
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceCross-linking of a polyketide synthase domain leads to a recyclable biocatalyst for chiral oxygen heterocycle synthesis†
Lisa Wagner ,
Theresa Roß,
Tim Hollmann and
Frank Hahn*
,
Theresa Roß,
Tim Hollmann and
Frank Hahn*
Department of Chemistry, Faculty of Biology, Chemistry and Earth Sciences, University of Bayreuth, Universitätsstraße 30, 95447 Bayreuth, Germany. E-mail: frank.hahn@uni-bayreuth.de
First published on 7th June 2021
Abstract
The potential of polyketide synthase (PKS) domains for chemoenzymatic synthesis can often not be tapped due to their low stability and activity in vitro. In this proof-of-principle study, the immobilisation of the heterocycle-forming PKS domain AmbDH3 as a cross-linked enzyme aggregate (CLEA) is described. The AmbDH3-CLEA showed good activity recovery, stability and recyclability. Repetitive reactions on the semi-preparative scale were performed with high conversion and isolated yield. Similar to that observed for the free enzyme, the aggregate retained substrate tolerance and the ability for kinetic resolution. This first example of a successful enzymatic PKS domain immobilisation demonstrates that cross-linking can in principle be applied to this type of enzyme to increase its applicability for chemoenzymatic synthesis.
Type I-polyketide synthases (type I-PKS) are multi-domain enzymes that biosynthesise the backbones of reduced polyketide natural products. Their biochemistry is related to that of fatty acid synthases, but type I-PKS have a much broader range of products due to their more flexible assembly and enzymatic mechanism.1,2 Their catalytic cycle is based on the precisely tuned interplay of the individual modules and domains, which is in turn controlled by protein–protein and protein–substrate interactions.3–5
Due to their origin from this specific environment, isolated PKS domains often exhibit rather low turnover frequency and low stability in in vitro experiments.5–7 On the other hand, PKS domains frequently show relaxed substrate specificity and, particularly in the case of domains that catalyse synthetically attractive reactions, an application in chemoenzymatic synthesis is thus conceivable.6,8–13 Various recent reports have highlighted the existing potential of PKS enzymes for this purpose.14–19
Immobilisation is an effective way to increase the practical value of enzymes by providing them with higher stability and recyclability.20,21 Besides the covalent and non-covalent attachment of enzymes to solid phases, the formation of cross-linked enzyme aggregates (CLEAs) or cross-linked enzyme crystals are established approaches with specific advantages.22 The production of CLEAs is operationally simple and combines purification of the protein with the formation of a heterogenous catalyst. CLEAs basing on well-characterised primary metabolic biocatalysts, such as hydroxynitrile lyases and carbohydrate-processing enzymes, have frequently been reported as well as some basing on secondary metabolic enzymes.22–25
Despite the potential of this technique, successful immobilisation or cross-linking of type I-PKS has not been reported.26 This might be due to various reasons such as their problematic handling, their naturally low activity in vitro or their complex enzymatic cycle that involves major spatial rearrangements. Apart from improving the chemoenzymatic performance of individual domains, their immobilisation would represent a sensible first step towards immobilisation of larger PKS constituents.
It should be noted that immobilised type III-PKS have been employed for the synthesis of plant natural product libraries.27–29 The analogy between type I and type III-PKS is however limited, because being single-domain synthases, the latter differ fundamentally from type I-PKS, both structurally and mechanistically. We regarded CLEA formation to be well-suited for PKS immobilisation as this method is mild, easily adaptable and minimally interfering with the pre-organised superstructure of proteins.
We have recently described the bifunctional dehydratase-cyclase domain AmbDH3 from the ambruticin type I-PKS as a new biocatalyst for the synthesis of saturated oxygen heterocycles via intramolecular oxa-Michael addition (Scheme 1).12,15,30 Besides its broad substrate-tolerance and high stereoselectivity, this enzyme is characterised by convenient scalability of its conversions and the ability to perform kinetic resolution of chiral tetrahydropyrans (THPs).
