
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceThe mechanism of enhanced photocatalytic activity for water-splitting of ReS2 by strain and electric field engineering†
Jing Pan‡
 ,
Wannian Zhang‡,
Xiaoyong Xu and
Jingguo Hu*
,
Wannian Zhang‡,
Xiaoyong Xu and
Jingguo Hu*
College of Physics Science and Technology, Yangzhou University, Yangzhou, 225002, China. E-mail: jghu@yzu.edu.cn
First published on 30th June 2021
Abstract
To enhance the photocatalytic water splitting performance of 2D ReS2, we theoretically propose a feasible strategy to engineer its band structure by applying strain or an electric field. Our calculated results show that the strains greatly tune the electronic structure of ReS2 especially band gap and band edge positions, because the strains significantly alter the crystal structure and then cause rearrangement of the surface charge. However, electric fields have little influence on band gap but obviously affect the band edge positions. This is because the electric fields have little effect on the crystal structure of ReS2 but easily produce an in-plane electric dipole moment. The shifts in band edge position mainly arise from competition between the surface charge and the in-plane electric dipole. For an applied strain, the shifts are dominated by rearrangement of surface charge; for an applied electric field, the shifts are determined by an induced electric dipole moment. Importantly, functionalized ReS2 with a bi-axial strain of −4% or an electronic field of −0.1 V Å−1 may be good candidates for water-splitting photocatalysts owing to their suitable band edge positions for water splitting, ideal band gaps, good stability, reduced electron–hole recombination and high carrier mobility. We hope our findings will stimulate experimental efforts to develop new photocatalysts based on functionalized ReS2.
1. Introduction
Photocatalytic water splitting using solar energy for hydrogen generation is a clean and “green” approach to solving environmental and energy problems.1–12 Efficient semiconductor photocatalysts for water splitting should satisfy the following requirements: (1) an ideal band gap for wide visible-light adsorption; (2) suitable band edge positions which should straddle water redox potentials; (3) fewer electron–hole pair recombination; (4) high carrier mobility; and (5) good stability.13–16 Traditional photocatalysts such as TiO2,17,18 ZnO,19,20 and KNbO3 have been reported to be promising photocatalysts.21,22 Nevertheless, their large band gaps greatly limit their visible-light absorption and result in poor photo-to-hydrogen conversion efficiency.Since graphene was first prepared, two-dimensional (2D) transition-metal materials have shown efficient photocatalytic properties because of their good surface to volume ratio, abundant chemically active sites, and low charge carrier diffusion distance.23–28 2D transition-metal dichalcogenides (TMDs),29–32 as represented by MoS2, have a direct band gap, strong visible-light absorption, excellent stability, and high electrocatalytic activity, so they are potential candidates for electronic, optoelectronic, and photovoltaic applications.33–35 Different from the traditional TMDs (e.g., MoS2 and WS2), ReS2 has a triclinic symmetry and displays a distorted 1T (1T′) octahedral crystal structure, showing a particular electronic and optical anisotropy with a high experimental anisotropic mobility ratio of 3.1.36–40 The electronic and dynamic decoupling in the interface with weak van der Waals forces enable it to retain a direct band gap (∼1.5 eV) almost independent of thickness.41 The weak coupling enables ReS2 to be easily prepared as nanosheets, exhibiting excellent Raman active modes and catalytic activity in the hydrogen evolution reaction.42 These advantages make ReS2 a potential candidate for photocatalytic water splitting.13 However, the conduction band minimum (CBM) of ReS2 is below the hydrogen redox potential of water, hindering its application in photocatalytic hydrogen generation.43,44 Therefore, it is necessary to tune the band energy alignment of ReS2 to enhance its photocatalytic activity for water splitting. In particular, mechanical strain or an external electric field are effective and practical ways to adjust the electronic properties of a 2D semiconductor.45–52 For example, Bai et al. have shown that single-layer ZnGeN2 could be effectively tuned to provide a better match with the redox potentials of water and to enhance light absorption in the visible-light region at a tensile strain of 5%.49 Edalati et al. suggested that CsTaO3 and LiTaO3 exhibited optical bandgap narrowing and ∼2.5 times enhancement of photocatalytic hydrogen generation by straining.50 Wang et al. reported that the biaxial strain tuned the bandgap and band alignment of a 2D CdS/g-C3N4 heterostructure for visible-light photocatalytic water splitting and accelerated the separation of photogenerated carriers and improved the photocatalytic activity.51 Dai et al. have described the ferroelectric photocatalysts could be greatly improved because the intrinsic internal electric field was beneficial for separation and migration of photogenerated carriers.52
In this work, we investigate the potential of 2D ReS2 for photocatalytic water splitting by strain and electric field functionalization. The first-principles calculations show that the strains greatly engineer the band gap and band alignment of ReS2. A progressive shrinking in the band gap can be observed upon increasing the applied strain for both tensile and compressive strain, and for both axial and bi-axial strain. This is because the strains induce significant geometric distortion and lead to charge rearrangement. Although the band gap of ReS2 is insensitive to an electric field, the band alignment depends greatly on an electric field because the electric field has little influence on the crystal structure but can produce an in-plane dipole moment. As a result, functionalized ReS2 with a bi-axial strain of −4% or an electronic field of −0.1 V Å−1 are considered good candidates for water-splitting photocatalysts.
