrate (1 cm × 1 cm, 0.5 mm thick) was dip-coated with a 3 wt% PMBA solution to introduce amino groups on its surface. PMBA was dissolved in ethyl acetate at 3 wt%. An acrylic substrate was immersed in the polymeric solution for a few seconds and dried overnight at 25 °C in a vacuum chamber. Amino groups protected by tert-butyloxycarbonyl (Boc) groups on the dip-coated acrylic substrate surface were deprotected with 4 M aqueous HCl (2 mL) overnight at 40 °C to expose amino groups on the outermost surface.Immobilization of AuNPs on acrylic substrate surface by electrostatic interactions
A colloidal solution of AuNPs (pH 5–6) was prepared as reported previously (ESI†).39 The immobilization of AuNPs was performed by immersing the substrate in a AuNPs colloidal solution and the solution was gently mixed for 30 min at 25 °C. The substrates were slowly drawn from the solution and dried at 100 °C overnight.Before and after drying the substrates, the immobilization stability of the AuNPs was examined by immersing the substrates in a washing solution and shaking it for 1 h at 25 °C. Absorption spectra of the substrates were measured with a spectrophotometer (V-630, JASCO, Tokyo, Japan). Two different aqueous washing solutions were used: 5 mM dithiothreitol (DTT) and 0.1 M HCl solutions. The names of the samples are summarized in Table 1.
| Sample name | Description |
|---|---|
| Boc-substrate | Substrate displaying Boc-protected amino groups |
| NH2-substrate | Substrate displaying amino groups |
| (Washing solution)–AuNP-substrate | A AuNP-substrate was washed with a washing solution before drying. |
| Δ–AuNP–NH2-substrate | After immobilization of AuNPs on a substrate, a substrate was heated and dried at 100 °C overnight. |
| (Washing solution)–Δ–AuNP–NH2-substrate | A AuNP-substrate was washed with a washing solution after drying at 100 °C. |
Surface morphology observation of AuNPs-immobilized substrates
Surface morphologies of the substrates were observed with a field-emission scanning electron microscope (FE-SEM, JSF-7500F, JEOL, Tokyo). All SEM images were collected at an acceleration voltage of 7 kV and an emission current of 10 μA.FT-IR measurements
FT-IR measurements of the substrates surface were performed with an FT-IR spectrometer (Nicolet™ iS50, Thermo Fisher Scientific), equipped with an MCT detector. The measurements were performed with a p-polarized IR beam and an optical set-up with a single reflection diamond ATR accessory (Golden Gate™, Specac, Orpington, UK).Immobilization of horseradish peroxidase on AuNPs-immobilized substrate40
AuNPs-immobilized substrates were washed three times with Milli-Q water and immersed in 2 mL of Milli-Q water containing 5 mM N-hydroxysuccinimide and 10 mM water-soluble carbodiimide and shaken for 30 min at 25 °C. The substrates were immersed in phosphate buffer (0.1 M, pH 7.4, 2 mL) containing 0.3 mg mL−1 horseradish peroxidase (HRP) and 0.15 M NaCl and shaken for 2 h at 25 °C. The substrates were washed three times with phosphate buffer (0.1 M, pH 7.4) containing 0.05 vol% Tween-20 and 0.15 M NaCl, and then washed twice with Milli-Q water. The enzymatic activity of HRP on the substrate surface was measured with a mixture of 0.1 mL phosphate buffer and a 0.9 mL ELISA-POD substrate kit containing 2,2′-azinobis[3-ethylbenzothiazoline-6-sulfonic acid]-diammonium salt (ABTS) (Nakarai Tesque, Kyoto, Japan). A 100 μL portion of the reaction solution was periodically withdrawn and transferred to a UV/vis microplate reader to measure the absorbance at 405 nm.Immobilization of SH-DNA on AuNPs-immobilized substrate41
Prior to DNA immobilization, 5′-thiolated DNA (19-mer, 84.3 nmol) was dissolved in 843 μL of sterile water. A DNA solution (62.5 μL) was well mixed with 0.1 M DTT solution in 50 mM phosphate buffer (pH 7.0, containing 1 M KCl) in a microtube. The reduced 5′-thiolated DNA was purified using a NAP-5 column (GE-Healthcare, Piscataway, NJ) with 50 mM phosphate buffer (pH 7.0, containing 1 M KCl). The DNA solution (500 μL) eluted from a NAP-5 column was diluted with phosphate buffer to a concentration of 2.5 μM 5′-thiolated DNA in 50 mM phosphate buffer containing 1 M KCl. A solution of reduced 5′-thiolated DNA (100 μL) was then loaded as a droplet onto the surface of a AuNPs-immobilized substrate. After incubation for 2 h at 25 °C, the substrates were washed three times with phosphate buffer (50 mM containing 1 M KCl, pH 7.0). A solution of the complementary DNA strand conjugated with fluorescein isothiocyanate (FITC) at the 5′ end (2 μM in 8× saline sodium citrate (SSC) buffer of 100 μL) was loaded onto the DNA-immobilized surface. The substrate with a fluorescent DNA solution was heated at 80 °C for 10 min and then incubated in a humidity chamber at 40 °C overnight. This procedure was used for the DNA hybridization. After washing the substrate with excess 8× SSC buffer, the substrate was immersed in fresh phosphate buffer (0.1 M, pH 7.6, 2.0 mL) for 10 min at 90 °C to dissociate the DNA duplex. The FITC-labeled DNA liberated in the supernatant was measured with a fluorescent spectrophotometer (λex = 495 nm, λem = 515 nm) at 25 °C.Results and discussion
Amino group functionalization of an acrylic substrate surface by dip-coating with PMBA
We prepared an acrylic substrate displaying amino groups on its surface, as reported previously. Briefly, a random acrylic copolymer, having Boc-protected amino groups (termed PMBA, Fig. 1), was synthesized by free-radical polymerization of methyl methacrylate and 2-(tert-butoxycarbonylamino)ethyl methacrylate (Boc-AEMA) (Fig. S1†). Mw and Mw/Mn of PMBA were 7.3 × 104 and 1.5, respectively. Elemental analysis revealed that the monomer composition ratio (MMA/Boc-AEMA) was 9![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 (Table S1†). Since our previous work demonstrated that dip-coating with PMBA having a 9:1 monomer ratio provided a large amount of amino groups on the outermost surface,36 this monomer ratio was adopted in the present study. An acrylic substrate was dip-coated with a 3 wt% PMBA solution in ethyl acetate and dried. The coated PMBA was not detached from a substrate surface in an aqueous solution. The Boc protecting groups induced surface segregation of amino groups in PMBA on the outermost surface because hydrophobic moieties of a polymer are preferentially segregated at the air/polymer interface to minimize its surface energy.36 After dip-coating and drying, amino groups were displayed on the surface by deprotection of the Boc groups with 4 M HCl aqueous solution. Prior to immobilization of AuNPs, a cleavable fluorescent compound (FITC–S–S–COOH, Fig. S2†) was used to quantify the surface density of amino groups on the surface of the substrate (Fig. S3†).36,38 FITC–S–S–COOH has a reactive carboxy group and a cleavable disulfide bond. FITC–S–S–COOH conjugated with amino groups on the surface in the presence of a condensation agent. Reduction of the disulfide bond released the fluorophore into the solution. The amount of the immobilized fluorescent compound was quantified with a conventional fluorescence spectrophotometer. The surface of the substrate dip-coated with PMBA displayed amino groups at a density of approximately 27 pmol cm−2. Assuming that the amino groups were displayed on the substrate surface in an evenly spaced manner, the spacing between amino groups was calculated to be 2.5 nm. SEM observations showed that there were no foreign particles on the surface of the dip-coated substrate and the surface remained smooth after dip-coating (Fig. S4†). Notably, the acrylic substrate remained transparent after dip-coating and deprotection (Fig. S5†).
