
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceBi(OTf)3-catalysed intramolecular cyclisation of unsaturated acetals†
Raphaël Sagetab,
Piotr Jaunky*b and
Elisabet Duñach *a
*a
aInstitut de Chimie de Nice, Université Cote d'Azur, CNRS, Parc Valrose, 06108 Nice cedex 2, France. E-mail: dunach@unice.fr; Web: http://web.univ-cotedazur.fr/labs/icn/fr
bV. MANE Fils, Centre de Recherche en Chimie Organique, 620 Route de Grasse, Le Bar-sur-Loup 06620, France. E-mail: piotr.jaunky@mane.com; Web: https://www.mane.com/
First published on 14th June 2021
Abstract
A series of highly functionalized carbocycles was efficiently prepared via the selective cyclisation of unsaturated acetals and ketals in the presence of only 1 mol% of Bi(III) or Fe(III) triflates as the catalysts at room temperature, with yields ranging from 60 to 90%. With Bi(OTf)3 catalysis, α,β-unsaturated ether carbocycles are formed selectively, whereas with the Fe(OTf)3 system, a cycloisomerisation to carbocyclic diethers is mainly obtained. This acetal/olefin cyclisation could be run at a multi-gram scale and compound 2c could be obtained on a 300 gram-scale with a yield of 69% after precipitation in hexane.
Introduction
In the context of C–C bond formation catalysed by Lewis acids, several fundamental processes have found important applications, namely the Friedel–Crafts arylations,1 aldol-type condensations,2 diene cyclisations,3 carbonyl-ene or Prins reactions,4 among others. Although the carbonyl-ene reaction is well documented, both under stoichiometric and catalytic conditions,5 related enol ether–olefin6 or acetal–olefin cyclisations have been much less developed.Among acetal–olefin coupling reactions, the Hosomi–Sakurai reaction involves the Lewis acid promoted allylation of an acetal with an allylsilane (generally allyltrimethylsilane) for the synthesis of homoallyl ethers,7 including several interesting examples in the field of glycochemistry.8 This process takes advantage of the nucleophilic activation of the allyl group by the β-stabilisation effect of the silicon atom. Besides the Hosomi–Sakurai allylation, examples of acetal-ene reactions involving non-activated double bonds are scarce. Coupling reactions have been reported under stoichiometric or over-stoichiometric amounts of Lewis acids such as TiCl4,9 SnCl4,10,11 SnCl2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 12 or EtAlCl2.13 To our knowledge, only two examples using PtCl2 or FeCl3 refer to catalytic processes.14,15
12 or EtAlCl2.13 To our knowledge, only two examples using PtCl2 or FeCl3 refer to catalytic processes.14,15
Interested by the catalytic activation of olefins by strong Lewis acids such as metallic triflates,16,17 we present here our results on novel aspects of the cyclisation of acetals and ketals bearing differently substituted olefins on the side-chain.
Results and discussion
We observed that in the presence of a low amount of a metal triflate, generally 1 mol%, the model unsaturated acetal 1a could undergo a selective cyclisation. Compound 1a was prepared via allylation of dimethyl malonate and further alkylation by 2-bromoacetaldehyde dimethyl acetal. Its catalytic reaction in the presence of a Lewis acid afforded the highly functionalised carbocycles 2a and/or 3a (Scheme 1). | ||
| Scheme 1 Intramolecular cyclisation of unsaturated acetals catalysed by metal triflates at 1 mol%. | ||
Acetal 1a was chosen for the optimisation of the cyclisation conditions. Table 1 summarises the results of the effect of several parameters. A first reaction was carried out using 1 mol% of Al(OTf)3 in nitromethane at room temperature (entry 1). The reaction was followed by GC and the complete conversion of 1a was reached after 3 h. Compounds 2a and 3a were formed as the main products in 75% combined yield in a 39:61 ratio, respectively. Unsaturated ether 2a was issued from a cyclisation involving the loss of methanol, whereas dimethoxy derivative 3a involves a cycloisomerisation process.
