
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAdsorption of lindane (γ-hexachlorocyclohexane) on nickel modified graphitic carbon nitride: a theoretical study†
Nguyen Thi Thu Ha a,
Pham Thi Beab and
Nguyen Ngoc Ha*a
a,
Pham Thi Beab and
Nguyen Ngoc Ha*a
aFaculty of Chemistry, Hanoi National University of Education, 136 Xuan Thuy Str., Hanoi, Vietnam. E-mail: hann@hnue.edu.vn
bFaculty of Natural Science and Technology, Taynguyen University, Daklak, Vietnam
First published on 14th June 2021
Abstract
Adsorption of lindane (HCH) on nickel modified graphitic carbon nitride (Ni-gCN) was investigated using a novel, accurate and broadly parametrized self-consistent tight-binding quantum chemical (GFN2-xTB) method. Two graphitic carbon nitride (gCN) models were used: corrugated and planar, which represent the material with different thicknesses. Electronic properties of the adsorbates and adsorbent were estimated via vertical ionization potential, vertical electron affinity, global electrophilicity index and the HOMO and LUMO. Adsorption energy and population analyses were carried out to figure out the nature of the adsorption process. The results reveal that the introduction of the nickel atom significantly influences the electronic properties of gCN, and results in the improvement of adsorption ability of gCN for lindane. Lindane adsorption on Ni-gCN is considered as chemisorption, which is primarily supported by the interaction of the nickel atom and chlorine atoms of HCH. The effect of solvents (water, ethanol, acetonitrile) was investigated via the analytical linearized Poisson–Boltzmann model. Due to the strong chemisorption, Ni-gCN can collect lindane from different solvents. The adsorption configurations of HCH on Ni-gCN were also shown to be thermally stable at 298 K, 323 K, 373 K, 473 K, and 573 K via molecular simulation calculations. The obtained results are useful for a better understanding of lindane adsorption on Ni-gCN and for the design of materials with high efficiency for lindane treatment based on adsorption-photocatalytic technology.
Introduction
Persistent organic pollutants (POPs) are a class of pollutants of global concern due to their bioaccumulative properties, high toxicity, and extensive damage to humans and biodiversity.1 Recognizing the global issue of POPs, the Stockholm Convention entered into force in 2001 with the aim of regulating or prohibiting a preliminary list of twelve POPs, reducing pollution, and gradually treating and destroying these compounds.2Among persistent organic pollutants, POP plant protection chemicals attract the attention of researchers. One of the most commonly used pesticides is lindane or γ-hexachlorocyclohexane (HCH).3 Because of its difficulties decomposing, HCH pollution in soil and groundwater remains a common environmental issue. Currently, there are many different methods for treating POPs, such as biodegradation method,4 advanced oxidation processes,5 combustion,6 etc. Among these methods, advanced oxidation processes, including photocatalytic degradation, have shown a high efficiency in decomposing pesticides in water.7 For any chemical reaction that occurs on the surface of a heterogeneous catalyst, the adsorption stage is a critical step. If the material does not have the ability to adsorb, it can not exhibit high catalytic performance. Because of the synergistic effect of adsorption, the photocatalytic decomposition of POPs will be extremely efficient. Therefore, the study of adsorption capacity is extremely important to serve as a basis for selecting the appropriate catalyst.
Graphitic carbon nitride (gCN) is a non-metallic polymer that is attracting interest in the fields of next-generation photocatalytic materials due to its appropriate band gap (approximately 2.7 eV).8 gCN based materials have also been widely investigated as an effective adsorbent for metal ions,9 flavonoids,10 dyes,11 fluorine-containing compounds12 and plant protection chemicals13–15 including HCH.16 The synergistic effect between adsorption and photocatalysis of gCN based materials has been mentioned in various studies. Xu et al. showed that increasing the surface area of gCN improves its photocatalytic activity for methylene blue degradation.17 According to Li et al., the enhanced photocatalytic activity of the gCN/ZnO composite may be due to the synergistic effect of photon acquisition and direct contact, i.e. the adsorption of organic dyestuff on the surface.18 The photocatalytic and adsorption performance of gCN has been greatly improved by different modification methods, including element doping. Jiang et al. demonstrated that doping alkali metal ions to gCN increases the surface area, reduces band gap, increases electron displacement, leading to increased efficiency for photocatalytic hydrogen production.19 Zhang et al. studied the NO2 adsorption capacity of gCN systems modified by different metals by DFT method.20 The results show that the NO2 adsorption capacity of the metal doped material increases significantly compared to the pristine gCN.
Nickel-based catalysts have been widely studied for photocatalytic reactions such as hydrogen production,21,22 carbon dioxide reduction,23–25 dye degradation.26–29 Nickel based catalysts are easy to fabricate, inexpensive, but exhibit relatively high activity compared with other transition and noble metal-based catalysts. For example, Shtyka et al. studied the photocatalytic reduction of CO2 to methanol over Pt, Pd, Ni, Cu/TiO2 catalysts. The authors showed that the Pt and Ni were the most active in terms of the formation rate of methanol.24 Ni doping can greatly increase the visible light photocatalytic performance of TiO2 photocatalyst through the introduction of impurity bands in the band gap of TiO2, and thus reduces the band gap.29,30 However, the application of nickel-based catalyst in the treatment of POPs is still very limited. There are only a few studies dealing with the use of nickel catalysts for POPs degradation.31,32
In this paper, we describe in great detail the adsorption performance of nickel doped graphitic carbon nitride (Ni-gCN) for HCH by theoretical calculations. Adsorption is the first step of the photocatalytic – adsorption process. Understanding the adsorption process will help clarify and explain the mechanism of reactions that take place on the photocatalytic surface.
Models and computational methods
Models
Graphitic carbon nitride is built from s-triazine or s-heptazine (tri-s-triazine) units bound via sp3 hybridization nitrogen atoms. In this work, we constructed two models of s-triazine graphitic carbon nitride: corrugated (cGN) and planar (pGN) models in which the boundary atoms were saturated by hydrogen atoms. Each model consists of 195 atoms and has a molecular formula of C75N100H20. It is reported in several studies that the corrugated form (cGN) has lower energy, and therefore, is more stable than the planar form.33–35 In the high symmetry planar structure – pGN, all atoms are fixed in a plane. Meanwhile, the corrugated (cGN) structure is characterized by the out-of-plane displacement of the atoms in order to reduce the electronic repulsion encountered by the lone pair electrons of nitrogen atoms. Thus, pGN exists only for multilayer g-C3N4 structures in which the van der Waals interactions with the low layers are strong enough to keep the top layer flat. On the contrary, in the single layer structures, g-C3N4 is stabilized by corrugation. Therefore, we use the pGN model to represent gCN with a large thickness, while the cGN model represents gCN with a thin layer structure. This idea of using the corrugated and planar models of gCN was mentioned in our previous study.36 A nickel atom was doped on the surface of both cGN and pGN to form Ni-cGN and Ni-pGN, respectively.Computational methods
All geometry and energy calculations were performed using an accurate and broadly parametrized self-consistent tight-binding quantum chemical method called GFN2-xTB (short for “Geometry, Frequency, Noncovalent, eXtended Tight-Binding”) implemented in xTB program.37 The accuracy of the GFN2-xTB method is benchmarked for a wide variety of systems.38–40 This method takes into account the contribution of halogen, hydrogen bonding and dispersion force (via D4 London model). In this work, geometry optimization was performed with an electronic temperature of 300 K, integral cutoff of 0.25 × 102, SCF convergence of 0.1 × 10−5 Ha and the wavefunction convergence of 0.1 × 10−3 e.The adsorption energy (Eads) was calculated as follow:
| Eads = Eadsorbate+substrate − Eadsorbate − Esubstrate | (1) |
The adsorption energy can be used to describe if the interaction between the HCH molecule and Ni-gCN is energetically favorable. Furthermore, to evaluate the nature of the interaction between HCH and Ni-gCN, a significant change in the geometrical parameters was analyzed. The population analysis giving the charge transfer, which is a rationale for the type of adsorption, was examined. Besides, the Wiberg bond order (BO) which is a measure of electron population overlap between two atoms, was also discussed.