:1 (Table S1†). Since our previous work demonstrated that dip-coating with PMBA having a 9:1 monomer ratio provided a large amount of amino groups on the outermost surface,36 this monomer ratio was adopted in the present study. An acrylic substrate was dip-coated with a 3 wt% PMBA solution in ethyl acetate and dried. The coated PMBA was not detached from a substrate surface in an aqueous solution. The Boc protecting groups induced surface segregation of amino groups in PMBA on the outermost surface because hydrophobic moieties of a polymer are preferentially segregated at the air/polymer interface to minimize its surface energy.36 After dip-coating and drying, amino groups were displayed on the surface by deprotection of the Boc groups with 4 M HCl aqueous solution. Prior to immobilization of AuNPs, a cleavable fluorescent compound (FITC–S–S–COOH, Fig. S2†) was used to quantify the surface density of amino groups on the surface of the substrate (Fig. S3†).36,38 FITC–S–S–COOH has a reactive carboxy group and a cleavable disulfide bond. FITC–S–S–COOH conjugated with amino groups on the surface in the presence of a condensation agent. Reduction of the disulfide bond released the fluorophore into the solution. The amount of the immobilized fluorescent compound was quantified with a conventional fluorescence spectrophotometer. The surface of the substrate dip-coated with PMBA displayed amino groups at a density of approximately 27 pmol cm−2. Assuming that the amino groups were displayed on the substrate surface in an evenly spaced manner, the spacing between amino groups was calculated to be 2.5 nm. SEM observations showed that there were no foreign particles on the surface of the dip-coated substrate and the surface remained smooth after dip-coating (Fig. S4†). Notably, the acrylic substrate remained transparent after dip-coating and deprotection (Fig. S5†).
Immobilization of AuNPs on the surface of the acrylic substrate
AuNPs were synthesized from tetraauric acid and citric acid (Au-colloidal solution). TEM observations revealed that the mean particle size of the as-synthesized AuNPs was 13 nm (Fig. 2a). Fig. 2b shows the absorption spectrum of the colloidal solution. The maximum absorption wavelength was 520 nm, which indicates the formation of AuNPs 15 nm in diameter.42 The aim of the present study was to immobilize AuNPs uniformly and stably on the surface of an acrylic substrate. Three different types of surfaces were examined for the AuNPs immobilization: a bare acrylic substrate (termed bare substrate), an acrylic substrate coated with PMBA without deprotection (termed Boc-substrate), and an acrylic substrate coated with PMBA and Boc-deprotected (displaying NH2 groups on the surface, termed NH2-substrate). These three substrate types were immersed in an Au-colloidal solution and then immersed in the washing solutions.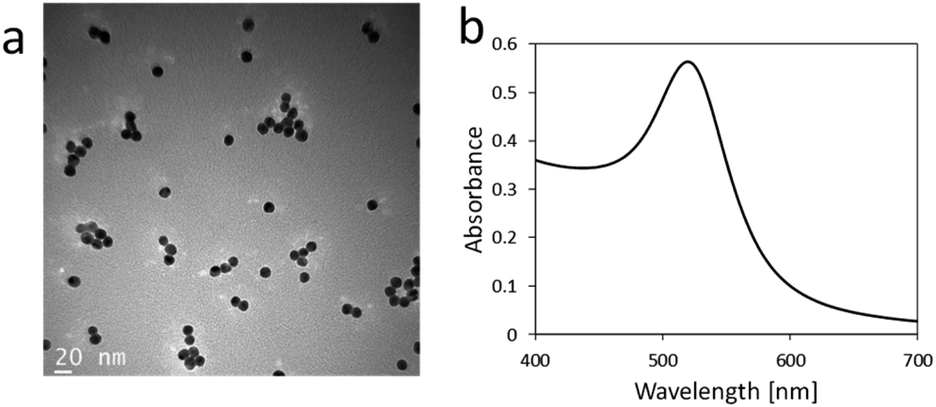 | ||
| Fig. 2 (a) TEM image of AuNPs synthesized in the present study. (b) Absorption spectrum of Au-colloidal solution. | ||
All the substrates tested were colored reddish or purplish on visual inspection, which was derived from the color of AuNPs (insets of Fig. 3a and S6†). The bare and Boc-substrates had a heterogeneous purplish color, indicating that AuNPs were aggregated on these surfaces. The NH2-substrate had a reddish and relatively homogeneous tint, indicating that AuNPs were uniformly immobilized without aggregation. The surfaces of the AuNPs prepared in the present study were covered with citric acid. The electrostatic interactions between these carboxy groups and the amino groups displayed on the surface of the substrate contributed to the uniform immobilization of AuNPs on the surface. The absorption spectra also confirmed the immobilization of the AuNPs (Fig. 3b).
 | ||
| Fig. 3 AuNPs-immobilized substrates. (a) SEM images of AuNPs-immobilized substrates. Insets are photographs of the substrates (see also Fig. S6†). (b) Absorption spectra of AuNPs-immobilized substrates. | ||
All the AuNPs-immobilized substrates had a strong absorption band at 500–600 nm. The AuNPs immobilized on the bare substrate had a red-shifted and broad absorption peak (peak at 564.5 nm, from an average of 4 substrates) compared with the original spectrum of the Au-colloidal solution (Fig. 2b). This result is consistent with aggregation of the AuNPs on the surface of a bare substrate. The Boc-substrate immobilized AuNPs had an absorption peak at 534.5 nm. The absorption spectrum of the NH2-substrate immobilizing AuNPs had a sharp and intense absorption at 531.5 nm, which was close to that of the Au-colloidal solution. Lopez-Perez et al. reported that electrostatic interaction of citrate-capped AuNPs and a cationic biopolymer red-shifted and broadened the absorption spectrum of AuNPs.43 The electrostatic interaction in the present study would also account for the slightly red-shifted and broadened absorption spectrum of AuNPs on the NH2-substrate.