| Entry | Catalyst (1 mol%) | Solvent | Reaction time (h) | Yield of cyclisation (%) | Selectivity 2a:3a |
|---|---|---|---|---|---|
| a General reaction conditions: 1a (0.5 mmol) in the corresponding solvent (5 mL) with the added catalyst (1 mol% unless otherwise stated) was stirred at 20 °C (unless otherwise stated). Reactions were followed by GC with 1,4-dichlorobenzene as the internal standard and stopped after complete consumption.b The reaction was run at 0 °C.c The cyclisation was carried out with 0.1 mol% of catalyst at 20 °C. | |||||
| 1 | Al(OTf)3 | MeNO2 | 3 | 75 | 39:61 |
| 2 | Cu(OTf)2 | MeNO2 | 0.5 | 69 | 41:59 |
| 3 | Mg(OTf)2 | MeNO2 | 6 | — | — |
| 4 | Fe(OTf)3 | MeNO2 | 0.17 | 81 | 40:60 |
| 5 | Bi(OTf)3 | MeNO2 | 0.17 | 77 | 42:58 |
| 6 | Al(OTf)3 | DCE | 1 | 63 | 66:34 |
| 7 | Cu(OTf)2 | DCE | 3 | 61 | 69:31 |
| 8 | Bi(OTf)3 | DCE | 0.17 | 69 | 78:22 |
| 9 | Fe(OTf)3 | DCE | 0.5 | 64 | 73:27 |
| 10 | Bi(OTf)3 | CH2Cl2 | 0.17 | 68 | 89:11 |
| 11 | Bi(OTf)3 | Toluene | 3 | 72 | 87:13 |
| 12b | Bi(OTf)3 | CH2Cl2 | 7 | 39 | 86:14 |
| 13c | Bi(OTf)3 | CH2Cl2 | 6 | 66 | 87:13 |
The same reaction with Cu(OTf)2 as the catalyst led to a complete conversion within 0.5 h, with a cyclisation yield of 69% and a ratio 2a:3a of 41:59 (entry 2). No reaction occurred with Mg(OTf)2 (entry 3) and starting 1a could be recovered. With Fe(OTf)3 and Bi(OTf)3 at 1 mol% (entries 4 and 5), the conversion of 1a was complete in less than 10 min with cyclisation yields of 77–81% and isomer ratios of 40:60 and 42:58, respectively.
These different catalysts were also tested in 1,2-dichloroethane (DCE) at room temperature. Al(OTf)3 and Cu(OTf)2 reacted smoothly in 1–3 h (entries 6 and 7). The most active catalysts resulted to be Bi(OTf)3 and Fe(OTf)3 (entries 8 and 9), both affording selectivities towards 2a in the range of 73–78%. Interestingly, in DCE the selectivity 2a : 3a was reversed as compared to that obtained in nitromethane.
The reaction catalysed by Bi(OTf)3 in DCE was slightly more efficient as compared to Fe(OTf)3 and led to a higher selectivity towards 2a. Similar results were obtained with Bi(OTf)3 in dichloromethane (DCM) as the solvent within 10 min reaction at 20 °C, with a selectivity of 89% for 2a (entry 10). The cyclisation of 1a was also possible with Bi(OTf)3 in toluene and resulted in 72% of 2a:3a with a ratio of 87:13 after 3 h at room temperature (entry 11).
At 0 °C, Bi(OTf)3 (1 mol%) led to a 85% conversion of 1a after 7 h, with a cyclisation yield of only 39% (entry 12). The cyclisation was also efficient with only 0.1 mol% of Bi(OTf)3 at room temperature, with a selectivity 2a:3a of 87:13, but it was only completed after 6 h, with some product degradation (entry 13). In some cases, diene 4a, issued from methanol elimination from 2a (and/or 3a) was also formed in low yields (see Scheme 2). Possibly, the polymerisation of the 1,3-diene 4a accounts for this product degradation.
 | ||
| Scheme 2 Proposed cyclisation mechanism of unsaturated acetal 1a with a metal triflate. | ||
The Lewis superacid character imparted by the triflate ligands was crucial for an efficient catalytic activity, since both BiCl3 and BiBr3, under the conditions of entry 10, failed to give any conversion of 1a.
For comparison, we also tested the use of triflic acid (HOTf) as the catalyst. At 1 mol% in nitromethane, 1a afforded 76% of 2a:3a with a ratio of 37:63 after 1 h at room temperature. This result indicated a relatively efficient protic acid cyclisation, although other protic acids (e.g. p-toluenesulfonic acid) afforded poor results. We continued the study with metal triflates as the catalysts, HOTf being toxic and its handling as a fuming compound being much less convenient.18
The results of Table 1 indicate a strong influence of the nature of the solvent in the outcome of the cyclisation. In a polar solvent such as nitromethane, the formation of 3a was favoured (entries 1, 2, 4 and 5), whereas in less polar solvents such as toluene or chlorinated solvents, compound 2a was formed with a higher selectivity (entries 6–13). Moreover, the several catalysts tested presented a strong difference in activity. Whereas in nitromethane, the complete conversion of 1a was reached in 10 min with Bi(OTf)3 and Fe(OTf)3, it needed 0.5 h with Cu(OTf)2 and 3 h with Al(OTf)3. No reaction of 1a with Mg(OTf)2 could be observed after 6 h.
