
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceComputational insights into Ir(III)-catalyzed allylic C–H amination of terminal alkenes: mechanism, regioselectivity, and catalytic activity†
Deng Pana,
Gen Luo *b,
Yang Yua,
Jimin Yanga and
Yi Luo*ac
*b,
Yang Yua,
Jimin Yanga and
Yi Luo*ac
aState Key Laboratory of Fine Chemicals, School of Chemical Engineering, Dalian University of Technology, Dalian 116024, China. E-mail: luoyi@dlut.edu.cn
bInstitutes of Physical Science and Information Technology, Anhui University, Hefei 230601, China. E-mail: luogen@ahu.edu.cn
cPetroChina Petrochemical Research Institute, Beijing, 102206, China
First published on 26th May 2021
Abstract
Computational studies on Ir(III)-catalyzed intermolecular branch-selective allylic C–H amination of terminal olefins with methyl dioxazolone have been carried out to investigate the mechanism, including the origins of regioselectivity and catalytic activity difference. The result suggests that the reaction proceeds through generation of active species, alkene coordination, allylic C–H activation, decarboxylation, migratory insertion, and protodemetalation. The presence of AgNTf2 could thermodynamically promote the formation of catalytically active species [Cp*Ir(OAc)]+. Both the weaker Ir–C(internal) bond and the closer interatomic distance of N⋯C(internal) in the key allyl-Ir(V)-nitrenoid intermediate make the migratory insertion into Ir–C(internal) bond easier than into the Ir–C(terminal) bond, leading to branch-selective allylic C–H amidation. The high energy barrier for allylic C–H activation in the Co system could account for the observed sluggishness, which is mainly ascribed to the weaker coordination capacity of alkenes to the triplet Cp*Co(OAc)+ and the deficient metal⋯H interaction to assist hydrogen transfer.
Introduction
Nitrogen-containing molecules are ubiquitous in natural products, synthetic intermediates, pharmaceuticals, and functional materials. Therefore, the construction of C–N bonds has been of fundamental importance for synthetic chemists in past decades.1 In this context, transition-metal-mediated C–H aminations have become vital methods,2,3 among which direct amination of the C–H bond is one of the most efficient ways, which could dramatically simplify synthetic routes, providing a straightforward and atom-economy methodology. Since the first report of transition-metal-catalyzed direct C–H amination,4 remarkable advances have been made in this area both experimentally and theoretically.5,6 Compared with chelate-assisted C–H amination, the allylic C–H amination is of great interest because the vinyl group is not only prevalent in large amounts of compounds but is also feasible for functionalization. Despite significant advances in this field, the precise control of the regioselectivity in the C–H amination reactions is still challenging.7–10Recently, Rovis and co-workers reported an elegant work of Ir(III)-catalyzed intermolecular allylic C–H amination of unactivated terminal olefins using alkyl oxazolones as aminating agents, featuring excellent regioselectivity (Scheme 1a).11 A putative catalytic cycle was also proposed on the basis of relevant control experiments (Scheme 2). The monoacetato Cp*Ir(III) (OAc)+(NTf2)− (I), generated from [Cp*IrCl2]2, AgNTf2, and LiOAc, was proposed as the catalytically active species. Alkene coordination gave the π-complex II, and metalation (C–H activation) formed allyl-Ir(III) species III. The subsequent oxidation of Ir(III) via N–O bond cleavage of oxazolone and CO2 extrusion (decarboxylation) produced the allyl-Ir(V)-nitrenoid species IV. Subsequently, the regioselective migratory insertion and protodemetalation gave the branched amidation product and regenerate the active species I.
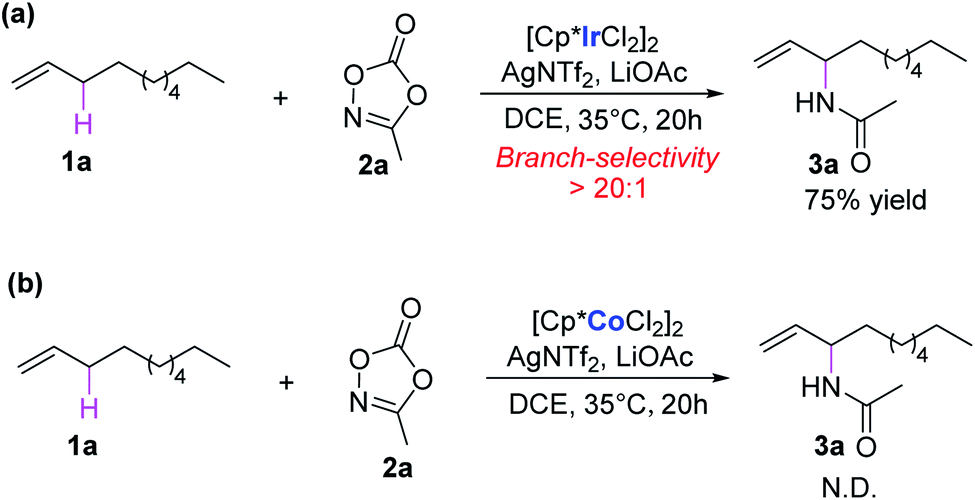 | ||
| Scheme 1 (a) Cp*Ir(III)- and (b) Cp*Co(III)-catalyzed allylic C–H amination of terminal alkene. | ||
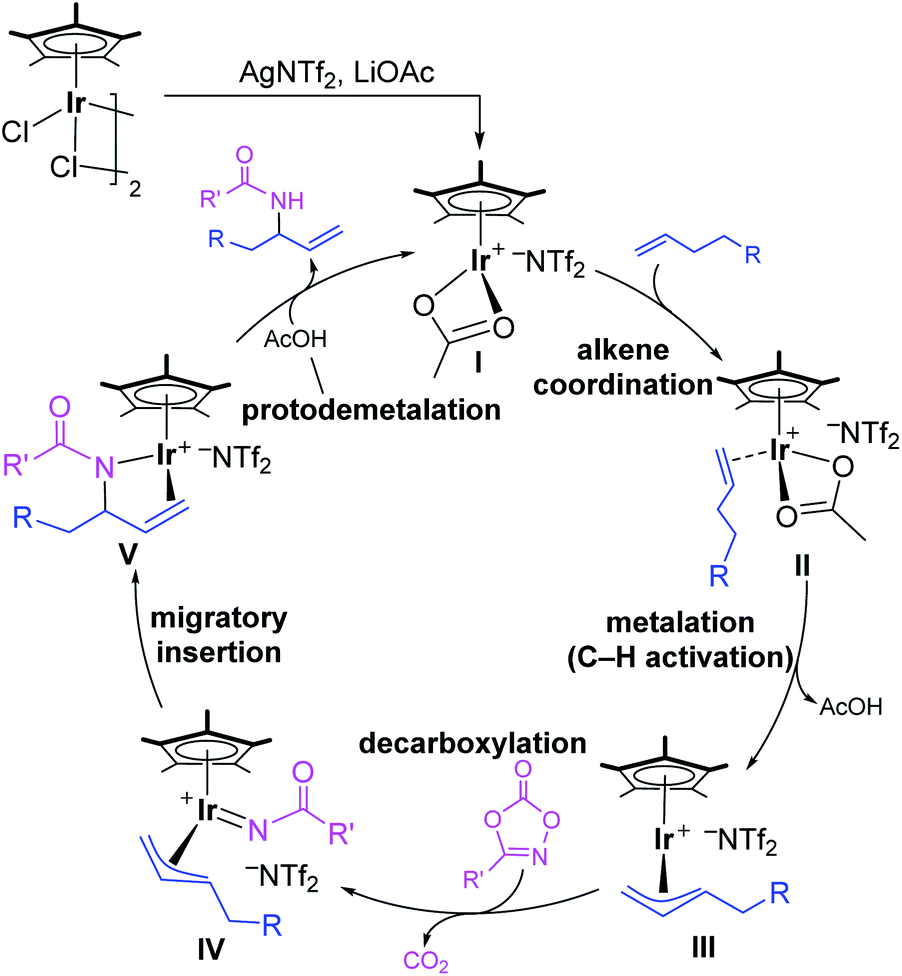 | ||
| Scheme 2 Plausible mechanism for Ir(III)-catalyzed allylic C–H amination of terminal alkene. | ||
Despite the above mechanistic suggestions, many details are still ambiguous. Such as the reasonability of the proposed mechanism, the structural information on the key intermediates and transition states, and the details of the energy profile. More importantly, the origin of regioselectivity for this important allylic C–H amination of unactivated terminal olefins remain unclear. In addition, it was also experimentally found that the reaction could not proceed when the analogue Cp*Co(III) was used as precatalyst (Scheme 1b). The origin of such distinct catalytic activity of these two similar precatalysts was also unknown. To clarify these issues, we performed density functional theory (DFT) calculations on the model reaction of Ir(III)/Co(III)-catalyzed intermolecular allylic C–H amination reaction of 1-decene (1a) with methyl dioxazolone (2a) (Scheme 1). The related mechanism has been carefully explored and some interesting results were obtained on the origins of regioselectivity and catalytic activity. The results reported here are expected to enrich the Ir(III)-catalyzed C–H amination of olefins and provides useful information for the development of new transition-metal-catalyzed regioselective C–H amination systems.
