
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDirect enantioseparation of axially chiral 1,1′-biaryl-2,2′-diols using amidine-based resolving agents†
Koichi Kodama *,
Fusato Takase and
Takuji Hirose
*,
Fusato Takase and
Takuji Hirose
Department of Applied Chemistry, Graduate School of Science and Engineering, Saitama University, Japan. E-mail: kodama@mail.saitama-u.ac.jp
First published on 19th May 2021
Abstract
Amidine-based optically active resolving agents for enantiomer separation of axially chiral 1,1′-biaryl-2,2′-diols have been developed. A strongly basic amidine bearing no substituents on its nitrogen atoms enables the formation of their diastereomeric salts upon being mixed with weakly acidic phenol derivatives. Enantiopure 1,1′-biaryl-2,2′-diols can be obtained in high yields after only one crystallization of their salts with the chiral amidine derived from dehydroabietic acid. X-ray crystallography revealed that the amidine moiety forms a salt with the phenol group and additional intermolecular NH/π interactions contribute to the efficient chiral recognition process.
Introduction
Among the various methods used for the industrial production of optically pure chiral compounds, optical resolution of a racemate into its enantiomers via diastereomeric salt formation, followed by recrystallization, is one of the most reliable and economical methods reported to date.1 Various chiral carboxylic acids and amines have been developed as acidic and basic resolving agents for this purpose. However, this method is sometimes not applicable to weak acids and bases because they cannot form their corresponding diastereomeric salts. To overcome this problem, chiral sulfonic acids and phosphoric acids have been developed as acidic resolving agents for the enantioseparation of weakly basic amines due to their strong acidities.2 On the other hand, strongly basic resolving agents used for the separation of weak acids are limited.Amidines have been comprehensively demonstrated to be stronger Brønsted bases than conventional amines. Several chiral amidines have been developed and applied as N-chelating ligands,3 asymmetric organocatalysts,4 and in the kinetic resolution of alcohols.5 Recently, chiral supramolecular structures constructed via salt formation using chiral amidines and carboxylic acids have been reported.6 Their robust supramolecular assembly has been attributed to the strong charge-assisted hydrogen bonds formed during the salt formation process. It is expected that such strongly basic chiral amidines can be applied toward the enantioseparation of weakly acidic compounds via diastereomeric salt formation.
The atropisomers of 1,1′-biaryl-2,2′-diols (1) are an important class of compounds with weakly acidic phenol moieties. Functionalized chiral biaryl derivatives can be applied as ligands in asymmetric synthesis,7 dopants for nematic liquid crystals to induce a helical arrangement, etc.8 Dobashi reported the NMR-based chiral recognition of 1,1′-binaphthyl-2,2′-diol (BINOL, 1a) using a twisted chiral bisamidine.9 Enantiopure 1a is useful not only as a chiral ligand in catalytic asymmetric reactions, but as a starting material for the synthesis of various chiral auxiliaries, such as BINAP.10 Although various methods to access enantiopure 1a have been reported including enantioselective oxidative coupling, enzymatic resolution, chemical resolution of its diastereomers with chiral auxiliaries,10 and complexation with chiral reagents,11 the practical enantioseparation of 1 from its racemate is still challenging.
In this paper, we report the efficient enantioseparation of 1 via diastereomeric salt formation using chiral amidine-based resolving agents (2).
Results and discussion
Enantioseparation of 1a by N,N′-disubstituted chiral amidine (2a)
We selected N,N′-disubstituted chiral amidine 2a as a resolving agent because resolving agents bearing aromatic groups generally result in dense molecular packing during the formation of their salt crystals. Amidine 2a in its optically active form has been previously reported and was easily synthesized from commercially available (S)-1-phenylethylamine (3) (Scheme 1).11 As previously reported, the IR spectrum of 2a in solid state exhibits an absorption band at 1637 cm−1, corresponding to the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N stretching vibrations.
N stretching vibrations.
 | ||
| Scheme 1 Synthesis of chiral amidine 2a. Reagents and conditions: (a) PhCOCl, NaOH, H2O, 0 °C to rt; (b) (COCl)2, 2,6-lutidine, dry CH2Cl2, 0 °C to rt; (c) 3, reflux. | ||
Sterically demanding 1a was selected as the first target axially chiral phenol. Equimolar amounts of rac-1a and 2a were mixed and crystallized from toluene to give colorless crystals. Analysis of the 1H NMR spectrum shows that the resulting crystals consisted of both 1a and 2a (84% yield based on half the amount of 1a used; molar ratio 1a![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2a = 1.8:1). The complex of 1a and 2a was decomposed and 1a was isolated via extraction. The enantiopurity of 1a was determined using chiral HPLC analysis, which was as high as 81% ee, and (S)-1a was found to be preferentially crystallized with 2a.
:2a = 1.8:1). The complex of 1a and 2a was decomposed and 1a was isolated via extraction. The enantiopurity of 1a was determined using chiral HPLC analysis, which was as high as 81% ee, and (S)-1a was found to be preferentially crystallized with 2a.
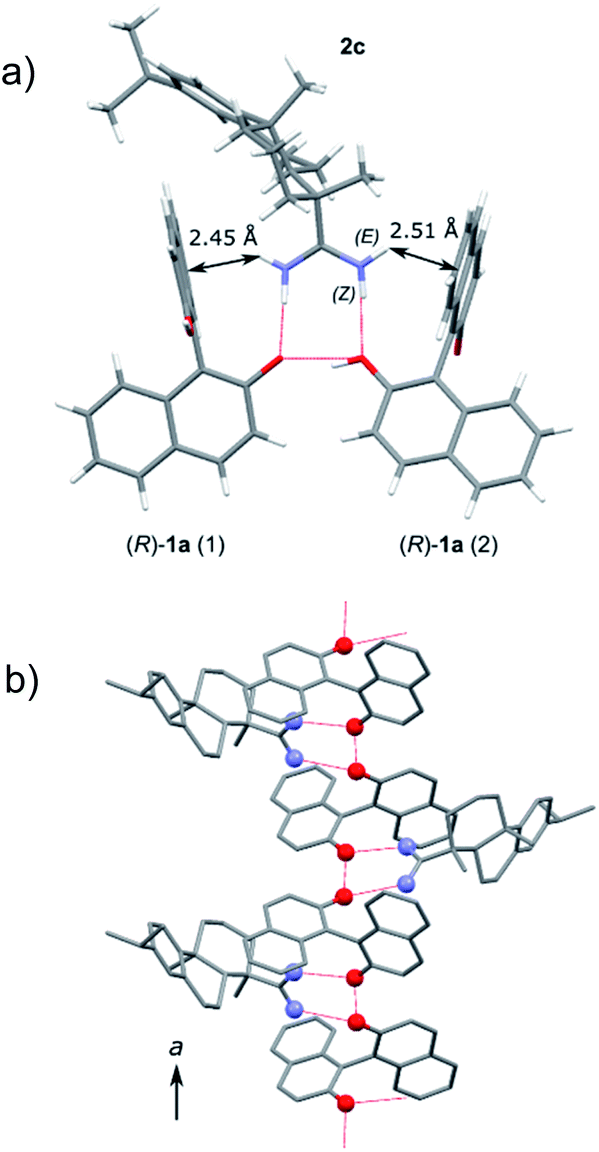
Platelet crystals suitable for X-ray crystallographic analysis were obtained during the crystallization step and it was found that 2a and 1a were co-crystallized with a molar ratio of (S)-1a:2a = 2:1 (Fig. 1). There are two independent (S)-1a molecules in the asymmetric unit cell. The 2a molecule was in the form of its protonated amidinium cation with an (E,E) configuration, which forms two hydrogen bonds with (S)-1a. The two N–H hydrogen atoms were combined with the same phenolate oxygen atom in (S)-1a to give the salt. Only one molecule of (S)-1a was ionized and the other (S)-1a molecule was not charged (Fig. 1a). The neutral (S)-1a molecule was incorporated by the formation of a hydrogen bond with the ionized (S)-1a molecule and these three molecules assemble to form an aggregate. All the naphthalene planes of (S)-1a were fixed by CH/π interactions with the neighboring aromatic CH of 2a, which should result in the good chiral recognition of the (S)-isomer of 1a (Fig. 1b).