For comparison, cyclisations of 1a with the reported PtCl2 or FeCl3 catalytic systems under the conditions of entry 10 were carried out. Whereas with FeCl3 no reaction occurred, in the presence of the PtCl2/AgOTf catalytic system, a conversion of 33% was obtained after 10 min with a yield of 2a of 13%; however, the reaction did not go to completion and the yield of 2a was of 37% after 8 h.
Concerning the cyclisation of 1a to 2a, the most efficient system was obtained with 1 mol% of Bi(OTf)3 in dichloromethane at room temperature, with a selectivity for 2a of 89% (entry 10). Under these conditions, 2a was formed in a cis/trans ratio of 15/85, as shown by NOE-NMR analysis.
Concerning the formation of 3a, we chose to further explore Fe(OTf)3 as the catalytic system. The best selectivities obtained in nitromethane were in the range of 60% (Table 1). In order to enhance the selectivity towards 3a, some cyclisations of 1a were run with 1 mol% of Fe(OTf)3 with added methanol. As depicted in Table 2, the addition of 2 or 4 equiv. of MeOH led to enhanced selectivities for 3a, up to 87% (entries 2 and 3). It is to note that using methanol as the solvent did not allow any cyclisation, possibly due to the deactivation of the catalyst (entry 4).
| Entry | MeOH | Cyclisation yield (%) | Selectivity 2a:3a |
|---|---|---|---|
| a General reaction conditions: 1a (0.5 mmol) in nitromethane (5 mL) with 1 mol% of Fe(OTf)3 and MeOH (0–4 equiv.) were stirred at 20 °C. Reactions were followed by GC with 1,4-dichlorobenzene as the internal standard and stopped after complete consumption of 1a.b Only MeOH (5 mL) was used as solvent. | |||
| 1 | — | 81 | 40:60 |
| 2 | 2 equiv. | 92 | 15:85 |
| 3 | 4 equiv. | 84 | 13:87 |
| 4b | Solvent | — | — |
Interestingly, for this novel acetal–olefin coupling, the experimental conditions can be tuned in order to direct the cyclisation of 1a either towards 2a or towards 3a, both catalytic systems affording chemoselectivities in the range of 80–90%.