The electronic properties of the adsorbent Ni-gCN and the adsorbate HCH were examined using vertical ionization potential (IP), vertical electron affinity (EA) and global electrophilicity index (GEI). The GEI is calculated as:
| GEI = (IP + EA)2/(8 × (IP − EA)) | (2) |
The GEI is considered as a metric for Lewis acidity which can provide information about the tend of electron transfer ability between species. The frontier molecular orbitals including the highest occupied molecular orbital (HOMO) and the lowest unoccupied molecular orbital (LUMO) were also analyzed to estimate the relative chemical reactivity of the studied systems.
Results and discussions
Geometrical structure and electronic properties of Ni-cCN and Ni-pGN
The optimized structures of Ni-cCN and Ni-pGN are presented in Fig. 1 and the calculated parameters are shown in Table 1. The interaction between Ni atom and cGN, pGN is characterized by the formation energy (Ef) which is defined as Ef = E (Ni-gCN) − E (Ni) − E (gCN).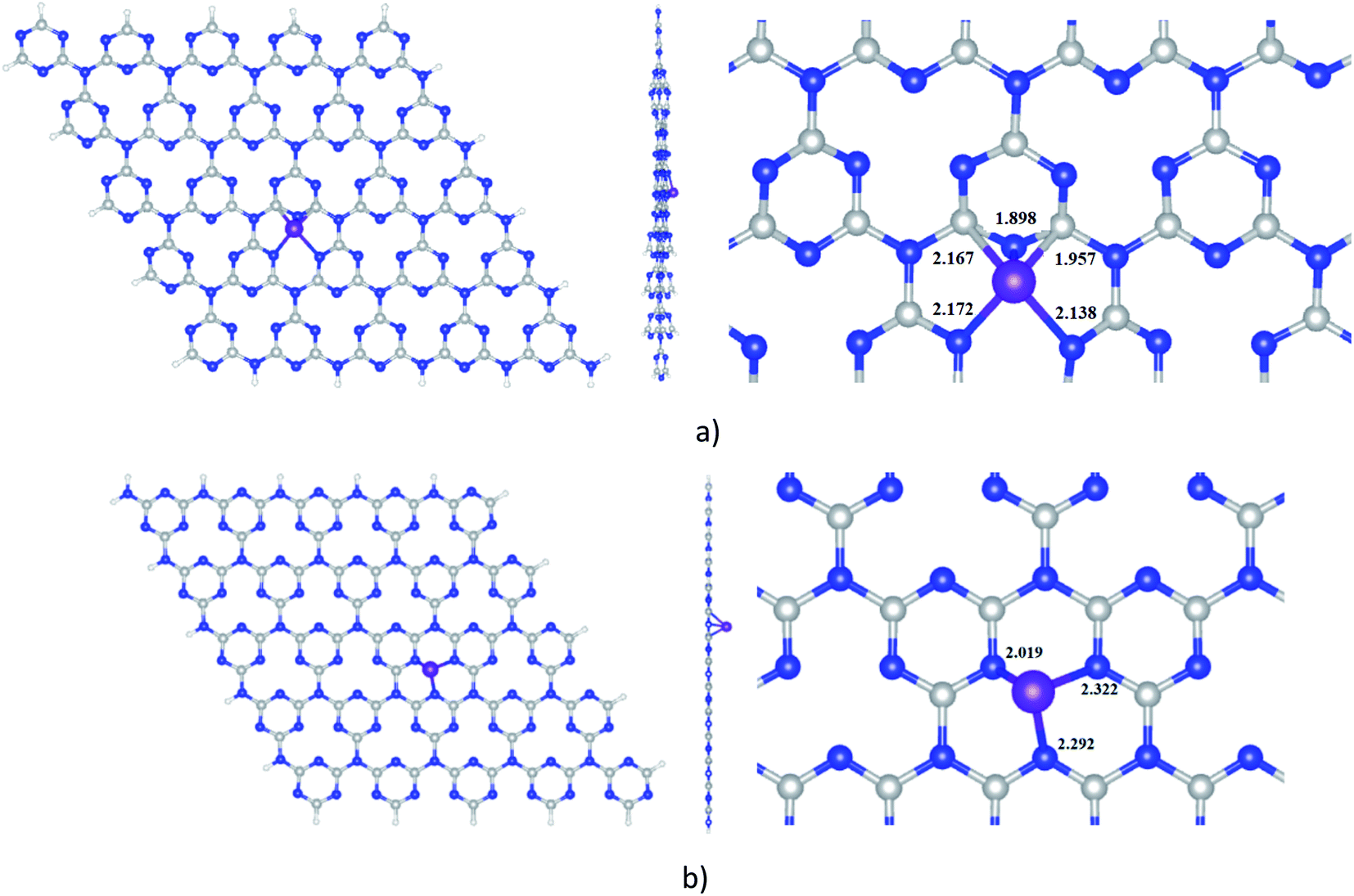 | ||
| Fig. 1 Optimized structures of Ni-cGN (a) and Ni-pGN (b) by the GFN2-xTB method; colours: grey: carbon, blue: nitrogen, white: hydrogen, violet: nickel; all key distances are in Å. | ||
| Parameter | Ni-cGN | Ni-pGN | cGN41 | pGN |
|---|---|---|---|---|
| Ef, kJ mol−1 | −447.1 | −312.9 | ||
| dmin (Ni-gCN), Å | 1.898 | 2.019 | ||
| q (Ni), e | −0.444 | −0.457 | ||
| BO (Ni-gCN) | 2.611 (BONi–N = 1.376, BONi–C = 0.808) | 1.841 (BONi–N = 1.140, BONi–C = 0.233) | ||
| IP, eV | 6.053 | 5.625 | 7.086 | 6.628 |
| EA, eV | 2.254 | 2.130 | 2.262 | 2.153 |
| GEI, eV | 2.270 | 2.151 | 2.264 | 2.154 |
The interaction between the Ni atom and the cGN as well as the pGN is energetically favourable due to the negative formation energy. When the Ni atom is doped on the surface of gCN, it chemically interacts with C and N atoms to form new chemical bonds Ni–N and Ni–C with the total bond order of 2.611 for cGN and 1.841 for pGN. The Ni atom more strongly interacts with the cGN structure than with the pGN expressed via the lower calculated Ef and higher BO. As a result, the distance from the Ni atom to the cGN (1.898 Å) is significantly smaller than that in the Ni-pGN structure (2.019 Å). A remarkable charge transfer from the cGN and pGN to the Ni atom is observed. The above-mentioned analysis clearly shows that the adsorption of Ni on both the corrugated and planar form of gCN can be considered as chemisorption, and thus, the properties of Ni-gCN are predicted to be entirely different from the pristine cCN or pGN.