Observations by SEM imaging (Fig. 3a), showed a surface morphology that was consistent with the above results. Whereas AuNPs were aggregated and heterogeneously distributed on the surfaces of bare and Boc substrates (no washing in Fig. 3a), a large amount of non-aggregated AuNPs were homogeneously distributed over the NH2 substrate before washing.
We washed the AuNPs-immobilized substrates with two different aqueous solutions (DTT solution and HCl solution) to investigate the immobilization stability of the AuNPs. Visual observations of the substrates (insets of Fig. 3a) revealed that the color of bare and Boc-substrates with immobilized AuNPs were clearly diluted after washing. SEM observations demonstrated that washing with DTT and HCl solutions detached large portions of AuNPs from the surfaces of the bare and Boc-substrates and that washing with DTT and HCl solutions seemed to induce aggregation of AuNPs on the surfaces. Washing with DTT solution also induced aggregation of AuNPs on the NH2-substrate but a small portion of AuNPs was detached. Washing with HCl solution detached a considerable portion of AuNPs from the NH2-substrate. The decrease and red shift of the absorption spectra of the substrates also indicated detachment and aggregation of AuNPs induced by washing with DTT and HCl solutions. The aggregation of AuNPs on the surfaces indicated migration of AuNPs induced by the washing procedure. These investigations revealed that the AuNPs were homogeneously immobilized on the surface of a NH2-substrate with a high density owing to electrostatic interactions.
Effect of immersion time on AuNPs immobilization
The immersion time in an Au-colloidal solution was investigated for the NH2-substrate. The immersion time was varied from 5 to 60 min. The immobilized of AuNPs was evaluated from the absorption spectra and SEM observations. Time course absorption spectra (Fig. 4a) showed that the peak around 525 nm increased as the immersion time was prolonged. Although there was a small red shift in the maximal absorption peak, the absorption spectrum after 60 min of immersion was comparable to that after 30 min of immersion. The small difference in the immobilization of AuNPs between 30 and 60 min immersion, suggests that the AuNPs aggregated to a small extent for the 60 min immersion. SEM observations (Fig. 4b) also revealed that the coverage of AuNPs on the surface increased as the immersion time increased. The substrate surfaces were densely covered with AuNPs after 30 and 60 min immersion. Some aggregates of AuNPs were found on the surface after 60 min of immersion, which agreed with the results of the absorption spectrum. Thus, immersion for 30 min in the Au-colloidal solution was sufficient for the AuNPs immobilization and used in the following experiments.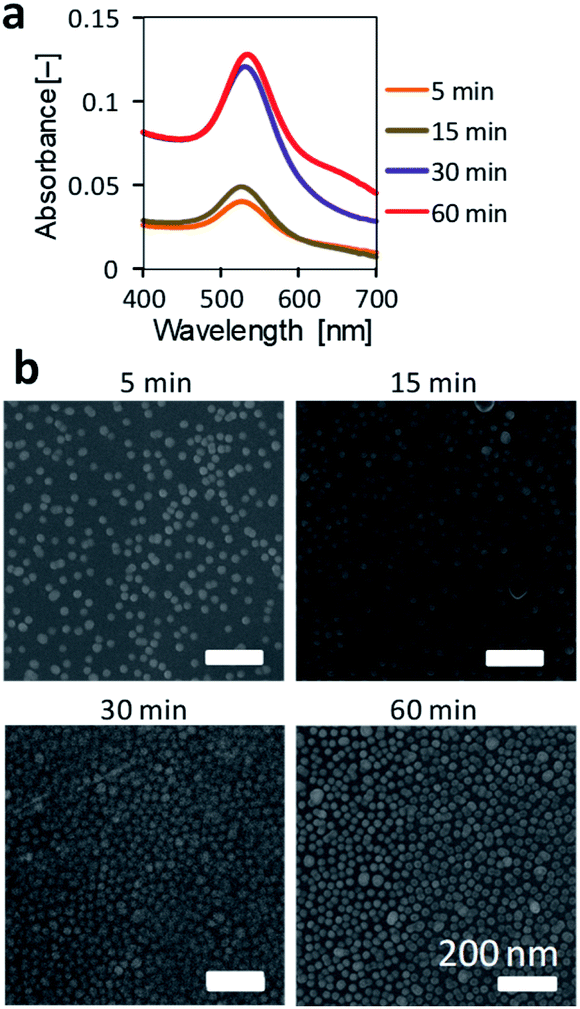 | ||
| Fig. 4 Effect of immersion time on the AuNPs immobilization on an NH2-substrate. (a) Absorbance spectra of NH2-substrates immobilizing AuNPs. (b) SEM observation of NH2-substrates immobilizing AuNPs. | ||
Formation of covalent bonds between AuNPs and a NH2-substrate
The above experiments are based on the electrostatic interactions of AuNPs immobilized on a NH2-substrate. Washing experiments revealed that the interaction between the AuNPs and the NH2-substrate surface was reversible. Hence, we attempted to increase the immobilization stability of the AuNPs. The AuNP-immobilized NH2-substrate was dried at 100 °C overnight to form amide bonds between carboxy groups of the AuNP and amino groups on the substrate (Δ–AuNP–NH2-substrate). We found that drying at 100 °C prevented detachment of AuNPs from the surface even after washing with DTT and HCl aqueous solutions (Fig. 5a and b). The washing procedures did not induce aggregation, which confirms that there was no migration of AuNPs. Indeed, there was little difference in the appearance of the Δ–AuNP–NH2-substrates between before and after washing (Fig. 5c).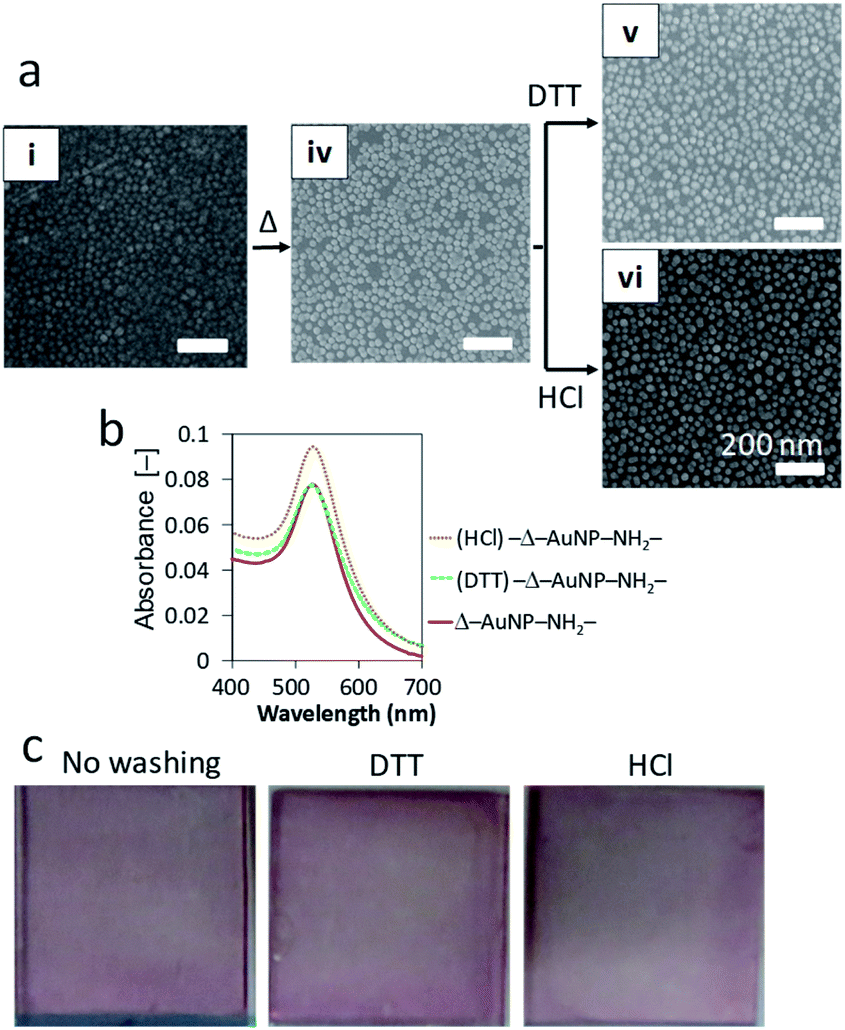 | ||