2. Computational model and methods
In our calculations, density functional theory (DFT) were adopted with the Vienna ab initio simulation package (VASP).53 The exchange correlation potential was the generalized gradient approximation (GGA) with Perdew–Burke–Ernzerhof (PBE).54,55 The frozen core projector augmented wave method was employed to describe the electron–ion interaction. The plane wave cutoff energy was 400 eV, the total change in energy for geometrical optimization was 1.0 × 10−5 eV, the atomic forces were less than 0.01 eV Å−1. 7 × 7 × 1 and 11 × 11 × 1 Monkhorst–Pack k-point mesh were respectively used for geometric optimization and the electronic structure calculations.As we know, monolayer ReS2 has almost the same electronic structure as multilayer ReS2.41 Thus, a 2 × 2 supercell of free-standing monolayer ReS2 was used as our model by cleaving the bulk ReS2 along (0001) direction. A vacuum space along the y-direction was 15 Å to separate the interactions between the neighbouring slabs. As shown in Fig. 1, uniaxial lattice strain (x-axial or z-axial) was imposed by changing the dimension of the supercell in the x- or z-directions. Bi-axial lattice strain was imposed by changing the dimension of the supercell in the x- and z-directions simultaneously. We calculated the percentage applied strain according to % strain = (a − a0)/a × 100%, where a and a0 are the lattice constants of the ReS2 monolayer before and after the application of strain. The strains were varied from −10% to 10% in steps of 2%, where the positive numbers represent tensile strains, and the negative numbers represent compressive strains. The positions of all the atoms in the supercell were fully relaxed but the lattice vectors were constrained. An external electric field intensity E⊥ was applied along the y-direction with a net electric-dipole moment, from −0.2 V Å−1 to 0.2 V Å−1 in steps of 0.05 V Å−1. Dipole corrections were applied to remove spurious dipole interactions between periodic images.56,57
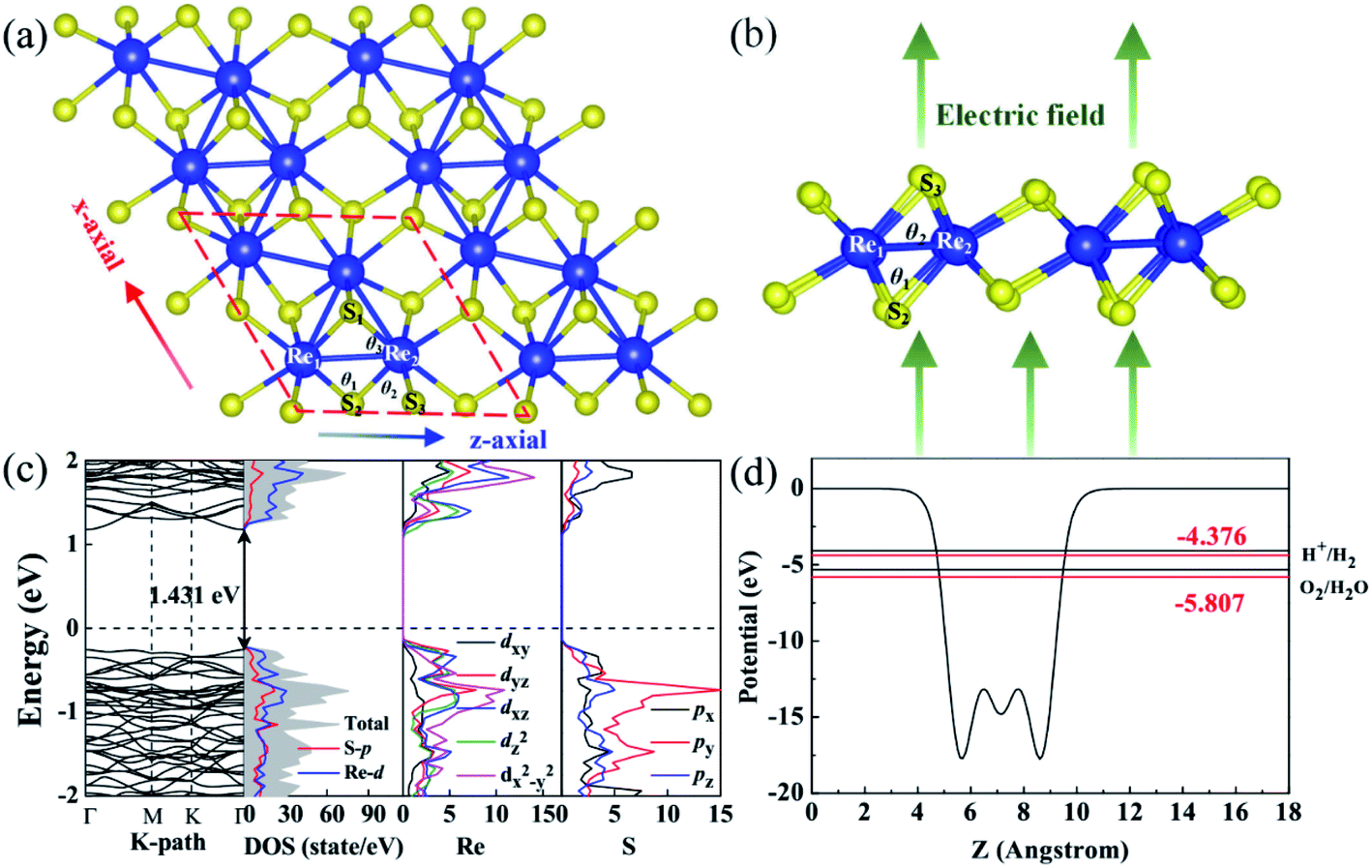 | ||
| Fig. 1 (a) Top and (b) side views of ML-ReS2; the blue and red arrows represent the directions of applied strain and the green arrows represent the direction of the electric field. (c) Band structure and atom-PDOS of ML-ReS2. (d) Work-function of ML-ReS2 (red line) and band alignments (denoting by the red solid lines) relative to the water redox potential positions (denoting by the black lines). | ||
3. Results and discussion
3.1 Pristine ReS2 monolayer
As shown in Fig. 1(a), the monolayer ReS2 retains a triclinic structure. The bond distances dRe–Re, dRe2–S2, dRe2–S3, and dS–S are 2.717 Å, 2.359 Å, 2.432 Å, and 3.807 Å, respectively. The bond angles of Re1–S2–Re2, S2–Re2–S2, and S1–Re2–S3 are defined as θ1, θ2, and θ3 with values 70.2°, 105.2°, and 80.9°, which are consistent with the experimental values (dRe–Re = 2.722 Å, dRe2–S2 = 2.360 Å, dRe2–S3 = 2.434 Å, dS–S = 3.806 Å, θ1 = 70.3°, θ2 = 105.1°, and θ3 = 81.1°).The band structure of ReS2 was reported in our previous calculation.29 ML-ReS2 is a direct-band-gap semiconductor, both the valence band maximum (VBM) and the conduction band minimum (CBM) are located at the Γ point, with a calculated band gap of 1.431 eV. Analysis of the partial density of states (PDOS) shows that the VBM originates mainly from hybridization among S-3px, Re-5dyz, and Re-5dxz orbitals; the CBM consists mainly of S-3pz and Re-5dxz and Re-5dz2 [see PDOS in Fig. 1(c)]. Though the VBM is below the water oxidation level, the CBM is located below the hydrogen redox potential [see Fig. 1(d)], the photocatalytic activity of hydrogen production in ReS2 is inhibited. These calculated results are in good agreement with the experimental results.44