The introduction of nickel into gCN led to a slight increase in the GEI of Ni-cGN and Ni-pGN compared to that of the pristine gCN. The global electrophilicity index, GEI, is used as a general, quantitative and base-independent metric of Lewis acidity.42 Since the GEI of Ni-cGN is greater than that of Ni-pGN, the former has higher Lewis acidity than the latter. The acidity of the adsorbent surface may influence the HCH adsorption capacity.
To figure out the location of strongly correlated and chemically active electrons in molecules, the fractional occupation density (FOD) calculations43 were performed for Ni-cGN and Ni-pGN. The electronic density and the FOD schemes of Ni-gCN are shown in Fig. 1 – ESI.† Although electrons are delocalized over the gCN surface, the strongly correlated and chemically active electrons are mainly localized on the Ni atom for both Ni-cGN and Ni-pGN systems. This finding is similar to the obtained results from our previous study on the Ag-, Fe-doped gCN.41 Fig. 2 illustrates the highest occupied molecular orbital (HOMO) and the lowest unoccupied molecular orbital (LUMO) of the Ni-cGN and Ni-pGN systems. Obviously, the HOMO is supported mainly by Ni atom, while the LUMO primarily localizes on the gCN surface. Along with the above discussed FOD calculations, it can be assumed that the Ni atom will play the role of the adsorption center when interacting with the HCH molecule.
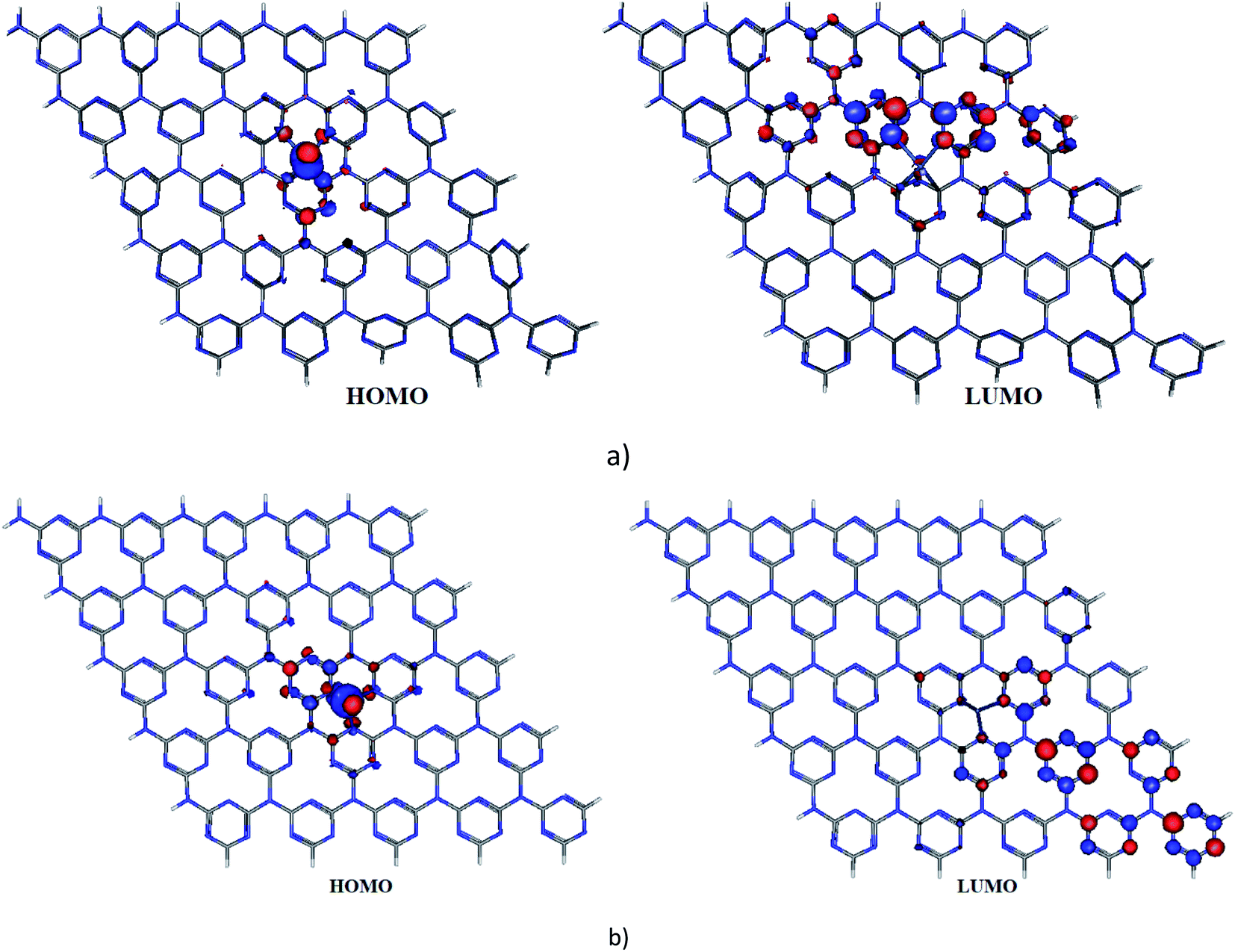 | ||
| Fig. 2 HOMO and LUMO of Ni-gCN depicted at an isovalue of 0.03 e A−3: (a) – Ni-cGN, (b) – Ni-pGN. | ||
Geometrical structure and electronic properties of HCH
It is well known that HCH has eight isomeric forms. The four common isomers of HCH are α-, β-, γ-, and δ-HCHs. Among these isomers, γ-HCH (also known as lindane) is usually the predominant isomer44 and thus, in this work we use γ-HCH as the model and it is simply denoted as HCH. The optimized structure of HCH is presented in Fig. 3a and the calculated geometrical and electronic parameters of HCH are shown in Table 2 with comparison to the references.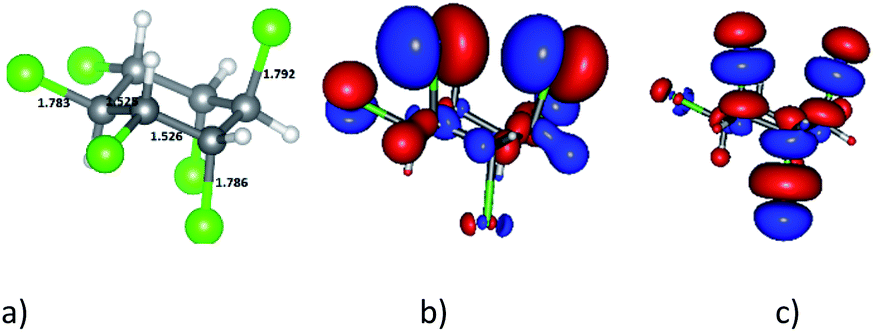 | ||
| Fig. 3 Optimized structure of HCH by GFN2-xTB method – (a); HOMO – (b) and LUMO – (c) of HCH depicted at isovalue of 0.03 e A−3, colours: grey: carbon, white: hydrogen, green: chlorine; all key distances are in Å. | ||
| Parameter | GFN2-xTB | Reference |
|---|---|---|
| a For CH3CHCl2.b For CH2ClCHCl2.c For cyclohexane.d Calculated at B3LYP/cc-pVDZ level of theory. | ||
| d(C–Cl), Å | 1.783; 1.786; 1.792 | 1.766a45, 1.776b46 |
| d(C–C), Å | 1.525; 1.526 | 1.540a45; 1.530b46 |
| d(C–H), Å | 1.094 | 1.101 (axial); 1.093 (equatorial)c47 |
| <H(a)CCl(e), degree | 106.41; 107.33; 106.41 | |
| <H(e)CCl(a), degree | 105.67; 106.54; 105.68 | |
| IP, eV | 9.681 | 9.87d48 |
| EA, eV | 0.150 | |
| GEI, eV | 1.267 | |
In the HCH structure, three chlorine atoms are located at the axial positions (a) and three chlorine atoms – at the equatorial (e) positions. Obviously, the results obtained by the GFN2-xTB method are consistent with the experimental and calculated data from references. The IP, EA and GEI of HCH are 9.681 eV, 0.150 eV and 1.267 eV, respectively. Since the GEI of HCH is lower than that of Ni-gCN, HCH is predicted to be an electron donor when interacting with Ni-gCN. The HOMO and LUMO of HCH are illustrated in Fig. 3b and c. It can be seen that the HOMO and LUMO of HCH are mainly located at the chlorine atoms. Therefore, the chlorine atoms are expected to be active centers when HCH interacts with Ni-gCN.
