
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceFacile synthesis and biological evaluation of tryptamine-piperazine-2,5-dione conjugates as anticancer agents†
Jiang-Ping Meng ,
Shi-Qiang Li,
Yan Tang,
Zhi-Gang Xu,
Zhong-Zhu Chen and
Li-Xia Gao*
,
Shi-Qiang Li,
Yan Tang,
Zhi-Gang Xu,
Zhong-Zhu Chen and
Li-Xia Gao*
National & Local Joint Engineering Research Center of Targeted and Innovative Therapeutics, IATTI, College of Pharmacy, Chongqing University of Arts and Sciences, Chongqing 402160, China. E-mail: LixiaGao@cqwu.edu.cn
First published on 17th August 2021
Abstract
A facile and efficient route to synthesize N-heterocyclic fused tryptamine-piperazine-2,5-dione conjugates was developed via a post-Ugi cascade reaction. The targeted compounds were prepared by means of a mild reaction and simple operation procedure, which could be applied to a broad scope of starting materials. Compound 6h was demonstrated to induce significant growth inhibition of AsPC-1 and SW1990 human pancreatic cancer cell lines (IC50 = 6 ± 0.85 μM). Our protocol allows for the construction of a structurally diverse compound library and paves a new avenue for the discovery of pancreatic cancer drug candidates.
Introduction
Multicomponent reactions (MCRs) have attracted considerable attention due to their wide application in the rapid construction of highly functionalized important chemical and biomedical heterocyclic skeletons.1 In particular, the Ugi reaction is well known as a versatile and highly efficient tool for the preparation of novel drug scaffolds from commercially available starting materials with high atom economy.2 Using alkynyls as Ugi inputs was a conventional strategy to induce new C–C bonds during the construction of heterocycles.3 In this regard, 2-azetidinones, commonly known as β-lactams, could be constructed by C–C bond formation employing C3 carbon in Ugi adducts with terminal or substituted alkynes (Scheme 1).4 From similar Ugi adducts, the Srivastava group reported that this reaction could occur at room temperature in the presence of NaH.5 The Van der Eycken group also published their findings that a γ-lactam core could be constructed under different conditions.4b,6 Besides the C–C bond formation, the Zhang group reported a C–N bond formation protocol to build piperazine-2,5-dione core through alkyne and amide groups (Scheme 1).7 However, the selective formation of piperazine-2,5-dione over the other β and γ-lactams still remains challenging and deserves further exploration.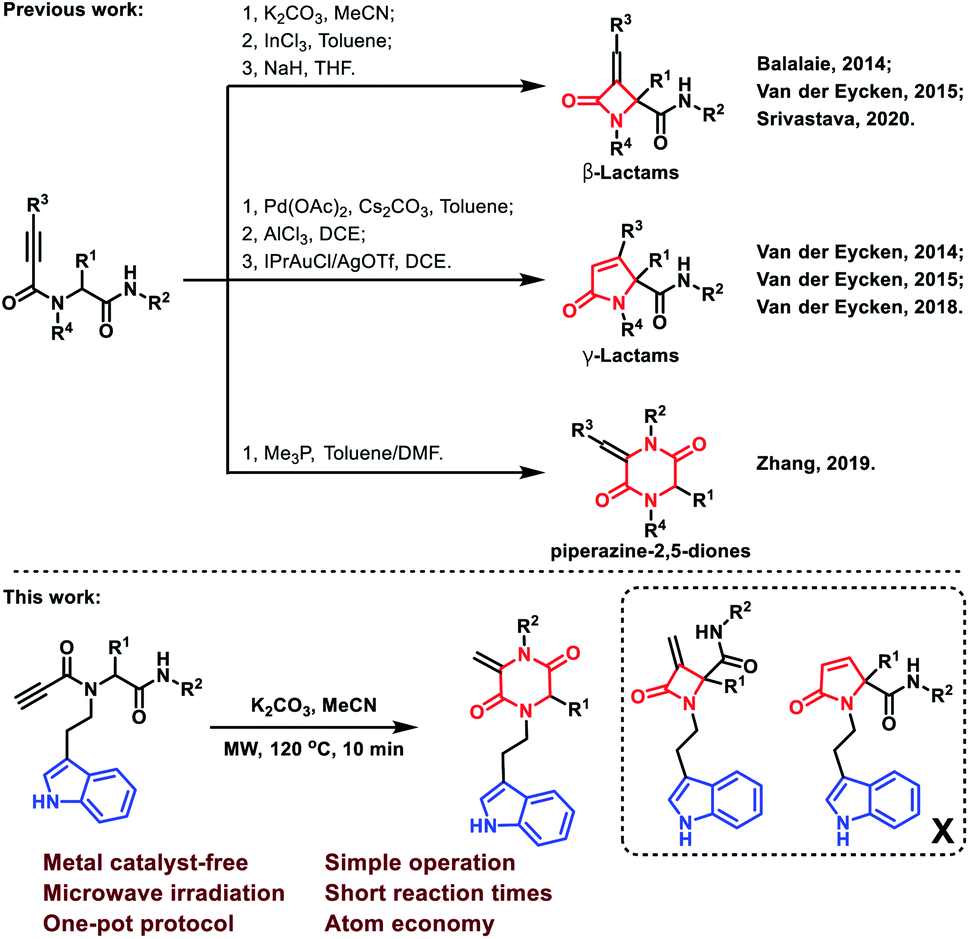 | ||
| Scheme 1 Terminal and substituted alkynes to construct lactams and piperazine-2,5-diones. | ||