3.2 Tuning band edge positions to improve the photocatalytic performance of ReS2 through applying uniaxial or bi-axial strain
As shown in Fig. 2, tensile strains, whether uniaxial or bi-axial, elongate the in-plane bond lengths, but slightly shorten the vertical S–S distance because the lattice vectors in the plane are fixed in the process of atomic relaxation. Compressive strains produce the opposite effect. Here dRe2–S3 increases with an increase in x-axial strain, while it is almost unchanged with an increase in z-axial strain because there are no components of dRe2–S3 in the z-direction. Similarly, dRe2–S3 and dRe–Re increase with an increase in z-axial strain, while they are almost unchanged with an increase in x-axial strain because there are no components of dRe–Re in the z-direction. As far as bond angle is concerned, the opposite sides of θ1 and θ3 are Re1–Re2 and S1–S3, respectively, which being parallel to z-axial and x-axial strains, will be elongated by tensile strains. Thus, θ1 increases with an increase in z-axial strain and θ3 increases with an increase in x-axial strain. The influence of bi-axial strain is found to be more pronounced than that of uniaxial strains because it comes from the common effect of x-axial and z-axial strains.
 | ||
| Fig. 2 The calculated structural parameters and formation energies of ReS2 as a function of (a) x-axial, (b) z-axial and (c) bi-axial strain. | ||
To further investigate the stability of ReS2, we calculated its formation energy, which is defined as:46 Ef = E2D/N2D − E3D/N3D, where E2D and E3D are the total energy of monolayer and bulk ReS2, and N2D and N3D are the number of atoms in monolayer and bulk ReS2. The formation energies of x-axial-strained ReS2 decrease with an increase in the strains and the decrease is sharper under compressive strains than under tensile strains. Additionally, all the formation energies have negative values and are smaller than that of pristine ReS2, indicating that monolayer ReS2 is stable. For z-axial and bi-axial strains, the formation energies decrease with an increase in the compressive strains but increase with an increase in the tensile strains, indicating that the systems are more stable under compressive strains than under tensile strains.
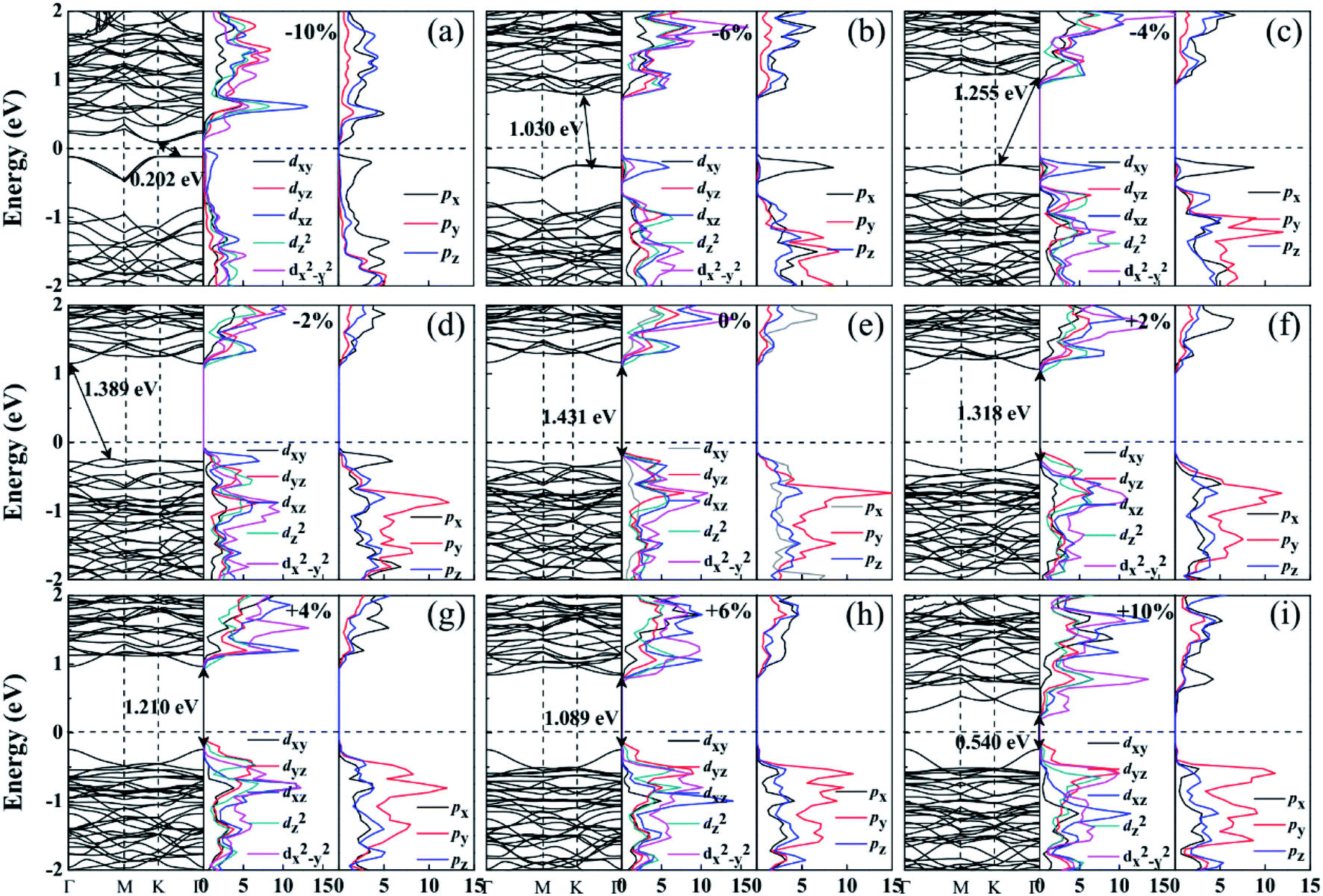 | ||
| Fig. 3 Band structure and PDOS of ML-ReS2 at x-axial strains of (a) −10%, (b) −6%, (c) −4%, (d) −2%, (e) 0%, (f) 2%, (g) 4%, (h) 6% and (i) 10%. | ||
For z-axial compressive strained ReS2, the energy of the CBM decreases with an increase in the compressive strain, thus, the band gap decreases from 1.431 to 0.688 eV when the compressive strain varies from 0 to −10% [see Fig. S1(a)–(d) in the ESI†].42 Under z-axial tensile strain, ReS2 changes to indirect-band-gap semiconductor, the VBM locates at the Γ point but its energy decreases, while the CBM relocates from the Γ point to the M point and then to the K point. The PDOS in Fig. S1(f)–(i)† indicates that the Re-5dxz states and S-3py states play important roles in the process. This is mainly induced by the changes in dS–S and dRe–Re. Thus, the band gap decreases from 1.431 eV to 0.783 eV as the tensile strains vary from 0 to 10%.