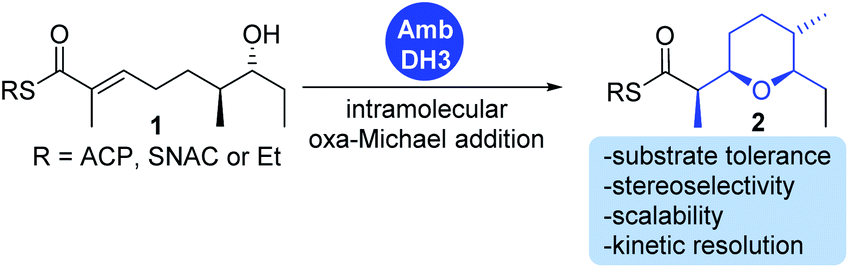 | ||
| Scheme 1 AmbDH3 catalyses intramolecular oxa-Michael addition leading to chiral oxygen heterocycles. ACP: acyl carrier protein; SNAC: N-acetylcysteamine. | ||
We thus chose AmbDH3 for a proof-of-principle study on the cross-linking of PKS domains that could ideally lead to an improved heterocyclisation catalyst.
The formation of CLEAs is achieved by cross-linking of free amino groups on the protein surface by bifunctional linkers, like glutaraldehyde (GA).22 A sufficient number of accessible residues is required, otherwise these need to be installed by artificial surface amination. Luckily, inspection of the AmbDH3 crystal structure (PDB-ID: 5O16) revealed the presence of various evenly distributed lysines and arginines on the enzyme surface, suggesting its suitability for CLEA formation (Fig. 1a).30 The active centre of AmbDH3 contains a catalytic dyad of His–Arg within an otherwise highly hydrophobic cavity, making disturbance of this region by the cross-linker rather unlikely.30 The near-surface localisation of the active site should also allow good accessibility for substrates with expectable limitations caused by the slowed diffusion through the cross-linking network.
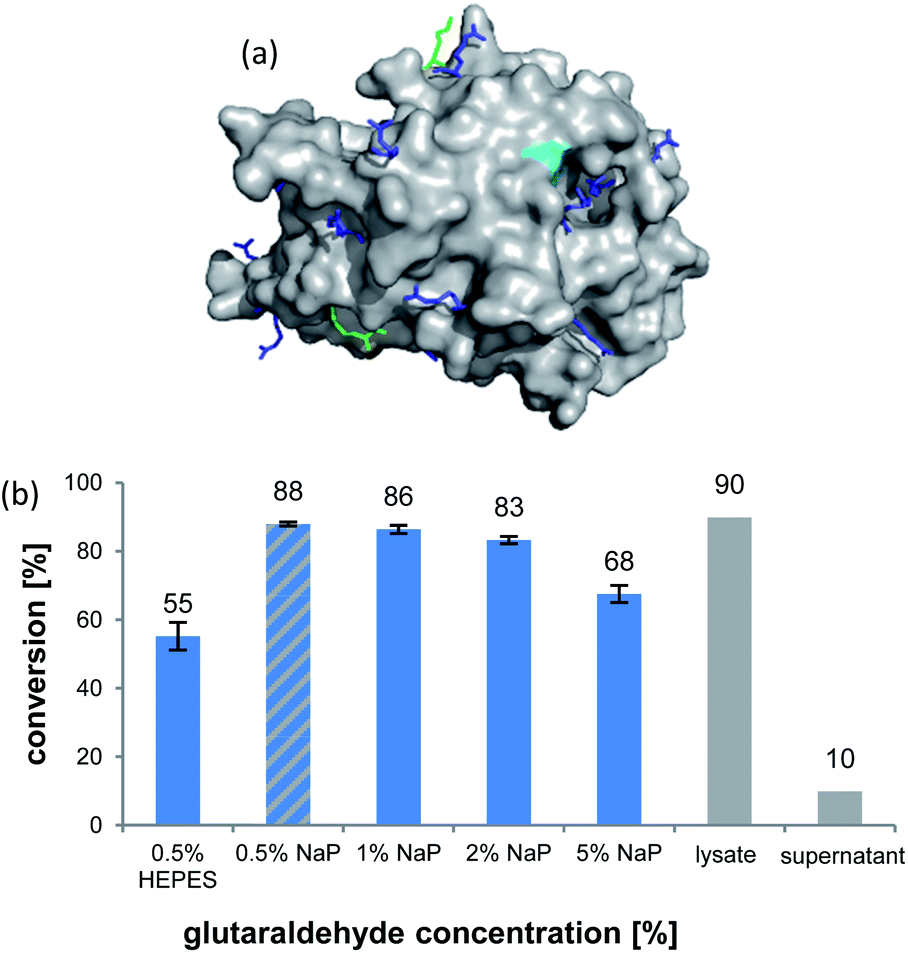 | ||
| Fig. 1 (a) Crystal structure of AmbDH3 with highlighted lysine (light green) and arginine (blue) surface residues and the active centre (turquoise). (b) Conversion of 1 by different preparations of AmbDH3-CLEA (blue bars) as well as the lysate and the supernatant from CLEA preparation (0.5% GA, NaP buffer; grey bars). Reaction conditions: 332 nmol (2 mM) of 1, CLEA/lysate from 4 mg ambDH3-expressing cells, 16 h, 30 °C. | ||
AmbDH3-CLEAs were prepared by precipitation of the enzyme with ammonium sulphate and cross-linking with GA in the same vessel.24 Two buffers and GA concentrations ranging from 0.5 to 5% were investigated (Fig. 1b and S1–S7†). The resulting aggregates were assessed regarding their ability to convert 1 into THP 2 in 16 h endpoint assays.15 All experiments were performed using an identical quantity of ambDH3-expressing cells for the preparation of the cell-free lysate or the