The piperazine-2,5-dione pharmacophore is presented as an important class of bioactive agents exhibiting anticancer, antibiotics and antibacterial activities.8 Therefore, a facile and economic protocol for the synthesis of new and diverse molecules bearing the piperazine-2,5-dione motif is highly demanded. The tryptamine substructure is presented in numerous naturally occurring and synthetic compounds, many of which exhibit important pharmacological activity.9 Antimigraine drugs, such as sumatriptan,10 naratriptan11 and eletriptan12 have revolutionized migraine drug discovery and spurred further research for more efficient and safer treatments through activation of 5-HT1D, 5-HT1B and 5-HT1F receptors with high affinities.13 In continuation of our endeavor towards the development of diversity-oriented synthesis of heterocyclic scaffolds through post-Ugi modifications,14 we envisioned the synthesis of tryptamine-piperazine-2,5-diones could be achieved by an Ugi cascade cyclization. To the best of our knowledge, no tryptamine-piperazine-2,5-diones have been reported as anticancer agents so far; therefore, our findings lay the foundation for the discovery and development of substituted tryptamines in cancer research. Herein, we report a post-Ugi base-catalyzed intramolecular nucleophilic cyclization for the synthesis of tryptamine-piperazine-2,5-diones for cancer treatment. As anticipated, the representative compound showed excellent selectivity and potent anti-proliferative activity against AsPC-1 and SW1990 human pancreatic cancer cell lines.15
Results and discussion
To investigate the potential of post-Ugi cyclization reactions for the construction of heterocycles, a highly functionalized precursor 5a was readily constructed through the Ugi four-component reaction from benzaldehyde 1a, propiolic acid 2, tryptamine 3 and benzyl isocyanide 4a (Scheme 1). The Ugi reaction proceeded in methanol at room temperature in high yield, so we focused on the reaction condition optimization of the subsequent cyclization of 5a.We screened various inorganic and organic bases in terms of their ability to promote the nucleophilic α-addition of amides (Table 1, entries 1–9). To our delight, tBuONa and NaOH allowed for the successful transformation from 5a to 6a in over 30% isolated yield; but in organic base systems, the targeted compound 6a was found in lower yields and compound 7a and 8a could be found from LC/MS analysis (entries 10–12). Interestingly, when K2CO3 was used, good to excellent yields were observed for the target product 6a. Particularly, when 5a was irradiated under microwave at 120 °C for 10 min in the presence of 2.0 equiv. of K2CO3, 6a was obtained in 92% yield (entry 19). Elevated reaction temperature or prolonged reaction time both diminished the product yield (entries 20 and 21). With the well-established reaction condition in hand, we turned our attention to further exploration instead of trying other conditions, including Me3P.7
|
|||||||
|---|---|---|---|---|---|---|---|
| Entry | Base | Solvt. | Temp. (°C) | Time (min) | Yield (%) | ||
| 6ab | 7ab | 8ab | |||||
| a Reaction conditions: 5a (0.3 mmol) and bases (2.0 eq.) in solvent (2.0 mL). MW = microwave irradiation.b Isolated yields. DABCO = 1,4-diazabicyclo[2.2.2]octane; DIPEA = N,N-diisopropylethylamine; DIPA = diisopropanolamine. | |||||||