In a first set of experiments, the cyclisation was extended to other substrates of type 1 using 1 mol% of Bi(OTf)3 in dichloromethane at room temperature (Table 3). The cyclisation of acetal 1b afforded the six-membered ring carbocycle 2b in 60% yield with a cis/trans ratio of 70:30 (entry 2). The ratio 2b:3b was 95:5. Unsaturated ketals reacted also efficiently. Thus, the ketal 1c gave access to 2c in 81% yield (entry 3). The cis/trans ratio in this case was 70/30. Isolated 2c could be crystallised in hexane and the X-ray analysis confirmed its cis-stereochemistry, in agreement with 2D-NMR.19
| Entry | Reaction time | Substrate | Main product | Yield | Ratio cis/trans |
|---|---|---|---|---|---|
| a General reaction conditions: 1 (6.9–20.8 mmol) in CH2Cl2 (69–104 mL) with Bi(OTf)3 (1 mol%) was stirred at 20 °C. Reactions were followed by GC with 1,4-dichlorobenzene as the internal standard and stopped after complete consumption of the starting material. Cyclised compounds were purified by column chromatography on silica-gel with cyclohexane/ethyl acetate mixtures as the eluents.b For 2d, the ratio of α,β- versus β,γ-double bond was 20:80.c No reaction occurred in CH2Cl2 and MeNO2 was used as the solvent. For 2i, the ratio terminal versus internal olefin was 56/44. |
|||||
| 1 | 10 min | 1a |  |
60% | 15/85 |
| 2 | 10 min | 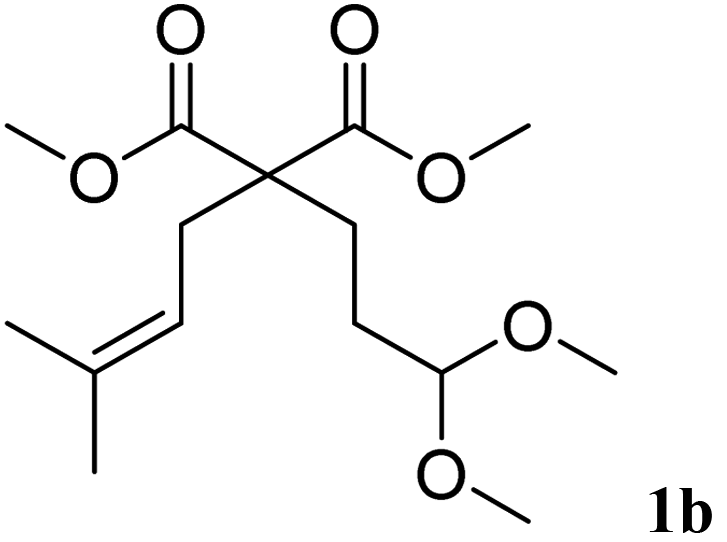 |
 |
60% | 70/30 |
| 3 | 10 min | 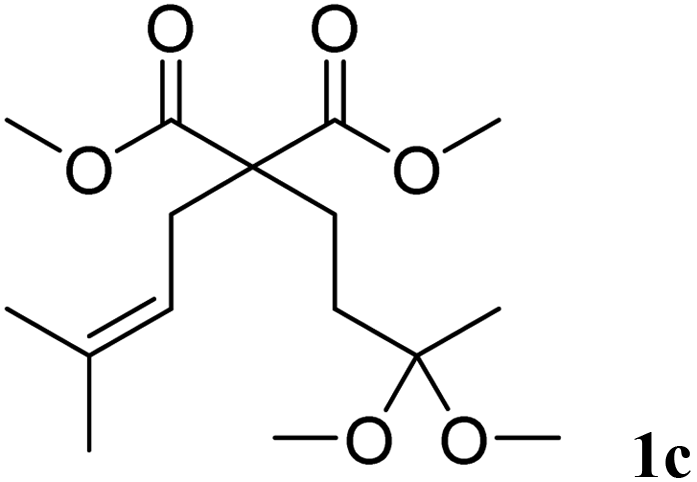 |
 |
81% | 70/30 |
| 4b | 10 min |  |
 |
75% | — |
| 5 | 10 min |  |
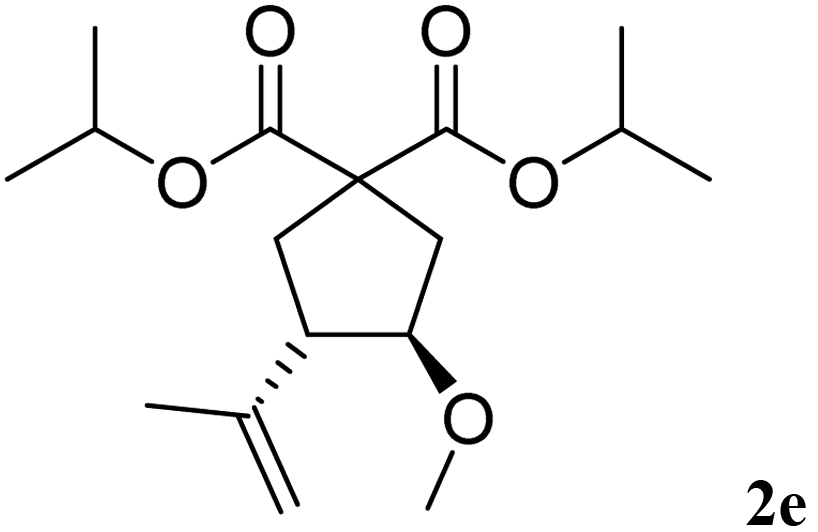 |
70% | 10/90 |
| 6 | 20 min | 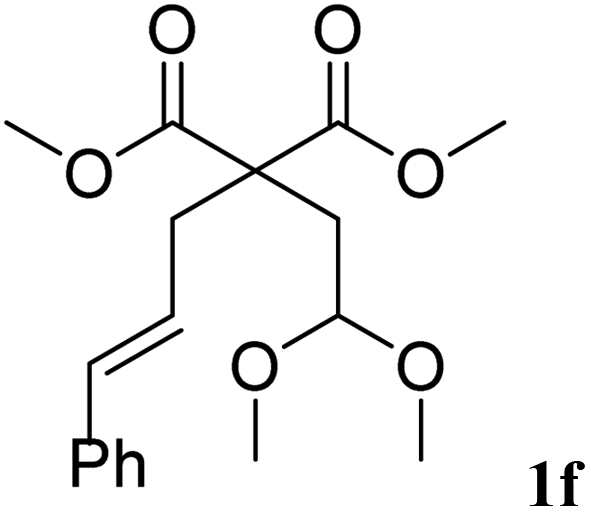 |
 |
85% | 30/70 |
| 7 | 20 min |  |
 |
91% | 35/65 |
| 8 | 20 min | 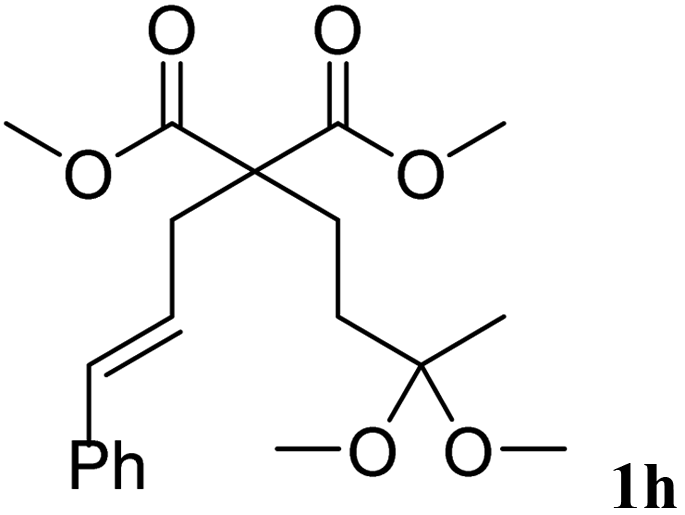 |
 |
80% | 65/35 |
| 9c | 24 h |  |
 |
2i: 19% | — |
| 5i: 49% | 60/40 | ||||
The 2,2-disubstituted olefin 1d afforded 2d in 75% yield, a ratio of 80/20 was found for the position of the double bond (entry 4).
The more hindered diisopropyl malonate 1e reacted efficiently affording 2e in 70% yield, with a cis/trans ratio of 10/90 (entry 5).
The cinnamyl acetal derivatives 1f and 1g (entries 6 and 7) gave the 5- and 6-membered rings 3f and 3g, in 85 and 91% yield, respectively. In these cases, the corresponding mono-methoxylated carbocycle 2 was not formed, and the cycloisomerisation compounds 3f and 3g were obtained chemospecifically. Analogous unsaturated ketal 1h reacted similarly and afforded 3h in 80% yield (entry 8). For 3f and 3g, the major isomer presented a trans-stereochemistry with selectivities of 65–70%. The main isomer of 3h was a cis carbocycle with a selectivity of 65%.