 | ||
| Fig. 1 Crystal structure of 2a·2(S)-1a. The oxygen and nitrogen atoms are represented using red and blue spheres. The dotted lines represent the hydrogen bonds. The arrows represent the CH/π interactions. | ||
Unfortunately, this complex formation process was not always reproducible and occasionally 1a was crystallized alone under the same conditions. The IR spectrum of a mixture of 1a and 2a in methanol showed that the absorption band at 1637 cm−1, corresponding to 2a, was unchanged upon the addition of 1a, which indicates no obvious salt formation (Fig. S1†). This can be attributed to the steric hindrance of the bulky chiral substituents of 2a around the amidine hydrogen-bonding site, which prevents salt formation with 1a.
Synthesis of N-non-substituted chiral amidines (2b and 2c)
In order to prepare salts with phenols more reliably, a series of chiral amidines without any substituents on their nitrogen atoms were designed. Such non-substituted amidines may be better candidates as resolving agents because of their reduced steric hindrance and increased number of hydrogen atoms available to form hydrogen bonds. Non-substituted amidines have been applied as ligands to detect carboxylic acids and inhibitors13 and recently used to construct supramolecular hydrogen-bonded organic frameworks (HOFs) by combining with carboxylic acids or sulfonic acids. Such salt-type HOFs with large and permanent porosities have been applied in selective gas absorption, enzyme encapsulation, etc.14 However, chiral non-substituted amidines are very rare with only a few examples have been reported.15Benzamidine, which is one of the simplest non-substituted amidines was studied as a model compound in our preliminary experiments to examine the salt formation ability of non-substituted amidines with phenols. When benzamidine was mixed with 4-nitrophenol, the resulting IR spectrum showed that the absorption peak at 1637 cm−1, corresponding to benzamidine, was shifted to 1654 cm−1, which suggested the formation of the amidinium salt, although a mixture of 4-nitrophenol and 2a showed no such a shift in the absorption peak (Fig. S2†). The 1:1 salt formed between benzamidine and 4-nitrophenol was also confirmed using X-ray crystallography (Fig. S3†).
One general and convenient synthetic route toward non-substituted amidines is to start from their corresponding carboxylic acids by way of their nitrile derivatives.16 In addition, the amidine functional group should be close to the asymmetric center to enhance the chiral recognition ability. Based on these concepts, non-substituted chiral amidine 2b derived from (S)-naproxen (5) was designed. Compound 5 is a commercially available chiral carboxylic acid, which has been used as an acidic resolving agent as well as a non-steroidal anti-inflammatory drug.17 Amidine 2b was synthesized from enantiopure 5 via nitrile derivative 7 (Scheme 2). Nitrile 7 was converted into its corresponding N-hydroxyamidine (8), followed by O-acetylation and hydrogenolysis to afford desired non-substituted amidine (2b). However, it was found that the enantiopurity of the intermediate 8 decreased to 51% ee and 2b showed no optical activity. This suggests that the base used during the synthesis induced partial racemization due to the abstraction of the acidic hydrogen atom on the stereogenic center and the strong basicity of the amidine itself caused complete racemization of 2b.
 | ||
| Scheme 2 Synthesis of chiral amidine 2b. Reagents and conditions: (a) (COCl)2, 0 to 40 °C; (b) 28% NH3 (aq.), toluene, 0 °C to rt; (c) PPh3, Et3N, CCl4, dry CH2Cl2, reflux; (d) NH2OH·HCl, Et3N, dry EtOH, dry DMF, 80 °C; (e) Ac2O, pyridine, THF, 0 °C to rt; (f) H2 (1 atm), cat. Pd/C, EtOH, rt. | ||
To avoid racemization, dehydroabietic acid (10) bearing a quaternary stereogenic center was selected as the starting chiral carboxylic acid. Carboxylic acid 10 is an abundant natural acid obtained from rosin, which has been used as a chiral source because of its rigid and hydrophobic nature.18 Non-substituted chiral amidine 2c was successfully prepared in moderate-to-high yield using a similar synthetic route via nitrile 12 (Scheme 3). The high stereochemical purity of 2c was confirmed by its narrow melting point range, 1H NMR analysis, and specific optical rotation.19 The IR spectrum of a mixture of 2c and 1a showed that the absorption peak corresponding to the CN stretching vibration at 1634 cm−1 derived from 2c disappears and a new peak at 1673 cm−1 appeared, which indicates that the amidinium salt of 2c was formed upon mixing with 1a (Fig. S4).†
 | ||
| Scheme 3 Synthesis of chiral amidine 2c. Reagents and conditions: (a) (COCl)2, cat. DMF, dry CH2Cl2, 0 °C to reflux; (b) 28% NH3 (aq.), toluene, 0 °C to rt; (c) PPh3, Et3N, CCl4, dry CH2Cl2, reflux; (d) NH2OH·HCl, Et3N, dry EtOH, reflux; (e) Ac2O, pyridine, THF, 0 °C to rt; (f) H2 (1 atm), cat. Pd/C, EtOH, rt. | ||
Enantioseparation of 1 by N-non-substituted chiral amidine (2c)
The enantioseparation of rac-1a with 2c via diastereomeric salt formation was investigated (Table 1). Crystallization of a mixture of equimolar amounts of rac-1a and 2c from toluene afforded the 1:1 salt of 1a and 2c in high yield (88%) and the enantiopurity of 1a was as high as 97% for the (R)-isomer (entry 1). Using ethyl acetate as the solvent further increased the yield up to 94% and pure (R)-1a salt was obtained by only one crystallization step (entry 2). Ethanol, a more polar solvent, also afforded the salt of enantiopure (R)-1a, albeit in lower yield (entry 3). As a control experiment, dehydroabietylamine, which is a primary amine with the same chiral skeleton as 2c, was mixed with rac-1a and recrystallized from toluene. However only 1a was precipitated, which clearly suggests that the amidine functional group of 2c was essential for enantioselective salt formation with (R)-1a.