| 1 | Cs2CO3 | MeCN | r.t. | 120 | 5 | Trace | Trace |
| 2 | Cs2CO3 | MeCN | MW 80 | 10 | 30 | Trace | Trace |
| 3 | Cs2CO3 | MeOH | MW 80 | 10 | 19 | 10 | 0 |
| 4 | Cs2CO3 | EtOH | MW 80 | 10 | 26 | <5 | 0 |
| 5 | Cs2CO3 | DCE | MW 80 | 10 | 11 | 10 | 0 |
| 6 | Cs2CO3 | Dioxane | MW 80 | 10 | 15 | 0 | 0 |
| 7 | Cs2CO3 | DMF | MW 80 | 10 | <10 | Trace | 0 |
| 8 | tBuONa | MeCN | MW 80 | 10 | 41 | 10 | 0 |
| 9 | NaOH | MeCN | MW 80 | 10 | 36 | 0 | 0 |
| 10 | DABCO | MeCN | MW 80 | 10 | 21 | <5 | 0 |
| 11 | DIPEA | MeCN | MW 80 | 10 | 0 | <5 | <5 |
| 12 | DIPA | MeCN | MW 80 | 10 | 0 | Trace | <5 |
| 13 | Et3N | MeCN | MW 80 | 10 | 10 | 0 | 0 |
| 14 | K2CO3 | MeCN | MW 80 | 10 | 46 | 0 | 0 |
| 15 | K2CO3 | DMF | MW 80 | 10 | 23 | 0 | <5 |
| 16 | K2CO3 | MeCN | MW 90 | 10 | 50 | 0 | 0 |
| 17 | K2CO3 | MeCN | MW 100 | 10 | 53 | 0 | 0 |
| 18 | K2CO3 | MeCN | MW 110 | 10 | 71 | 0 | 0 |
| 19 | K2CO3 | MeCN | MW 120 | 10 | 92 | 0 | 0 |
| 20 | K2CO3 | MeCN | MW 130 | 10 | 83 | 0 | 0 |
| 21 | K2CO3 | MeCN | MW 120 | 20 | 82 | 0 | 0 |
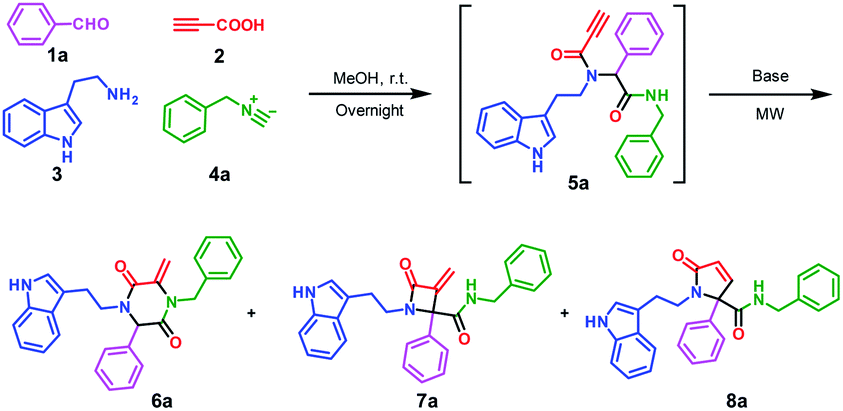
Encouraged by the excellent yield of 6a, we envisioned that crude 5a could be directly subjected to the cyclization reaction without further purification. To verify our hypothesis, the Ugi reaction solvent was removed under a gentle stream of nitrogen to give crude 5a, which was confirmed by LC/MS. Then, crude 5a was dissolved in MeCN and treated in microwave irradiation under basic conditions. This two-step one-pot cascade reaction delivered 6a in 71% yield. Then, we investigated the scope of this cyclization reaction by varying starting materials (Scheme 2). In all cases, initial Ugi products 5a–j were obtained in good yields; after removal of the reaction solvent, 5a–j were used directly without purification to give tryptamine-piperazine-2,5-diones 6a–j in 57–75% yields. Aromatic aldehyde bearing electron donating or withdrawing groups were all well tolerated; however, primary aliphatic aldehyde couldn't give the corresponding product 6k. Due to the limited source of tryptamines, only 2-(1H-indol-3-yl)ethan-1-amine 3 was tested and this unprotected indole group still provided the Ugi product in good yields. It is noteworthy that although the Ugi adduct 5 was not purified by column chromatography, the usage of the crude product has no discernible impact on the overall yield. In comparison with Balalaie's method4a for the construction of β-lactams, the only difference is alkyl amines in our method instead of aromatic amines in the Ugi inputs. The electro-withdraw substituents of aromatic amine in the Ugi skeletons would promote the intramolecular nucleophilic addition of carbanion to a triple bond to form the β-lactam skeleton. However, in our reaction system, alkyl tryptamine group would perform the intramolecular nucleophilic α-addition to form the piperazine-2,5-dione ring. In addition, metal catalysts are needed to activate terminal alkynes, which is why only trace of pyrrolidine-2,5-dione 8a was found by LC/MS.