CLEA, respectively. Cross-linking with 0.5% GA in sodium phosphate (NaP) buffer resulted in a CLEA that gave similar conversion as the expression lysate and a markedly higher value than one resulting from cross-linking in HEPES buffer. A 16 h incubation with the supernatant from the precipitation step served as a control experiment and showed only very low conversion of 1 into 2, indicating that most of the remaining enzymatic activity is bound in the CLEA. A GA concentration above 2% in NaP buffer led to a marked decrease of conversion. As preparation with 0.5, 1 or 2% GA gave no apparent difference in physical stability of the resulting CLEAs, cross-linking with 0.5% GA in NaP buffer for 2 h was used to form the AmbDH3-CLEAs in all subsequent experiments.
To more precisely describe the outcome of the immobilisation process, we determined the activity recovery in the CLEA. The enzymatic activity of the individual fractions from CLEA preparation was determined and set into relation as previously described (Table 1, Fig. S8, Tables S1 and S2†).24 81% of the lysate enzymatic activity were recovered. As obvious from the immobilisation yield and the immobilisation efficiency, both, incomplete binding of enzyme activity and a slightly lower activity of the cross-linked compared to the free enzyme, contributed to this incomplete recovery by a relevant degree. A reduced enzyme activity after cross-linking is well-described and can be attributed to disturbance of protein conformation, blockage of the active centres or diffusion restrictions.20,22 Aggregates with a higher activity could be obtained in the future by optimising the preparation procedure for AmbDH3-CLEA, for example by screening other cross-linking agents or fine-tuning of the molar ratio of cross-linker to enzyme.
Attempts to determine the amount of AmbDH3 in the CLEA did not give absolutely precise values, but allowed the estimation that ≤60% of the AmbDH3 in the lysate were bound in AmbDH3-CLEA (Fig. S9†). Together with the observed activity recovery this suggests that relevant parts of the free AmbDH3 in the lysate were inactive and that active AmbDH3 is enriched by the precipitation step during CLEA formation.
