
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceCoupling selective laser sintering and supercritical CO2 foaming for 3D printed porous polyvinylidene fluoride with improved piezoelectric performance
Cheng Yang,
Ning Chen,
Xingang Liu,
Qi Wang and
Chuhong Zhang *
*
State Key Laboratory of Polymer Materials Engineering, Polymer Research Institute of Sichuan University, Chengdu, 610065, China
First published on 9th June 2021
Abstract
In this study, a facile strategy coupling selective laser sintering (SLS) and supercritical carbon dioxide (ScCO2) foaming technology is proposed to prepare a three-dimensional porous polyvinylidene fluoride (PVDF) with an improved piezoelectric output. The effects of foaming conditions including temperature and pressure on foam morphology, crystallization behavior and piezoelectric properties have been systematically studied. It is found that indeed the mechanical stretching foaming process greatly improves the produced content up to 76.2% of the β-phase in PVDF. The piezoelectric output of the PVDF foam with the highest open-circuit voltage could go up to 8 V (4.5 times printed parts), which could light up 4 LED lights and charge 4.7 μF 50 V capacitor to 3.51 V in 275 s. This study provides a feasible approach to 3D porous material fabrication for achieving high-performance piezoelectric materials and demonstrates the promising potential of energy harvesters and smart sensors.
Introduction
The environmental problems caused by the excessive use of fossil energy have made the research of piezoelectric devices based on mechanical energy increasingly popular.1,2 However, low-frequency, random and discrete mechanical energy in nature is still difficult to efficiently collect and use, and has become a major obstacle for the development and promotion of piezoelectric devices.Piezoelectric materials have been widely used in power-to-electricity conversion.3 Polyvinylidene fluoride (PVDF), as a typical piezoelectric material, has the highest piezoelectric coefficient among polymers (d33 ≈ −20 to −34 PC/N).4 There are three different crystalline phases in PVDF, namely α, β, and γ phase.5 Among them, the β-phase has the highest dipole moment due to its all-trans conformation (TTT), and exhibits best piezoelectric, pyroelectric and ferroelectric properties.6,7 Compared to the stable α crystal, under normal processing conditions, the β crystal is thermally metastable and can only be obtained under certain specific crystallization conditions.8 At present, three methods have been proposed for increasing the β-phase content: cold stretching, solid-phase extrusion, and grinding.5,9 Besides benefiting from the excellent piezoelectric properties, good flexibility, and machinability, PVDF is also used in the fields of sensing and energy storage.10–12 However, the existing research and application of PVDF in these fields are limited to two-dimensional (2D) thin films. In contrast, compared to piezoelectric film materials, three-dimensional (3D) devices have the advantages of larger output power and stable energy storage.13
In addition, reports have shown that porous structures have a strong strain response and regular charge distribution after poling, facilitating the conversion of force to electricity.14–18 The methods for preparing porous PVDF generally involve the hard template method, sol–gel method, and solution method.14,15,19 Seung et al.20 prepared PVDF nano cell arrays through a template-assisted method, which were applied in nanogenerators. The porous PVDF nanogenerator delivered high rectified power density (0.17 MW cm−3), and its piezoelectric potential and current increased 5.2 and 6 times, respectively, in comparison to the 2D PVDF film. Zhang et al.21 used the sol–gel method to prepare mesoporous PVDF at room temperature. This method can be summarized as first filling the PVDF network with the polydimethylsiloxane (PDMS) elastomer and then removing the PDMS to create pores. However, these methods are complicated and time-consuming. Therefore, it is urgently needed to develop a simple and effective method for preparing 3D PVDF parts with a controllable pore structure.
On the one hand, selective laser sintering (SLS), as a branch of 3D printing, use powder as a raw material to design parts with complex structures. In addition, SLS does not require support during the molding process, eliminating complicated post-processing procedures and improving the production efficiency.22,23 In recent years, SLS has also been applied to the piezoelectric field.24 Apart from the capability of fabricating 3D structures, holes formed upon sintering powder particles also contribute to an improved piezoelectric output.
On the other hand, the supercritical fluid (SCF) foaming technology has been used in the preparation of porous PVDF piezoelectric device lately.25 Supercritical CO2 (ScCO2) can interact with the C–F bond of PVDF, facilitating the dissolution of CO2 as a blowing agent. The dissolved CO2 can plasticize the amorphous phase and reduce the melting temperature of the crystalline phase.26,27 Moreover, the pore growth along with foaming can be considered as a slow stretching process, which can effectively increase the β-phase content in the matrix.28,29 However, the SCF technology used to prepare microporous PVDF by far is limited to few 2D films.30,31
In this study, we propose a facile strategy for coupling SLS printing and SCF foaming technology to prepare 3D porous PVDF. ScCO2 was used as a physical blowing agent and plasticizer to foam printed parts in the mold for regulating the porous structure of PVDF. In addition, compared with the traditional pressing plate and coating method, SLS could design not only complex macrostructures but also special internal pores in printed parts, which is beneficial for the diffusion of CO2 and the subsequent foaming efficiency. The effects of foaming conditions on the crystal form, crystallinity, the melting behavior of PVDF, and the piezoelectric properties of final foamed parts were also studied.
Experimental
Materials
The PVDF particle (FR906), with a density of 1.79 g cm−3 and a melting point (Tm) of 172 °C, was purchased from Shanghai San'ai fu New Materials Technology Co. Ltd., China.CO2 with a purity of 99.5% was provided by Taiyuan Natural Gas Company, Chengdu, China.