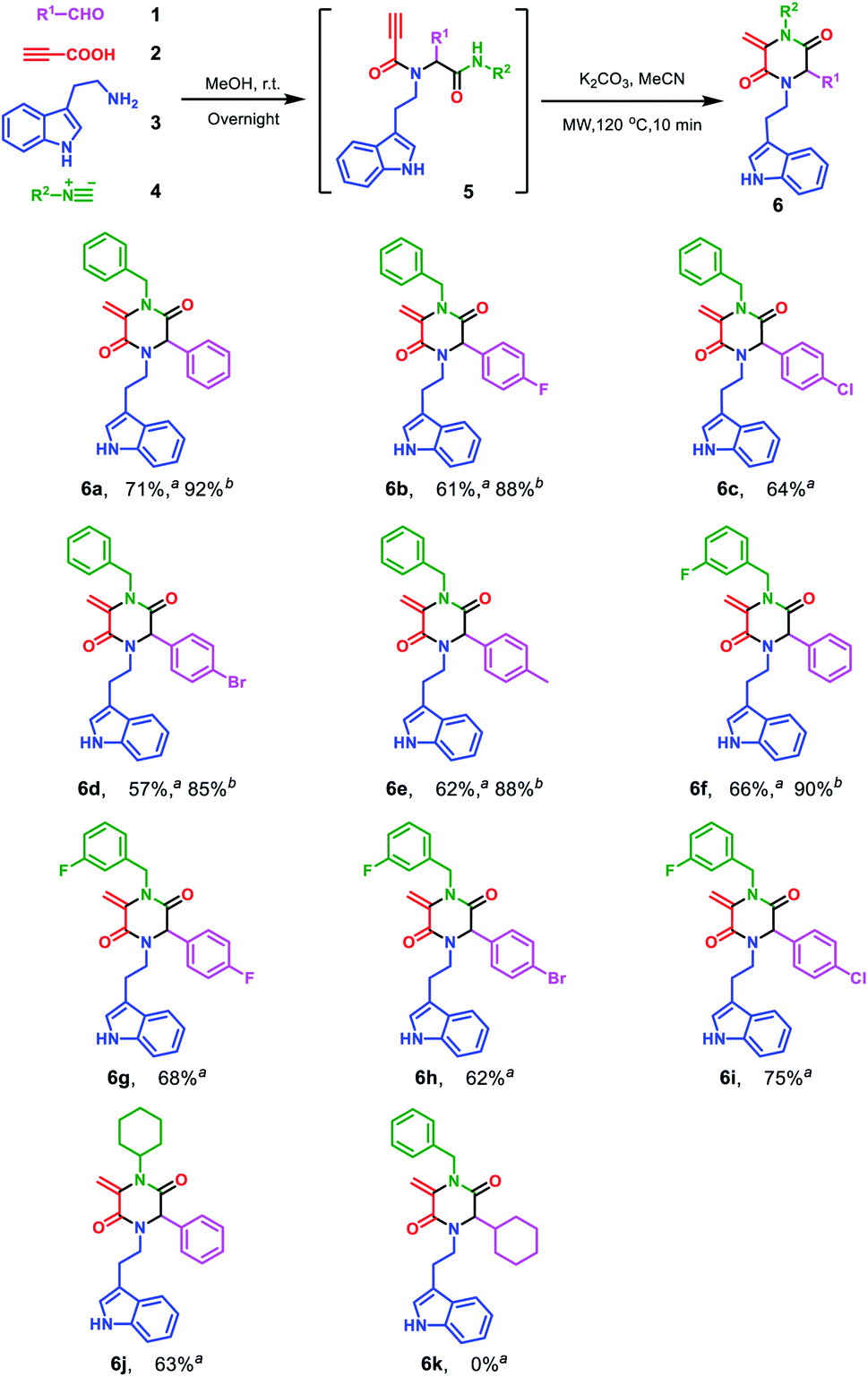 | ||
| Scheme 2 Scope of the Ugi cascade route leading to tryptamine-piperazine-2,5-diones 6a–j. Reaction conditions: 1 (1.0 mmol), 2 (1.0 mmol), 3 (1.0 mmol) and 4 (1.0 mmol) in MeOH (2.0 mL) under air; After workup, added K2CO3 (2.0 mmol) in MeCN (3.0 mL), microwave irradiation in a sealed tube at 120 °C, for 10 min. aIsolated yields with one-pot two-step protocol; bIsolated yields of only cyclization step. | ||
To evaluate the potential to develop a drug candidate from the synthesized compounds, a thymidine incorporation assay was used to measure cancer cell viability upon drug treatments at the concentration of 20 μM. Totally 10 cancer cell lines, including MHCC97H, MHCC97L, MDA-MB-231, MCF-7, UM1, Cal-27, HCT116, SW620, AsPC-1 and HPAC, were selected for initial screening (see ESI† for details), which are some of the difficult-to-inhibit cell lines in the National Cancer Institute's 60 human tumour cell lines panel.16 Interestingly, we found that 6h has selective inhibitory activities on pancreatic cancer cell lines AsPC-1 and HPAC (Fig. 1A and S1†). The results verified our assumption and proved that the tryptamine-piperazine-2,5-dione 6 could be a starting point towards the discovery of potent anticancer drugs. The compound 6h has not cytotoxic to normal pancreatic duct cells at 6 μM (Fig. S2†). In addition, 6h showed excellent drug activity compared with clinical drugs for pancreatic cancer (Fig. S3†). Compound 6h, bearing a bromo group at the para-position of phenyl ring, showed better potency than the chloro analogue 6i. However, compound 6d, lacking a fluoro group on the isocyanide starting material, exhibited much lower anti-proliferative activity. Taken together, these results further proved that substitution groups of aromatic ring of the aldehyde and the substituents of benzyl group played key roles in altering the compounds inhibitory potency against pancreatic cancer cells. The information of structure–activity relationships (SAR) provided us a guideline to improve the compounds anticancer activity in the future during structural modification.