Determination of the Michaelis–Menten parameters was complicated by practical issues like the small reaction scale and a limited solubility of the substrate 1 in the reaction buffer, leading to large errors. The Km and kcat/Km values of the AmbDH3-CLEA (4.4 ± 2.8 mM and 5.67 ± 4.87 s−1 mM−1) were in the same range as those of the free enzyme (8.2 ± 3.1 mm and 28.2 ± 14.8 s−1 mm−1) and suggested slower catalysis as well as similar specificity of the CLEA compared to the free enzyme (Fig. S10–S17 and Table S3†). With respect to the structure of AmbDH3 this suggests that the diffusion into the active centre and its flexibility is not severely affected by the cross-linking process. A time course experiment showed a steady increase of conversion to the final value of 81% during an overnight incubation (Fig. S18†). Of these, 58% were already achieved after the initial 5 h reaction, which represents a common time frame for a biocatalytic reaction.
AmbDH3-CLEA was subjected to various storage conditions and the effect on the conversion determined (Table 2, Fig. S20 and S21†). While the CLEA stored for 7 days at 20 °C or 4 °C, respectively, transformed 1 into 2 by a similar degree as the freshly prepared one, freeze-thawing to −20 or −80 °C had a slightly negative influence on the conversion. The AmbDH3-CLEA was also applied in a recycling experiment in which recovered aggregate was repetitively inserted into further reaction-wash cycles. The CLEA showed activity in all cycles with a conversion gradually decreasing from 86% to 55% (Fig. 2 and S22–S31†). It could not be clarified under these experimental setting if the decline in conversion was caused by diminishing activity of the CLEA itself or by loss of minor proportions of the CLEA in the workup steps of the individual cycles. These results suggest a certain stability of AmbDH3-CLEA and allow the conclusion that the aggregate retains a relevant part of its activity upon long-term storage, freeze-thawing and repetitive exposure to the conditions of a standard conversion experiment.
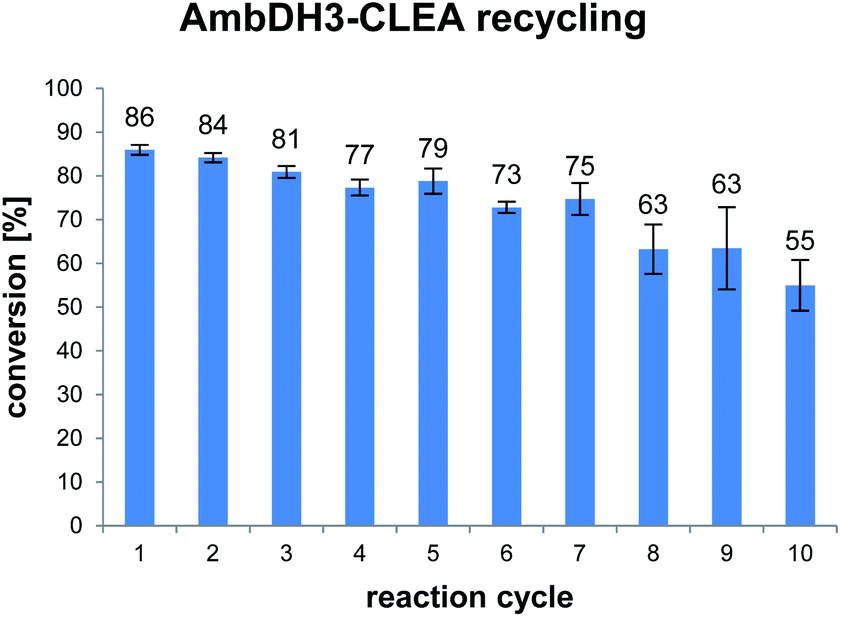 | ||
| Fig. 2 Conversion of 1 into 2 in ten subsequent reaction cycles using recovered AmbDH3-CLEA. The CLEA was washed with buffer after each cycle. Reaction conditions: 332 nmol (2 mM) of 1, 1.9 × 10−3 u AmbDH3-CLEA, 30 °C, 16 h. | ||
To clarify the performance under conditions more relevant for chemoenzymatic synthesis, we scaled up the reaction between AmbDH3-CLEA and 1 to 33 μmol (10 mg) starting material and doubled the substrate concentration to 4 mM (Fig. 3 and S32–S36†). Virtually quantitative conversion into homochiral 2 was observed in five repetitive reaction cycles, suggesting that the declining conversions over the analytical scale incubations were an artefact from the handling of small CLEA amounts. The average isolated yield of the five pooled reactions was 94%, which is significantly above the ∼80%, which were observed for purified AmbDH3 or the expression lysate on the same reaction scale.15 The co-precipitation of enzyme, 1 and 2 posed a major problem in the homogenous reaction with the free enzyme on reaction scales of 33 μmol 1 or above. A 2 h incubation with proteinase prior to extractive work-up of the homogenous reaction was necessary to dissolve this co-precipitate and obtain satisfactory isolated yields. The stability of AmbDH3-CLEA and its recoverability effectively eliminate the need for this measure in the heterogenous semi-preparative scale reaction and should thus also enable simplified workup on the preparative reaction scale.