|
||||
|---|---|---|---|---|
| Entry | Phenol | Solvent (mL) | Yieldb (%) | eec (%) |
| a Rac-1 and 2c (0.25 mmol) were used.b The yield is based on half the amount of the initial salt.c The ee was determined using HPLC analysis. The absolute configuration of the major enantiomer is shown in the parentheses. | ||||
| 1 | 1a | Toluene (8.5) | 88 | 97(R) |
| 2 | 1a | AcOEt (29.5) | 94 | >99(R) |
| 3 | 1a | EtOH (15.5) | 34 | >99(R) |
| 4 | 1b | Toluene (4.0) | 66 | Rac |
| 5 | 1b | Toluene/AcOEt (1.5/0.15) | Not crystallized | — |
| 6 | 1c | Toluene | Gelated | — |
| 7 | 1c | AcOEt (4.5) | 58 | >99(R) |
| 8 | 1c | EtOH (0.6) | 30 | 80(R) |
| 9 | 15 | Toluene/hexane (0.2/0.6) | Not crystallized | — |

Prompted by this result, the scope for the enantioseparation of other axially chiral 1,1′-biaryl-2,2′-diols was investigated (Table 1). Binaphthalene-based diol 1b bearing two bromine atoms also afforded a 1:1 salt with 2c. However, only racemic 1b was obtained from the salt and the enantioseparation was not successful (entry 4). On the other hand, 6,6′-dimethyl-2,2′-biphenol (1c), which is one of the simplest axially chiral biphenols, was crystallized with 2c from ethyl acetate to afford pure (R)-1c salt in a good yield (entry 7). Crystallization from ethanol again decreased the separation efficiency, which was attributed to the polar solvent reducing the efficiency of hydrogen bond formation and decreasing the rate of salt formation (entry 8). A previously reported method for the enantioseparation of 1c requires its derivatization into a phosphoric acid derivative, followed by diastereomeric salt formation with cinchonidine, a toxic alkaloid amine.20 To the best of our knowledge, this is the first example of a simple and direct enantiomeric separation of rac-1c using crystallization. On the other hand, the salt of 2c with monophenol 15 bearing another aliphatic hydroxy group on the asymmetric center was not crystallized (entry 9). This result indicates that the two phenolic hydroxy groups in 1,1′-biaryl-2,2′-diols cooperatively interact with the amidine moiety in 2c to form their corresponding salts.
A needle-like crystal was obtained from an ethanol solution of a mixture of (R)-1a and 2c, which was subjected to X-ray crystallography (Fig. 2). The 1:1 salt was formed between (R)-1a and 2c, and one hydroxy group in (R)-1a was ionized. The two amidinium hydrogen atoms in the (Z)-configuration of 2c form hydrogen bonds with the oxygen atoms of two molecules of 1a. The two oxygen atoms are combined via a OH–O hydrogen bond. The other two amidinium hydrogen atoms in the (E)-configuration form cationic NH–π interactions with the naphthalene rings in 1a (Fig. 2a).21 Two (R)-1a molecules are tightly fixed by these intermolecular interactions with 2c and they are helically arranged to construct a one-dimensional hydrogen-bonding network along the a-axis (Fig. 2b). Because both 1a and 2c are not flexible molecules, the efficient packing of the naphthalene moieties in 1a and the large hydrocarbon moiety in 2c also contribute to the highly stereoselective crystallization of one diastereomeric salt 2c·(R)-1a.
 | ||
| Fig. 2 Crystal structure of 2c·(R)-1a. The oxygen and nitrogen atoms are represented using red and blue spheres. The dotted lines represent the hydrogen bonds. The arrows represent the NH/π interactions. | ||
Conclusions
Chiral amidines have been developed for the enantioseparation of weakly acidic phenol derivatives via diastereomeric salt formation. Atropisomers of axially chiral 1,1′-binaphthyl-2,2′-diol (1a) and 6,6′-dimethyl-2,2′-biphenol (1c) have been separated in high yield and with high enantiopurity using a non-substituted chiral amidine (2c) derived from dehydroabietic acid (10), a commercially available chiral source. IR spectra and X-ray crystallography has shown that hydrogen bonds and additional NH–π interactions with the amidinium group played an important role in the chiral recognition process. Based on the reliable amidinium-phenolate salt formation, further design of chiral amidines as resolving agents will expand the scope for enantioseparation of different chiral phenols, which is now under investigation.Experimental
General and materials
All the 1H and 13C NMR spectra were measured using 300, 400, or 500 MHz spectrometers. IR spectra were reported in reciprocal centimeters. Melting points are uncorrected. Optical rotation values were measured with a polarimeter. All commercially available reagents and solvents were purchased and used as received unless noted. Dry THF was freshly distilled from sodium under a nitrogen atmosphere. Dry CH2Cl2 and dry CCl4 were distilled after drying over CaCl2 and stored with 4 Å molecular sieves under a nitrogen atmosphere. Dry triethylamine was distilled after drying over KOH and stored with KOH under a nitrogen atmosphere. Dry toluene was distilled from sodium under a nitrogen atmosphere and stored with sodium under a nitrogen atmosphere. Dry EtOH was distilled from sodium under a nitrogen atmosphere and stored with molecular sieves 4 Å under a nitrogen atmosphere. Dehydroabietic acid (10) was supplied from Arakawa Chemical Ind, Ltd. and purified by repeated recrystallization of its salt with 2-aminoethanol.22 The enantiomeric excess of the compounds was determined by chiral HPLC analysis (Daicel Chiralcel OD-3 column 4.6 × 250 mm or Chiralpak AS-3 column 2.1 × 250 mm) with UV detection at 254 nm.(S)-N-(1-Phenylethyl)benzamide (4)12b
To a vigorously stirred mixture of (S)-1-phenylethylamine (3) (2.81 g, 23.2 mmol) and NaOH (1.29 g, 32.2 mmol) in H2O (25 mL) was added dropwise benzoylchloride (3.62 g, 25.8 mmol) over 10 min at 0 °C. After the suspension was stirred for 2 h at room temperature, the white precipitate was filtered, washed several times with H2O and then dried in vacuo. The desired product 4 (4.24 g, 18.8 mmol, 81%) was obtained as a white solid. Mp: 118.5–120.3 °C. [α]17D = +7.2° (c 0.251, MeOH). 1H NMR (300 MHz, CDCl3): δ (ppm) 7.84–7.70 (m, 2H), 7.58–7.22 (m, 8H), 6.42–6.18 (br, 1H), 5.44–5.26 (m, 1H), 1.62 (d, J = 6.9 Hz, 3H). IR (KBr): ν (cm−1) 3451, 3331, 1634, 1523, 1490, 1319, 758, 700.(S,S)-N,N′-Bis(1-phenylethyl)benzamidine (2a)12b
A solution of 4 (0.903 g, 4.01 mmol) and 2,6-lutidine (0.647 g, 6.04 mmol) in dry CH2Cl2 (10 mL) was cooled to 0 °C. Oxalyl chloride (0.543 g, 4.28 mmol) diluted with dry CH2Cl2 (5 mL) was slowly added to the solution over 30 min. Stirring was continued at 0 °C for 30 min, and the solution was allowed to warm to room temperature and stirred for 30 min. 3 (0.490 g, 4.04 mmol) diluted with dry CH2Cl2 (5 mL) was slowly added to the solution over 30 min at room temperature. The reaction mixture was refluxed for 24 h, and then concentrated under reduced pressure. The residue was dissolved in AcOEt (20 mL) and extracted with 1 N HCl aq. (10 mL × 15). The aqueous phase was basified with 6 N NaOH aq. and extracted with CHCl3 (5 mL × 5). The organic phase was dried over anhydrous Na2SO4, filtered and concentrated under reduced pressure. The residue was suspended in hexane, and the resulting solid was collected by filtration. The crude product (0.530 g) was recrystallized from EtOH (0.8 mL) and the desired product 2a (0.441 g, 1.34 mmol, 33%) was obtained as colorless crystals. Mp: 124.3–125.0 °C. [α]24D = −48.5° (c 1.00, CHCl3). 1H NMR (300 MHz, DMSO-d6): δ (ppm) 7.52–6.72 (m, 15H), 6.70–6.56 (m, 1H), 5.26–5.02 (m, 1H), 4.12–3.96 (m, 1H), 1.49 (d, J = 7.2 Hz, 3H), 1.17 (d, J = 6.3 Hz, 3H). IR (KBr): ν (cm−1) 3060, 2955, 2877, 1637, 1599, 1494, 1484, 1451, 1361, 1349, 1309, 1268, 1142, 1090, 766, 699.(S)-2-(6-Methoxy-2-naphthyl)propionamide (6)23
Oxalyl chloride (3.0 mL) and DMF (3 drops) were added to (S)-2-(6-methoxy-2-naphthyl)propanoic acid (5) (2.32 g, 10.1 mmol) under a nitrogen atmosphere at 0 °C, and the resulting solution was refluxed for 2 h. After the excess of oxalyl chloride was distilled off, dry toluene (20 mL) and 28% NH3 aq. (4 mL) were added at 0 °C. After the resulting suspension was stirred at room temperature for 1 h, the white precipitate formed was filtered, washed several times with H2O and then dried in vacuo. The desired product 6 (2.21 g, 9.63 mmol, 96%, >99% ee) was obtained as a white solid, which was used for next step without further purification. Mp: 177.0–179.0 °C. [α]22D = +33.3° (c 0.195, MeOH). 1H NMR (400 MHz, CDCl3): δ (ppm) 7.80–7.66 (m, 3H), 7.44–7.34 (m, 1H), 7.20–7.08 (m, 2H), 3.92 (s, 3H), 3.74 (q, J = 7.2 Hz, 1H), 1.61 (d, J = 7.2 Hz, 3H). IR (KBr): ν (cm−1) 3348, 3195, 2983, 2898, 1660, 1606, 1505, 1486, 1461, 1403, 1309, 1267, 1228, 1217, 1173, 1114, 1027, 927, 894, 854, 814. HPLC analysis (Daicel Chiralcel OD-3, hexane/2-propanol = 80:20, 1.0 mL min−1, 254 nm UV detector; tr(S) = 10.3 min, tr(R) = 16.4 min).