Computational details
All calculations were conducted using the Gaussian 16 program package.12 The hybrid B3LYP functional13 corrected with the empirical dispersion term (known as Grimme-D3 with the zero-damping scheme)14 was used for geometry optimizations of all stationary points. In the optimizations, the 6-31G(d,p) basis set was used for C, H, N, and O atoms, and the SDD pseudopotential basis sets15 were considered for metal atoms. Geometrical parameters for the stationary points were fully optimized without any constraints. Vibrational frequency analyses were performed on the optimized geometries at the same level of theory to characterize the stationary points as local minima (no imaginary frequency) or transition states (only one imaginary frequency). Intrinsic reaction coordinate (IRC) calculations were used to verify that the transition state connects with appropriate reactant and product. To obtain more accurate energies, single-point calculations were further performed at the higher level of B3LYP-D3/def2-TZVP16 together with the SMD solvation model17 for considering the dichloroethane solvation effect. The free energies in solution reported in this study include the free-energy corrections calculated by the vibrational frequency analyses. In addition, the minimum-energy crossing point (MECP) was optimized using the code developed by Harvey and co-workers.18 Three-dimensional structures were showed using the CYLview software.19Results and discussion
Formation of active species
It was experimentally found that the precatalyst [Cp*IrCl2]2 was ineffective without adding additional AgNTf2. This drove us to investigate the role of AgNTf2 and the formation of active species. Under the experimental conditions,11 various Ir(III) species (e.g., Cp*Ir(OAc)2, Cp*Ir(OAc)(NTf2), [Cp*Ir(OAc)]+ + X− (X− = OAc− and NTf2−), and [Cp*Ir(NTf2)]+ + OAc−) may exist in the presence of precatalyst [Cp*IrCl2]2, AgNTf2, and LiOAc. As shown in Scheme 3, in the absence of AgNTf2, the formation of neutral Cp*Ir(OAc)2 is endergonic by 16.3 kcal mol−1 (eqn (1)). The dissociation of an anion OAc− from Cp*Ir(OAc)2 to give cationic [Cp*Ir(OAc)]+ together with OAc− is further endergonic (ΔG = 31.2 kcal mol−1, eqn (2)), suggesting that it is an energetically inaccessible process. It is worth noting that, when AgNTf2 was introduced, the formation of neutral Cp*Ir(OAc) (NTf2) is less endergonic (ΔG = 9.3 kcal mol−1, eqn (3)) and the dissociation of NTf2− from Cp*Ir(OAc) (NTf2) to give cationic [Cp*Ir(OAc)]+ is only endergonic by 8.7 kcal mol−1 (eqn (4)). In addition, the formation of [Cp*Ir(OAc)]+ or [Cp*Ir(NTf2)]+ via two molecules of LiOAc and AgNTf2 was also calculated and was ruled out due to the relatively high reaction free energy (ΔG = 37.2 kcal mol−1, eqn (5); ΔG = 42.0 kcal mol−1, eqn (6)). Taken together with the above calculations, we can conclude that the cationic [Cp*Ir(OAc)]+ species should be the active species involved in further catalytic cycle. And, AgNTf2 play an important role in thermodynamically promoting the formation of [Cp*Ir(OAc)]+. These computational results could provide a preliminary understanding on the role of AgNTf2 additive and the formation of active species [Cp*Ir(OAc)]+.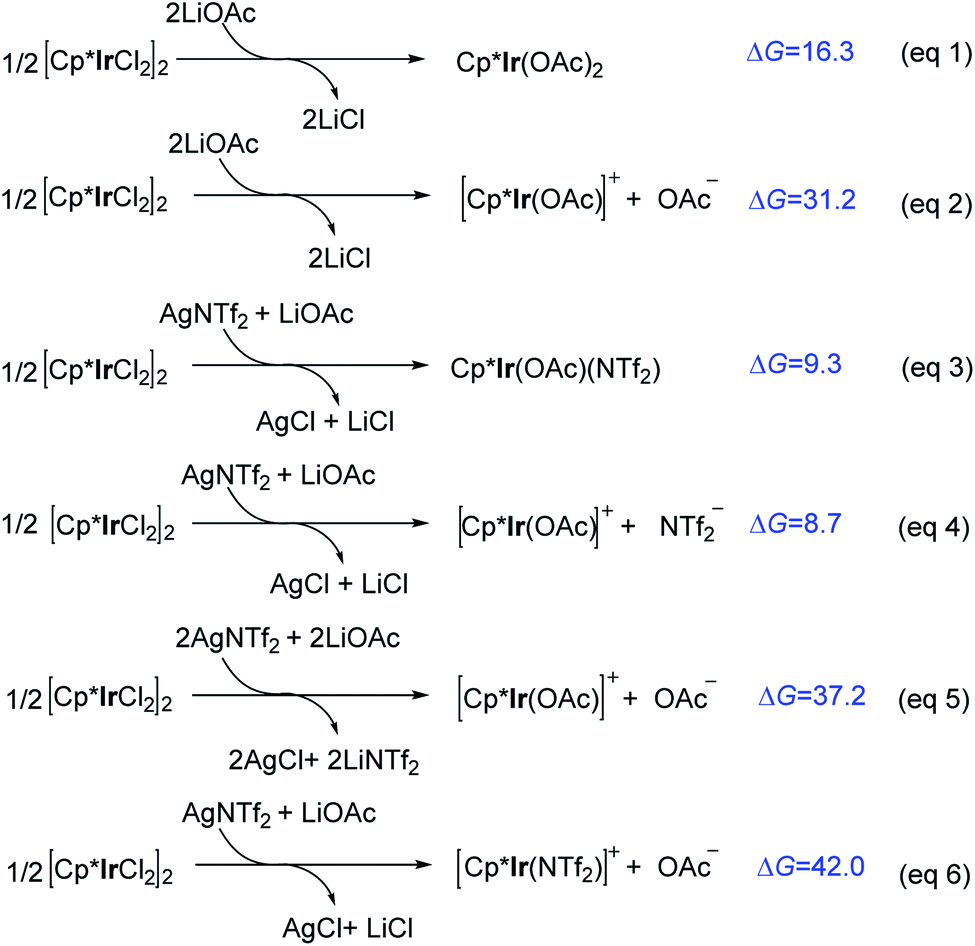 | ||
| Scheme 3 Calculated reaction free energies (ΔG, kcal mol−1) for the formation of various Ir(III) species. | ||
Matalation (allylic C–H activation) and decarboxylation