The effect of the bi-axial strain on ReS2 is the result of the common effect of x-axial and z-axial strain on ReS2. Thus, bi-axial-strained ReS2 is an indirect-band-gap semiconductor and the band gap decreases (from 1.431 eV to 0.452 eV) with an increase in the strains, whether compressive or tensile. ReS2 displays metallic properties at 10% bi-axial compressive strain as there are a few bands across the Fermi level which mainly originate from hybridization of Re-5dx2−y2, 5dxz, and S-3px. Clearly, for the bi-axial compressive strain, Re-5dxz and S-3px play a leading role near the Fermi level. While for the bi-axial tensile strain, Re-5dx2−y2 and S-3py play important roles near the Fermi level [see Fig. S2†].
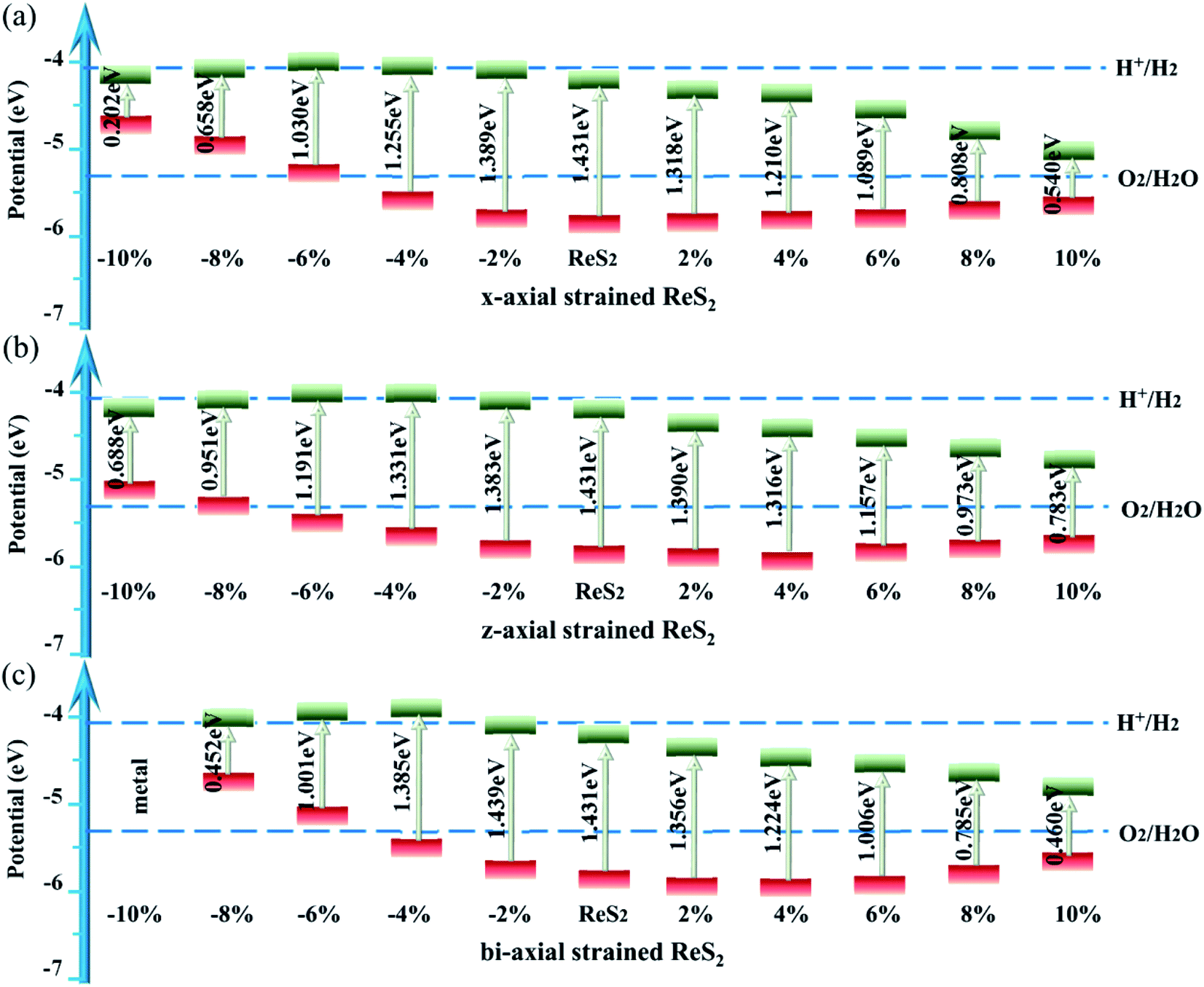 | ||
| Fig. 4 Calculated band alignments and band gaps of ReS2 at different (a) x-axial, (b) z-axial and (c) bi-axial strains in comparison with water redox potentials. | ||
As we know, the work function is greatly determined by the crystal structure which is related to the surface charge distribution and the electron surface barrier caused by an internal electron field related to the surface electric dipole distribution. There is competition between the surface charge and the surface electric dipole. The in-plane strains cause severe distortion of the crystal structure, then influence the surface charge distribution, and thus decisively affect the work function. However, the in-plane strain induces little geometric distortion in the vertical direction, therefore inducing a very small dipole moment perpendicular to the surface component μ⊥,60 and then slightly affecting the work function. For example, for ReS2 with a bi-axial strain of −4%, the induced dipole density μ⊥ is only −0.006 D nm−2 [see the Table S1 in the ESI†],42 little affecting the work function. Thus, the strain-induced rearrangement of surface charge plays an important role, leading to a great change in the work function of −0.382 eV. According to ΔV⊥ = −ΔW⊥, where ΔV⊥and ΔW⊥ respectively refer to the band edge shifts and the opposite direction. The band edge position upshifts by 0.382 eV [see the Table in the ESI†].42 As a result, the CBM of ReS2 straddles the water reduction potential, and ReS2 satisfies the demands of water splitting.
3.3 Tuning band edge positions to improve the photocatalytic performance of ReS2 through applying an electric field
We further attempted to tune the band structure of ReS2 by applying an electric field. The vertical electric fields are applied with different intensities along the y-direction. Fig. 5(a) shows that the formation energies of ReS2, defined as Ef = E2D/N2D − E3D/N3D,46 have negative values, indicating that the systems have good stability under different vertical electric fields. | ||
| Fig. 5 (a) Formation energy, (b) average electrostatic potential and (c) work function of ReS2 under different electric fields. | ||