The possibility of preparing 7-membered rings was tested with substrate 1i, which afforded 19% of the expected 2i, together with 49% of an unexpected cyclisation compound 5i. The formation of lactone 5i can be explained by the further transformation of 2i, involving the cyclisation of one of the ester groups on the tertiary position of the double bond. This type of ester/olefin cyclisation to lactones has already been observed in related Lewis acid-catalysed reactions.3,20
Noteworthy, this acetal/olefin cyclisation could be run at a multi-gram scale. Moreover, cyclohexane derivative 2c could be obtained on a 300 gram-scale with 69% yield after precipitation in hexane.
Concerning the stereoselectivity in these reactions with Bi(OTf)3 as the catalyst in dichloromethane, we can observe that whereas the cis-isomer was favoured in six-membered ring carbocycles 2b and 2c, the five-membered rings 2a and 2e gave mainly the trans-isomer. The cinnamyl-derived 3f and 3g afforded the trans-isomers preferentially, both for 5 or 6-membered rings, whereas the more hindered 3h led to a preferential cis-stereoselectivity. In order to better understand the differences in selectivity, theoretical DFT calculations were carried out comparing the relative energies of 1a, 1b and 2a, 2b on their cis/trans forms, as well as the energies of their carbocation intermediates (see above, Mechanistic considerations). The calculated data indicated a very small energy difference (less than 4.5 kJ mol−1) between cis/trans in both 5 or 6-membered rings. Again, a very small difference in the activation energy for the cyclisation of the corresponding cations (less than 8.5 kJ mol−1) was obtained (see the ESI† for details). This small energy differences may explain the results obtained for the reaction selectivity. In the related literature on glycosylation reactions with several nucleophiles, it has already been observed that small changes in the nature of the nucleophiles could reverse the stereochemical outcome of the reactions.8
Examples of the cycloisomerisation of 1 to 3 using Fe(OTf)3 as the catalyst at 1 mol% in nitromethane, in the presence of 2 equiv. of methanol are presented in Table 4.