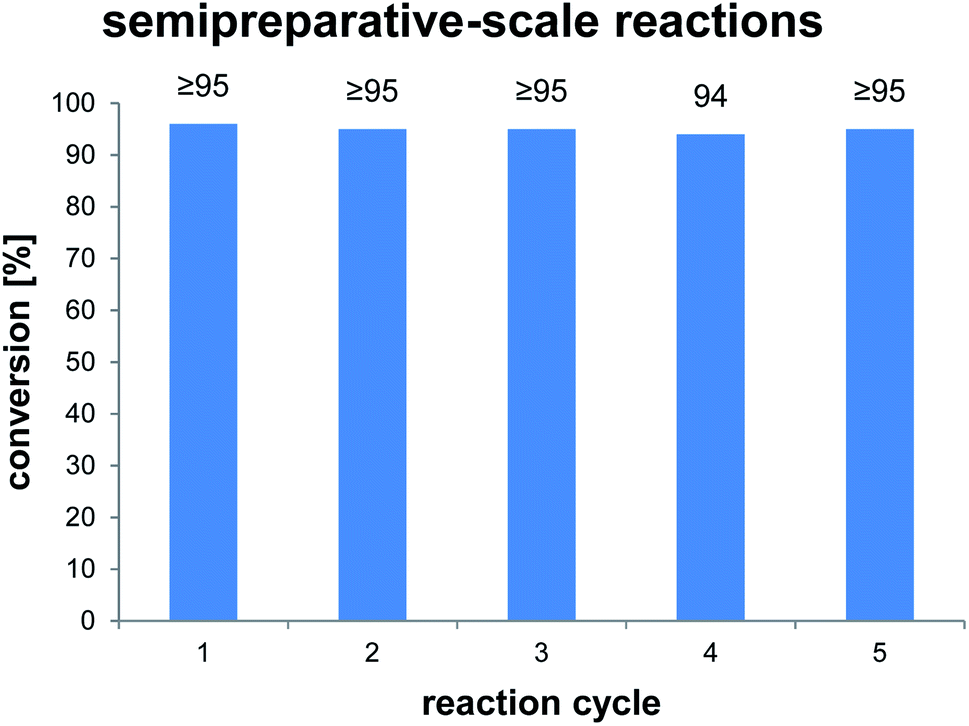 | ||
| Fig. 3 Conversions of semi-preparative scale reactions of AmbDH3-CLEA with 1. The conversion was determined by 1H NMR spectroscopy. Reaction conditions: 33 μmol (4 mM) of 1, 0.19 u AmbDH3-CLEA, 30 °C, 16 h. | ||
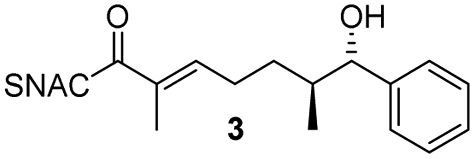
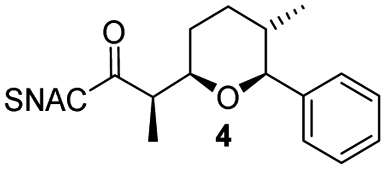
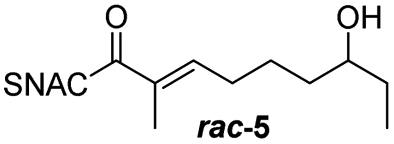
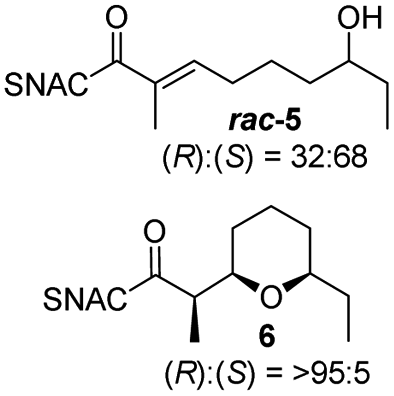
Finally, we investigated the substrate tolerance of the AmbDH3-CLEA. We incubated the aggregate with the substrate surrogates 3 and rac-5 and determined conversion and isolated yield (Table 3 and Fig. S37–S41†). AmbDH3-CLEA converted 3 stereospecifically and with 80% conversion into phenyl-THP 4. In the reaction with rac-5, AmbDH3-CLEA converted only one stereoisomer, (R)-5, into (R)-6 and thus enabled one-step resolution of a chiral THP with three stereocentres. The conversions of 3 and (R)-5 by AmbDH3-CLEA is lower than in the analogous reactions with the free enzyme,15 which is consistent with the finding that the CLEA does not show full activity recovery.