According to previous studies and above discussions, the cationic species [Cp*Ir(OAc)]+ (Ir-1) with unsaturated coordination environment should be the active species, which promotes the alkene coordination and further activation. As shown in Fig. 1, the coordination of 1-decene (1a) to [Cp*Ir(OAc)]+ (Ir-1) could form the more stable π-complex Ir-2. This step is exergonic by 4.3 kcal mol−1. To achieve allylic C–H activation, Ir-2 firstly isomerizes to Ir-2′ via changing the coordination mode of acetate from κ2 to κ1 and forming an agostic interaction Ir⋯H–C instead.20 Such an isomerization step via Ir-TSiso has an energy barrier of 10.9 kcal mol−1 and is endothermic by 10.2 kcal mol−1. Then, C–H activation in Ir-2′ takes place through a traditional concerted metalation deprotonation (CMD) transition state Ir-TS3 to give the η3-allyl-Ir(III) intermediate Ir-4. The relative activation energy for C–H activation process is 19.6 kcal mol−1, which should be kinetically reasonable under the experimental condition (35 °C).11 Starting from Ir-4, the coordination of methyl-1,4,2-dioxazol-5-one (2a) to the Ir(III) center of η3-allyl-Ir(III) intermediate Ir-4 is exergonic by 0.8 kcal mol−1, giving the intermediate Ir-5 for further C–N bond formation. Subsequently, the decarboxylation (CO2 extrusion) easily takes place via Ir-TS6 to yield the allyl-Ir(V)-nitrenoid Ir-7. The step has a low energy barrier of 4.0 kcal mol−1 and is highly exothermic by 21.5 kcal mol−1 (relative to the precatalyst and substrates). Such low energy barrier could be ascribed to the unique binding feature between the dioxazole moiety and the Ir center.21 By comparison, the CO2 extrusion from free methyl dioxazolone (2a) was calculated and encountered a high energy barrier (33.6 kcal mol−1, Fig. S4†).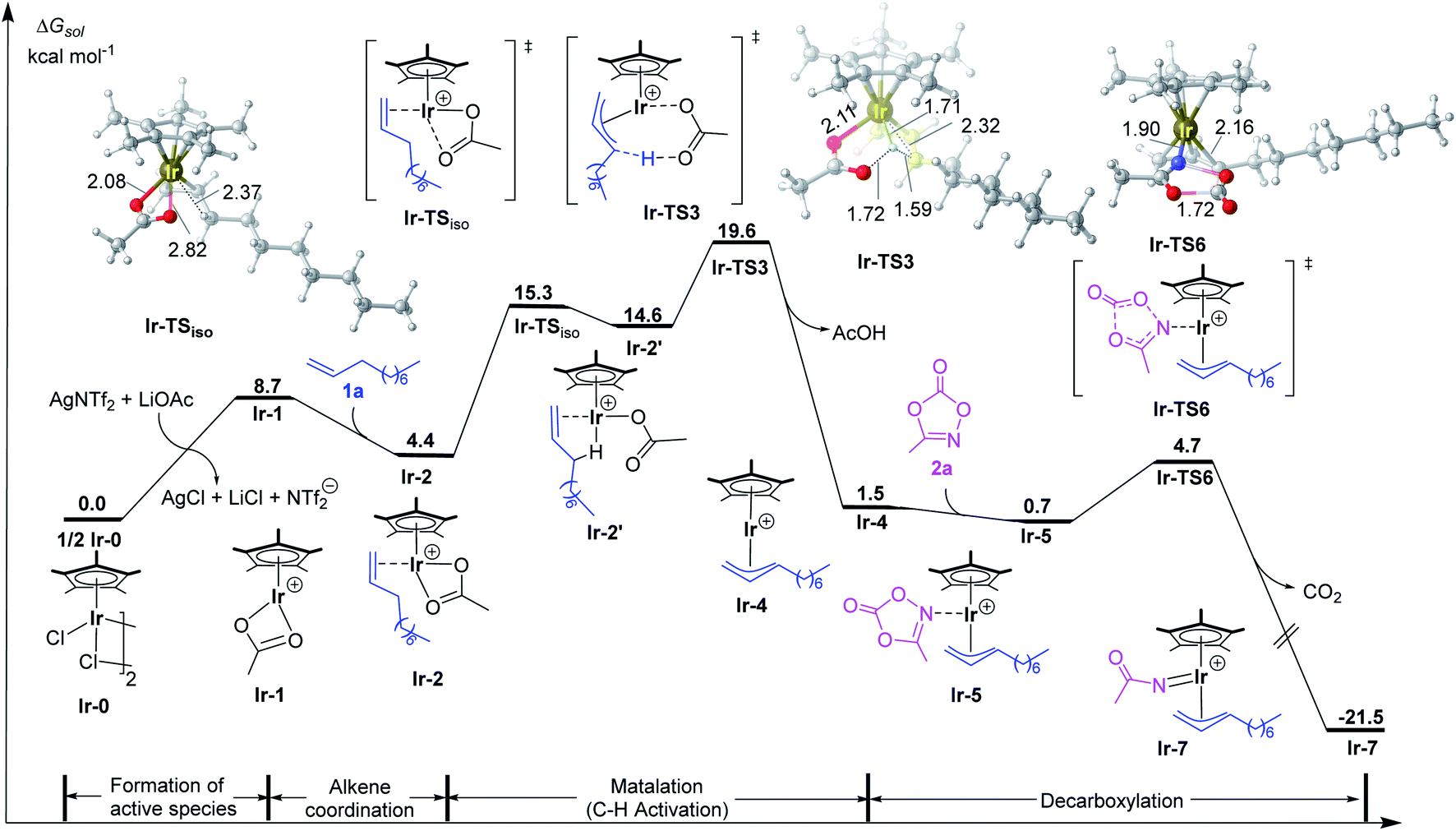 | ||
| Fig. 1 Calculated free energy profiles for allylic C–H activation by the Ir(III) catalyst. Bond distances shown in the structures are given in angstroms. | ||
Migratory insertion (C–N formation) and protodemetalation
After the formation of allyl-Ir(V)-nitrenoid intermediate Ir-7, there are two potential sites for migratory insertion (Fig. 2), viz., terminal (C1, leading to the linear amidation product) or internal position (C3, leading to the branched amidation product). As we can see from Fig. 2, the migratory insertion takes place at the C3 site (via Ir-TS8, black line) is more favorable than C1 site (via Ir-TS8′, blue line) by 3.5 kcal mol−1 and is an irreversible process, which is well in line with the regioselectivity observed experimentally.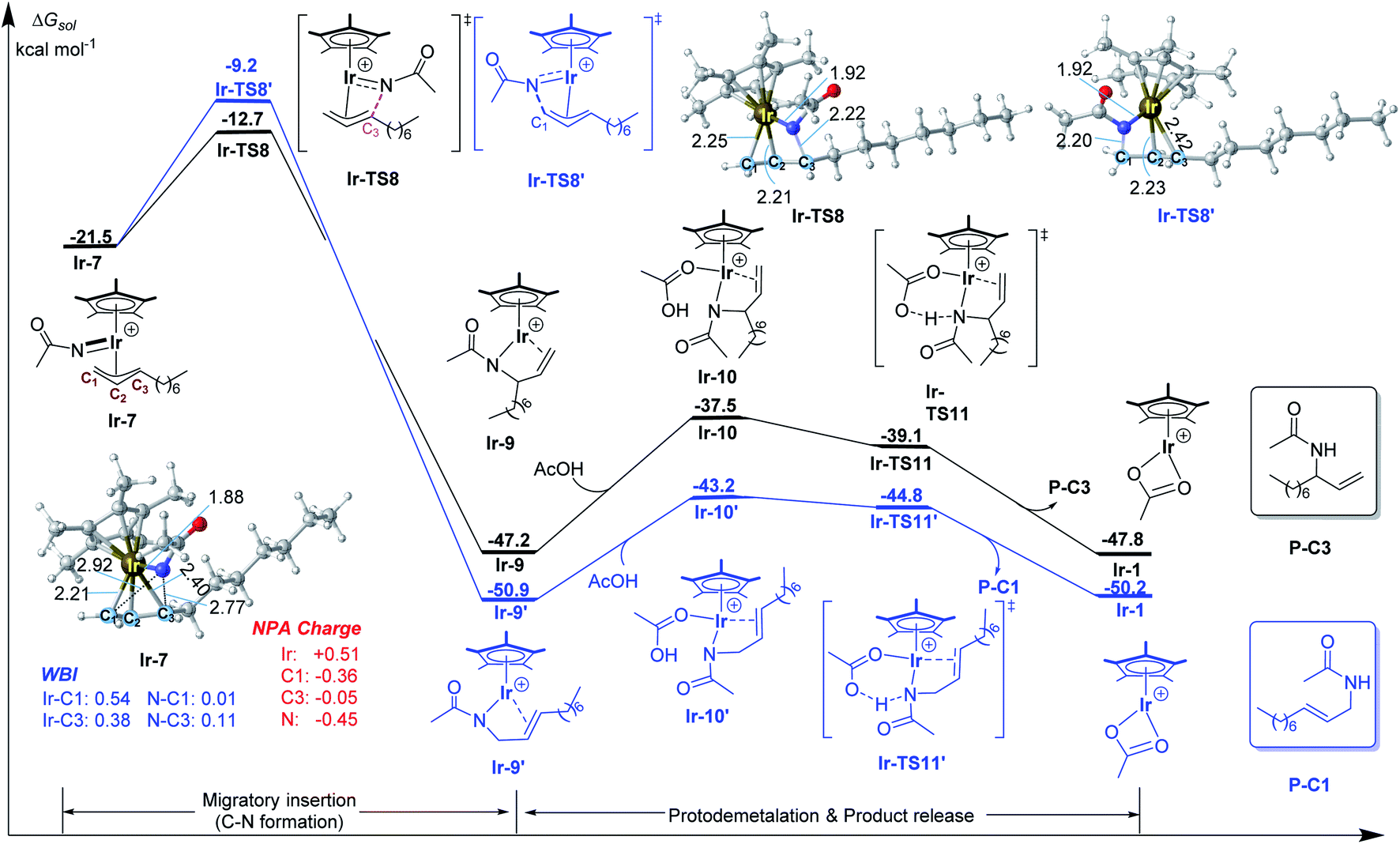 | ||
| Fig. 2 Calculated free energy profiles for C–N bond formation by the Ir(III) catalyst. Bond distances shown in the structures are given in angstroms. | ||
Starting from the preferred migratory insertion product Ir-9, the coordination of a molecule of AcOH, which is formed in the C–H activation step (Fig. 1), could form the adduct Ir-10 and then the protodemetalation of amination product takes place via a barrierless transition state Ir-TS11 to give the final branched product P-C3 and regenerates the active species Ir-1. As discussed above, the computational results generally support the proposed mechanism, which mainly involves the generation of the active species [Cp*Ir(OAc)]+ with the aid of AgNTf2, alkene coordination, allylic C–H activation (matalation) giving η3-allyl-Ir(III) intermediate, decarboxylation forming the key Ir(V)-nitrenoid species, regioselective migratory insertion (C–N formation), and the final protodemetalation producing the amidation product and regeneration of the active species.