| Entry | Reaction time | Substrate | Product | Yield | Ratio cis/trans |
|---|---|---|---|---|---|
| a General reaction conditions: 1 (8.7–10.4 mmol) in MeNO2 (100–150 mL) with Fe(OTf)3 (1 mol%) and 2 equiv. of MeOH were stirred at 20 °C. Reactions were followed by GC with 1,4-dichlorobenzene as the internal standard and stopped after complete consumption of the starting material. Compounds 3 were purified by column chromatography on silica-gel with cyclohexane/ethyl acetate mixtures as the eluents. | |||||
| 1 | 10 min | 1a |  |
78% | 60/40 |
| 2 | 1 h |  |
 |
51% | 83/17 |
| 3 | 10 min |  |
 |
72% | 65/35 |
| 4 | 2 h | 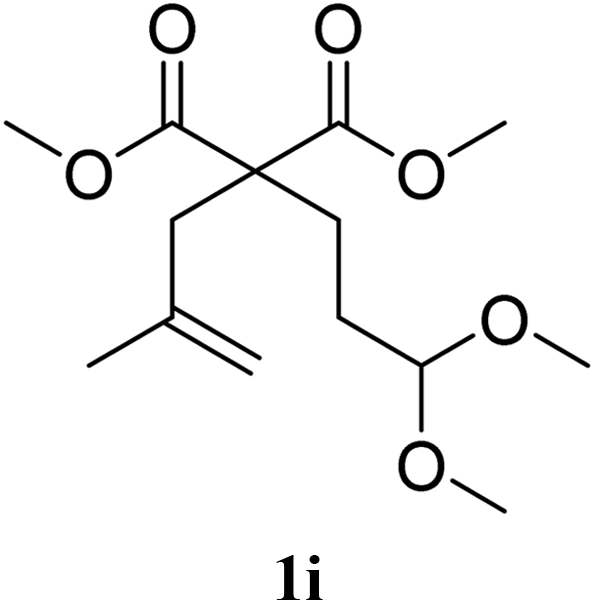 |
 |
64% | 84/16 |
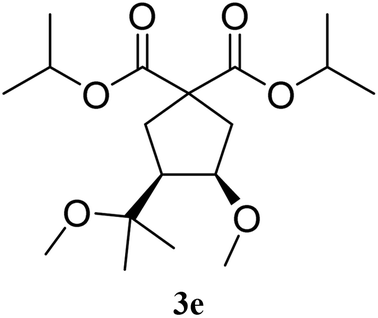
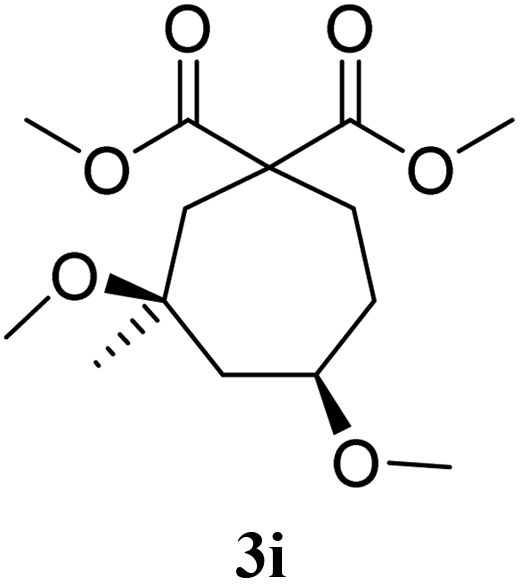
With Fe(OTf)3 catalysis, compound 3a was obtained from 1a after 10 minutes at room temperature, with a yield of 78%. The cis/trans ratio for 3a was of 60/40, according to NOE analysis (entry 1). The prenyl derivative 1b gave the 6-membered ring 3b with a moderate yield of 51%, and a cis/trans ratio of 83/17 (entry 2). The analogous diisopropyl malonate 3e was obtained with 72% yield and a cis/trans selectivity of 65/35 (entry 3). The 7-membered ring 3i was obtained with 64% yield and a cis/trans ratio of 84/16 (entry 4).
In the examples developed with Fe(OTf)3 catalysis in nitromethane, all the compounds were selective towards the cis-isomer.