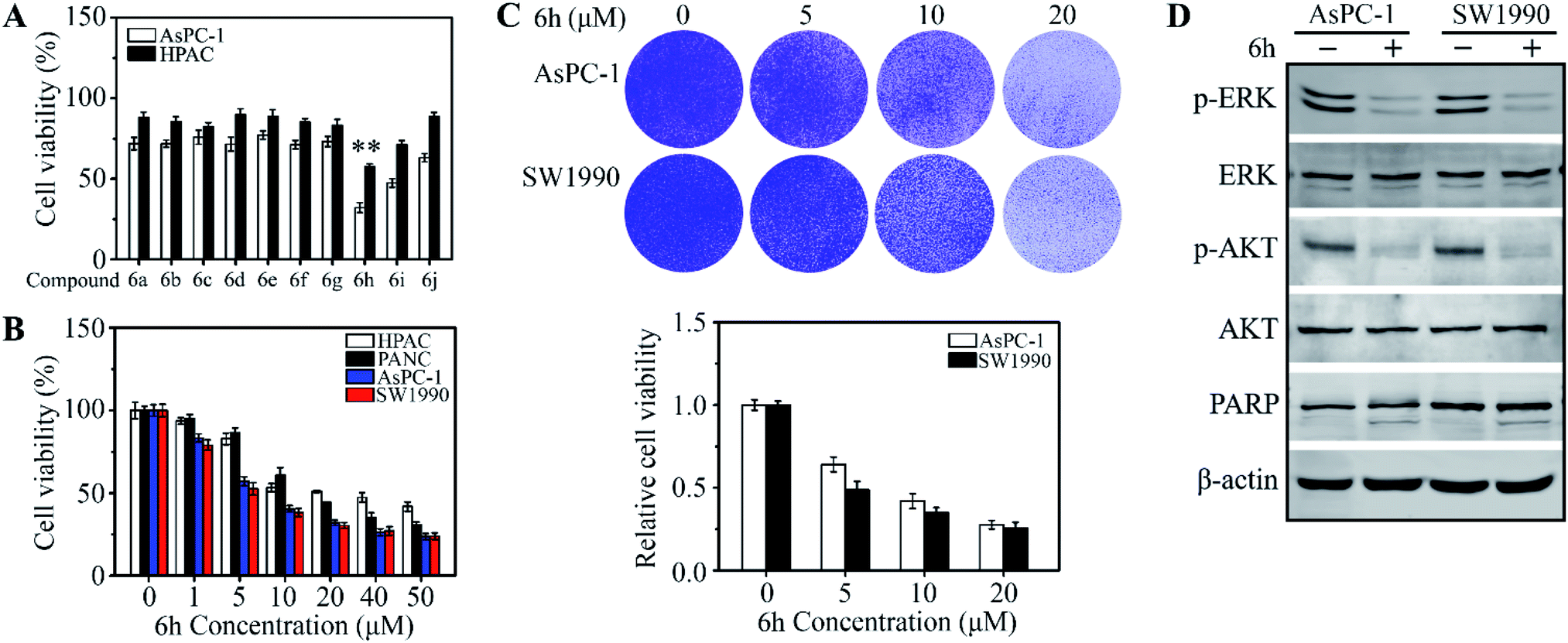 | ||
| Fig. 1 6h inhibits cell viability and induces apoptosis in pancreatic cancer cells. (A and B) The effect of compounds 6a–j on cell viability in pancreatic cancer cell lines. In (A), AsPC-1 and HPAC cells were treated with the 20 μM concentrations of 6a–j for 48 hours, and cell viability was detected by MTT assays. In (B), 5 ×103 AsPC-1, PANC-1, SW1990 and PANC-1 cells were seeded into 96-well-plates and treated with 6 μM 6h DMSO for 48 hours, In (C), AsPC-1 and SW1990 cells were determined the growth ability. AsPC-1 and SW1990 cells were treated with 6 μM 6h for 48 hours, and the viability cells were staining by crystal violet. The crystal violet staining pictures are shown in the up panel and quantitative data are shown in the down panel. In (D), western blot assay detected the cell proliferation and apoptosis protein expression *p < 0.05; **p < 0.01. | ||
To determine the effective dose range of 6h, four cell lines, AsPC-1, PANC-1, SW1990 and HPAC, were treated with 6h at drug concentrations from 1 μM to 50 μM for 48 hours. These results indicated that pancreatic cancer cells have different sensitivity to 6h. We selected two cells (AsPC-1, SW1990) that were more sensitive to 6h for subsequent experiments (Fig. 1B). Compound 6h exhibited dose-dependent anticancer activities and selectivity in AsPC-1 and HPAC cell lines with an IC50 = 6 ± 0.85 μM. These data were in line with the cell growth assays data when AsPC-1 and SW1990 cells were treated with 6h at different drug concentrations (Fig. 1C). We next determined cell growth inhibition rate with western blot at 6 μM of 6h. Oncogenic ERK and AKT phosphorylation were significantly suppressed upon drug treatment for 48 h and these results suggested that 6h induced AsPC-1 and SW1990 cell apoptosis (Fig. 1D). To study the possible mechanisms that induced cell death, we examined the expression of apoptosis-related protein in the presence or absence of 6h. A significantly increased apoptotic rate was found following 6h exposure (Fig. 1D), which was accompanied by increased levels of cleaved PARP. These data proved that compound 6h can inhibit human pancreatic cell proliferation and induce cell apoptosis.