(S)-2-(6-Methoxy-2-naphthyl)propionitrile (7)24
To a stirred solution of 6 (0.300 g, 1.31 mmol) in dry CH2Cl2 (15 mL), PPh3 (0.420 g, 1.60 mmol), dry triethylamine (0.161 g, 1.59 mmol) and dry CCl4 (0.246 g, 1.60 mmol) were added under a nitrogen atmosphere. The solution was refluxed for 20 h. After the reaction was quenched with H2O, the organic layer was separated, washed with 1 N HCl aq., dried over anhydrous Na2SO4, filtered and concentrated under reduced pressure. The crude product (0.875 g) was purified by silica gel column chromatography (eluent: CHCl3). The desired product 7 (0.244 g, 1.15 mmol, 88%, >99% ee) was obtained as a pale yellow solid. Mp: 98.7–100.0 °C. [α]17D = −28.9° (c 1.00, CHCl3). 1H NMR (300 MHz, CDCl3): δ (ppm) 7.84–7.68 (m, 3H), 7.46–7.34 (m, 1H), 7.22–7.08 (m, 2H), 4.04 (q, J = 7.2 Hz, 1H), 3.93 (s, 3H), 1.72 (d, J = 7.2 Hz, 3H). IR (KBr): ν (cm−1) 3065, 3020, 2992, 2963, 2942, 2905, 2840, 2240, 1916, 1777, 1712, 1632, 1604, 1506, 1483, 1448, 1419, 1393, 1376, 1356, 1260, 1214, 1188, 1165, 1085, 1024, 960, 927, 891, 856. HPLC analysis (Daicel Chiralpak AS-3, hexane/2-propanol = 99.5:0.5, 0.3 mL min−1, 254 nm UV detector; tr(S) = 34.6 min, tr(R) = 41.2 min).
(S)-N-Hydroxy-2-(6-methoxy-2-naphthyl)propionamidine (8)
To a stirred solution of 7 (1.14 g, 5.40 mmol) in dry EtOH (3 mL) and dry DMF (3 mL), H2NOH·HCl (1.13 g, 16.3 mmol) and dry triethylamine (1.65 g, 16.3 mmol) were added, and then the solution was refluxed under a nitrogen atmosphere for 10 h. After cooling to room temperature, the solution was concentrated under reduced pressure. The residue was dissolved in AcOEt (40 mL) and washed with H2O/sat. NaCl aq. = 1/1. The organic phase was dried over anhydrous Na2SO4, filtered and concentrated under reduced pressure. The crude product (0.893 g) was purified by silica gel column chromatography (eluent: CHCl3/MeOH = 30/1, v/v). The desired product 8 (0.272 g, 1.11 mmol, 21%, 51% ee) was obtained as a white solid. Mp: 142.0–143.5 °C (21% ee). [α]18D = −10.5° (c 0.506, MeOH) (21% ee). 1H NMR (300 MHz, CDCl3): δ (ppm) 7.80–7.64 (m, 3H), 7.46–7.36 (m, 1H), 7.20–7.04 (m, 2H), 4.50–4.26 (br, 2H), 3.92 (s, 3H), 3.76 (q, J = 7.2 Hz, 1H), 1.58 (d, J = 7.2 Hz, 3H). 13C NMR (100 MHz, CDCl3): δ (ppm) 157.7, 156.6, 136.9, 133.8, 129.2, 128.9, 127.5, 126.2, 125.6, 119.1, 105.7, 55.3, 41.8, 18.0. IR (KBr): ν (cm−1) 3479, 3372, 3268, 3055, 2976, 2936, 1663, 1637, 1607, 1586, 1505, 1486, 1460, 1449, 1418, 1390, 1266, 1231, 1216, 1192, 1174, 1160, 1124, 1078, 1026, 921, 889, 849. HPLC analysis (Daicel Chiralcel OD-3, hexane/2-propanol = 80:20, 1.0 mL min−1, 254 nm UV detector; tr(S) = 10.5 min, tr(R) = 13.1 min).
(S)-N-Acetoxy-2-(6-methoxy-2-naphthyl)propionamidine (9)
To a stirred solution of 8 (0.267 g, 1.10 mmol) in THF (5 mL), pyridine (0.111 g, 1.40 mmol) and acetic anhydride (0.135 g, 1.32 mmol) were added at 0 °C, and then the solution was stirred at room temperature for 1 h. After the solvent was distilled off, the residue was dissolved in CHCl3 (50 mL), and washed with 1 N HCl aq., sat. NaHCO3 aq. and sat. NaCl aq. The organic phase was dried over anhydrous Na2SO4, filtered and concentrated under reduced pressure. The desired product 9 (0.301 g, 1.05 mmol, 96%, 51% ee) was obtained as a white solid, which was used for next step without further purification. Mp: 105.0–108.0 °C. 1H NMR (300 MHz, CDCl3): δ (ppm) 7.80–7.68 (m, 3H), 7.50–7.38 (m, 1H), 7.22–7.08 (m, 2H), 4.66–4.42 (br, 2H), 4.04–3.84 (m, 4H), 2.19 (s, 3H), 1.67 (d, J = 7.2 Hz, 3H). 13C NMR (100 MHz, CDCl3): δ (ppm) 168.8, 160.3, 157.9, 135.4, 133.9, 129.2, 128.9, 127.6, 126.2, 125.5, 119.3, 105.7, 55.4, 41.3, 19.9, 17.7. IR (KBr): ν (cm−1) 3449, 3332, 3196, 3060, 2962, 2936, 2841, 1744, 1627, 1505, 1486, 1464, 1439, 1418, 1392, 1371, 1266, 1227, 1173, 1119, 1027, 1010, 957, 927, 885, 855, 814. HPLC analysis (Daicel Chiralcel OD-3, hexane/2-propanol = 80:20, 1.0 mL min−1, 254 nm UV detector; tr(S) = 13.8 min, tr(R) = 30.7 min).