When comparing the selectivities with both Bi(OTf)3 and Fe(OTf)3 systems for the same starting substrates, differences appear in the cases of the major products for the reactions of 1a and 1e. Here again, the literature indicates in related examples that the nature of the solvent (CH2Cl2 and nitromethane) as well as the nature of the catalyst could strongly influence the isomer ratios.21
Mechanistic considerations
The acetal–olefin cyclisation of malonate derivatives of type 1 was catalysed by Bi(OTf)3 in dichloromethane to afford generally monomethylated derivatives 2. In addition, 1 could also afford dimethoxy-substituted carbocycles of type 3 with Fe(OTf)3/MeOH catalytic system in nitromethane. A common first part of the mechanism is proposed to proceed through the initial activation of the acetal group by the highly electrophilic Lewis acid. As depicted in Scheme 2 in the case of 1a, the activation of the acetal is followed by the formation of an intermediate carbocation A, by loss of one methoxy group.22 Follows the intramolecular nucleophilic addition of the double bond generating intermediate carbocation B. The ring-closing step (C–C bond formation) is controlled by the stability of B, and the ring size depends on the olefin substitution. Intermediate B may react according to the nature of the solvent: in a polar solvent such as nitromethane, we suggest that the MeO− group eliminated in the first step stays coordinated to the Bi(III) centre and can therefore attack on carbocation B, leading to the formation of 3a. In less polar solvents such as DCE or DCM, intermediate B mainly evolves through a selective proton elimination to 2a. The cyclised compound 2a can also form diene 4a, observed as a by-product, via further methanol elimination. In polar solvents, the ion-pair in intermediates A and B should be better stabilised as compared to their stabilisation in less polar ones. In the case of the reactivity of cinnamyl derivatives such as 1f–1h, the higher stabilisation of intermediates B due to the resonance effect of the phenyl group may facilitate the attack of the methoxy group, to selectively form compounds 3.This process can be mechanistically related to the carbonyl-ene cyclisation of unsaturated aldehydes, which leads to the corresponding homoallylic alcohols.4 However, the carbonyl-ene reaction has only been efficiently developed with unsaturated aldehydes and presents a very low interest with unsaturated ketones, due to low yields and reaction reversibility.23
Scheme 3 illustrates the comparison between the reactivity of the carbonyl-ene and the ketal-ene processes with the same catalytic system. Whereas ketone 6 reacted with 1 mol% of Bi(OTf)3 in dichloromethane to give alcohol 7 in 23% yield after 48 h at room temperature, the corresponding ketal 1c afforded the analogous 2c with a yield of 81% in only 10 min at the same temperature. The results indicated that the ketal activation was highly facilitated and that the cyclisation of 1c to 2c was much more efficient than the cyclisation of ketone 6 to alcohol 7.
 | ||
| Scheme 3 Comparison between the reactivity of ketal 1c and the corresponding ene-reaction with ketone 6. | ||
DFT calculations on the relative energy of the formation of the carbocation intermediates of type A from acetals (and ketals) and the analogous carbocations issued from carbonyl compounds were carried out (Scheme 2). In order to simplify the calculations, the Lewis acid was replaced by a proton and the substituents of the acetal and the carbonyl compounds by methyl or ethyl groups. The results indicated a much higher stabilisation of intermediates A for acetals of ketals as compared to analogous carbonyl compound intermediates, in agreement with the experimental results (Scheme 3). Thus, whereas the addition of a proton to an aldehyde or ketone led to a stabilisation of −11 and −30 kJ mol−1, respectively, the proton addition to the corresponding dimethyl acetal or ketal gave stabilisation energies of −101 and −150 kJ mol−1, respectively. The formation of methanol in the case of acetal/ketal protonation may partly explain this efficient stabilisation. The simulation of the cyclisation of 1a to 2a by a proton gave an activation energy of 63–69 kJ mol−1. Experimentally, with Bi(OTf)3 as the catalyst, the activation energy for the cyclisation of 1a was evaluated to be of 151 kJ mol−1 after kinetic data of reactions run at 0, 10 and 20 °C. An important energy difference was found in this case for the DFT process calculated for a proton cyclisation as compared to the experimental data in which the cyclisation is run with Bi(OTf)3, a much more bulky Lewis acid catalyst. Details of these calculations are to be found in the ESI.†
Conclusions
In conclusion, an efficient and highly catalytic intramolecular acetal–olefin cyclisation of compounds of type 1 was developed, by using only 1 mol% of Bi(OTf)3 in low polarity solvents such as dichloromethane. The coupling reaction selectively afforded α,β-unsaturated ethers of type 2. Bismuth(III) triflate resulted to be a powerful catalyst: the activation of the acetal group takes place readily and the reactions were generally completed in 10–20 min at room temperature. The present transformation provides a simple and selective procedure to access highly functionalised carbocycles in good yields.By adapting the reaction conditions, carbocyclic diethers of type 3 could also be prepared in a cycloisomerisation process, in particular by using a 1 mol% Fe(OTf)3-based catalytic system in polar aprotic solvents such as nitromethane, under very mild conditions.
Cycloisomerisations leading to 5-, 6-, and also 7-membered ring highly substituted carbocycles were obtained.