| Fig. 5 (a) SEM observation of AuNP–NH2-substrates before and after heat treatment (Δ). (b) Absorbance spectra of Δ–AuNP–NH2-substrates after washing. (c) Photographs of Δ–AuNP–NH2-substrates before and after washing. | ||
FT-IR measurements showed an absorbance peak at 1620 cm−1 only for the Δ–AuNP–NH2-substrate (Fig. 6), which suggested the formation of amide bonds.44,45 The relatively small and broad absorbance peak would be due to the small amount of amide bonds present only on the substrate surface. Hence, drying at 100 °C led to the formation of an amide bond between a carboxy group of a AuNP and an amino group on the surface of the substrate. Although Lopez-Perez et al. reported that the covalent linkage (amide formation) between citrate-capped AuNPs and NH2 groups of a biopolymer largely shifted the absorption wavelength typical of AuNPs,43 we did not observe a significant change in the absorption spectrum before and after heat treatment. This would be because only a limited portion (limited contact area) of carboxy groups on AuNPs involved in the amide formation. When the Δ–AuNP–NH2-substrate was stored in the dry state at room temperature for 5 days, there observed no significant difference in the absorbance spectra. The amide bond formation contributes to the high stability of the immobilized AuNPs. The following experiments were performed using a Δ–AuNP–NH2-substrate.
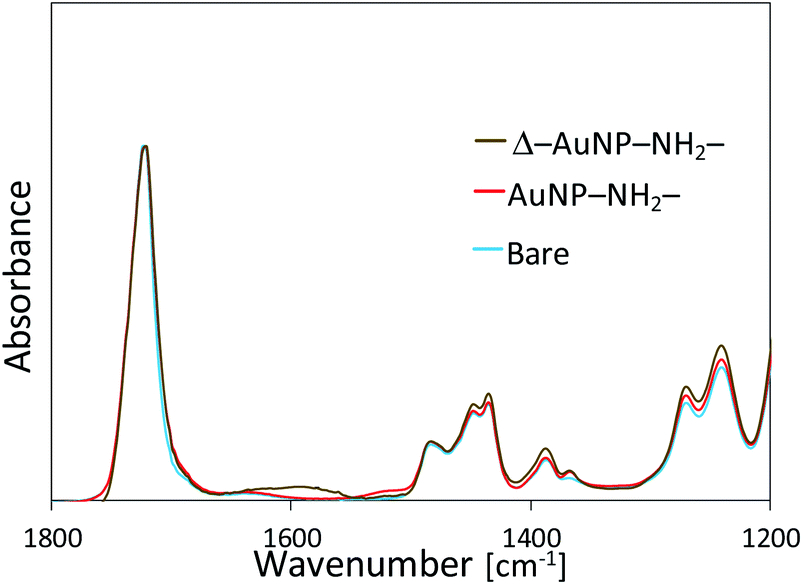 | ||
| Fig. 6 FT-IR spectra of AuNPs-immobilized substrates before and after heat treatment. | ||
Control of AuNPs immobilization by the surface-displayed amino groups
In our previous study, we reported that the amount of amino groups displayed on the acrylic substrate surface can be controlled by mixing solutions of PMBA and PMMA for dip-coating.36 Because the amount of surface-displayed amino groups is proportional to the weight ratio of PMBA in the mixture, the amount of surface-displayed amino groups and the mean spacing of amino groups were estimated (Table 2).| PMBA/PMMA (weight ratio) in a polymer solution | Surface density of NH2 groups [pmol cm−2] | Estimated mean spacing of NH2 groups [nm] | Immobilized AuNPsb (/μm2) |
|---|---|---|---|
| a Surface density of the NH2 groups is proportional to the PMBA content in a polymer mixture.36b Number of AuNPs was counted from the SEM images (Fig. 7). | |||
| PMBA/PMMA = 1/0 | 27 | 2.5 | 943 |
| PMBA/PMMA = 1/9 | 2.7 | 7.8 | 556 |
| PMBA/PMMA = 1/19 | 1.4 | 11 | 303 |
| PMBA/PMMA = 1/99 | 0.27 | 25 | 49 |
We investigated control of the AuNPs immobilization by varying the amount of surface-displayed amino groups. Substrate samples were prepared by dip-coating with mixed solutions of PMBA/PMMA (1:0, 1:9, 1:19, and 1:99 w/w). The substrates were immersed in the Au-colloidal solution and dried at 100 °C overnight. The amount of immobilized AuNPs decreased as the PMBA content decreased in the PMBA/PMMA solution (Fig. 7). Although the number of immobilized AuNPs decreased as the PMBA/PMMA ratio decreased, the number of immobilized AuNPs was not directly proportional to the surface density of NH2 groups (Table 2). Since the mean diameter of a AuNP was 13 nm and the estimated mean spacing of NH2 groups was less than 13 nm except for the PMBA/PMMA ratio of 1:99, more than one NH2 group were used to immobilize a single AuNP when the PMBA/PMMA ratio was 1:0, 1:9, and 1:19. When the PMBA/PMMA ratio was 1:19, the amount of immobilized AuNPs was obviously small, suggesting that multipoint binding is required for the AuNP immobilization. Hence, the non-stoichiometric behavior was observed towards AuNPs immobilization.