Interaction between HCH and Ni-cGN and Ni-pGN
When HCH interacts with Ni-gCN, there are a number of adsorption positions. The favourable adsorption configurations of HCH on Ni-gCN (HCH/Ni-gCN) were preliminary determined using the iMTD-GC algorithm. The iMTD-GC workflow generates conformer/rotamer ensembles by extensive metadynamic sampling (MTD) based on, with an additional genetic z-matrix crossing (GC) step at the end.49 The adsorption configurations obtained from iMTD-GC calculation were then fully refined by the GFN2-xTB method. Fig. 4 and 5 illustrate three favorable adsorption configurations of HCH on Ni-cGN and on Ni-pGN, respectively. In the first adsorption configurations, HCH interacts with the Ni active site via two Cl atoms at the equatorial positions (denoted as HCH-ee/Ni-cGN and HCH-ee/Ni-pGN). In the second configurations, the Ni atom is bound with one Cl atom at axial position and one other Cl atom at equatorial position (denoted as HCH-ae/Ni-cGN and HCH-ae/Ni-pGN). In the third configurations, the Ni atom is bound with two Cl atoms at axial positions (denoted as HCH-aa/Ni-cGN and HCH-aa/Ni-pGN). The adsorption energies are determined to be −268.66 kJ mol−1, −281.96 kJ mol−1 and −286.37 kJ mol−1 for HCH-ee/Ni-cGN, HCH-ae/Ni-cGN and HCH-aa/Ni-cGN, respectively. Therefore, the HCH-aa/Ni-cGN is considered as the most favourable adsorption configuration of HCH on Ni-cGN.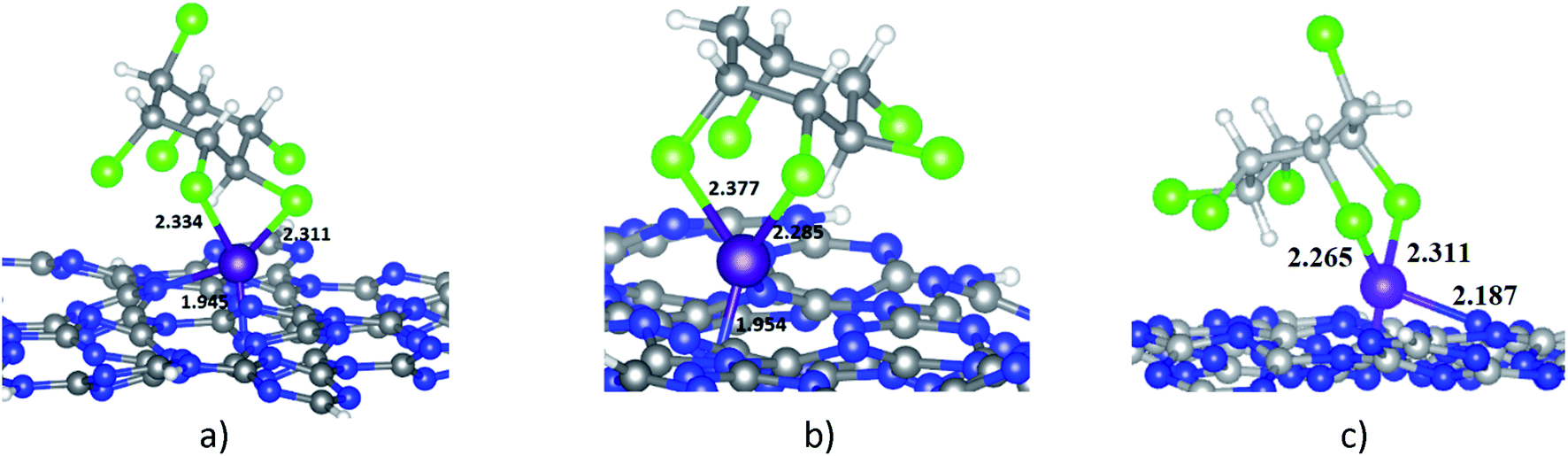 | ||
| Fig. 4 The favourable adsorption configurations of HCH on Ni-cGN: (a) HCH-ee/Ni-cGN; (b) HCH-ae/Ni-cGN; (c) HCH-aa/Ni-cGN colours: grey: carbon, blue: nitrogen, white: hydrogen, violet: nickel, green: chlorine; all key distances are in Å. | ||
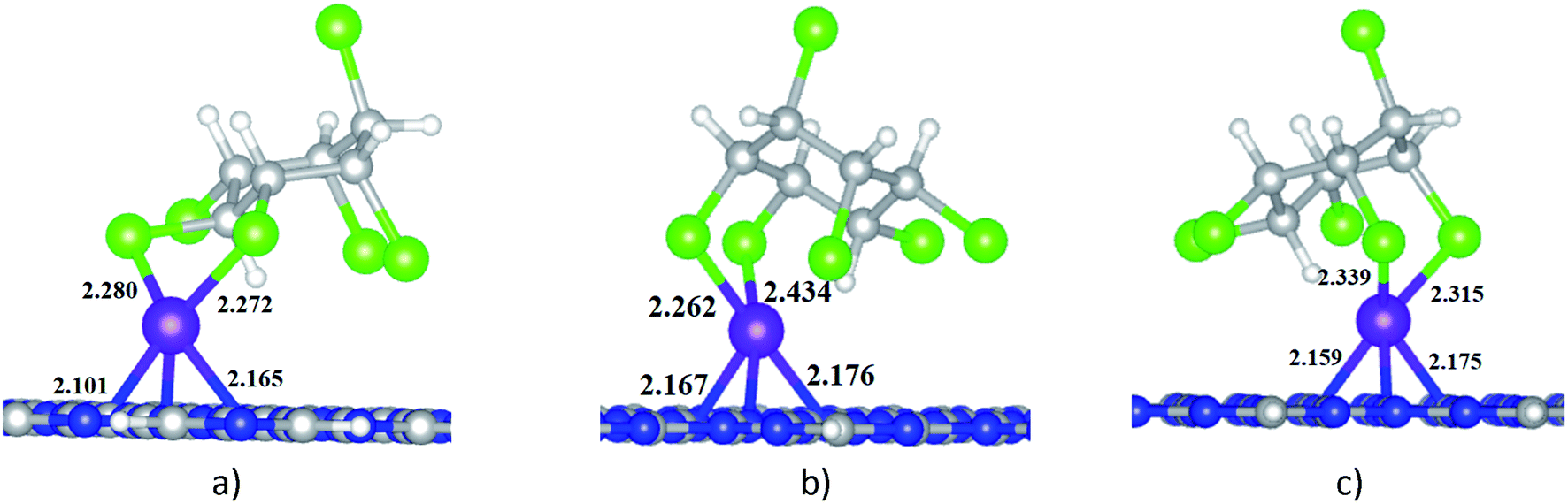 | ||
| Fig. 5 The favourable adsorption configurations of HCH on Ni-pGN: (a) HCH-ee/Ni-pGN; (b) HCH-ae/Ni-pGN; (c) HCH-aa/Ni-pGN; colours: grey: carbon, blue: nitrogen, white: hydrogen, violet: nickel, green: chlorine; all key distances are in Å. | ||
Similarly, the adsorption energies corresponding to the formation of the HCH-ee/Ni-pGN, HCH-ae/Ni-pGN and HCH-aa/Ni-pGN configurations are −272.87 kJ mol−1, −282.52 kJ mol−1 and −298.53 kJ mol−1, respectively. Thus, the formation of HCH-aa/Ni-pGN is more preferable than the formation of the HCH-ee/Ni-pGN and HCH-ae/Ni-pGN.
The most energetically favorable adsorption configurations HCH-aa/Ni-cGN and HCH-aa/Ni-pGN will be simply refered as HCH/Ni-cGN and HCH/Ni-pGN, respectively in the next discussion for more details to figure out the nature of the adsorption process.
The parameters calculated for HCH/Ni-cGN and HCH/Ni-pGN are shown in Tables 3 and 4. The adsorption of HCH on pristine cGN and pGN is also introduced for comparison.
| Parameter | HCH-aa/Ni-cGN | HCH/cGN |
|---|---|---|
| a Wiberg bond orders: summarized from chemical bonds formed between atoms of HCH and atoms of adsorbent with order >0.10. | ||
| Eads, kJ mol−1 | −286.37 | −91.86 |
| dmin (HCH-adsorbent), Å | 2.265 (dNi–Cl) | 2.367 (dH–N) |
| q (HCH), e | +0.488 | 0.000 |
| q (Ni), e | −0.624 | — |
| BOa (HCH-adsorbent) | 0.885 | 0 |
| BOa (Ni) | 2.893 (BONi–Cl = 0.885) | — |
| Parameter | HCH-aa/Ni-pGN | HCH/pGN |
|---|---|---|