Origin of regioselectivity
To have a better understanding of the regioselectivity, the structure of Ir-7 was carefully examined (Fig. 2). It is found that the Ir–C1 bond is stronger than Ir–C3 bond, reflected by the shorter bond distance of Ir–C1 (2.21 vs. 2.40 Å of Ir–C3) and the larger Wiberg bond indexes (WBI) for Ir–C1 (0.54 vs. 0.38 for Ir–C1). This suggests that the migratory insertion into Ir–C3 bond is easier than Ir–C1 bond. In addition, the interatomic distance N⋯C3 (2.77 Å) is shorter than N⋯C1 (2.92 Å), which will also support more favorable N–C3 formation compared with N–C1. Therefore, both the weaker Ir–C3 bond and the shorter interatomic distance of N⋯C3 make the migratory insertion into Ir–C3(internal) bond easier than Ir–C1(terminal) bond, leading to branch-selective allylic C–H amidation.For a comparison, the migratory insertion in the case of Rh catalyst was also calculated. As displayed in Fig. 3, although the migratory insertion takes place at the C3 site (via Rh-TS8) is more favorable than C1 site (via Rh-TS8′) by 2.9 kcal mol−1, the energy difference is smaller than that in Ir case (3.5 kcal mol−1), which is in general agreement with the observed lower regioselectivity in Rh system (2.9![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 for Rh vs. > 20:1 for Ir).11 In Rh-7, similar to Ir case, the Rh–C1 (WBI: 0.48) bond is stronger than Rh–C3 (WBI: 0.32) bond, which could account for the superiority of migratory insertion into Rh–C3. To be noted, in contrast to Ir-7, the bond index of N–C3 (0.11) is similar to N–C1 (0.10) in Rh-7, which will lower the regioselectivity in comparison with Ir system. The smaller difference in NPA charge between N and C3 in Rh-7 than that in Ir-7 could be also account for the observed different regioselectivity.
:1 for Rh vs. > 20:1 for Ir).11 In Rh-7, similar to Ir case, the Rh–C1 (WBI: 0.48) bond is stronger than Rh–C3 (WBI: 0.32) bond, which could account for the superiority of migratory insertion into Rh–C3. To be noted, in contrast to Ir-7, the bond index of N–C3 (0.11) is similar to N–C1 (0.10) in Rh-7, which will lower the regioselectivity in comparison with Ir system. The smaller difference in NPA charge between N and C3 in Rh-7 than that in Ir-7 could be also account for the observed different regioselectivity.
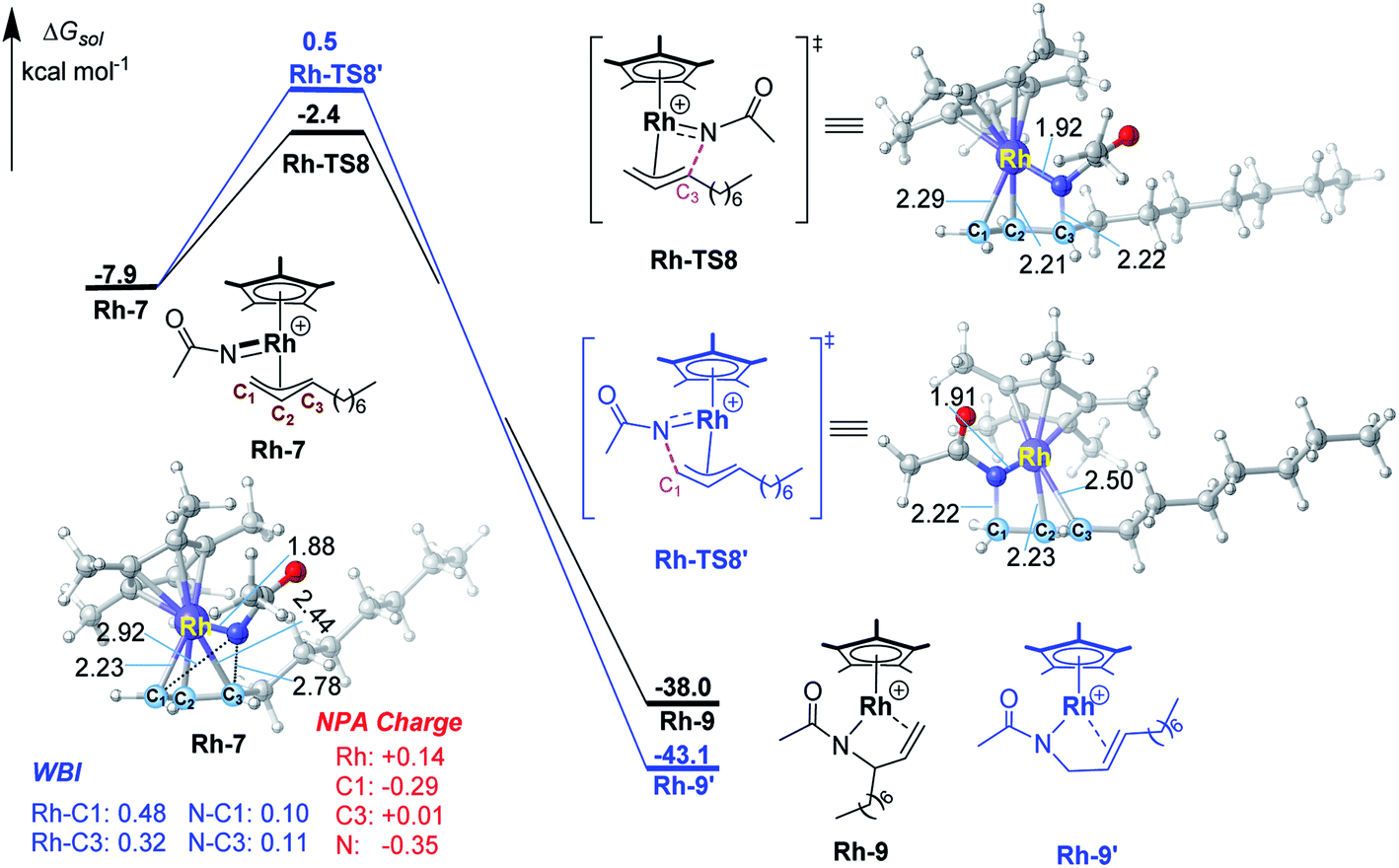 | ||
| Fig. 3 Calculated free energy profiles for migratory insertion by the Rh(III) catalyst. Bond distances shown in the structures are given in angstroms. | ||
Catalytic activity of different catalyst