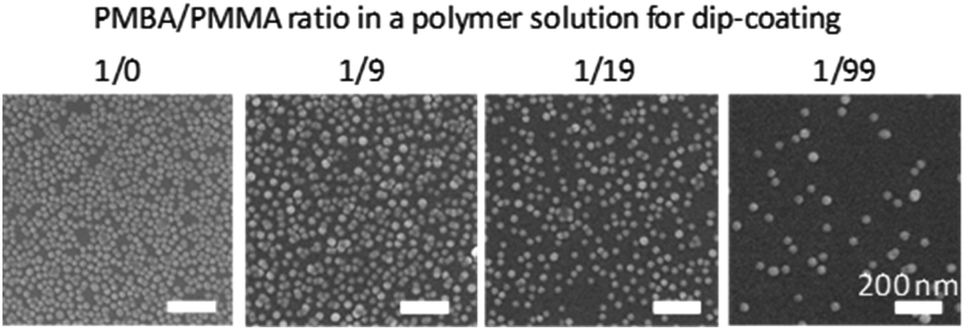 | ||
| Fig. 7 SEM observation of Δ–AuNP–NH2-substrates prepared with various ratios of PMBA/PMMA in a polymer solution. | ||
Immobilization of horseradish peroxidase (HRP) on AuNPs-immobilized substrates
Gold-deposited surfaces have been used to immobilize biomolecules and ligands for electrochemistry, biosensing, and diagnostics; thus, the AuNPs-immobilized substrate might be used to immobilize biomolecules. Here, we attempted to immobilize horseradish peroxidase (HRP) on a AuNPs-immobilized substrate. As mentioned above, carboxy groups derived from citrate were displayed on the immobilized AuNPs. Because HRP has two externally exposed amino groups, the carboxy group of the AuNPs could conjugate with N-hydroxysuccinimide (NHS) in the presence of a condensation agent (water-soluble carbodiimide) and then the amino groups of HRP reacted with the NHS ester to immobilize HRP on the AuNPs-immobilized substrate (Fig. 8a). Hydrogen peroxide and ABTS were used as substrates to measure the HRP activity. Fig. 8b shows the ABTS oxidation catalyzed by the HRP immobilized on the substrates. All the substrates underwent the HRP immobilization process. The bare substrates had negligible HRP activity. The NH2-substrate also showed negligible HRP activity. These results suggest that there was negligible nonspecific adsorption of HRP on bare and NH2-substrates. The Δ–AuNP–NH2-substrate had remarkable activity, indicating successful immobilization of active HRP on the substrate surface. It should be noted that the HRP immobilization did not cause a significant change in absorbance spectrum (around 525 nm).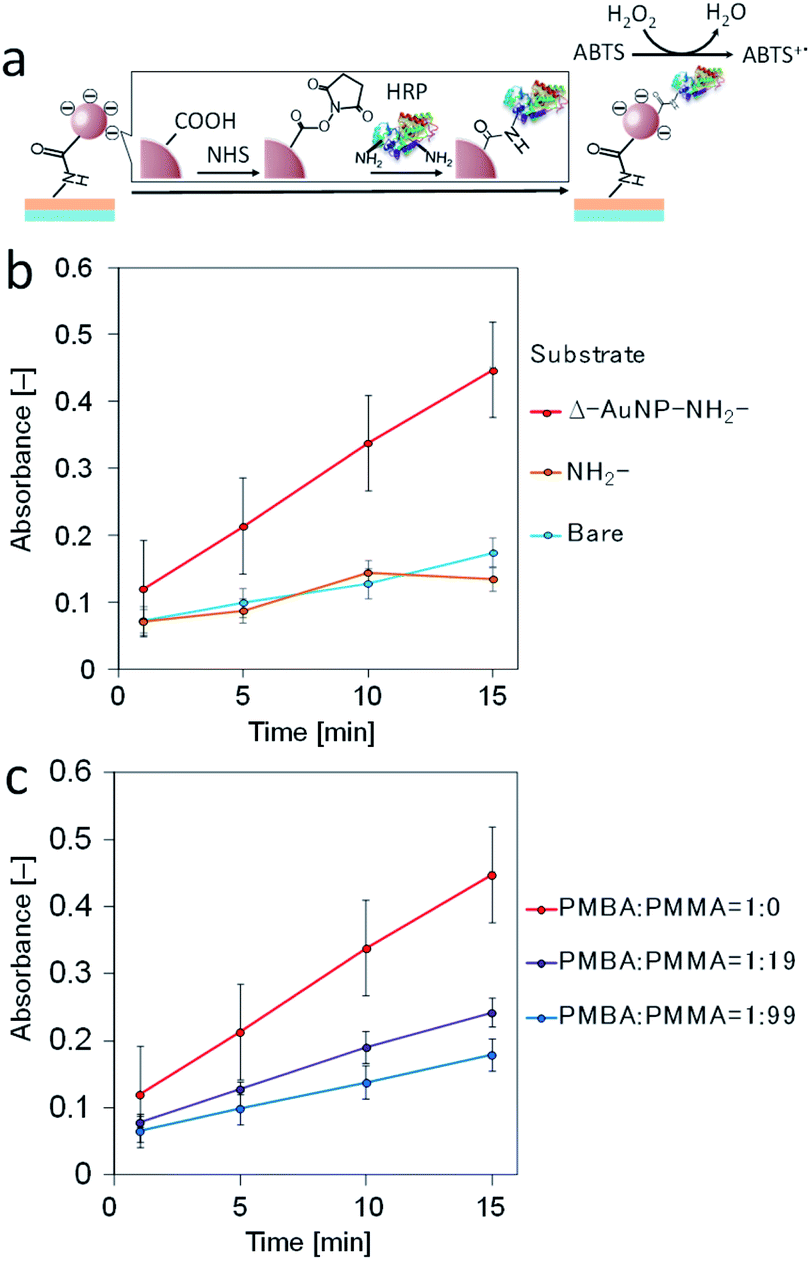 | ||
| Fig. 8 (a) Schematic illustration of the immobilization of horseradish peroxidase (HRP) on a AuNPs-immobilized substrate. (b) ABTS oxidation catalyzed by HRP immobilized on a substrate. (c) Effect of the PMBA content on the ABTS oxidation catalyzed by HRP immobilized on a substrate. | ||
In the above investigation, we demonstrated the density control of the immobilized AuNPs by varying the PMBA content in the dip-coating solution. Here, we attempted to control the HRP immobilization by controlling the amount of immobilized AuNPs. The PMBA/PMMA ratios (1:0, 1:19, and 1:99 in weight) in a polymer mixture were used. The HRP activity decreased as the PMBA content decreased (Fig. 8c). These results indicate that the amount of immobilized HRP decreased as the amount of AuNPs on the substrate surface decreased.