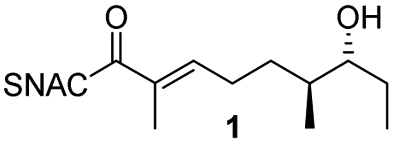
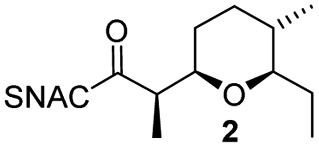
In summary, we performed a proof-of-principle study for PKS domain cross-linking on the example of the THP-forming AmbDH3. AmbDH3-CLEA showed a good activity recovery, stability towards a couple of treatments and could be recycled at least ten times. Similar to the free enzyme, AmbDH3-CLEA exhibited substrate tolerance and kinetic resolution ability in semi-preparative scale reactions as well as higher isolated yield and simpler work-up in reactions with the standard substrate surrogate 1. Ultimately, a heterocyclisation catalyst with improved properties was obtained.
Our work shows that type I PKS domains can be converted into an active, immobilised form with synthetically useful properties by cross-linking. It lays the foundation for applying this method to other PKS domains and larger PKS components, such as entire modules.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
Funding from the DFG (Emmy Noether Research Group, HA 5841/2-1 and a DFG-Sachbeihilfe, HA 5841/4-1) and the Deutsche Bundesstiftung Umwelt (doctoral scholarship to L. W.) is gratefully acknowledged. We thank the “Zentrale Analytik” at the Faculty BCG, University of Bayreuth as well as the “Nordbayerisches NMR-Zentrum”.Notes and references
- C. Hertweck, Angew. Chem., Int. Ed., 2009, 48, 4688–4716 CrossRef CAS PubMed.
- A. T. Keatinge-Clay, Chem. Rev., 2017, 117, 5334–5366 CrossRef CAS PubMed.
- T. Robbins, Y.-C. Liu, D. E. Cane and C. Khosla, Curr. Opin. Struct. Biol., 2016, 41, 10–18 CrossRef CAS PubMed.
- K. J. Weissman and R. Müller, ChemBioChem, 2008, 9, 826–848 CrossRef CAS PubMed.
- M. Klaus and M. Grininger, Nat. Prod. Rep., 2018, 35, 1070–1081 RSC.
- W. D. Fiers, G. J. Dodge, D. H. Sherman, J. L. Smith and C. C. Aldrich, J. Am. Chem. Soc., 2016, 138, 16024–16036 CrossRef CAS PubMed.
- W. D. Fiers, G. J. Dodge, Y. Li, J. L. Smith, R. A. Fecik and C. C. Aldrich, Chem. Sci., 2015, 6, 5027–5033 RSC.
- G. Berkhan, C. Merten, C. Holec and F. Hahn, Angew. Chem., Int. Ed., 2016, 55, 13589–13592 CrossRef CAS PubMed.