| a Wiberg bond orders: summarized from chemical bonds formed between atoms of HCH and atoms of adsorbent with order >0.10. | ||
| Eads, kJ mol−1 | −298.53 | −50.69 |
| dmin (HCH-adsorbent), Å | 2.315 (dNi–Cl) | 2.935 (dCl–N) |
| q (HCH), e | +0.485 | +0.002 |
| q (Ni), e | −0.795 | — |
| BOa (HCH-adsorbent) | 0.797 | 0 |
| BOa (Ni) | 2.706 (BONi–Cl = 0.782) | — |
The adsorption energy of HCH on the pristine cGN is determined to be −91.86 kJ mol−1. The minimal distance from HCH to the cGN surface is 2.367 Å. Adsorption of HCH on pristine cGN is referred to as physisorption. There is no charge transfer observed between HCH and cGN. Meanwhile, the adsorption energy is greatly decreased as a Ni atom is doped to cGN. A significant charge transfer of 0.465 e from HCH to Ni-cGN is observed, which results in the decreasing of the atomic charge on the Ni atom compared to that in the initial Ni-cGN structure (see Table 1). This finding is consistent with the above discussed on the GEI values of HCH and Ni-cGN. The HCH molecule forms new chemical bonds with Ni-cGN. The total bond order between HCH and Ni-cGN is 0.832, which is mainly contributed by the interaction of the Ni atom with two Cl atoms. Therefore, the adsorption of HCH on Ni-cGN may be considered as chemisorption.
Likewise, the interaction between HCH and Ni-pGN releases more energy than that between HCH and the pristine pGN. The minimal distance from HCH to the Ni-pGN and the pristine pGN is 2.262 Å and 2.935 Å, respectively. A notable charge transfer between HCH is observed (+0.482 e), while there is almost no charge transfer between HCH and the pGN (+0.002 e). The total bond order between HCH and Ni-pGN is 0.750. Thus, the adsorption of HCH on Ni-pGN is considered as chemisorption, while the adsorption of HCH on the pristine pGN is physisorption.
It is noted that the adsorption energy of HCH on Ni-cGN is very close to that of HCH on Ni-pGN. Meanwhile, the adsorption energy of HCH on the pristine cGN is significantly lower than that of HCH on the pristine pGN. This finding reveals that, for unmodified gCN materials, the thickness may strongly affect adsorption activity. The thinner the material is, the higher its HCH adsorption capacity. But when denatured by Ni, the adsorption capacity of materials is less influenced by thickness, since adsorption occurs on the metal active site. However, it should be emphasized that, the thickness will influence the optical properties of the material36,50 and thus, influence the photocatalytic HCH conversion efficiency.
To estimate the transition states (if any) of the HCH adsorption process on Ni-gCN, the meta-dynamics reaction path finder51 implemented in GFN2-xTB programs was performed. This method is based on a simple metadynamics bias potential adding a repulsive potential on the reactant structure and an attractive potential on the product structure. The calculated results show that the relative energies of fourteen configurations during the adsorption of HCH on Ni-cGN continuously decrease from the initial to the final configuration (see Fig. 2 – ESI†). Thus, the adsorption of HCH on Ni-cGN does not involve a transition state, i.e., the adsorption process is not influenced by the kinetic factors.