Fig. 6 displays the electronic structures of ReS2 under different external electric fields. ReS2 maintaining direct-band-gap semiconductor properties. For a positive electric field, the Re-5d and S-3p components of the CBM are slightly enlarged [see Fig. 6(d)–(f)], for a negative electric field, the Re-5d component of the VBM is slightly enlarged [see Fig. 6(a)–(c)]. Thus the band gap decreases slightly with an increase in the electric fields, changing from 1.431 to 1.411 eV, and the minimum value of 1.411 eV is found under electric fields of −0.2 V Å−1 and 0.2 V Å−1.
 | ||
| Fig. 6 Band structure and PDOS of ML-ReS2 under vertical electric fields of (a) −0.2 V Å−1, (b) −0.15 V Å−1, (c) −0.1 V Å−1, (d) 0.1 V Å−1, (e) 0.15 V Å−1 and (f) 0.2 V Å−1. | ||
Though the electric field has little influence on the band gap, it produces an in-plane dipole moment and then greatly affects the average electrostatic potential of ReS2. As shown in the charge difference density in the inset of Fig. 7, surface charge transfer depends on the direction of E⊥: for a negative electric field, the charge transfers from the upper side to the lower side, whereas, for a positive electric field, the charge transfers from the lower side to the upper side. The Bader charge analysis also quantitatively shows that there are 0.036e charges transferred from the upper side to the lower side for E⊥ = −0.1 V Å,59 there are 0.143e charges transfer from the lower side to the upper side for E⊥ = 0.1 V Å−1. Simultaneously, a vertical electric field alters the surface charge concentration of ReS2. Fig. 5(b) shows the electrostatic potential ![[V with combining macron]](https://www.rsc.org/images/entities/i_char_0056_0304.gif) (y) under different electric fields, which displays a sawtooth behavior. For a negative electric field, (y) increases linearly with an increasing vacuum layer, while the opposite is true for a positive field. Thus, the work function on both sides can be defined as W = mE⊥ + Φw,61,62 where Φw is the work function of ReS2 without the external electric field, and m is the slope of the line relating to vacuum lengths. Fig. 5(c) shows that the work function varies linearly with the electric field, and the change in work function (ΔW⊥ arises from the electric-field-induced dipole density, expressed as ΔW⊥ = μ⊥/Aε0, where A is the surface area per molecule, ε0 is the dielectric constant of the vacuum). As shown in Table 1, when E⊥ is −0.1 V Å−1, the induced dipole density is −1.382 D nm−2, and the resultant change in work function is 0.521 eV, which is in agreement with the energy shift calculated from the average electrostatic potential.
(y) under different electric fields, which displays a sawtooth behavior. For a negative electric field, (y) increases linearly with an increasing vacuum layer, while the opposite is true for a positive field. Thus, the work function on both sides can be defined as W = mE⊥ + Φw,61,62 where Φw is the work function of ReS2 without the external electric field, and m is the slope of the line relating to vacuum lengths. Fig. 5(c) shows that the work function varies linearly with the electric field, and the change in work function (ΔW⊥ arises from the electric-field-induced dipole density, expressed as ΔW⊥ = μ⊥/Aε0, where A is the surface area per molecule, ε0 is the dielectric constant of the vacuum). As shown in Table 1, when E⊥ is −0.1 V Å−1, the induced dipole density is −1.382 D nm−2, and the resultant change in work function is 0.521 eV, which is in agreement with the energy shift calculated from the average electrostatic potential.