2-(6-Methoxy-2-naphthyl)propionamidine (2b)
A suspension of 9 (0.720 g, 2.51 mmol) and 10% Pd–C (0.173 g) in EtOH (30 mL) was stirred under a hydrogen atmosphere at room temperature for 2 h. The solid was filtered off and the filtrate was concentrated under reduced pressure. The residue (0.752 g) was recrystallized from MeOH (8.5 mL) to give the acetate salt of the product as a solid. The separated filtrate was concentrated under reduced pressure and the residue was recrystallized from EtOH/H2O (4 mL/0.5 mL). The combined solid was dissolved in CHCl3 (30 mL) and 1 N NaOH aq. (20 mL) was added. The aqueous layer was separated and extracted with CHCl3 (15 mL × 5). The combined organic layer was washed with sat. NaCl aq., dried over anhydrous Na2SO4, filtered and concentrated under reduced pressure. The desired product 2b (0.597 g, 2.07 mmol, 83%, racemic) was obtained as a white solid. Mp: 125.0–129.0 °C. [α]26D = 0° (c 1.00, CHCl3). 1H NMR (300 MHz, CDCl3): δ (ppm) 7.80–7.62 (m, 3H), 7.40–7.30 (m, 1H), 7.22–7.04 (m, 2H), 3.92 (s, 3H), 3.73 (q, J = 7.2 Hz, 1H), 1.58 (d, J = 7.2 Hz, 3H). IR (KBr): ν (cm−1) 3321, 3163, 2966, 1684, 1635, 1606, 1505, 1485, 1455, 1436, 1392, 1264, 1216, 1163, 1030, 927, 891, 853.Dehydroabietyl amide (11)25
To a stirred solution of dehydroabietic acid (10) (2.01 g, 6.09 mmol) in dry CH2Cl2 (3 mL), DMF (3 drops) and oxalyl chloride (1.5 mL) were added under a nitrogen atmosphere at 0 °C, and the solution was refluxed for 2 h. After the volatile components were distilled off, dry toluene (10 mL) and 28% NH3 aq. (3 mL) were added at 0 °C. The resulting suspension was stirred at room temperature for 2 h, and then was added to AcOEt (100 mL) and H2O (50 mL). The organic layer was separated and washed with sat. NaHCO3 aq. and sat. NaCl aq. The organic phase was dried over anhydrous Na2SO4, filtered and concentrated under reduced pressure. The desired product 11 (1.95 g, 6.50 mmol, 97%) was obtained as a pale yellow solid, which was used for next step without further purification. Mp: 153.0–156.0 °C. [α]25D = +41.1° (c 1.00, CHCl3). 1H NMR (300 MHz, CDCl3): δ (ppm) 7.22–7.12 (m, 1H), 7.04–6.96 (m, 1H), 6.90–6.82 (m, 1H), 5.90–5.60 (br, 1H), 5.50–5.20 (br, 1H), 2.96–2.85 (m, 2H), 2.85–2.73 (m, 1H), 2.39–2.25 (m, 1H), 2.16–2.04 (m, 1H), 1.90–1.40 (m, 7H), 1.29 (s, 3H), 1.26–1.14 (m, 9H). IR (KBr): ν (cm−1) 3428, 3328, 2927, 2867, 1629, 1575, 1498, 1456, 1383, 1362, 1085, 1036, 905, 883, 822.Dehydroabietyl cyanide (12)26
To a stirred solution of 11 (5.49 g, 18.3 mmol) in dry CH2Cl2 (100 mL), PPh3 (7.20 g, 27.4 mmol), dry triethylamine (2.78 g, 27.4 mmol) and dry CCl4 (4.23 g, 27.5 mmol) were added under a nitrogen atmosphere. The solution was refluxed for 17 h. After the reaction was quenched with H2O, the organic layer was separated, washed with 1 N HCl aq., dried over anhydrous Na2SO4, filtered and concentrated under reduced pressure. The crude product (14.8 g) was purified by silica gel column chromatography (eluent: hexane/CHCl3 = 1/1, v/v). The desired product 12 (4.77 g, 16.9 mmol, 93%) was obtained as a white solid. Mp: 77.0–80.0 °C. [α]26D = +37.0° (c 1.00, CHCl3). 1H NMR (300 MHz, CDCl3): δ (ppm) 7.20–7.10 (m, 1H), 7.06–6.92 (m, 1H), 6.92–6.86 (m, 1H), 3.10–2.92 (m, 2H), 2.92–2.70 (m, 1H), 2.40–2.24 (m, 1H), 2.14–1.66 (m, 8H), 1.42 (s, 3H), 1.22 (d, J = 6.9 Hz, 6H), 1.18 (s, 3H). 13C NMR (100 MHz, CDCl3): δ (ppm) 146.2, 145.4, 134.2, 127.0, 126.6, 124.2, 124.0, 46.8, 37.5, 37.4, 37.3, 37.2, 33.5, 29.8, 25.2, 23.9, 21.7, 18.9, 17.7. IR (KBr): ν (cm−1) 3010, 2930, 2930, 2863, 2225, 1612, 1496, 1458, 1419, 1383, 889, 819. MS (MALDI-TOF) m/z calcd for C20H27N + Na+: 304.204 [M + Na]+; found: 304.245.N-Hydroxy-dehydroabietyl amidine (13)
To a stirred solution of 12 (4.76 g, 16.9 mmol) in dry EtOH (35 mL), H2NOH·HCl (5.89 g, 84.8 mmol) and dry triethylamine (8.58 g, 84.8 mmol) were added, and then the solution was refluxed under a nitrogen atmosphere for 28 h. After cooling to room temperature, the solution was concentrated under reduced pressure. The residue was dissolved in AcOEt (100 mL) and washed with H2O and sat. NaCl aq. The organic layer was dried over anhydrous Na2SO4, filtered and concentrated under reduced pressure. The crude product (5.76 g) was purified by silica gel column chromatography (eluent: hexane/AcOEt = 2/1, v/v). The desired product 13 (1.66 g, 5.29 mmol, 31%) was obtained as a white solid. Mp: 89.7–91.7 °C. [α]24D = +86.2° (c 1.00, CHCl3). 1H NMR (300 MHz, CDCl3): δ (ppm) 7.22–7.12 (m, 1H), 7.04–6.94 (m, 1H), 6.94–6.84 (m, 1H), 4.80–4.48 (br, 2H), 2.98–2.74 (m, 3H), 2.45–2.28 (m, 1H), 2.00–1.56 (m, 8H), 1.34–1.18 (m, 12H). 13C NMR (100 MHz, CDCl3): δ (ppm) 160.0, 147.2, 145.9, 134.8, 127.0, 124.0, 123.9, 46.3, 42.8, 38.3, 37.5, 37.4, 33.5, 30.0, 25.5, 24.0, 20.1, 18.7, 16.0. IR (KBr): ν (cm−1) 3500, 3399, 3255, 2930, 1651, 1575, 1497, 1457, 1382, 1362, 1230, 1197, 1173, 1140, 1074, 923, 822. MS (MALDI-TOF) m/z calcd for C20H30N2O + H+: 315.244 [M + H]+; found: 315.242.N-Acetoxy-dehydroabietyl amidine (14)