Analysis of the HOMO, LUMO energies of the nickel doped cGN systems and HCH (Fig. 6a) clearly shows that the gap between the LUMO of the Ni-cGN/Ni-pGN and the HOMO of HCH is larger than that between the HOMO of the Ni-cGN/Ni-pGN and the LUMO of HCH. As a result, the interaction between the Ni-cGN/Ni-pGN HOMO and the LUMO of HCH is more energetically favorable than the interaction between the Ni-cGN/Ni-pGN LUMO and the HOMO of organic molecule. Consequently, the HOMO of the adsorption configurations is mainly supported by the HOMO of the nickel doped cGN systems (Fig. 6b and c).
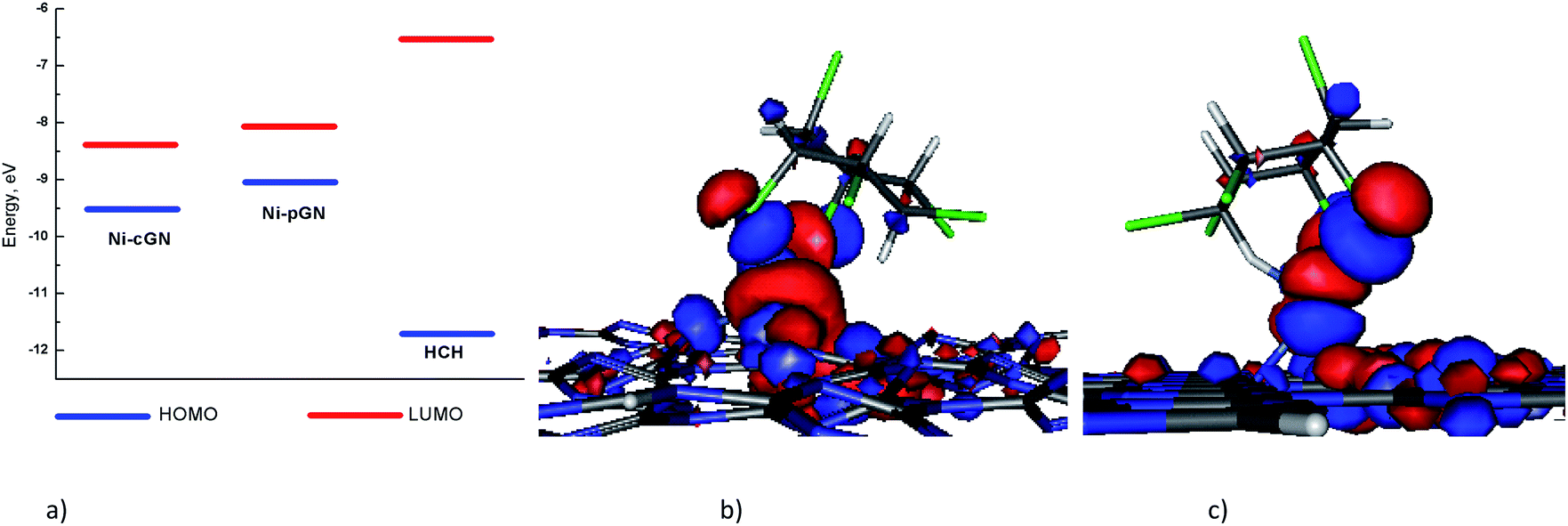 | ||
| Fig. 6 (a) Energy diagram of HOMOs and LUMOs of Ni-cGN, Ni-pGN and HCH; the HOMO of the adsorption configurations: (b) HCH-aa/Ni-cGN and (c) HCH-aa/Ni-pGN depicted at isovalue of 0.03 e Å−3. | ||
Thus, the doping of the nickel atom on the surface of both cGN and pGN greatly increases the adsorption ability for HCH. This is important for the next conversion steps of HCH because when chemically adsorbed, the C–Cl bonds will be strongly activated, that is, weakened and easily broken.
Effect of solvents on the adsorption of HCH on Ni-cGN and Ni-pGN
In fact, the adsorption process often takes place in solutions. Therefore, it is necessary to take into account the effect of solvents on the HCH adsorption on cGN based material. In this study, the influence of water solvent was included via an analytical linearized Poisson–Boltzmann (ALPB) solvation model. We considered three solvents: water, ethanol and acetonitrile (Table 5), in which the solubility of HCH is different.52| Parameter | HCH-aa/Ni-cGN | HCH-aa/Ni-pGN |
|---|---|---|
| a Wiberg bond orders: summarized from bonds formed between atoms of HCH and atoms of adsorbent with order >0.10. | ||
| In water | ||
| Eads, kJ mol−1 | −305.38 | −302.96 |
| dmin (HCH-adsorbent), Å | 2.294 (dNi–Cl) | 2.233 (dCl–N) |
| q (HCH), e | +0.574 | +0.557 |
| q (Ni), e | −0.684 | −0.820 |
| BOa (HCH-adsorbent) | 0.923 | 0.829 |
| BOa (Ni) | 2.980 (BONi–Cl = 0.923) | 2.713 (BONi–Cl = 0.829) |
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) |
||
| In ethanol | ||
| Eads, kJ mol−1 | −306.13 | −300.48 |
| dmin (HCH-adsorbent), Å | 2.291 (dNi–Cl) | 2.327 (dCl–N) |
| q (HCH), e | +0.578 | +0.562 |
| q (Ni), e | −0.685 | −0.806 |
| BOa (HCH-adsorbent) | 0.937 | 0.836 |
| BOa (Ni) | 2.981 (BONi–Cl = 0.937) | 2.737 (BONi–Cl = 0.836) |
|
||
| In acetonitrile | ||
| Eads, kJ mol−1 | −274,69 | −279,99 |
| dmin (HCH-adsorbent), Å | 2.283 (dNi–Cl) | 2.321 (dCl–N) |
| q (HCH), e | +0.529 | +0.518 |
| q (Ni), e | −0.651 | −0.770 |
| BOa (HCH-adsorbent) | 0.903 | 0.814 |
| BOa (Ni) | 2.952 (BONi–Cl = 0.903) | 2.705 (BONi–Cl = 0.814) |
Obviously, the adsorption of HCH in all three studied solvents is energetically preferable due to the negative values of the calculated Eads. The geometrical parameters, charge on atoms and the bond orders between Ni active center and HCH in the adsorption configurations in the presence of solvents are moderately changed compared to that in the vacuum. However, the adsorption process is still chemical in nature. The calculated Eads are as follows: in water ≈ in ethanol < in vacuum < in acetonitrile. This is due to the different solubility of HCH in different solvents: HCH is poorly soluble in water and ethanol, but highly soluble in acetonitrile. Even so, HCH is still “collected” and adsorbed on the nickel doped gCN surface due to the strong interaction with the metal active site. This finding indicates that nickel doped gCN is a potential material for the HCH adsorption from solutions.