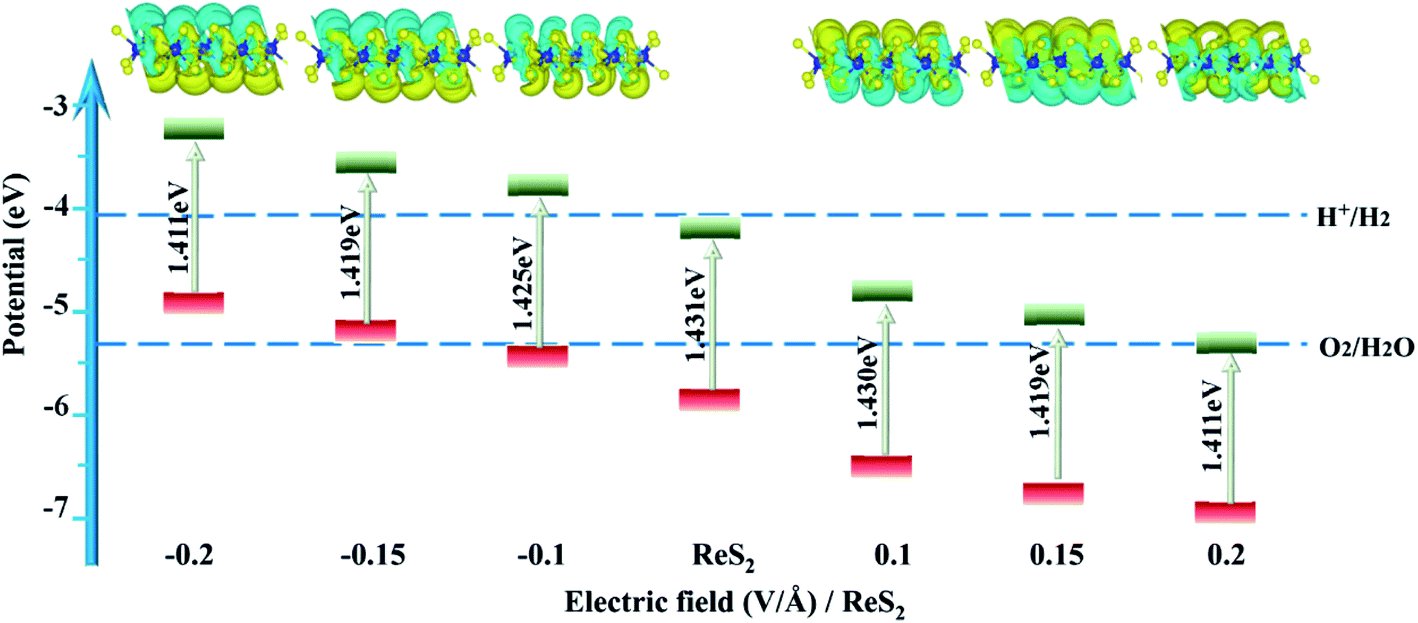 | ||
| Fig. 7 The band alignments and band gaps of ReS2 at different external electric fields. The inset shows the charge density difference of ReS2; yellow and green colors represent charge accumulation and charge depletion, respectively. The isovalue is 0.003 e Å−3. | ||
| E⊥ | μ⊥/A | ΔW⊥ | Eg | EVBM | ECBM | Ef |
|---|---|---|---|---|---|---|
| −0.2 | −2.767 | −1.043 | 1.411 | −4.743 | −3.332 | −0.8 |
| −0.15 | −2.074 | −0.782 | 1.419 | −5.012 | −3.593 | −0.9 |
| −0.1 | −1.382 | −0.521 | 1.425 | −5.279 | −3.854 | −0.6 |
| 0 | 0 | 0 | 1.431 | −5.807 | −4.376 | −1.1 |
| 0.1 | 1.389 | 0.523 | 1.430 | −6.330 | −4.900 | −1.2 |
| 0.15 | 2.081 | 0.784 | 1.419 | −6.580 | −5.161 | −0.9 |
| 0.2 | 2.777 | 1.047 | 1.411 | −6.835 | −5.424 | −1.7 |
Fig. 7 shows the band edges of ReS2 with different electric fields. ReS2 obviously upshifts with increasing negative electric fields, and the reduction capacity increases, while it downshifts with increasing positive electric fields, and the oxidation capacity increases. It is worth mentioning that the applied electric field can not only tune the band edge but also accelerate electron–hole separation and enhance activity carrier mobility. For ReS2 with an applied electric field of −0.1 V Å−1, the band edge upshifts by 0.521 eV and straddles the water redox potential.
4. Conclusions
To summarize, based on DFT calculations, we applied an external strain or an electric field to engineer the electronic structure of ReS2 to meet the requirements of hydrogen production through photocatalytic water splitting. The in-plane strains have a great influence on the band gap and band alignment originating from great changes in the crystal structure. An increase in the strain decreases the band gap of ReS2, and the band edge positions downshift with an increase in the tensile strains and upshift with an increase in compressive strains, where the shifts come mainly from rearrangement of surface charge. While the external electronic fields slightly influence the band gap of ReS2 but greatly affect its band alignment. The band edge position shifts come from an external-electronic-field-induced dipole moment. Importantly, ReS2 with a bi-axial strain of −4% or an electric field of −0.1 V Å−1 can be considered strong candidates for photocatalytic water splitting because they possess suitable band edge positions for water splitting, ideal band gaps for visible-light absorption, good stability, reduced electron–hole recombination, and high carrier mobility. Notably, applying strain or an electric field is a feasible means of functionalization of single-layered and multilayered nanostructures for better photocatalytic water splitting.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work is supported by the NSFC (11774302, 21903014 and 12074332) and the Qinglan Project of Jiangsu Province of Yangzhou University. The authors are grateful for access to the computational resources at YZU.References
- R. Schloegl, Nat. Mater., 2008, 7, 772 CrossRef CAS PubMed.