Although [Cp*IrCl2]2 as a precatalyst works for such kind of C–H amination, the analogue [Cp*CoCl2]2 failed to produce any amidation products under the same experimental conditions. To elucidate the origin of such a reactivity difference, we carried out DFT calculations for the case of [Cp*CoCl2]2. Firstly, similar to the case of [Cp*IrCl2]2, the free energy of the formation of active species [Cp*Co(OAc)]+ was calculated. The result shows that the triplet is the ground state of [Cp*CoCl2]2 and is more stable than its singlet by 5.1 kcal mol−1. In the case of triplet, the formation free energy of [Cp*Co(OAc)]+ is 6.8 kcal mol−1 (Fig. 4), which is lower than that the case of Ir (8.7 kcal mol−1). Therefore, the difference of catalytic activity between the two cases (Ir and Co systems) cannot be explained by the formation of active species.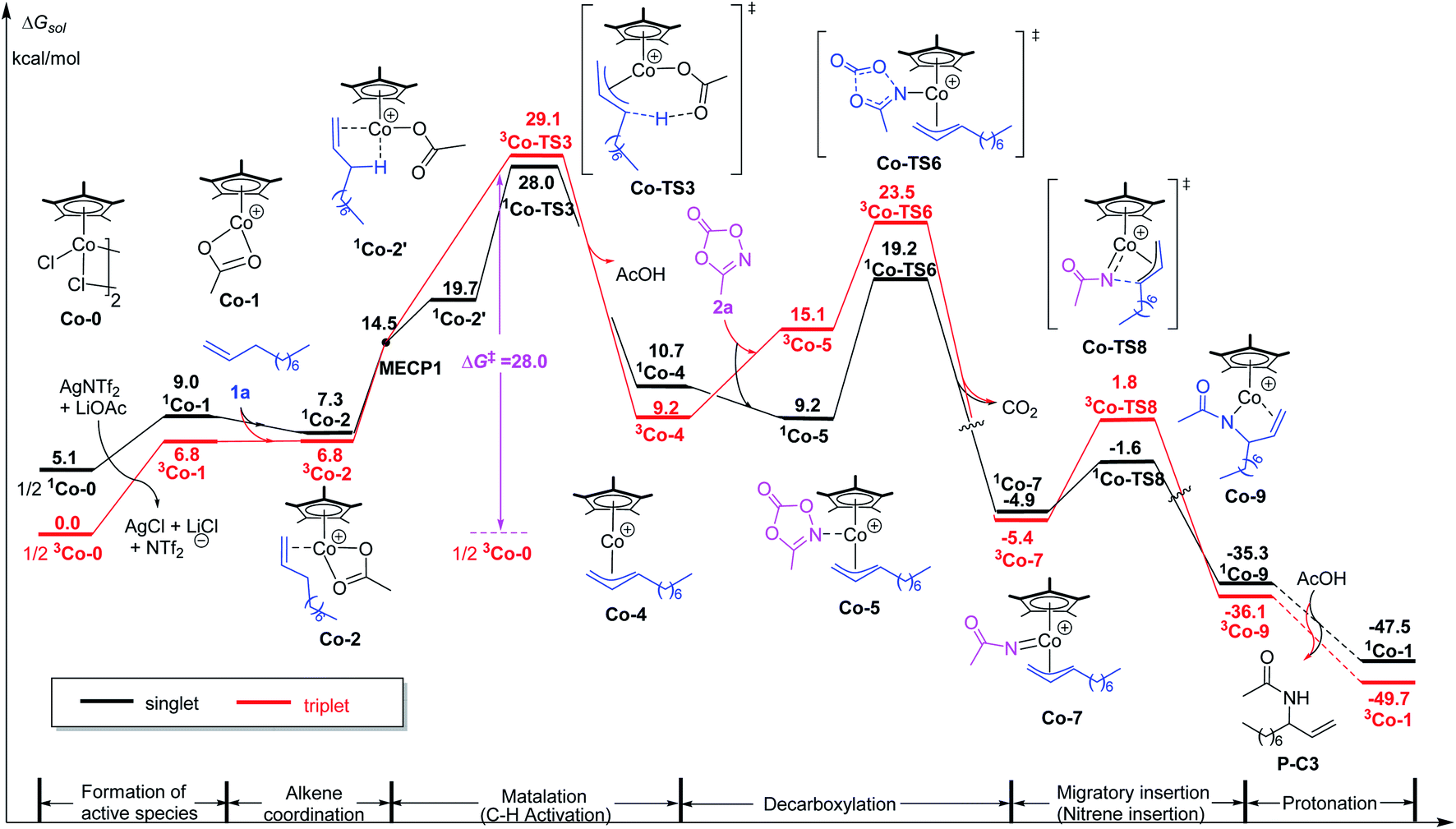 | ||
| Fig. 4 Calculated free energy profiles for intermolecular allylic C–H amidation of 1-decene by the Co(III) catalyst. | ||
Subsequently, the energy profile of Cp*Co(III)-catalyzed terminal alkene C–H amidation reaction was further calculated. The results show that Cp*Co(III)-catalyzed process is similar to that of Cp*Ir(III). It is also worth noting that Co-catalyzed process follows a two-state reactivity scenario (Fig. 4). Unsurprisingly, the allylic C–H activation of terminal olefin have an energy barrier of 28.0 kcal mol−1, which is significantly higher than that for Ir case (ΔG‡ = 19.6 kcal mol−1, Fig. 1). The high energy barrier of allylic C–H activation for Co catalyst could account for its catalytic sluggishness. Such a high energy barrier could be ascribed to the relatively unstable alkene-coordinating π-complex and the less stable transition state 1Co-TS3 in comparison with Ir system.
As for the alkene-coordinating π-complex, the coordination of an alkene to 3Co-1 giving 3Co-2 is an isoenergetic process (Fig. 4), but it is an exothermic process for Ir system (ca 4.3 kcal mol−1, Fig. 1) which could effectively lower the energy platform. To shed light on the different relative stability of the two π-complexes Ir-2 and 3Co-2, distortion/interaction analysis22 has been conducted (Fig. 5). In such analysis, each π-complex structure was divided into two fragments, viz., the coordinating alkene part (fragment sub) and the remaining metal moiety (fragment cat-M). ΔEdist is the energy required to distort separated fragments from their equilibrium geometry into the olefin-coordinated π-complexes. This energy is regarded as the destabilizing factor from geometry distortion. ΔEint denotes the interaction energy between the fragments in the π-complex. The energy ΔE‡ is defined as ΔE‡ = ΔEdist(cat) + ΔEdist(sub) + ΔEint. As shown in Fig. 5, although the distortion energies of the two fragments in Ir-2 (ΔEdist(cat-Ir) = 6.6 kcal mol−1 and ΔEdist(sub) = 7.6 kcal mol−1) are larger than that in 3Co-2 (ΔEdist(cat-Co) = 1.1 kcal mol−1 and ΔEdist(sub) = 1.7 kcal mol−1), the interaction between the two fragments in Ir-2 (ΔEint = −35.0 kcal mol−1) is significantly stronger than that in 3Co-2 (ΔEint = −18.3 kcal mol−1) by −16.7 kcal mol−1, making Ir-2 more stable than 3Co-2. Therefore, it is the discriminatory strength of interaction that causes the different relative stability of the two π-complexes Ir-2 and 3Co-2.