- D. H. Kwan and P. F. Leadlay, ACS Chem. Biol., 2010, 5, 829–838 CrossRef CAS PubMed.
- S. K. Piasecki, C. A. Taylor, J. F. Detelich, J. Liu, J. Zheng, A. Komsoukaniants, D. R. Siegel and A. T. Keatinge-Clay, Chem. Biol., 2011, 18, 1331–1340 CrossRef CAS PubMed.
- J. Wunderlich, T. Roß, M. Schröder and F. Hahn, Org. Lett., 2020, 22, 4955–4959 CrossRef CAS PubMed.
- G. Berkhan and F. Hahn, Angew. Chem., Int. Ed., 2014, 53, 14240–14244 CrossRef CAS PubMed.
- P. Pöplau, S. Frank, B. I. Morinaka and J. Piel, Angew. Chem., Int. Ed., 2013, 52, 13215–13218 CrossRef PubMed.
- S. Sundaram, H. J. Kim, R. Bauer, T. Thongkongkaew, D. Heine and C. Hertweck, Angew. Chem., Int. Ed., 2018, 57, 11223–11227 CrossRef CAS PubMed.
- T. Hollmann, G. Berkhan, L. Wagner, K. H. Sung, S. Kolb, H. Geise and F. Hahn, ACS Catal., 2020, 10, 4973–4982 CrossRef CAS.
- C. C. Aldrich, L. Venkatraman, D. H. Sherman and R. A. Fecik, J. Am. Chem. Soc., 2005, 127, 8910–8911 CrossRef CAS PubMed.
- A. Pinto, M. Wang, M. Horsman and C. N. Boddy, Org. Lett., 2012, 14, 2278–2281 CrossRef CAS PubMed.
- E. S. Sattely, M. A. Fischbach and C. T. Walsh, Nat. Prod. Rep., 2008, 25, 757–793 RSC.
- S. Friedrich and F. Hahn, Tetrahedron, 2015, 71, 1473–1508 CrossRef CAS.
- C. Garcia-Galan, Á. Berenguer-Murcia, R. Fernandez-Lafuente and R. C. Rodrigues, Adv. Synth. Catal., 2011, 353, 2885–2904 CrossRef CAS.
- R. A. Sheldon, Adv. Synth. Catal., 2007, 349, 1289–1307 CrossRef CAS.
- R. A. Sheldon, Catalysts, 2019, 9, 261 CrossRef CAS.
- M. R. Bennett, M. L. Thompson, S. A. Shepherd, M. S. Dunstan, A. J. Herbert, D. R. M. Smith, V. A. Cronin, B. R. K. Menon, C. Levy and J. Micklefield, Angew. Chem., Int. Ed., 2018, 57, 10600–10604 CrossRef CAS PubMed.
- M. Frese and N. Sewald, Angew. Chem., Int. Ed., 2015, 54, 298–301 CrossRef CAS PubMed.
- M. Ismail, M. Frese, T. Patschkowski, V. Ortseifen, K. Niehaus and N. Sewald, Adv. Synth. Catal., 2019, 361, 2475–2486 CrossRef CAS.
- L. Betancor and H. R. Luckarift, Biotechnol. Genet. Eng. Rev., 2010, 27, 95–114 CrossRef CAS PubMed.
- B. Ku, J. Cha, A. Srinivasan, S. J. Kwon, J.-C. Jeong, D. H. Sherman and J. S. Dordick, Biotechnol. Prog., 2006, 22, 1102–1107 CrossRef CAS PubMed.
- S. J. Kwon, M.-Y. Lee, B. Ku, D. H. Sherman and J. S. Dordick, ACS Chem. Biol., 2007, 2, 419–425 CrossRef CAS PubMed.
- M. I. Kim, S. J. Kwon and J. S. Dordick, Org. Lett., 2009, 11, 3806–3809 CrossRef CAS PubMed.
- K. H. Sung, G. Berkhan, T. Hollmann, L. Wagner, W. Blankenfeldt and F. Hahn, Angew. Chem., Int. Ed., 2018, 57, 343–347 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1ra03692k |
| This journal is © The Royal Society of Chemistry 2021 |