Conclusion
In conclusion, a series of tryptamine-piperazine-2,5-dione derivatives was designed and synthesized via Ugi cascade reaction. Microwave-assisted synthesis protocol delivered target compounds in high purity and excellent yield. The nonlinear SAR studies showed that the substitution groups of aldehyde and benzyl group in isocyanide played vital roles in altering the compounds inhibitory potency against pancreatic cancer cells. Furthermore, 6h significantly decreased the phosphorylation of oncogenic proteins ERK and AKT; and meanwhile increased levels of cleaved PARP in AsPC-1 and SW1990 cells. Exemplified by compound 6h, our work presented in this paper paved a new avenue for the discovery of pancreatic cancer drug.Conflicts of interest
The authors declare no competing financial interest.Acknowledgements
The authors would like to thank the Science and Technology Research Program of Chongqing Municipal Education Commission (KJQN202001347 and KJZD-M201801301) and the Natural Science Foundation Project of CQ CSTSC (cstc2019jcyj-msxmX0012, cstc2019jcyj-msxmX0202, cstc2020jcyj-msxmX0733 and cstc2018jszx-cyzdX0023). We would also like to thank Ms H. Z. Liu and J. Xu for obtaining the LC/MS, HRMS and NMR data.Notes and references
- For selected reviews, see: (a) A. Dömling and I. Ugi, Angew. Chem., Int. Ed., 2000, 39, 3168–3210 CrossRef; (b) G. R. Kuethe, Chem. Rev., 2006, 106, 2875–2911 CrossRef PubMed; (c) A. Dömling, W. Wang and K. Wang, Chem. Rev., 2012, 112, 3083–3135 CrossRef PubMed; (d) T. Vlaar, B. U. Maes, E. Ruijter and R. V. Orru, Angew. Chem., Int. Ed., 2013, 52, 7084–7097 CrossRef CAS PubMed; (e) B. H. Rotstein, S. Zaretsky, V. Rai and A. K. Yudin, Chem. Rev., 2014, 114, 8323–8359 CrossRef CAS PubMed; (f) S. Brauch, S. S. Van Berkel and B. Westermann, Chem. Soc. Rev., 2013, 42, 4948–4962 RSC.