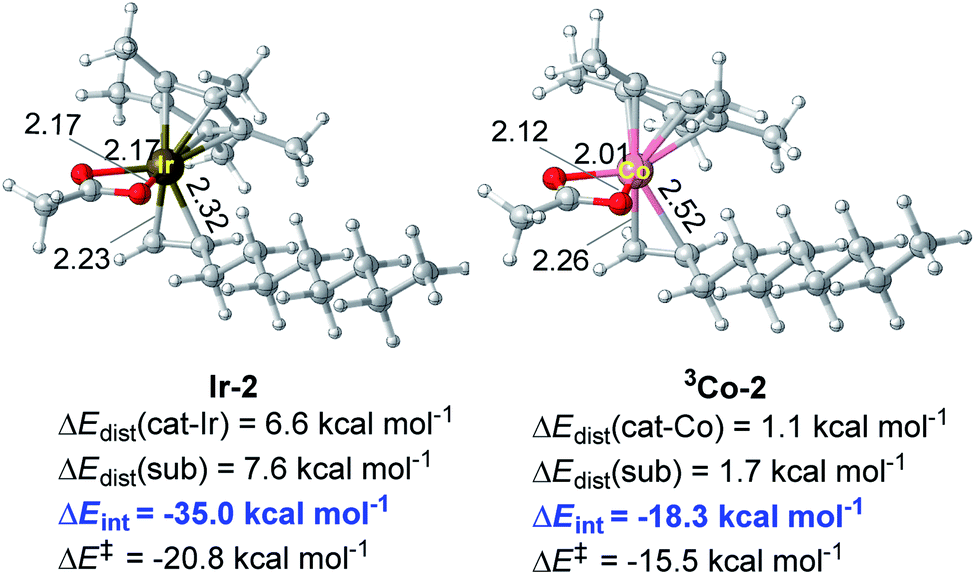 | ||
| Fig. 5 Optimized structures of π-complexes Ir-2 and 3Co-2 and their distortion/interaction analysis. Bond distances shown in the structures are given in angstroms (Å). | ||
The frontier molecular orbitals of the two π-complexes could also provide valuable information to explain their relative stability (Fig. 6). In the case of Ir-2, there is strong orbital interaction between the metal center and alkene part. For instance, the HOMO-1 orbital is the σ binding orbital which is composed of the d orbital of Ir and π-bonding orbital of the alkene. The HOMO orbital is the back donation π-binding orbital, which is composed of the d orbital of Ir and π*-bonding orbitals of alkene moiety. The electrons in HOMO and HOMO-1 orbitals are localized in the d orbital of the metal atom and alkene moiety in Ir-2. However, in 3Co-2, only a very small number of electrons in SOMO and SOMO-1 orbitals are localized in the Co-d orbital and alkene moiety. This may be due to the high spin of 3Co-2, which makes the metal center has fewer empty d orbitals, so that the d orbital cannot accept more electrons. Meanwhile, the single electron in Co-d orbital has the Pauli repulsion to the alkene electrons. Therefore, the coordination capacity of alkene in 3Co-2 is weaker than that in Ir-2.
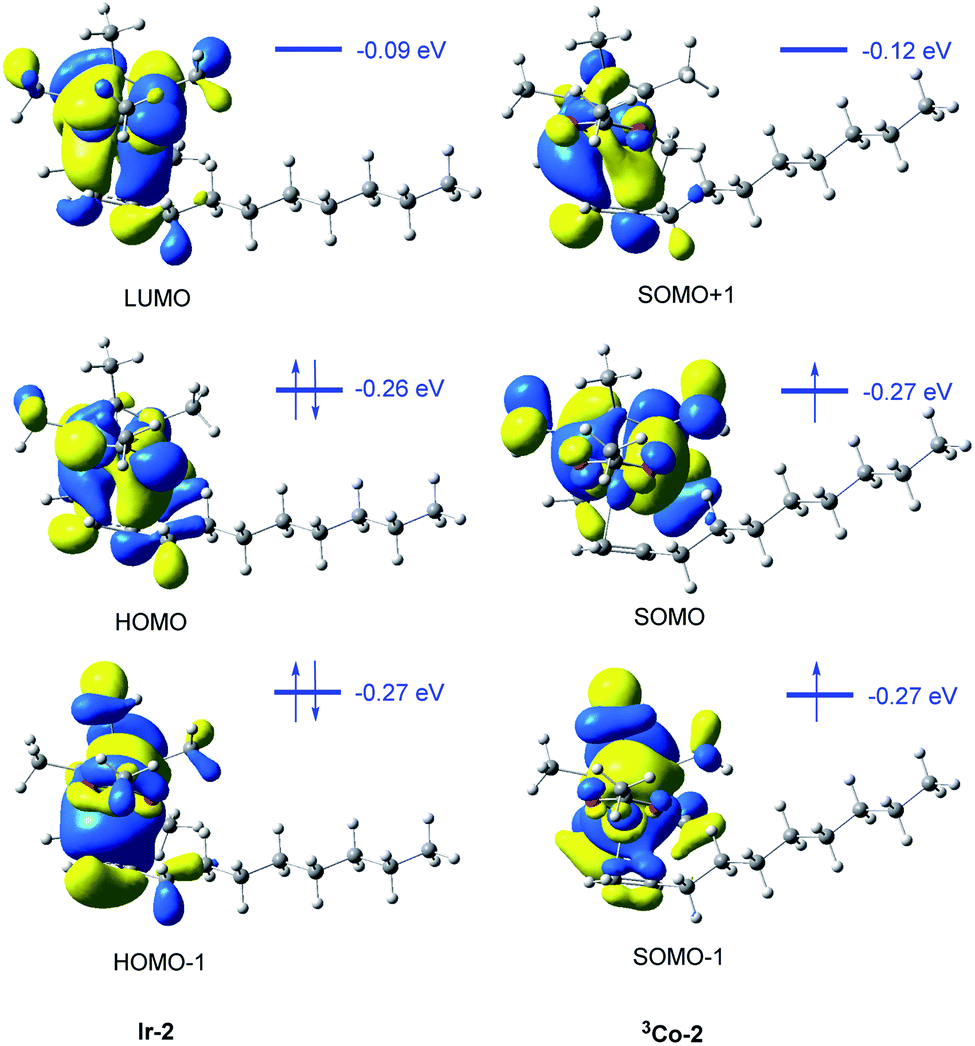 | ||
| Fig. 6 Frontier molecular orbital analysis of alkene-coordinating π-complexes Ir-2 and 3Co-2. | ||
As for the C–H activation transition state, the singlet 1Co-TS3 is slightly lower than 3Co-TS3 by 1.1 kcal mol−1 and the spin-crossing process occurs before C–H activation. The net energy barrier from π-complex 3Co-2 to 1Co-TS3 is 21.2 kcal mol−1, while the energy barrier in Ir system is 15.2 kcal mol−1 (Ir-2 → Ir-TS3, Fig. 1). To get more information on the difference of C–H activation process in two systems, the structures of Ir-TS3 and 1Co-TS3 are carefully compared (Fig. 7). As displayed in Fig. 7, it is found that the C3–H bond is more activated in Ir-TS3 than that in 1Co-TS3, manifested by the longer bond distance (d(C3⋯H) = 1.59 Å in Ir-TS3 vs. d(C3⋯H) = 1.40 Å in 1Co-TS3) and smaller bond index (WBI(C3⋯H) = 0.32 in Ir-TS3 vs. WBI(C3⋯H) = 0.36 in 1Co-TS3). Importantly, the metal center plays a vital role in hydrogen transfer process. The results show that although the atomic radius of Ir is larger than Co, the distance of Ir⋯H in Ir-TS3 (1.71 Å) is shorter than d(Co⋯H) in 1Co-TS3 (1.86 Å), suggesting that the stronger metal⋯H interaction exists in Ir-TS3 compared to 1Co-TS3. The WBI values of metal⋯H (WBI(Ir⋯H) = 0.31 in Ir-TS3 vs. WBI(Co⋯H) = 0.14 in 1Co-TS3) and the tortile angles of C3–H–O1 (128.5° in Ir-TS3 vs. 147.5° in 1Co-TS3) could further illustrate the aforementioned reactivity difference in the two catalytic systems.
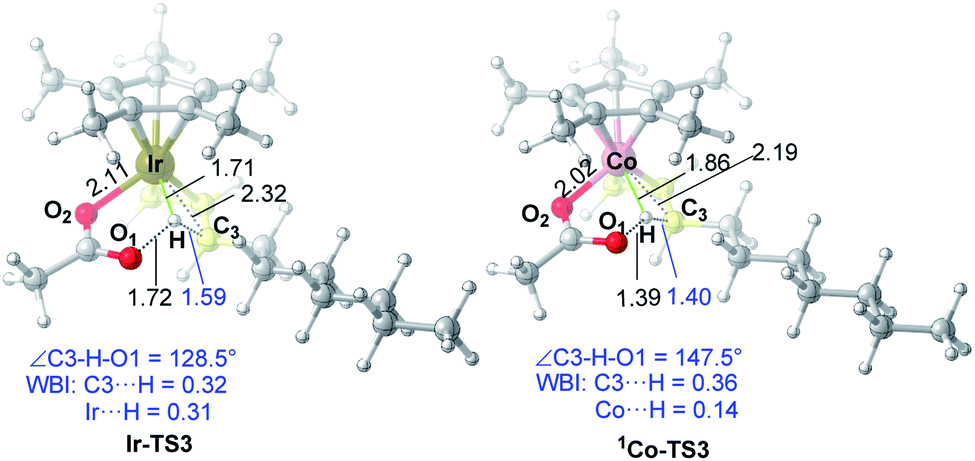 | ||
| Fig. 7 Optimized transition state structures of Ir-TS3 and 1Co-TS3. The bond distances shown in the structures are given in angstroms (Å). | ||
On the basis of above discussion, the high catalytic activity of Ir catalyst is mainly attributed to the relative stability of alkene-coordinating π-complex and the stronger metal⋯H interaction assisting hydrogen transfer in comparison with Co system. In addition, Ir-catalyzed C–H activation process is exothermic, while it is an endothermic process for Co system. Consequently, Cp*Ir(III)-catalyzed allylic C–H activation of terminal alkene is more favorable than Cp*Co(III) both thermodynamically and kinetically.