Experimental section
General cyclisation procedure: the unsaturated acetal 1 (1 equiv.) was added to the appropriate solvent (DCM, DCE or MeNO2, 0.1 M). Bi(OTf)3 or Fe(OTf)3 (0.01 equiv.) were added to the solution and the mixture was stirred at room temperature. After full conversion of 1, the reaction was quenched with neutral activated aluminum oxide. The solution was then filtered off and the solvent removed under reduced pressure. The oily residue was purified by flash-chromatography through silica-gel with cyclohexane/AcOEt mixtures as the eluents or by distillation. See the ESI† for further details.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
The authors acknowledge the University Côte d'Azur, the CNRS and V. MANE FILS for financial support of this work. We are grateful to Dr M. Gaysinski for NMR assistance.Notes and references
- M. Rueping and B. J. Nachtsheim, Beilstein J. Org. Chem., 2010, 6, 6, DOI:10.3762/bjoc.6.6
.
- R. Mahrwald, Modern Aldol Reactions, Wiley-VCH, Weinheim (GE), 2004, vol. 1 and 2, ISBN 3-527-30714-1 Search PubMed
- F. Grau, A. Heumann and E. Duñach, Angew. Chem., Int. Ed., 2006, 45, 7285–7289 CrossRef CAS PubMed
- B. B. Snider, Acc. Chem. Res., 1980, 13, 426–432 CrossRef CAS
- K. Mikami and M. Shimizu, Chem. Rev., 1992, 92, 1021–1050 CrossRef CAS
- L. Lempenauer, G. Lemière and E. Duñach, Adv. Synth. Catal., 2019, 361, 5284–5304 CrossRef CAS
- A. Hosomi, M. Endo and H. Sakurai, Chem. Lett., 1976, 5, 941–942 CrossRef
-
(a) J. R. Krumper, W. A. Salamant and K. A. Woerpel, J. Org. Chem., 2009, 74, 8039–8050 CrossRef CAS PubMed
- W. H. Bunnelle, D. W. Seamon, D. L. Mohler, T. F. Ball and D. W. Thompson, Tetrahedron Lett., 1984, 25, 2653–2654 CrossRef CAS
- W. S. Johnson and R. B. Kinnel, J. Am. Chem. Soc., 1966, 88, 3861–3862 CrossRef CAS PubMed
- T. A. Blumenkopf, M. Bratz, A. Castañeda, G. C. Look, L. E. Overman, D. Rodriguez and A. S. Thompson, J. Am. Chem. Soc., 1990, 112, 4386–4399 CrossRef CAS
- W. S. Johnson, A. Van der Gen and J. J. Swoboda, J. Am. Chem. Soc., 1967, 89, 170–172 CrossRef CAS
- D. Berger and L. E. Overman, Synlett, 1992, 10, 811–813 Search PubMed
- A. Ladépêche, E. Tam, J. E. Ancel and L. Ghosez, Synthesis, 2004, 9, 1375–1380 Search PubMed
- K. Miura, H. Izumi, H. Kinoshita, J. Ichikawa and A. Hosomi, Chem. Lett., 2009, 38, 1204–1205 CrossRef CAS
- S. Kobayashi, M. Sugiura, H. Kitagawa and W. W.-L. Lam, Chem. Rev., 2002, 102, 2227–2302 CrossRef CAS PubMed
- S. Antoniotti, V. Dalla and E. Duñach, Angew. Chem., Int. Ed., 2010, 49, 7860–7888 CrossRef CAS PubMed
- B. Bouguerne, P. Hoffmann and C. Lherbet, Synth. Commun., 2010, 40, 915–926 CrossRef CAS
- See details of the X-ray of 2c in the ESI.†.
-
(a) B. Cacciuttolo, S. Poulain-Martini, F. Fontaine-Vive, M. A. H. Abdo, H. El-Kashef and E. Duñach, Eur. J. Org. Chem., 2014, 7458–7468 CrossRef CAS
- J. C. Kendale, E. M. Valentín and K. A. Woerpel, Org. Lett., 2014, 16, 3684–3687 CrossRef CAS PubMed
- P. O. Adero, H. Amarasekara, P. Wen, L. Bohé and D. Crich, Chem. Rev., 2018, 118, 8242–8284 CrossRef CAS PubMed
- P. Tremel, C. Iacobucci, L. Massi, S. Olivero, J. F. Gal and E. Duñach, New J. Chem., 2015, 39, 7453–7458 RSC
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2084106. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d1ra03686f |
| This journal is © The Royal Society of Chemistry 2021 |