- (a) W. Zhang, Green Chem., 2009, 11, 911–920 RSC; (b) A. Singh and A. Kumar, RSC Adv., 2015, 5, 2994–3004 RSC; (c) L. Banfi, A. Basso, C. Chiappe, F. De Moliner, R. Riva and L. Sonaglia, Org. Biomol. Chem., 2012, 10, 3819–3829 RSC.
- (a) Z. Xu, F. De Moliner, A. P. Cappelli and C. Hulme, Angew. Chem., Int. Ed., 2012, 51, 8037–8040 CrossRef CAS PubMed; (b) W. Liao, S. Li, J. Wang, Z. Zhang, Z. Yang, D. Xu, C. Xu, H. Lan, Z. Chen and Z. Xu, ACS Comb. Sci., 2016, 18, 65–69 CrossRef CAS PubMed.
- (a) E. Ghabraie, S. Balalaie, S. Mehrparvar and F. Rominger, J. Org. Chem., 2014, 79, 7926–7934 CrossRef CAS PubMed; (b) Z. Li, U. K. Sharma, Z. Liu, N. Sharma, J. N. Harvey and E. V. Van der Eycken, Eur. J. Org. Chem., 2015, 2015, 3957–3962 CrossRef CAS.
- S. P. Singh, S. Tripathi, A. Yadav, R. Kant, H. K. Srivastava and A. K. Srivastava, Chem. Commun., 2020, 56, 12789–12792 RSC.
- (a) N. Sharma, Z. Li, U. K. Sharma and E. V. Van der Eycken, Org. Lett., 2014, 16, 3884–3887 CrossRef CAS PubMed; (b) Z. Li, L. Song, L. Van Meervelt, G. Tian and E. V. Van der Eycken, ACS Catal., 2018, 8, 6388–6393 CrossRef CAS; (c) C. Liu, G. Wang, Y. Wang, K. Van Hecke, O. P. Pereshivko and V. A. Peshkov, Tetrahedron Lett., 2018, 59, 1823–1827 CrossRef CAS.
- Y.-K. Xi, H. Zhang, R.-X. Li, S.-Y. Kang, J. Li and Y. Li, Chem.–Eur. J., 2019, 25, 3005–3009 CrossRef CAS PubMed.
- (a) Z. Fu, Y. Hou, C. Ji, M. Ma, Z. Tian, M. Deng, L. Zhong, Y. Chua and W. Li, Bioorg. Med. Chem., 2018, 26, 2061–2072 CrossRef CAS PubMed; (b) S. Liao, X. Qin, D. Li, Z. Tu, J. Li, X. Zhou, J. Wang, B. Yang, X. Lin, J. Liu, X. Yang and Y. Liu, Eur. J. Med. Chem., 2014, 83, 236–244 CrossRef CAS PubMed; (c) D. Kumar, S. K. Gupta, A. Ganeshpurkar, G. Gutti, S. Krishnamurthy, G. Modi and S. K. Singh, Eur. J. Med. Chem., 2018, 150, 87–101 CrossRef CAS PubMed; (d) M. Luo, G. Tang, J. Ju, L. Lu and H. Huang, Nat. Prod. Res., 2016, 30, 138–143 CrossRef CAS PubMed.