Conclusions
In summary, Cp*Ir(III)-catalyzed allylic C–H amination reaction of 1-decene with methyl dioxazolone has been investigated by DFT calculations. The computational results suggest that the reaction mainly includes the following steps: the formation of active species [Cp*Ir(OAc)]+, alkene coordination, allylic C–H activation (matalation) giving η3-allyl-Ir(III) intermediate, decarboxylation of methyl dioxazolone to form the key Ir(V)-nitrenoid species, regioselective migratory insertion (C–N formation), and the final protodemetalation to give the amidation product and regenerate the active species. The results show that AgNTf2 plays an important role in the formation of active species [Cp*Ir(OAc)]+. As for the regioselectivity, in the key Ir(V)-nitrenoid species, both the weaker Ir–C(internal) bond and the shorter interatomic distance of N⋯C(internal) make the migratory insertion into Ir–C (internal) bond easier than Ir–C (terminal), leading to branch-selective allylic C–H amidation. By comparison of Cp*Ir(III)- and Cp*Co(III)-catalyzed processes, it is obvious that the higher catalytic activity of Ir complex is mainly attributed to the relative stability of alkene-coordinating π-complex and the stronger metal⋯H interaction assisting hydrogen transfer in comparison with Co system. The analyses of geometric parameters, frontier molecular orbital, bond indexes, and distortion-interaction offered insights into such catalytic systems. The result presented here could add better understanding to the mechanism, regioselectivity, and catalytic activity of transition-metal-catalyzed allylic C–H amination and is expected to be helpful for further development of regioselective C–H amination reactions.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was partly supported by the grants from NSFC (U1862115, 21674014, 22003001) and the Innovation and Entrepreneurship Project of Overseas Returnees in Anhui Province (2020LCX006). We gratefully appreciate accesses to the Network and Information Center of Dalian University of Technology and the High-performance Computing Platform of Anhui University for computational resources.References
- (a) K. Shin, H. Kim and S. Chang, Acc. Chem. Res., 2015, 48, 1040–1052 CrossRef CAS PubMed; (b) P. Ruiz-Castillo and S. L. Buchwald, Chem. Rev., 2016, 116, 12564–12649 CrossRef CAS PubMed; (c) R. Ma and M. C. White, J. Am. Chem. Soc., 2018, 140, 3202–3205 CrossRef CAS PubMed; (d) M. D. Karkas, Chem. Soc. Rev., 2018, 47, 5786–5865 RSC; (e) K. Muniz, Acc. Chem. Res., 2018, 51, 1507–1519 CrossRef CAS PubMed; (f) Y. Zhao and W. Xia, Chem. Soc. Rev., 2018, 47, 2591–2608 RSC; (g) O. I. Afanasyev, E. Kuchuk, D. L. Usanov and D. Chusov, Chem. Rev., 2019, 119, 11857–11911 CrossRef CAS PubMed.
- (a) J. L. Roizen, M. E. Harvey and J. Du Bois, Acc. Chem. Res., 2012, 45, 911–922 CrossRef CAS PubMed; (b) M.-L. Louillat and F. W. Patureau, Chem. Soc. Rev., 2014, 43, 901–910 RSC; (c) T. Xiong and Q. Zhang, Chem. Soc. Rev., 2016, 45, 3069–3087 RSC; (d) H. Kim and S. Chang, Acc. Chem. Res., 2017, 50, 482–486 CrossRef CAS PubMed; (e) Y. Park, Y. Kim and S. Chang, Chem. Rev., 2017, 117, 9247–9301 CrossRef CAS PubMed; (f) X. Qi, Y. Li, R. Bai and Y. Lan, Acc. Chem. Res., 2017, 50, 2799–2808 CrossRef CAS PubMed.
- (a) J. F. Hartwig, Acc. Chem. Res., 2008, 41, 1534–1544 CrossRef CAS PubMed; (b) J. F. Hartwig, Nature, 2008, 455, 314–322 CrossRef CAS PubMed.
- (a) R. Breslow and S. H. Gellman, J. Chem. Soc., Chem. Commun., 1982, 1400–1401 RSC; (b) R. Breslow and S. H. Gellman, J. Am. Chem. Soc., 1983, 105, 6728–6729 CrossRef CAS.
- (a) P. Dydio, H. M. Key, H. Hayashi, D. S. Clark and J. F. Hartwig, J. Am. Chem. Soc., 2017, 139, 1750–1753 CrossRef CAS PubMed; (b) K. Shing, Y. Liu, B. Cao, X. Chang, T. You and C. Che, Angew. Chem., Int. Ed., 2018, 57, 11947–11951 CrossRef CAS PubMed; (c) A. Nasrallah, V. Boquet, A. Hecker, P. Retailleau, B. Darses and P. Dauban, Angew. Chem., Int. Ed., 2019, 58, 8192–8196 CrossRef CAS PubMed; (d) R. Singh and A. Mukherjee, ACS Catal., 2019, 9, 3604–3617 CrossRef CAS; (e) R. J. Harris, J. Park, T. A. F. Nelson, N. Iqbal, D. C. Salgueiro, J. Bacsa, C. E. MacBeth, M. H. Baik and S. B. Blakey, J. Am. Chem. Soc., 2020, 142, 5842–5851 CrossRef CAS PubMed; (f) J. Lee, S. Jin, D. Kim, S. H. Hong and S. Chang, J. Am. Chem. Soc., 2021, 143, 5191–5200 CrossRef CAS PubMed.
- (a) E. N. Bess, R. J. DeLuca, D. J. Tindall, M. S. Oderinde, J. L. Roizen, J. Du Bois and M. S. Sigman, J. Am. Chem. Soc., 2014, 136, 5783–5789 CrossRef CAS PubMed; (b) T. M. Figg, S. Park, J. Park, S. Chang and D. G. Musaev, Organometallics, 2014, 33, 4076–4085 CrossRef CAS; (c) G. Manca, E. Gallo, D. Intrieri and C. Mealli, ACS Catal., 2014, 4, 823–832 CrossRef CAS; (d) S. H. Park, J. Kwak, K. Shin, J. Ryu, Y. Park and S. Chang, J. Am. Chem. Soc., 2014, 136, 2492–2502 CrossRef CAS PubMed; (e) X. Zhang, H. Xu and C. Zhao, J. Org. Chem., 2014, 79, 9799–9811 CrossRef CAS PubMed; (f) K. Hou, D. A. Hrovat and X. Bao, Chem. Commun., 2015, 51, 15414–15417 RSC; (g) M. J. Ajitha, K.-W. Huang, J. Kwak, H. J. Kim, S. Chang and Y. Jung, Dalton Trans., 2016, 45, 7980–7985 RSC; (h) M. Anand, R. B. Sunoj and H. F. Schaefer III, ACS Catal., 2016, 6, 696–708 CrossRef CAS; (i) C. Kong, N. Jana, C. Jones and T. G. Driver, J. Am. Chem. Soc., 2016, 138, 13271–13280 CrossRef CAS PubMed; (j) J. Wang, C. Zhao, Y. Weng and H. Xu, Catal. Sci. Technol., 2016, 6, 5292–5303 RSC; (k) H. Xu, X. Zhang, Z. Ke and C. Zhao, RSC Adv., 2016, 6, 29045–29053 RSC; (l) Y. Zhou and X. Bao, Org. Lett., 2016, 18, 4506–4509 CrossRef CAS PubMed; (m) D. Fujita, H. Sugimoto, Y. Shiota, Y. Morimoto, K. Yoshizawa and S. Itoh, Chem. Commun., 2017, 53, 4849–4852 RSC; (n) B. E. Haines, T. Kawakami, K. Kuwata, K. Murakami, K. Itami and D. G. Musaev, Chem. Sci., 2017, 8, 988–1001 RSC; (o) P. F. Kuijpers, M. J. Tiekink, W. B. Breukelaar, D. L. J. Broere, N. P. van Leest, J. I. van der Vlugt, J. N. H. Reek and B. de Bruin, Chem.–Eur. J., 2017, 23, 7945–7952 CrossRef CAS PubMed; (p) Z. Li, D. J. Burnell and R. J. Boyd, J. Phys. Chem. B, 2017, 121, 10859–10868 CrossRef CAS PubMed; (q) J. Wang, K. Zheng, B. Lin and Y. Weng, RSC Adv., 2017, 7, 34783–34794 RSC; (r) X. Zhang, J. Organomet. Chem., 2017, 832, 1–8 CrossRef CAS; (s) D. Gao and X. Bao, Org. Chem. Front., 2018, 5, 1471–1482 RSC; (t) A. Varela-Alvarez, B. E. Haines and D. G. Musaev, J. Organomet. Chem., 2018, 867, 183–192 CrossRef CAS; (u) P. Li and Z. Cao, Organometallics, 2018, 38, 343–350 CrossRef; (v) Y. Hwang, H. Jung, E. Lee, D. Kim and S. Chang, J. Am. Chem. Soc., 2020, 142, 8880–8889 CrossRef PubMed; (w) Y. Yu, G. Luo, J. Yang and Y. Luo, Catal. Sci. Technol., 2019, 9, 1879–1890 RSC; (x) Y. Yu, G. Luo, J. Yang and Y. Luo, Catal. Sci. Technol., 2020, 10, 1914–1924 RSC; (y) Y. Yu, G. Luo, J. Yang and Y. Luo, Chin. J. Chem., 2020, 38, 1526–1532 CrossRef CAS.