- A. Kudo and Y. Miseki, Chem. Soc. Rev., 2009, 38, 253–278 RSC.
- S. Chu and A. Majumdar, Nature, 2012, 488, 294–303 CrossRef CAS PubMed.
- A. Fujishima and K. Honda, Nature, 1972, 238, 37–38 CrossRef CAS.
- T. Su, Q. Shao, Z. Qin, Z. Guo and Z. Wu, ACS Catal., 2018, 8, 2253–2276 CrossRef CAS.
- W. W. Zhong, Z. P. Wang, N. Gao, L. A. Huang, Z. P. Lin, Y. P. Liu, F. Q. Meng, J. Deng, S. F. Jin, Q. H. Zhang and L. Gu, Angew. Chem., Int. Ed., 2020, 59, 22743–22748 CrossRef CAS PubMed.
- S. J. Shen, Z. P. Lin, K. Song, Z. P. Wang, L. G. Huang, L. H. Yan, F. Q. Meng, Q. H. Zhang, L. Gu and W. W. Zhong, Angew. Chem., Int. Ed., 2021, 60, 12360–12365 CrossRef CAS.
- Z. Lin, S. Shen, Z. Wang and W. Zhong, iScience, 2021, 24, 102469 CrossRef PubMed.
- B. Fang, J. H. Kim, M.-S. Kim and J.-S. Yu, Acc. Chem. Res., 2013, 46, 1397–1406 CrossRef CAS PubMed.
- S. Yu, S. Song, R. Li and B. Fang, Nanoscale, 2020, 12, 19536–19556 RSC.
- B. Z. Fang, M. Kim, J. H. Kim and J. S. Yu, Langmuir, 2008, 24, 12068–12072 CrossRef CAS PubMed.
- Y. Xing, B. Fang, A. Bonakdarpour, S. Zhang and D. P. Wilkinson, Int. J. Hydrogen Energy, 2014, 39, 7859–7867 CrossRef CAS.
- M. Faraji, M. Yousefi, S. Yousefzadeh, M. Zirak, N. Naseri, T. H. Jeon, W. Choi and A. Z. Moshfegh, Energy Environ. Sci., 2019, 12, 59–95 RSC.
- G. Liao, J. Fang, Q. Li, S. Li, Z. Xu and B. Fang, Nanoscale, 2019, 11, 7062–7096 RSC.
- G. Liao, C. Li, X. Li and B. Fang, Cell Rep. Phys. Sci., 2021, 2, 100355 CrossRef.
- Y. Liu, S. Shen, Z. Li, D. Ma, G. Xu and B. Fang, Mater. Charact., 2021, 174, 111031 CrossRef CAS.
- M. Ni, M. K. H. Leung, D. Y. C. Leung and K. Sumathy, Renewable Sustainable Energy Rev., 2007, 11, 401–425 CrossRef CAS.
- S. U. Khan, M. Al-Shahry and W. B. Ingler Jr, Science, 2002, 297, 2243–2245 CrossRef CAS PubMed.
- J. Liqiang, Q. Yichun, W. Baiqi, L. Shudan, J. Baojiang, Y. Libin, F. Wei, F. Honggang and S. Jiazhong, Sol. Energy Mater. Sol. Cells, 2006, 90, 1773–1787 CrossRef.
- T. Xu, L. Zhang, H. Cheng and Y. Zhu, Appl. Catal., B, 2011, 101, 382–387 CrossRef CAS.
- Y. Huan, X. Wang, W. Hao and L. Li, RSC Adv., 2015, 5, 72410–72415 RSC.
- G. Liao, Y. Gong, L. Zhang, H. Gao, G.-J. Yang and B. Fang, Energy Environ. Sci., 2019, 12, 2080–2147 RSC.
- A. H. Castro Neto, F. Guinea, N. M. R. Peres, K. S. Novoselov and A. K. Geim, Rev. Mod. Phys., 2009, 81, 109–162 CrossRef CAS.
- K. S. Novoselov, A. K. Geim, S. V. Morozov, D. Jiang, M. I. Katsnelson, I. V. Grigorieva, S. V. Dubonos and A. A. Firsov, Nature, 2005, 438, 197–200 CrossRef CAS PubMed.
- S. Stankovich, D. A. Dikin, G. H. Dommett, K. M. Kohlhaas, E. J. Zimney, E. A. Stach, R. D. Piner, S. T. Nguyen and R. S. Ruoff, Nature, 2006, 442, 282–286 CrossRef CAS PubMed.
- Z. P. Wang, Z. P. Lin, J. Deng, S. J. Shen, F. Q. Meng, J. T. Zhang, Q. H. Zhang, W. W. Zhong and L. Gu, Adv. Energy Mater., 2021, 11, 7 Search PubMed.
- L. Lu, S. Zou and B. Fang, ACS Catal., 2021, 11, 6020–6058 CrossRef CAS.
- C. Wang, M. Zhu, Z. Cao, P. Zhu, Y. Cao, X. Xu, C. Xu and Z. Yin, Appl. Catal., B, 2021, 291, 120071 CrossRef CAS.
- Q. H. Wang, K. Kalantar-Zadeh, A. Kis, J. N. Coleman and M. S. Strano, Nat. Nanotechnol., 2012, 7, 699–712 CrossRef CAS PubMed.
- M. Chhowalla, H. S. Shin, G. Eda, L. J. Li, K. P. Loh and H. Zhang, Nat. Chem., 2013, 5, 263–275 CrossRef PubMed.
- Z. Lin, B. Xiao, Z. Wang, W. Tao, S. Shen, L. Huang, J. Zhang, F. Meng, Q. Zhang, L. Gu and W. Zhong, Adv. Funct. Mater., 2021, 2102321 CrossRef.