Thermal stability of the adsorption configurations of HCH on Ni-cGN and Ni-pGN
The thermal stability of the adsorption profiles is also a property of concern. While computational studies are conducted at 0 K, the actual adsorption-catalytic experiments often take place at room temperature or at higher temperatures up to hundreds of Celsius degrees. An adsorption-photocatalytic process is rarely carried out at very high temperatures. Therefore, we analyzed the thermal stability of the adsorption configurations at room temperature (298 K) (Fig. 7a), and at some higher temperatures (323 K, 373 K, 473 K, 573 K) (Fig. 7b) using molecular dynamics (MD) simulations. In this work, the MD simulations were performed in NVT ensemble using xTB program.37 The total simulation time was 50 ps with a time step of 4 fs.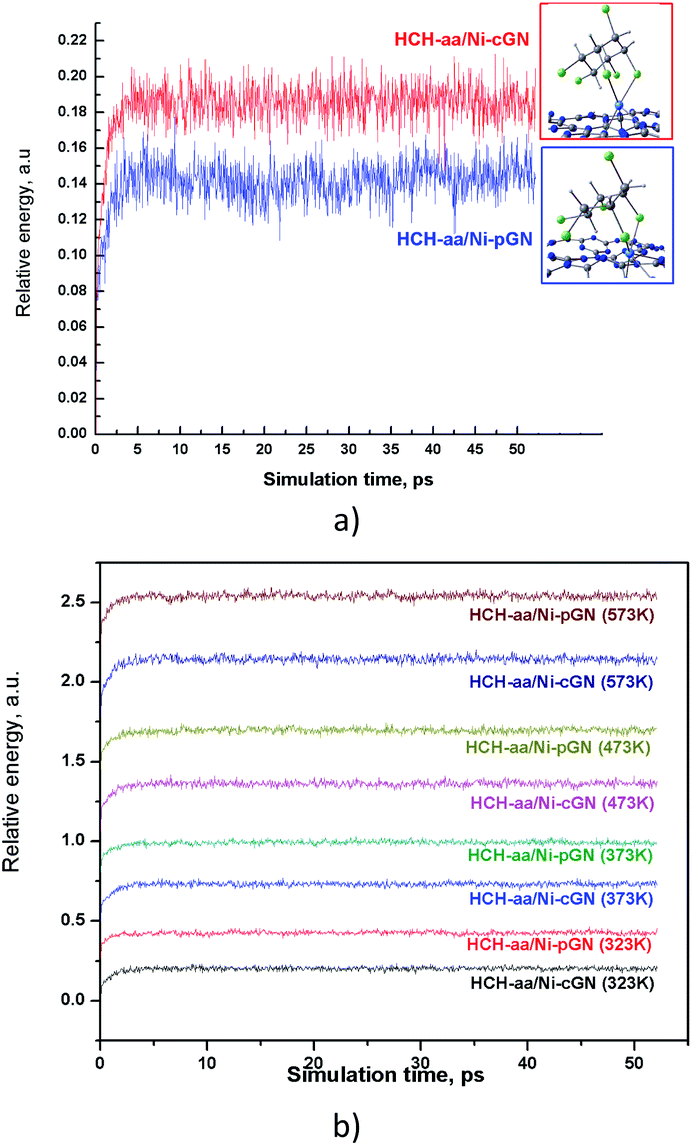 | ||
| Fig. 7 Temperature profiles predicted for HCH-aa/Ni-pGN and HCH-aa/Ni-cGN: (a) at 298 K and; (b) at higher temperatures. | ||
Obviously, the adsorption configuration of HCH on Ni doped gCN has at least thermal stability up to 300 °C. The thermal stability of HCH/Ni-gCN configurations is attributed to the strong binding of HCH via formation of chemical bonds with Ni atom. This is very convenient for the next step, converting HCH into a less toxic compound on the Ni active site.
Conclusions
The adsorption of HCH on Ni-cGN and Ni-pGN was investigated using a combination of GFN2-xTB method and molecular dynamics simulation. The results show that the adsorption of HCH on Ni-cGN and Ni-pGN is chemisorption. The most preferred adsorption configuration corresponds to the formation of the bonds between the Ni atom and the Cl atoms at the axial positions. The adsorption capacity of Ni doped gCN is less influenced by the thickness of the materials, but is more affected by the presence of solvents. The HCH adsorption capacity of Ni-gCN in water and in ethanol was higher than in acetonitrile due to the lower solubility of HCH in the aforementioned solvents. Furthermore, the adsorption configurations of HCH on Ni-cGN as well as on Ni-pGN were shown to be thermal stable at least up to 300 °C due to the strong chemical interaction between HCH and the adsorbents. These findings can be used to gain a deeper understanding of lindane adsorption on cGN based materials and to design high-efficiency materials for lindane treatment using adsorption-photocatalytic technology.Author contributions
Conceptualization: Nguyen Ngoc Ha; data curation: Nguyen Thi Thu Ha, Pham Thi Be, formal analysis: Nguyen Thi Thu Ha, and Pham Thi Be, investigation: Nguyen Thi Thu Ha, Nguyen Ngoc Ha, Pham Thi Be; methodology: Nguyen Ngoc Ha and Nguyen Thi Thu Ha; project administration: Nguyen Ngoc Ha; visualization: Pham Thi Be; writing – original draft: Nguyen Thi Thu Ha; writing – review & editing: Nguyen Ngoc Ha.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research is funded by the Ministry of Science and Technology under grant number ĐTĐL.CN-66/19. The authors would like to express their deep gratitude to Prof. Le Minh Cam, Hanoi National University of Education for promoting research ideas and in-depth discussions on the paper.References
- Persistent Organic Pollutants (POPs) and Pesticides | The Caribbean Environment Programme (CEP), https://www.unep.org/cep/persistent-organic-pollutants-pops-and-pesticides, accessed 15 April 2021 Search PubMed.
- UNTC, https://treaties.un.org/Pages/ViewDetails.aspx?src=IND%26mtdsg_no=XXVII-15%26chapter=27%26clang=_en, accessed 15 April 2021.
- N. Nayyar, N. Sangwan, P. Kohli, H. Verma, R. Kumar, V. Negi, P. Oldach, N. K. Mahato, V. Gupta and R. Lal, Rev. Environ. Health, 2014, 29, 49–52 CAS.
- K. Takagi, J. Pestic. Sci., 2020, 45, 119–123 CrossRef CAS PubMed.
- M. A. Oturan and J. J. Aaron, Crit. Rev. Environ. Sci. Technol., 2014, 44, 2577–2641 CrossRef CAS.
- L. Smith-Hansen and K. H. Jørgensen, Fire Saf. J., 1994, 23, 51–66 CrossRef CAS.
- S. Devipriya and S. Yesodharan, Sol. Energy Mater. Sol. Cells, 2005, 86, 309–348 CrossRef CAS.
- M. Ismael, J. Alloys Compd., 2020, 846, 156446 CrossRef CAS.
- C. Shen, C. Chen, T. Wen, Z. Zhao, X. Wang and A. Xu, J. Colloid Interface Sci., 2015, 456, 7–14 CrossRef CAS PubMed.
- X. Ding, J. Zhu, Y. Zhang, Q. Xia, W. Bi, X. Yang and J. Yang, Talanta, 2016, 154, 119–126 CrossRef CAS PubMed.
- B. Zhu, P. Xia, W. Ho and J. Yu, Appl. Surf. Sci., 2015, 344, 188–195 CrossRef CAS.
- T. Yan, H. Chen, X. Wang and F. Jiang, RSC Adv., 2013, 3, 22480–22489 RSC.