Immobilization of 5′-thiolated DNA (SH-DNA)
Thiol-containing substances are widely used to modify a gold substrate because of its unique interaction with a gold surface. We used this interaction to immobilize 5′-thiolated DNA (SH-DNA, 20-mer) on the Δ–AuNP–NH2-substrate (Fig. 9a). The immobilized SH-DNA was quantified by fluorescently labeled complementary DNA (FITC-DNA). After the duplex formation of SH-DNA and FITC-DNA, the complementary strand was liberated to the solution by heating, and the fluorescence was measured. Fig. 9b shows that SH-DNA was immobilized only on the Δ–AuNP–NH2-substrate and that the amount of the immobilized SH-DNA was 24 pmol cm−2, which was comparable with our previous reports.36,41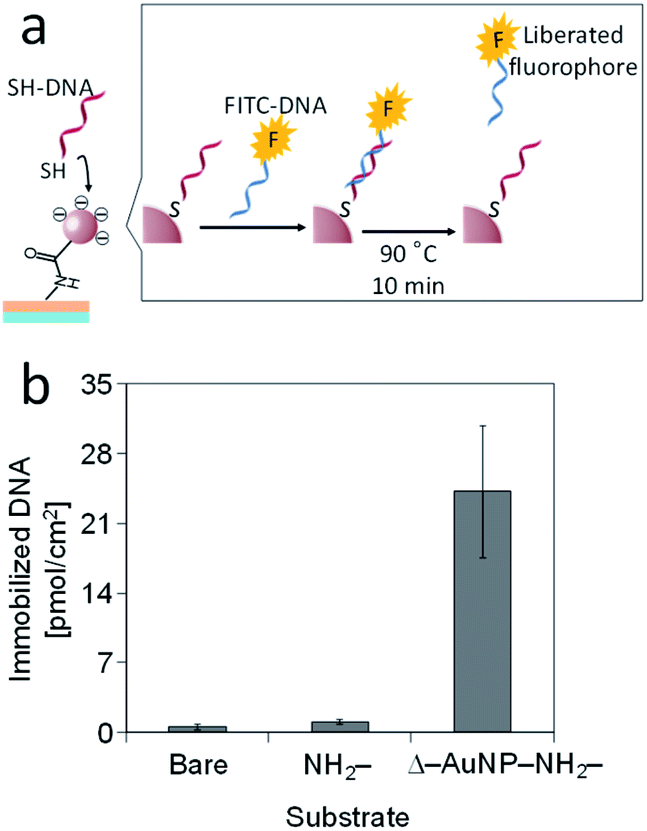 | ||
| Fig. 9 (a) Schematic illustration of the immobilization of SH-DNA on a AuNPs-immobilized substrate. (b) Immobilization of SH-DNA on bare, NH2- and AuNPs-immobilized substrates. | ||
Non-thiolated DNA was also used for the DNA immobilization instead of SH-DNA and the duplex formation with FITC-DNA was evaluated. We did not observe FITC-DNA liberated from the bare, NH2– and Δ–AuNP–NH2-substrates, meaning no DNA duplex formation on the substrates in the absence of SH-DNA.
Simultaneous immobilization of HRP and SH-DNA
Co-immobilization of multiple functional molecules and nanosized assemblies extends the potential of surface engineering, especially in sensing, biotechnology and catalyst engineering.46–48 Finally, we attempted to immobilize both molecules on the same substrate surface (Fig. 10a). First, HRP was immobilized on a Δ–AuNP–NH2-substrate and the HRP activity was measured before immobilization of SH-DNA (Fig. 10b). Then, SH-DNA was immobilized on the surface of the substrate and quantified by FITC-DNA to reveal that SH-DNA was immobilized on the surface at 70 pmol cm−2 (Fig. 10c). Again, the HRP activity was measured. Fig. 10b shows the ABTS oxidation catalyzed by immobilized HRP before and after the immobilization of SH-DNA. Both HRP and SH-DNA were successfully immobilized on the same substrate surface. However, the oxidation of ABTS before immobilization of SH-DNA was 3 times as fast as that after the immobilization of SH-DNA. Because SH-DNA and ABTS are negatively charged, electrostatic repulsions between them likely prevent access of ABTS to the substrate surface, resulting in slow oxidation of ABTS.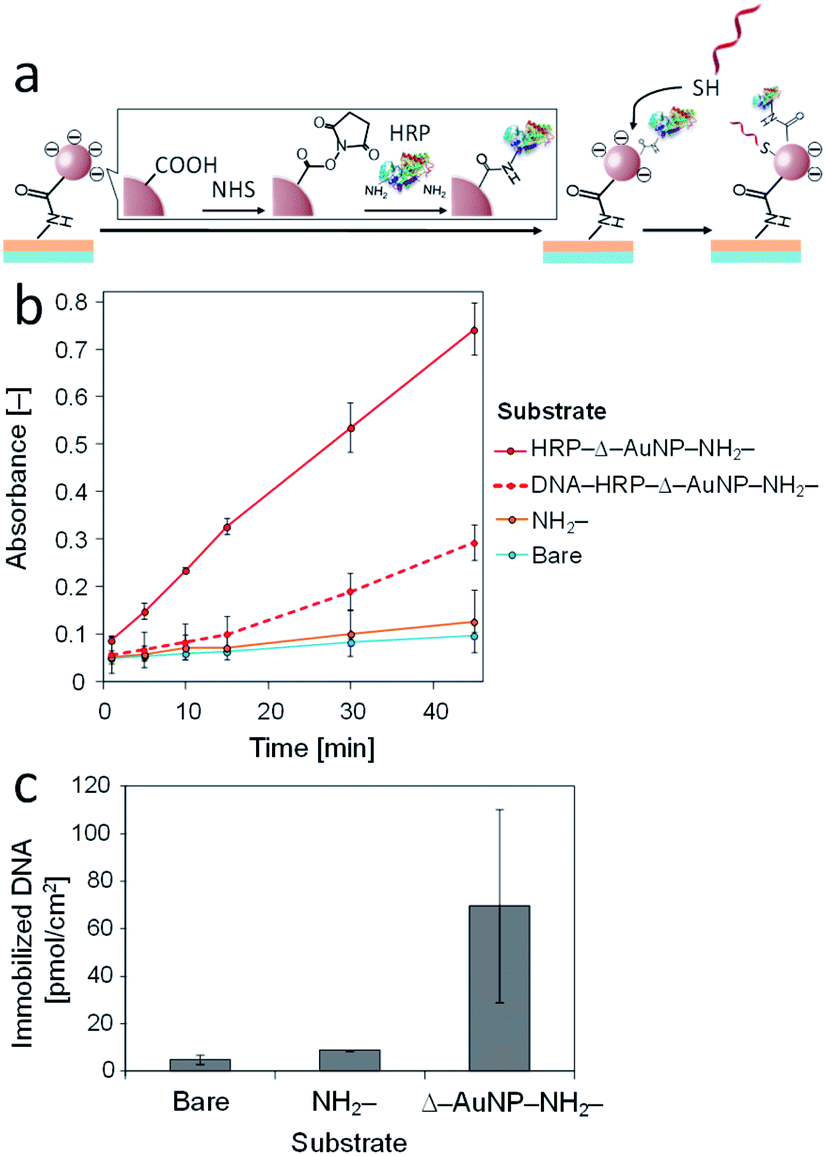 | ||
| Fig. 10 (a) Schematic illustration of simultaneous immobilization of SH-DNA and HRP on a AuNPs-immobilized substrate. (b) ABTS oxidation catalyzed by HRP immobilized on a substrate. (c) SH-DNA immobilized on a substrate. | ||
The amount of the SH-DNA immobilized in the presence of HRP (70 pmol cm−2) was greater than that in the absence of HRP (24 pmol cm−2). The surface of a Δ–AuNP–NH2-substrate was negatively charged owing to the carboxy groups on the AuNPs and SH-DNA being negatively charged, which inhibited access of SH-DNA to the surface. To immobilize HRP on the surface, a portion of the carboxy groups were transformed to amide. HRP (pI 7.2) is electrically neutral under the present conditions (pH 7.4).49 This effect reduced the electrostatic repulsion between the surface and SH-DNA, resulting in a large amount of the SH-DNA immobilizing in the presence of HRP. It should be noted that we did not observe any DNA immobilization on the HRP-immobilized surface when non-thiolated DNA was used instead of SH-DNA.
Conclusions