To a stirred solution of 13 (1.64 g, 5.21 mmol) in THF (44 mL), pyridine (0.503 g, 6.36 mmol) and acetic anhydride (0.638 g, 6.25 mmol) were added at 0 °C, and then the solution was stirred at room temperature for 1 h. After the solvent was distilled off, the residue was dissolved in CHCl3 (50 mL), and washed with 1 N HCl aq., sat. NaHCO3 aq. and sat. NaCl aq. The organic layer was dried over anhydrous Na2SO4, filtered and concentrated under reduced pressure. The desired product 14 (1.69 g, 4.73 mmol, 91%) was obtained as a white solid, which was used for next step without further purification. Mp: 125.8–127.8 °C. [α]24D = +66.6° (c 1.00, CHCl3). 1H NMR (300 MHz, CDCl3): δ (ppm) 7.24–7.12 (m, 1H), 7.06–6.98 (m, 1H), 6.92–6.86 (m, 1H), 4.86–4.64 (br, 2H), 2.98–2.74 (m, 3H), 2.44–2.28 (m, 1H), 2.20 (s, 3H), 1.98–1.62 (m, 8H), 1.31 (s, 3H), 1.26 (s, 3H), 1.22 (d, J = 6.9 Hz, 6H). 13C NMR (100 MHz, CDCl3): δ (ppm) 169.7, 163.5, 147.0, 145.9, 134.6, 127.0, 124.0, 124.0, 46.4, 43.5, 38.1, 37.9, 37.4, 33.5, 30.0, 25.5, 24.0, 20.4, 20.2, 18.6, 16.0. IR (KBr): ν (cm−1) 3501, 3378, 2957, 2869, 1743, 1626, 1582, 1497, 1459, 1384, 1364, 1231, 1008, 939, 882, 822. MS (MALDI-TOF) m/z calcd for C22H32N2O2 + Na+: 379.236 [M + Na]+; found: 379.217.Dehydroabietyl amidine (2c)
A suspension of 14 (1.68 g, 4.72 mmol) and 10% Pd–C (0.614 g) in EtOH (60 mL) was stirred under a hydrogen atmosphere at room temperature for 1 day. Pd–C was filtered off and the filtrate was concentrated under reduced pressure. The residue was suspended in hexane, and the resulting solid was collected by filtration. The residue (1.44 g) was dissolved in CHCl3 (30 mL) and sat. NaHCO3 aq. (70 mL) was added. The aqueous layer was extracted with CHCl3 (15 mL × 3). The combined organic layer was washed with sat. NaCl aq., dried over anhydrous Na2SO4, filtered and concentrated under reduced pressure. The desired product 2c (1.22 g, 4.09 mmol, 87%) was obtained as a white solid. Mp: 76.7–79.7 °C. [α]25D = +54.7° (c 1.00, CHCl3). 1H NMR (300 MHz, CDCl3): δ (ppm) 7.24–7.12 (m, 1H), 7.08–6.96 (m, 1H), 6.94–6.84 (m, 1H), 3.00–2.76 (m, 3H), 2.44–2.30 (m, 1H), 1.92–1.38 (m, 8H), 1.27, (s, 3H), 1.25 (s, 3H), 1.22 (d, J = 7.2, 6H). 13C NMR (100 MHz, CDCl3): δ (ppm) 174.8, 147.0, 146.0, 134.5, 127.0, 124.1, 124.0, 46.8, 45.6, 38.2, 38.1, 37.3, 33.5, 29.9, 25.3, 24.0, 20.5, 19.0, 16.8. IR (KBr): ν (cm−1) 3345, 2958, 2869, 1634, 1577, 1497, 1459, 1382, 1363, 1201, 1171, 822. MS (MALDI-TOF) m/z calcd for C20H30N2 + H+: 299.249 [M + H]+; found: 299.241.(Rac)-6,6′-dibromo-1,1′-bi-2-naphthol (1b)27
Rac-1,1′-bi-2-naphthol (1a) (1.00 g, 3.49 mmol) was dissolved in dry CH2Cl2 (40 mL) under a nitrogen atmosphere. After the mixture was cooled to −10 °C, bromine (1.51 g, 9.45 mmol) diluted with CH2Cl2 (4 mL) was added dropwise over 30 min and the solution was stirred for an additional 2.5 h. After the solution was gradually warmed to room temperature and stirred for another 1 h, the reaction was quenched with sat. Na2SO3 aq. (25 mL). The aqueous phase was extracted with CHCl3 (15 mL × 3) and the combined organic phase was washed with sat. NaCl aq. dried over anhydrous MgSO4, filtered and concentrated under reduced pressure. The crude product (1.59 g) was recrystallized from toluene/heptane (7 mL/5 mL) and the desired product 1b (1.11 g, 2.51 mmol, 72%) was obtained as colorless needles. Mp: 206.0–207.0 °C. 1H NMR (300 MHz, CDCl3): δ (ppm) 8.06 (d, J = 1.8 Hz, 2H), 7.90 (d, J = 9.0 Hz, 2H), 7.46–7.32 (m, 4H), 6.97 (d, J = 9.0 Hz, 2H), 5.00 (s, 2H). IR (KBr): ν (cm−1) 3451, 2952, 1612, 1586, 1502, 1466, 1407, 1382, 1350, 1319, 1268, 1216, 1162, 1145, 1125, 1066, 951, 930, 876, 811.(Rac)-6,6′-dimethyl-2,2′-bisphenol (1c)28
To a solution of 4,6-di-tert-butyl-4-methylphenol (1.01 g, 4.60 mmol) in CH2Cl2 (9 mL), CuCl (45.5 mg, 0.460 mmol) and N,N,N′,N′-tetramethylethylenediamine (80.1 mg, 0.689 mmol) were added. The suspension was stirred under air at room temperature for 8 h. After addition of H2O (20 mL) to the reaction mixture, the whole was extracted with CHCl3 (10 mL × 3). The organic phase was washed with sat. NaCl aq. and dried over anhydrous Na2SO4, filtered and concentrated under reduced pressure. The crude product (1.19 g) was purified by silica gel column chromatography (eluent: hexane/CHCl3 = 1/1, v/v) to afford (rac)-3,3′,5,5′-tetra-tert-butyl-6,6′-dimethyl-2,2′-bisphenol (0.594 g, 1.35 mmol, 59%) as a white solid. Mp: 244.0–245.3 °C. 1H NMR (300 MHz, CDCl3): δ (ppm) 7.39 (s, 2H), 4.80 (s, 2H), 2.00 (s, 6H), 1.42 (s, 18H), 1.40 (s, 18H). IR (KBr): ν (cm−1) 3504, 2991, 2959, 2909, 2871, 1560, 1470, 1414, 1395, 1362, 1332, 1280, 1254, 1233, 1196, 1167, 1116, 1033, 927.To a solution of rac-3,3′,5,5′-tetra-tert-butyl-6,6′-dimethyl-2,2′-bisphenol (0.303 g, 0.691 mmol) in dry toluene (5 mL) was added AlCl3 (39.2 mg, 0.294 mmol) in small portions at 0 °C under a nitrogen atmosphere. The suspension was stirred at 50 °C for 18 h, and AlCl3 (99.3 mg, 0.745 mmol) was added to the suspension at 0 °C. The suspension was stirred at 50 °C for 3 h, and AlCl3 (58.8 mg, 0.441 mmol) was added to the suspension at 0 °C. The suspension was stirred at 50 °C for 3 h, and AlCl3 (58.4 mg, 0.438 mmol) was added to the suspension at 0 °C. After the suspension was stirred at 50 °C for 13 h, the suspension was cooled to 0 °C and carefully quenched by addition of H2O (14 mL) and 3 N HCl aq. (56 mL). The organic phase was separated and the aqueous phase was extracted with Et2O (10 mL × 3). The combined organic phase was extracted with 3 N NaOH aq. (10 mL × 3). The aqueous phase was acidified with 6 N HCl aq. and extracted with Et2O (10 mL × 3). The organic phase was dried over anhydrous Na2SO4, filtered and concentrated under reduced pressure. The desired product 1c (0.145 g, 0.677 mmol, 98%) was obtained as a white solid. Mp: 159.7–162.7 °C. 1H NMR (300 MHz, CDCl3): δ (ppm) 7.32–7.18 (m, 2H), 7.00–6.86 (m, 4H), 4.66 (s, 2H), 2.01 (s, 6H). IR (KBr): ν (cm−1) 3464, 3413, 3034, 2973, 2915, 1605, 1575, 1465, 1378, 1335, 1281, 1261, 1180, 1090, 1025, 1006, 947, 883.