- (a) W. Zhang, N.-X. Wang and Y. Xing, Synlett, 2015, 26, 2088–2098 CrossRef CAS; (b) X. Yang, G. Shan, L. Wang and Y. Rao, Tetrahedron Lett., 2016, 57, 819–836 CrossRef CAS; (c) J. C. K. Chu and T. Rovis, Angew. Chem., Int. Ed., 2018, 57, 62–101 CrossRef CAS PubMed; (d) T. G. Saint-Denis, R.-Y. Zhu, G. Chen, Q. F. Wu and J.-Q. Yu, Science, 2018, 359, eaao4798 CrossRef PubMed; (e) T. Knecht, S. Mondal, J. H. Ye, M. Das and F. Glorius, Angew. Chem., Int. Ed., 2019, 58, 7117–7121 CrossRef CAS PubMed; (f) J. S. Burman, R. J. Harris, C. M. B. Farr, J. Bacsa and S. B. Blakey, ACS Catal., 2019, 9, 5474–5479 CrossRef CAS.
- (a) F. Liron, J. Oble, M. M. Lorion and G. Poli, Eur. J. Org. Chem., 2014, 2014, 5863–5883 CrossRef CAS; (b) C. C. Pattillo, J. I. Strambeanu, P. Calleja, N. A. Vermeulen, T. Mizuno and M. C. White, J. Am. Chem. Soc., 2016, 138, 1265–1272 CrossRef CAS PubMed; (c) R. Ma and M. C. White, J. Am. Chem. Soc., 2018, 140, 3202–3205 CrossRef CAS PubMed; (d) R. Wang, Y. Luan and M. Ye, Chin. J. Chem., 2019, 37, 720–743 CrossRef CAS.
- M. S. Chen and M. C. White, J. Am. Chem. Soc., 2004, 126, 1346–1347 CrossRef CAS PubMed.
- (a) A. Archambeau and T. Rovis, Angew. Chem., Int. Ed., 2015, 54, 13337–13340 CrossRef CAS PubMed; (b) Y. Shibata, E. Kudo, H. Sugiyama, H. Uekusa and K. Tanaka, Organometallics, 2016, 35, 1547–1552 CrossRef CAS; (c) J. S. Burman and S. B. Blakey, Angew. Chem., Int. Ed., 2017, 56, 13666–13669 CrossRef CAS PubMed; (d) T. A. F. Nelson and S. B. Blakey, Angew. Chem., Int. Ed., 2018, 57, 14911–14915 CrossRef CAS PubMed.
- H. Lei and T. Rovis, J. Am. Chem. Soc., 2019, 141, 2268–2273 CrossRef CAS PubMed.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, X. Li, M. Caricato, A. V. Marenich, J. Bloino, B. G. Janesko, R. Gomperts, B. Mennucci, H. P. Hratchian, J. V. Ortiz, A. F. Izmaylov, J. L. Sonnenberg, D. Williams-Young, F. Ding, F. Lipparini, F. Egidi, J. Goings, B. Peng, A. Petrone, T. Henderson, D. Ranasinghe, V. G. Zakrzewski, J. Gao, N. Rega, G. Zheng, W. Liang, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell, J. A. Montgomery Jr, J. E. Peralta, F. Ogliaro, M. J. Bearpark, J. J. Heyd, E. N. Brothers, K. N. Kudin, V. N. Staroverov, T. A. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. P. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, J. M. Millam, M. Klene, C. Adamo, R. Cammi, J. W. Ochterski, R. L. Martin, K. Morokuma, O. Farkas, J. B. Foresman and D. J. Fox, Gaussian 16, Revision A.03, Gaussian, Inc., Wallingford, CT, 2016 Search PubMed.
- (a) A. D. Becke, J. Chem. Phys., 1993, 98, 1372–1377 CrossRef CAS; (b) C. Lee, W. Yang and R. G. Parr, Phys. Rev. B: Condens. Matter Mater. Phys., 1988, 37, 785–789 CrossRef CAS PubMed; (c) A. D. Becke, Phys. Rev. A: At., Mol., Opt. Phys., 1988, 38, 3098–3100 CrossRef CAS PubMed.
- S. Grimme, J. Antony, S. Ehrlich and H. Krieg, J. Chem. Phys., 2010, 132, 154104 CrossRef PubMed.
- (a) M. Kaupp, P. V. Schleyer, H. Stoll and H. Preuss, J. Chem. Phys., 1991, 94, 1360–1366 CrossRef CAS; (b) A. Henglein, J. Phys. Chem., 1993, 97, 5457–5471 CrossRef CAS; (c) U. Steinbrenner, A. Bergner, M. Dolg and H. Stoll, Mol. Phys., 1994, 82, 3–11 CrossRef CAS.
- F. Weigend and R. Ahlrichs, Phys. Chem. Chem. Phys., 2005, 7, 3297–3305 RSC.
- A. V. Marenich, C. J. Cramer and D. G. Truhlar, J. Phys. Chem. B, 2009, 113, 4538–4543 CrossRef CAS PubMed.
- J. N. Harvey, M. Aschi, H. Schwarz and W. Koch, Theor. Chem. Acc., 1998, 99, 95–99 Search PubMed.
- C. Y. Legault, CYLview, 1.0b, Université de Sherbrooke, 2009, http://www.cylview.org Search PubMed.
- Although some studies have shown that the acetate isomerization step is often more energy demanding than the C–H cleavage, the energy barrier of acetate isomerization via Ir-TSiso is lower than that of the following C–H activation step (via Ir-TS3) in current study. See refs: (a) A. de Aguirre, S. Díez-González, F. Maseras, M. Martín and E. Sola, Organometallics, 2018, 37, 2645–2651 CrossRef CAS; (b) D. L. Davies, S. A. Macgregor and A. I. Poblador-Bahamonde, Dalton Trans., 2010, 39, 10520–10527 RSC.
- (a) Y. Park, J. Heo, M.-H. Baik and S. Chang, J. Am. Chem. Soc., 2016, 138, 14020–14029 CrossRef CAS PubMed; (b) S. Y. Hong, Y. Hwang, M. Lee and S. Chang, Acc. Chem. Res., 2021 DOI:10.1021/acs.accounts.1c00198.
- (a) F. Bickelhaupt and K. Houk, Angew. Chem., Int. Ed., 2017, 56, 10070–10086 CrossRef CAS PubMed; (b) I. Fernández and F. Bickelhaupt, Chem. Soc. Rev., 2014, 43, 4953–4967 RSC; (c) F. Liu, Y. Liang and K. Houk, Acc. Chem. Res., 2017, 50, 2297–2308 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1ra03842g |
| This journal is © The Royal Society of Chemistry 2021 |