In the present study, we prepared a plastic substrate displaying amino groups on its surface by simple dip-coating with a functional polymer solution and succeeded in immobilizing AuNPs on the surface with a high density. A heat treatment formed amide bonds between the carboxy groups on AuNPs and amino groups on the substrate surface. These covalent bonds stabilized the immobilized AuNPs, and the resulting substrate was resistant to washing with acid and thiol-containing aqueous solutions. The acrylic substrates with immobilized AuNPs were transparent with a red tint. The surface density of amino groups was controlled on the basis of the composition of a functional polymer solution used for dip-coating, which in turn controlled the surface density of immobilized AuNPs. We finally succeeded in immobilizing biomolecules onto the AuNPs-immobilized plastic surface by two different chemistries. The enzyme (HRP) was immobilized on the AuNPs by amide formation and 5′-thiolated DNA was immobilized on the AuNPs by S–Au interactions. These reactions allowed for simultaneous immobilization of two different kinds of biomolecules onto a plastic substrate, which would extend the potential of (bio)catalysts immobilized on a surface.50 This AuNPs-immobilized on a plastic substrate is transparent, lightweight, disposable, and capable of readily immobilizing biomolecules, suggesting great potential in biosensing and diagnostics.Author contributions
M. Matsumoto and T. M. planed the study framework and wrote the manuscript. M. Matsumoto, K. K. and M. H. did the experiments. M. Matsui did the FTIR measurements. M. Matsumoto and K. M. did the microscope observations. All authors have given approval to the final version of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This study was financially supported by JSPS KAKENHI Grant Numbers 20H02542, 20H04711 and 19H05458. The authors thank the Edanz Group (https://www.edanzediting.com/ac) for editing a draft of this manuscript.Notes and references
- Y. C. Yeh, B. Creran and V. M. Rotello, Nanoscale, 2012, 4, 1871–1880 RSC.
- K. Sugawa, T. Akiyama, H. Kawazumi and S. Yamada, Langmuir, 2009, 25, 3887–3893 CrossRef CAS PubMed.
- J. H. Lee, B. S. Park, H. G. Ghang, H. Song and S. Y. Yang, ACS Appl. Mater. Interfaces, 2018, 10, 13397–13405 CrossRef CAS PubMed.
- A. Svard, J. Neilands, E. Palm, G. Svensater, T. Bengtsson and D. Aili, ACS Appl. Nano Mater., 2020, 3, 9822–9830 CrossRef.
- Y. Z. Fu, R. Yuan, L. Xu, Y. Q. Chai, X. Zhong and D. P. Tang, Biochem. Eng. J., 2005, 23, 37–44 CrossRef CAS.
- S. Garabagiu and G. Mihailescu, J. Electroanal. Chem., 2011, 659, 196–200 CrossRef CAS.
- H. Gehan, L. Fillaud, M. M. Chehimi, J. Aubard, A. Hohenau, N. Felidj and C. Mangeney, ACS Nano, 2010, 4, 6491–6500 CrossRef CAS PubMed.
- S. Karabel Ocal, J. Patarroyo, N. B. Kiremitler, S. Pekdemir, V. F. Puntes and M. S. Onses, J. Colloid Interface Sci., 2018, 532, 449–455 CrossRef CAS PubMed.
- H. Y. Lin, C. T. Chen and Y. C. Chen, Anal. Chem., 2006, 78, 6873–6878 CrossRef CAS PubMed.
- G. Jie, B. Liu, H. Pan, J. J. Zhu and H. Y. Chen, Anal. Chem., 2007, 79, 5574–5581 CrossRef CAS PubMed.
- M. Ben Haddada, M. Huebner, S. Casale, D. Knopp, R. Niessner, M. Salmain and S. Boujday, J. Phys. Chem. C, 2016, 120, 29302–29311 CrossRef CAS.
- N. Bhalla, D. Lee, S. Sathish and A. Q. Shen, Nanoscale, 2017, 9, 547–554 RSC.
- R. K. Gupta, D. Y. Kusuma, P. S. Lee and M. P. Srinivasan, ACS Appl. Mater. Interfaces, 2011, 3, 4619–4625 CrossRef CAS PubMed.
- R. K. Gupta, G. Ying, M. P. Srinivasan and P. S. Lee, J. Phys. Chem. B, 2012, 116, 9784–9790 CrossRef CAS PubMed.
- P. Sengupta and B. L. V. Prasad, ACS Omega, 2018, 3, 4242–4251 CrossRef CAS PubMed.