(Rac)-1-[hydroxy(phenyl)methyl]-2-naphthol (1d)29
To a stirred suspension of Mg turnings (0.304 g, 12.5 mmol) in dry THF (4 mL) under a nitrogen atmosphere was added dropwise a solution of bromobenzene (1.96 g, 12.5 mmol) in dry THF (9 mL) over 1.5 h at room temperature. After formation of the Grignard reagent has started, the suspension was stirred at room temperature for 30 min, and then refluxed for 1 h. After cooling with an ice bath, 2-hydroxy-1-naphthaldehyde (0.861 g, 5.00 mmol), which was dissolved in dry THF (5.5 mL), was added dropwise to the mixture over 30 min and the suspension was stirred for 2 h at room temperature. The reaction was quenched with sat. NH4Cl aq. (5 mL) and H2O (15 mL) and the whole was extracted with CHCl3 (10 mL × 3). The organic phase was washed with sat. NaCl aq., dried over anhydrous Na2SO4, filtered and concentrated under reduced pressure. The crude product was purified by silica gel column chromatography (eluent: hexane/AcOEt = 4/1, v/v). The desired product 1d (1.23 g, 4.91 mmol, 98%) was obtained as a white solid. Mp: 118.5–120.3 °C. 1H NMR (300 MHz, CDCl3): δ (ppm) 9.21 (s, 1H), 7.82–7.62 (m, 3H), 7.50–7.12 (m, 8H), 6.82 (d, J = 2.6 Hz, 1H), 2.92 (d, J = 2.6 Hz, 1H). IR (KBr): ν (cm−1) 3363, 3029, 1625, 1602, 1521, 1469, 1455, 1411, 1326, 1265, 1226, 1154, 1065, 1011, 939, 830.Enantiomer separation of phenols (1) with chiral amidines (2)
Equimolar amounts of 2 and rac-1 (0.250 mmol) were dissolved in MeOH or CHCl3, and the solution was concentrated under reduced pressure to give a salt. An appropriate solvent was added to the salt with heating until a homogeneous solution was formed. The solution was gradually cooled to room temperature and left at the temperature for several days to induce crystallization. The salt 1·2 was collected by filtration and dried in vacuo at room temperature. The yield was calculated based on a half amount of rac-1 initially used. A part of the salt was dissolved in ethyl acetate, and the organic phase was washed with 1 N HCl aq. to remove 2. The organic phase was dried over anhydrous Na2SO4, filtered and concentrated under reduced pressure. The residue was purified by silica gel preparative TLC to give 1. The enantiomeric excess of 1 was determined by chiral HPLC analysis. 1a (column: Chiralcel OD-3, eluent: 2-propanol/hexane = 1/9, flow rate: 0.5 mL min−1, detection: 254 nm, tr(S) = 29.0 min, tr(R) = 30.7 min). 1b (column: Chiralcel OD-3, eluent: 2-propanol/hexane = 1/9, flow rate: 1.0 mL min−1, detection: 254 nm, tr(1st) = 15.6 min, tr(2nd) = 35.0 min). 1c (column: Chiralcel OD-3, eluent: 2-propanol/hexane = 1/9, flow rate: 0.5 mL min−1, detection: 254 nm, tr(S) = 17.6 min, tr(R) = 32.1 min).X-ray crystallographic analysis
Single crystals suitable for X-ray diffraction analysis were prepared by slow evaporation of the saturated solutions of the salt. X-ray crystallographic data were collected on a Bruker Smart APEX II diffractometer with graphite monochromated Mo Kα radiation. Data collections were carried out at 150 K. The structures were solved by a direct method (SIR 2014) and refined by SHELXL-2013 or SHELXL-2018 programs.30 Crystallographic information files have been deposited with the Cambridge Structural Database. Deposition number 2057487–2057489 contains the supplementary crystallographic data for this paper.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by JSPS KAKENHI Grant Number JP20K05458.Notes and references
- (a) J. Jacques, A. Collet and S. H. Wilen, Enantiomers, Racemates, and Resolutions, Wiley & Sons, New York, 1981 Search PubMed; (b) D. Kozma, Optical Resolutions via Diastereomeric Salt Formation, CRC Press, London, 2002 Search PubMed; (c) R. Siedlecka, Tetrahedron, 2013, 69, 6331 CrossRef CAS.
- J. Jacques, C. Fouquey and R. Viterbo, Tetrahedron Lett., 1971, 12, 4617 CrossRef.
- (a) A. Bertogg and A. Togni, Organometallics, 2006, 25, 622 CrossRef CAS; (b) T. S. Brunner, P. Benndorf, M. T. Gamer, N. Knofel, K. Gugau and P. W. Roesky, Organometallics, 2016, 35, 3474 CrossRef CAS.
- (a) M. Ostendorf, S. van der Neut, F. P. J. T. Rutjes and H. Hiemstra, Eur. J. Org. Chem., 2000, 105 CrossRef CAS; (b) A. Singh, R. A. Yoder, B. Shen and J. N. Johnston, J. Am. Chem. Soc., 2007, 129, 3466 CrossRef CAS PubMed; (c) J. Jin, X. Li, X. Luo, J. S. Fossey and W. Deng, J. Org. Chem., 2017, 82, 5424 CrossRef CAS PubMed.
- (a) X. Li, H. Jiang, E. W. Uffman, L. Guo, Y. Zhang, X. Yang and V. B. Birman, J. Org. Chem., 2012, 77, 1722 CrossRef CAS; (b) X. Yang, V. D. Bumbu, P. Liu, X. Li, H. Jiang, E. W. Uffman, L. Guo, W. Zhang, X. Jiang, K. N. Houk and V. B. Birman, J. Am. Chem. Soc., 2012, 134, 17605 CrossRef CAS PubMed.
- (a) J. Tanabe, D. Taura, H. Yamada, Y. Furusho and E. Yashima, Chem. Sci., 2013, 4, 2960 RSC; (b) W. Makiguchi, J. Tanabe, H. Yamada, H. Iida, D. Taura, N. Ousaka and E. Yashima, Nat. Commun., 2015, 6, 7236 CrossRef CAS PubMed.