- C. Z. Wang, X. D. Shao, J. Pan, J. G. Hu and X. Y. Xu, Appl. Catal., B, 2020, 268, 7 Search PubMed.
- D. Voiry, J. Yang and M. Chhowalla, Adv. Mater., 2016, 28, 6197–6206 CrossRef CAS PubMed.
- Q. Lu, Y. Yu, Q. Ma, B. Chen and H. Zhang, Adv. Mater., 2016, 28, 1917–1933 CrossRef CAS PubMed.
- D. Wang, Y. Liu, X. Meng, Y. Wei, Y. Zhao, Q. Pang and G. Chen, J. Mater. Chem. A, 2017, 5, 21370–21377 RSC.
- D. A. Chenet, O. B. Aslan, P. Y. Huang, C. Fan, A. M. van der Zande, T. F. Heinz and J. C. Hone, Nano Lett., 2015, 15, 5667–5672 CrossRef CAS PubMed.
- E. Liu, Y. Fu, Y. Wang, Y. Feng, H. Liu, X. Wan, W. Zhou, B. Wang, L. Shao, C. H. Ho, Y. S. Huang, Z. Cao, L. Wang, A. Li, J. Zeng, F. Song, X. Wang, Y. Shi, H. Yuan, H. Y. Hwang, Y. Cui, F. Miao and D. Xing, Nat. Commun., 2015, 6, 6991 CrossRef CAS PubMed.
- J. Hamalainen, M. Mattinen, K. Mizohata, K. Meinander, M. Vehkamaki, J. Raisanen, M. Ritala and M. Leskela, Adv. Mater., 2018, 30, e1703622 CrossRef PubMed.
- L. Hart, S. Dale, S. Hoye, J. L. Webb and D. Wolverson, Nano Lett., 2016, 16, 1381–1386 CrossRef CAS PubMed.
- W. W. Zhong, B. B. Xiao, Z. P. Lin, Z. P. Wang, L. G. Huang, S. J. Shen, Q. H. Zhang and L. Gu, Adv. Mater., 2021, 33, 7 CrossRef PubMed.
- S. Tongay, H. Sahin, C. Ko, A. Luce, W. Fan, K. Liu, J. Zhou, Y. S. Huang, C. H. Ho, J. Yan, D. F. Ogletree, S. Aloni, J. Ji, S. Li, J. Li, F. M. Peeters and J. Wu, Nat. Commun., 2014, 5, 3252 CrossRef PubMed.
- M. Rahman, K. Davey and S.-Z. Qiao, Adv. Funct. Mater., 2017, 27, 1606129 CrossRef.
- Y. Li, Y.-L. Li, B. Sa and R. Ahuja, Catal. Sci. Technol., 2017, 7, 545–559 RSC.
- X. Xu, H. Zhao, R. Wang, Z. Zhang, X. Dong, J. Pan, J. Hu and H. Zeng, Nano Energy, 2018, 48, 337–344 CrossRef CAS.
- X. Liu and Z. Li, J. Phys. Chem. Lett., 2015, 6, 3269–3275 CrossRef CAS.
- H. Chen, C. Tan, K. Zhang, W. Zhao, X. Tian and Y. Huang, Appl. Surf. Sci., 2019, 481, 1064–1071 CrossRef CAS.
- B. You, M. T. Tang, C. Tsai, F. Abild-Pedersen, X. Zheng and H. Li, Adv. Mater., 2019, 31, e1807001 CrossRef PubMed.
- J. Qi, X. Li, X. Qian and J. Feng, Appl. Phys. Lett., 2013, 102, 173112 CrossRef.
- Y. Bai, G. Luo, L. Meng, Q. Zhang, N. Xu, H. Zhang, X. Wu, F. Kong and B. Wang, Phys. Chem. Chem. Phys., 2018, 20, 14619–14626 RSC.
- K. Edalati, K. Fujiwara, S. Takechi, Q. Wang, M. Arita, M. Watanabe, X. Sauvage, T. Ishihara and Z. Horita, ACS Appl. Energy Mater., 2020, 3, 1710–1718 CrossRef CAS.
- G. Wang, F. Zhou, B. Yuan, S. Xiao, A. Kuang, M. Zhong, S. Dang, X. Long and W. Zhang, Nanomaterials, 2019, 9, 244 CrossRef CAS PubMed.
- B. Dai, Y. CHen, S. M. Hao, H. Huang, J. Kou, C. Lu, Z. Lin and Z. Xu, J. Phys. Chem. Lett., 2020, 11, 7407–7416 CrossRef CAS PubMed.
- G. Kresse and J. Furthmüller, Phys. Rev. B: Condens. Matter Mater. Phys., 1996, 54, 11169 CrossRef CAS PubMed.
- P. E. Blochl, Phys. Rev. B: Condens. Matter Mater. Phys., 1994, 50, 17953–17979 CrossRef PubMed.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865 CrossRef CAS PubMed.
- J. Neugebauer and M. Scheffler, Phys. Rev. B: Condens. Matter Mater. Phys., 1992, 46, 16067–16080 CrossRef CAS PubMed.
- L. Bengtsson, Phys. Rev. B: Condens. Matter Mater. Phys., 1999, 59, 12301 CrossRef CAS.
- S. Yang, D. Prendergast and J. B. Neaton, Nano Lett., 2012, 12, 383–388 CrossRef CAS PubMed.
- W. Tang, E. Sanville and G. Henkelman, J. Phys.: Condens. Matter, 2009, 21, 084204 CrossRef CAS PubMed.
- J. Pan, Z. Wang, Q. Chen, J. Hu and J. Wang, Nanoscale, 2014, 6, 13565–13571 RSC.
- M. Topsakal, H. H. Gürel and S. Ciraci, J. Phys. Chem. C, 2013, 117, 5943–5952 CrossRef CAS.
- R. Gholizadeh and Y.-X. Yu, J. Phys. Chem. C, 2014, 118, 28274–28282 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1ra03821d |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2021 |