- R. Sardar, T. B. Heap and J. S. Shumaker-Parry, J. Am. Chem. Soc., 2007, 129, 5356–5357 CrossRef CAS PubMed.
- A. Mandl, S. L. Filbrun and J. D. Driskell, Bioconjugate Chem., 2017, 28, 38–42 CrossRef CAS PubMed.
- S. A. C. Carabineiro, L. M. D. R. S. Martins, M. Avalos-Borja, J. G. Buijnsters, A. J. L. Pombeiro and J. L. Figueiredo, Appl. Catal., A, 2013, 467, 279–290 CrossRef CAS.
- H. Shiigi, Y. Yamamoto, H. Yakabe, S. Tokonami and T. Nagaoka, Chem. Commun., 2003, 1038–1039, 10.1039/b212226j.
- X. M. Lin, H. M. Jaeger, C. M. Sorensen and K. J. Klabunde, J. Phys. Chem. B, 2001, 105, 3353–3357 CrossRef CAS.
- S. T. Liu, T. Zhu, R. S. Hu and Z. F. Liu, Phys. Chem. Chem. Phys., 2002, 4, 6059–6062 RSC.
- K. Bhattacharjee, K. Biswas and B. L. V. Prasad, J. Phys. Chem. C, 2020, 124, 23446–23453 CrossRef CAS.
- H. X. He, H. Zhang, Q. G. Li, T. Zhu, S. F. Y. Li and Z. F. Liu, Langmuir, 2000, 16, 3846–3851 CrossRef CAS.
- E. S. Kooij, H. Wormeester, E. A. M. Brouwer, E. van Vroonhoven, A. van Silfhout and B. Poelsema, Langmuir, 2002, 18, 4401–4413 CrossRef CAS.
- A. J. Downard, E. S. Q. Tan and S. S. C. Yu, New J. Chem., 2006, 30, 1283–1288 RSC.
- C. F. Chen, S. D. Tzeng, M. H. Lin and S. Gwo, Langmuir, 2006, 22, 7819–7824 CrossRef CAS PubMed.
- M. Ben Haddada, J. Blanchard, S. Casale, J. M. Krafft, A. Vallee, C. Methivier and S. Boujday, Gold Bull., 2013, 46, 335–341 CrossRef.
- K. C. Grabar, P. C. Smith, M. D. Musick, J. A. Davis, D. G. Walter, M. A. Jackson, A. P. Guthrie and M. J. Natan, J. Am. Chem. Soc., 1996, 118, 1148–1153 CrossRef CAS.
- X. D. Xu, Y. C. Wang and Z. F. Liu, J. Cryst. Growth, 2005, 285, 372–379 CrossRef CAS.
- K. Kimura, H. Yao and S. Sato, Synth. React. Inorg., Met.-Org., Nano-Met. Chem., 2006, 36, 237–264 CrossRef CAS.
- W. H. Chen, Y. T. Tseng, S. Hsieh, W. C. Liu, C. W. Hsieh, C. W. Wu, C. H. Huang, H. Y. Lin, C. W. Chen, P. Y. Lin and L. K. Chau, RSC Adv., 2014, 4, 46527–46535 RSC.
- Y. Yamanoi, T. Yonezawa, N. Shirahata and H. Nishihara, Langmuir, 2004, 20, 1054–1056 CrossRef CAS PubMed.
- O. Seitz, M. M. Chehimi, E. Cabet-Deliry, S. Truong, N. Felidj, C. Perruchot, S. J. Greaves and J. F. Watts, Colloids Surf., A, 2003, 218, 225–239 CrossRef CAS.
- J. Nagel, P. Chunsod, C. Zimmerer, F. Simon, A. Janke and G. Heinrich, Mater. Chem. Phys., 2011, 129, 599–604 CrossRef CAS.
- V. D'Britto, S. Tiwari, V. Purohit, P. P. Wadgaonkar, S. V. Bhoraskar, R. R. Bhonde and B. L. V. Prasad, J. Mater. Chem., 2009, 19, 544–550 RSC.
- A. Shimomura, T. Nishino and T. Maruyama, Langmuir, 2013, 29, 932–938 CrossRef CAS PubMed.
- S. Yamamoto, S. Kitahata, A. Shimomura, K. Tokuda, T. Nishino and T. Maruyama, Langmuir, 2015, 31, 125–131 CrossRef CAS PubMed.
- S. Shiota, S. Yamamoto, A. Shimomura, A. Ojida, T. Nishino and T. Maruyama, Langmuir, 2015, 31, 8824–8829 CrossRef CAS PubMed.
- Y. Eguchi, T. Kato, T. Tanaka and T. Maruyama, Chem. Commun., 2017, 53, 5802–5805 RSC.
- M. Hara, S. Kitahata, K. Nishimori, K. Miyahara, K. Tokuda, T. Nishino and T. Maruyama, Polym. J., 2019, 51, 489–499 CrossRef CAS.
- K. Miyahara, R. Sakai, M. Hara and T. Maruyama, Colloid Polym. Sci., 2019, 297, 927–931 CrossRef CAS.
- R. Coradeghini, S. Gioria, C. P. Garcia, P. Nativo, F. Franchini, D. Gilliland, J. Ponti and F. Rossi, Toxicol. Lett., 2013, 217, 205–216 CrossRef CAS PubMed.
- G. Lopez-Perez, R. Prado-Gotor, J. A. Fuentes-Rojas and M. J. Martin-Valero, J. Mol. Liq., 2020, 302, 112381 CrossRef CAS.
- B. L. Frey and R. M. Corn, Anal. Chem., 1996, 68, 3187–3193 CrossRef CAS.
- T. Maruyama, S. Katoh, M. Nakajima, H. Nabetani, T. P. Abbott, A. Shono and K. Satoh, J. Membr. Sci., 2001, 192, 201–207 CrossRef CAS.
- X. P. Deng and J. Lahann, Macromol. Rapid Commun., 2012, 33, 1459–1465 CrossRef CAS PubMed.
- S. Rigo, G. Gunkel-Grabole, W. Meier and C. G. Palivan, Langmuir, 2019, 35, 4557–4565 CrossRef CAS PubMed.
- L. Sola, D. Brambilla, A. Mussida, R. Consonni, F. Damin, M. Cretich, A. Gori and M. Chiari, ChemRxiv, 2020 DOI:10.26434/chemrxiv.13135769.v1.
- J. L. Zhang, F. Zhang, H. J. Yang, X. L. Huang, H. Liu, J. Y. Zhang and S. W. Guo, Langmuir, 2010, 26, 6083–6085 CrossRef CAS PubMed.
- X. Deng, C. Friedmann and J. Lahann, Angew. Chem., Int. Ed., 2011, 50, 6522–6526 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1ra03902d |
| This journal is © The Royal Society of Chemistry 2021 |