- Y. Wang and K. Maruoka, Org. Process Res. Dev., 2007, 11, 628 CrossRef CAS.
- R. Holzwarth, R. Bartsch, Z. Cherkaoui and G. Solladié, Eur. J. Org. Chem., 2005, 3536 CrossRef CAS.
- (a) Y. Dobashi, K. Kobayashi and A. Dobashi, Tetrahedron Lett., 1998, 39, 93 CrossRef CAS; (b) Y. Dobashi, K. Kobayashi, N. Sato and A. Dobashi, Tetrahedron Lett., 1998, 39, 2985 CrossRef CAS.
- (a) J. M. Brunel, Chem. Rev., 2005, 105, 857 CrossRef CAS PubMed; (b) J. M. Brunel, Chem. Rev., 2007, 107, PR1 CrossRef CAS.
- (a) K. Yoshizawa, S. Toyota and F. Toda, Tetrahedron, 2004, 60, 7767 CrossRef CAS; (b) W. Ha and Z. Shan, Tetrahedron: Asymmetry, 2006, 17, 854 CrossRef CAS; (c) X. Hu, Z. Shan and Q. Chang, Tetrahedron: Asymmetry, 2012, 23, 1327 CrossRef CAS.
- (a) T. Hasegawa, K. Morino, Y. Tanaka, H. Katagiri, Y. Furusho and E. Yashima, Macromolecules, 2006, 39, 482 CrossRef CAS; (b) P. Benndorf, C. Preuß and P. W. Roesky, J. Organomet. Chem., 2011, 696, 1150 CrossRef CAS.
- (a) L. Sebo, B. Schweizer and F. Diederich, Helv. Chim. Acta, 2000, 83, 80 CrossRef CAS; (b) T. Kusukawa, K. Matsumoto, H. Nakamura, W. Iizuka, K. Toyama and S. Takeshita, Org. Biomol. Chem., 2013, 11, 3692 RSC; (c) T. Kusukawa, H. Aramoto, T. Umeda and Y. Kojima, Tetrahedron, 2019, 75, 1293 CrossRef CAS.
- (a) S. A. Boer, M. Morshedi, A. Tarzia, C. J. Doonan and N. G. White, Chem.–Eur. J., 2019, 25, 10006 CrossRef CAS; (b) G. Xing, I. Bassanetti, S. Bracco, M. Negroni, C. Bezuidenhout, T. Ben, P. Sozzani and A. Comotti, Chem. Sci., 2019, 10, 730 RSC; (c) W. Liang, F. Carraro, M. B. Solomon, S. G. Bell, H. Amenitsch, C. J. Sumby, N. G. White, P. Falcaro and C. J. Doonan, J. Am. Chem. Soc., 2019, 141, 14298 CrossRef CAS PubMed; (d) S. A. Boer, P. Wang, M. J. MacLachlan and N. G. White, Cryst. Growth Des., 2019, 19, 4829 CrossRef CAS.
- (a) R. Roger and D. G. Neilson, J. Chem. Soc., 1959, 688 RSC; (b) D. F. Ewing and D. G. Neilson, J. Chem. Soc., 1965, 770 RSC; (c) J. C. Barnes and T. J. R. Weakley, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 1998, 54, 1170 CrossRef.
- B. D. Judkins, D. G. Allen, T. A. Cook, B. Evans and T. E. Sardharwala, Synth. Commun., 1996, 26, 4351 CrossRef CAS.
- K. Kodama, N. Hayashi, M. Fujita and T. Hirose, RSC Adv., 2014, 4, 25609 RSC.
- (a) J. Aires-de-Sousa, S. Prabhakar, A. M. Lobo, A. M. Rosa, M. J. S. Gomes, M. C. Corvo, D. J. Williams and A. J. P. White, Tetrahedron: Asymmetry, 2001, 12, 3349 CrossRef; (b) F. Li, S. Li, P. Zhang, Z. Huang, W. Zhang, J. Gong and Z. Yang, Chem. Commun., 2016, 52, 12426 RSC; (c) K. Kodama, N. Hayashi, Y. Yoshida and T. Hirose, Tetrahedron, 2016, 72, 1387 CrossRef CAS.
- F. F. Fleming, Z. Zhang, Q. Wang and O. W. Steward, Angew. Chem., Int. Ed., 2004, 43, 1126 CrossRef CAS PubMed.
- S. Kanoh, N. Tamura, M. Motoi and H. Suda, Bull. Chem. Soc. Jpn., 1987, 60, 2307 CrossRef CAS.
- (a) M. R. Davis and D. A. Dougherty, Phys. Chem. Chem. Phys., 2015, 17, 29262 RSC; (b) A. F. Pozharskii, O. V. Dyablo, O. G. Pogosova, V. A. Ozeryanskii, A. Filarowski, K. M. Vasilikhina and N. A. Dzhangiryan, J. Org. Chem., 2020, 85, 12468 CrossRef PubMed.
- G. Zhang, Y. Liao, Z. Wang, H. Nohira and T. Hirose, Tetrahedron: Asymmetry, 2003, 14, 3297 CrossRef CAS.
- (a) F. A. J. Kerdesky, C. D. W. Brooks, K. I. Hulkower, J. B. Bouska and R. L. Bell, Bioorg. Med. Chem., 1997, 5, 393 CrossRef CAS PubMed; (b) T. Scheidt, H. Land, M. Anderson, Y. Chen, P. Berglund, D. Yi and W. D. Fessner, Adv. Synth. Catal., 2015, 357, 1721 CrossRef CAS; (c) J. Otevrel, D. Svestka and P. Bobal, Org. Biomol. Chem., 2019, 17, 5244 RSC.
- (a) T. V. RajanBabu and A. L. Casalnuovo, J. Am. Chem. Soc., 1992, 114, 6265 CrossRef CAS; (b) R. Y. Liu, M. Bae and S. L. Buchwald, J. Am. Chem. Soc., 2018, 140, 1627 CrossRef CAS PubMed; (c) J. Otevrel, D. Svestka and P. Bobal, RSC Adv., 2020, 10, 25029 RSC.
- S. Manner, M. Vahermo, M. E. Skogman, S. Krogerus, P. M. Vuorela, J. Y. Kauhaluoma, A. Fallarero and V. M. Moreira, Eur. J. Med. Chem., 2015, 102, 68 CrossRef CAS PubMed.
- K. M. Lambert, J. M. Bobbitt, S. A. Eldirany, L. E. Kissane, R. K. Sheridan, Z. D. Stempel, F. H. Sternberg and W. F. Bailey, Chem.–Eur. J., 2016, 22, 5156 CrossRef CAS PubMed.
- Q. Sun, Z. Dai, X. Meng and F. Xiao, Chem. Mater., 2017, 29, 5720 CrossRef CAS.
- (a) E. Alberico, S. Karandikar and S. Gladiali, ChemCatChem, 2010, 2, 1395 CrossRef CAS; (b) Y. Kubota, S. Shirakawa, T. Inoue and K. Maruoka, Tetrahedron Lett., 2012, 53, 3739 CrossRef CAS.
- N. Meisinger, L. Roiser, U. Monkowius, M. Himmelsbach, R. Robiette and M. Waser, Chem.–Eur. J., 2017, 23, 5137 CrossRef CAS PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 2015, 71, 3 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Fig. S1–S4 and Table S1, experimental detail, copies of 1H, 13C NMR, and IR spectra, HPLC chromatograms. CCDC 2057487–2057489. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d1ra03546k |
| This journal is © The Royal Society of Chemistry 2021 |