- A. Watcharenwong, A. Kaeokan, R. Rammaroeng, P. Upama and P. Kajitvichyanukul, in IOP Conference Series: Earth and Environmental Science, Institute of Physics Publishing, 2017, vol. 78, p. 012012 Search PubMed.
- S. Vigneshwaran, J. Preethi and S. Meenakshi, Int. J. Biol. Macromol., 2019, 132, 289–299 CrossRef CAS PubMed.
- X. Liang, J. Fan, D. Liang, Y. Xu, Y. Zhi, H. Hu and X. Qiu, J. Colloid Interface Sci., 2021, 582, 70–80 CrossRef CAS PubMed.
- S. C. Tan and H. K. Lee, Microchim. Acta, 2020, 187, 1–10 CrossRef PubMed.
- H. Y. Xu, L. C. Wu, H. Zhao, L. G. Jin and S. Y. Qi, PLoS One, 2015, 10(11), e0142616 CrossRef PubMed.
- X. Li, M. Li, J. Yang, X. Li, T. Hu, J. Wang, Y. Sui, X. Wu and L. Kong, J. Phys. Chem. Solids, 2014, 75, 441–446 CrossRef CAS.
- J. Jiang, S. Cao, C. Hu and C. Chen, Cuihua Xuebao, 2017, 38, 1981–1989 CAS.
- H. ping Zhang, A. Du, N. S. Gandhi, Y. Jiao, Y. Zhang, X. Lin, X. Lu and Y. Tang, Appl. Surf. Sci., 2018, 455, 1116–1122 CrossRef.
- Y. Xu and R. Xu, Appl. Surf. Sci., 2015, 351, 779–793 CrossRef CAS.
- K. Mori, T. Itoh, H. Kakudo, T. Iwamoto, Y. Masui, M. Onaka and H. Yamashita, Phys. Chem. Chem. Phys., 2015, 17, 24086–24091 RSC.
- D. Mateo, J. Albero and H. García, Appl. Catal., B, 2018, 224, 563–571 CrossRef CAS.
- O. Shtyka, R. Ciesielski, A. Kedziora, W. Maniukiewicz, S. Dubkov, D. Gromov and T. Maniecki, Top. Catal., 2020, 63, 113–120 CrossRef CAS.
- E. A. Kozlova, M. N. Lyulyukin, D. V. Markovskaya, A. V. Bukhtiyarov, I. P. Prosvirin, S. V. Cherepanova and D. V. Kozlov, Top. Catal., 2020, 63, 121–129 CrossRef CAS.
- A. Akbari, Z. Sabouri, H. A. Hosseini, A. Hashemzadeh, M. Khatami and M. Darroudi, Inorg. Chem. Commun., 2020, 115, 107867 CrossRef CAS.
- K. N. Devi, S. A. Devi, W. J. Singh and K. J. Singh, J. Mater. Sci.: Mater. Electron., 2021, 32, 8733–8745 CrossRef CAS.
- R. Pol, M. Guerrero, E. García-Lecina, A. Altube, E. Rossinyol, S. Garroni, M. D. Baró, J. Pons, J. Sort and E. Pellicer, Appl. Catal., B, 2016, 181, 270–278 CrossRef CAS.
- B. Guan, J. Yu, S. Guo, S. Yu and S. Han, Nanoscale Adv., 2020, 2, 1352–1357 RSC.
- M. Alijani and N. N. Ilkhechi, Silicon, 2018, 10, 2569–2575 CrossRef CAS.
- H. Tian, J. Li, Z. Mu, L. Li and Z. Hao, Sep. Purif. Technol., 2009, 66, 84–89 CrossRef CAS.
- J. L. Rodríguez, M. A. Valenzuela, H. Tiznado, T. Poznyak, I. Chairez and D. Magallanes, J. Nanopart. Res., 2017, 19, 1–11 CrossRef.
- D. T. Vodak, K. Kim, L. Iordanidis, P. G. Rasmussen, A. J. Matzger and O. M. Yaghi, Chem.–Eur. J., 2003, 9, 4197–4201 CrossRef CAS PubMed.
- L. M. Azofra, D. R. MacFarlane and C. Sun, Phys. Chem. Chem. Phys., 2016, 18, 18507–18514 RSC.
- J. Gracia and P. Kroll, J. Mater. Chem., 2009, 19, 3013–3019 RSC.
- N. T. T. Ha, P. T. Be, P. T. Lan, N. T. Mo, L. M. Cam and N. N. Ha, RSC Adv., 2021, 11, 16351–16358 RSC.
- User Guide to Semiempirical Tight Binding — xtb doc 6.2 documentation, https://xtb-docs.readthedocs.io/en/latest/contents.html, accessed 28 May 2021 Search PubMed.
- C. Bannwarth, S. Ehlert and S. Grimme, J. Chem. Theory Comput., 2019, 15, 1652–1671 CrossRef CAS PubMed.
- S. Grimme, C. Bannwarth and P. Shushkov, J. Chem. Theory Comput., 2017, 13, 1989–2009 CrossRef CAS PubMed.
- C. Bannwarth, E. Caldeweyher, S. Ehlert, A. Hansen, P. Pracht, J. Seibert, S. Spicher and S. Grimme, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2021, 11, e1493 CAS.
- T. T. H. Nguyen, M. C. Le and N. N. Ha, Mol. Simul., 2021, 47, 10–17 CrossRef CAS.
- A. R. Jupp, T. C. Johnstone and D. W. Stephan, Dalton Trans., 2018, 47, 7029–7035 RSC.
- C. A. Bauer, A. Hansen and S. Grimme, Chem.–Eur. J., 2017, 23, 6150–6164 CrossRef CAS PubMed.
- G. Chen, in Encyclopedia of Toxicology, Elsevier, 3rd edn, 2014, pp. 874–876 Search PubMed.
- K. Hellwege and A. Hellwege, Group II: Atomic and Molecular Physics Volume 7: Structure Data of Free Polyatomic Molecules, Springer-Verlag, Berlin, 1976th edn, 1976 Search PubMed.
- P. Huisman and F. C. Mijlhoff, J. Mol. Struct., 1974, 21, 23–27 CrossRef CAS.
- Structure of Free Polyatomic Molecules - Basic Data, ed. K. Kuchitsu, Springer, Berlin, 1998, DOI:10.1007/978-3-642-45748-7.
- X. Yang, T. Imasaka, A. Li and T. Imasaka, J. Am. Soc. Mass Spectrom., 2016, 27, 1999–2005 CrossRef CAS PubMed.
- P. Pracht, F. Bohle and S. Grimme, Phys. Chem. Chem. Phys., 2020, 22, 7169–7192 RSC.
- C. Ren, Y. Zhang, Y. Li, Y. Zhang, S. Huang, W. Lin and K. Ding, J. Phys. Chem. C, 2019, 123, 17296–17305 CrossRef CAS.
- S. Grimme, J. Chem. Theory Comput., 2019, 15, 2847–2862 CrossRef CAS PubMed.
- W. H. Organization and I. P. on Chemical Safety, Companion volume to: Environmental health criteria, 1991 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1ra03797h |
| This journal is © The Royal Society of Chemistry 2021 |