- (a) I. Sosič, M. Anderluh, M. Sova, M. Gobec, I. Mlinarič Raščan, A. Derouaux, A. Amoroso, M. Terrak, E. Breukink and S. Gobec, J. Med. Chem., 2015, 58, 9712–9721 CrossRef PubMed; (b) B. M. Trost, W.-J. Bai, C. Hohn, Y. Bai and J. J. Cregg, J. Am. Chem. Soc., 2018, 140, 6710–6717 CrossRef CAS PubMed; (c) M. Ishikura, T. Abe, T. Choshi and S. Hibino, Nat. Prod. Rep., 2015, 32, 1389–1471 RSC.
- Z. Wojnarowska, J. Knapik, M. Rams-Baron, A. Jedrzejowska, M. Paczkowska, A. Krause, J. Cielecka-Piontek, M. Jaworska, P. Lodowski and M. Paluch, Mol. Pharmaceutics, 2016, 13, 1111–1122 CrossRef CAS PubMed.
- H. E. Connor, W. Feniuk, D. T. Beattie, P. C. North, A. W. Oxford, D. A. Saynor and P. P. Humphrey, Cephalalgia, 1997, 17, 145–152 CrossRef CAS PubMed.
- M. Capi, M. Curto, L. Lionetto, F. de Andrés, G. Gentile, A. Negro and P. Martelletti, Ther. Adv. Neurol. Disord., 2016, 9, 414–423 CrossRef CAS PubMed.
- P. J. Goadsby, Mol. Diagn. Ther., 1998, 10, 271–286 CAS.
- (a) G.-T. Song, N. McConnell, Z.-Z. Chen, X.-F. Yao, J.-H. Huang, J. Lei, H.-K. Lin, H. Li and Z.-G. Xu, Adv. Synth. Catal., 2018, 360, 3655–3661 CrossRef CAS; (b) Z.-Z. Chen, S.-Q. Li, Y.-J. Zhang, D.-Y. Tang, J.-P. Meng, J. Lei, H. Li and Z.-G. Xu, Org. Lett., 2018, 20, 7811–7815 CrossRef CAS PubMed; (c) J. Lei, Y. Li, L.-J. He, Y.-F. Luo, D.-Y. Tang, W. Yan, H.-K. Lin, H. Li, Z.-Z. Chen and Z.-G. Xu, Org. Chem. Front., 2020, 7, 987–992 RSC; (d) J. Lei, Y. Li, J. Xu, D.-Y. Tang, J.-W. Shao, H. Li, Z.-Z. Chen and Z.-G. Xu, Green Chem., 2020, 22, 3716–3720 RSC.
- (a) L. Gummidi, N. Kerru, P. Awolade, A. Raza, A. K. Sharma and P. Singh, Bioorg. Med. Chem. Lett., 2020, 30, 127544 CrossRef CAS PubMed; (b) J. S. Kumar, B. Thirupataiah, R. Medishetti, A. Ray, S. D. Bele, K. A. Hossain, G. S. Reddy, R. K. Edwin, A. Joseph, N. Kumar, G. G. Shenoy, C. M. Rao and M. Pal, Eur. J. Med. Chem., 2020, 201, 112335 CrossRef CAS PubMed; (c) J. S. Kumar, G. S. Reddy, R. Medishetti, A. Ray, S. D. Bele, K. A. Hossain, B. Thirupataiah, R. K. Edwin, P. Behera, A. Joseph, G. G. Shenoy, C. M. Rao and M. Pal, J. Mol. Struct., 2021, 1240, 130534 CrossRef; (d) S. Cascioferro, G. Li Petri, B. Parrino, B. El Hassouni, D. Carbone, V. Arizza, U. Perricone, A. Padova, N. Funel, G. J. Peters, G. Cirrincione, E. Giovannetti and P. Diana, Molecules, 2020, 25, 329 CrossRef CAS PubMed; (e) X. Qin and X. Cui, 3 Biotech, 2020, 10, 187 CrossRef PubMed.
- (a) N. A. Shafakat Ali, B. Ahmad Dar, V. Pradhan and M. Farooqui, Mini-Rev. Med. Chem., 2013, 13, 1792–1800 CrossRef CAS PubMed; (b) J. Xu, H.-B. Tan, Y.-J. Zhang, D.-Y. Tang, F. Zhan, H. Li, Z.-Z. Chen and Z.-G. Xu, Sci. Rep., 2020, 10, 9281 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details and spectroscopic characterization information. See DOI: 10.1039/d1ra03740d |
| This journal is © The Royal Society of Chemistry 2021 |