Preparation of PVDF powder
The PVDF powder was dried in an oven at 80 °C and then mixed with 0.5 wt% nanosilica in a high-speed mixer for 5 min at a speed of 20![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 rpm to improve its fluidity. The PVDF powder was sieved with a 100-mesh sieve to obtain a powder with applicable particle size, so that it could be used for laser sintering.
000 rpm to improve its fluidity. The PVDF powder was sieved with a 100-mesh sieve to obtain a powder with applicable particle size, so that it could be used for laser sintering.
Preparation of porous PVDF parts
The PVDF powder prepared above was spread using a roller to make the surface smooth. After that, the sealing door of the cavity was closed, and was filled with nitrogen to make the oxygen content less than 5.5 vol%, which is the space used by the selective laser sintering printer for powder preheating, sintering, molding, and cooling. Then, the powder bed was preheated to 155 °C, and the laser was turned on to selectively sinter the powder according to the set route. During the sintering process, the laser power was 40 W, the scanning distance was 0.1 mm, the scanning speed was 9600 mm s−1, and the thickness of the powder layer was 0.1 mm. Finally, the parts were taken out after cooling to room temperature. The PVDF-sintered part was put into a steel mold, and then the mould was put into an autoclave. After saturating with carbon dioxide at 45 °C and 7 MPa for 30 min, the autoclave was quickly heated to the corresponding temperature within 30 min, and then depressurized and cooled to obtain foamed parts. The specific preparation process is shown in Fig. 1.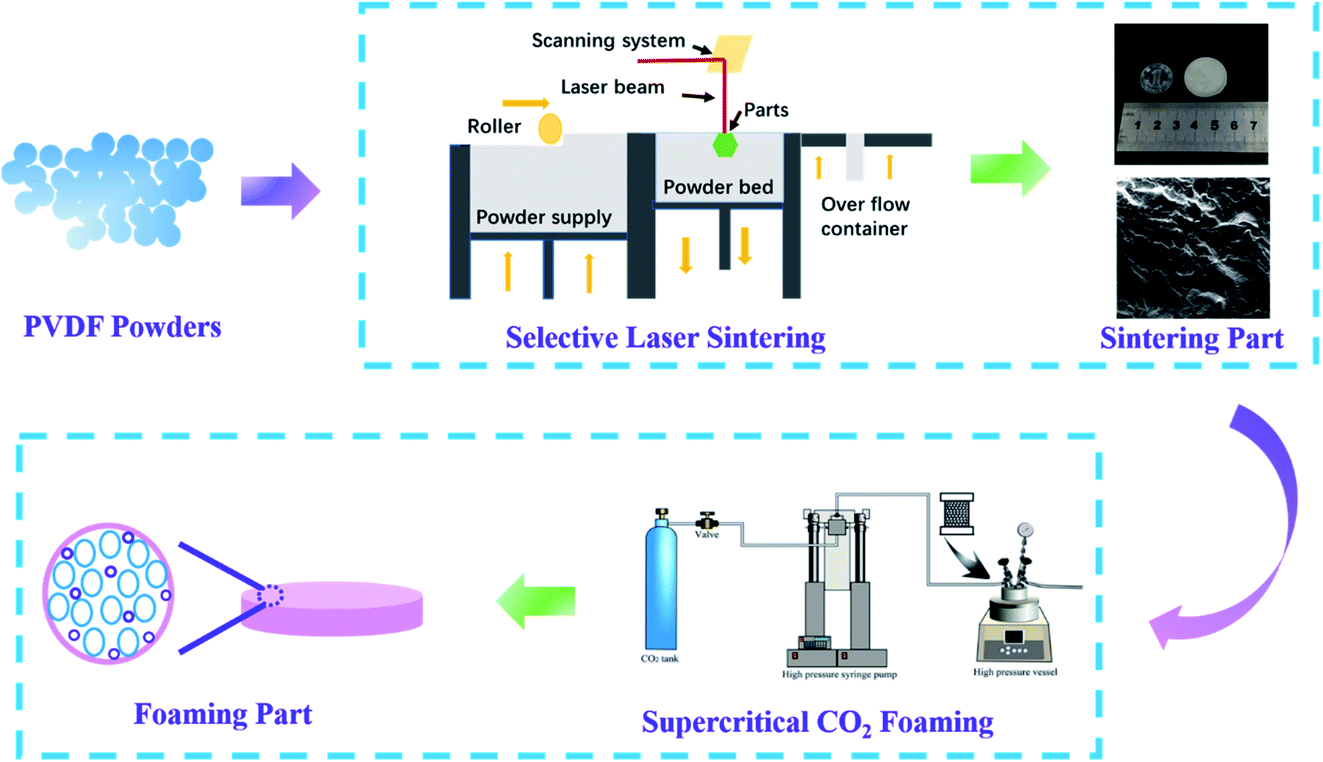 | ||
| Fig. 1 Preparation process of PVDF laser-sintered foam parts. | ||
Characterization
The particle size and the size distribution of each powder were measured by a particle size measuring device (Model S3500, Microtrac, USA). The powder was dispersed in an ethanol solvent, and ultrasonicated for about 5 min to obtain a uniformly dispersed suspension.The morphology of the powder, the morphology of the cross-section after SLS sintering, and the morphology of the cross-sectional foam were analyzed by SEM (INSPECTF, FEI, Japan). The sample was immersed in liquid nitrogen for 8 min and then quenched. For the test, the gold spray treatment was performed in advance, and the acceleration voltage was 20 kV. The Nano-Measurer software was used to calculate the average cell diameter and cell density. The formula used for calculating cell density is as follows:32
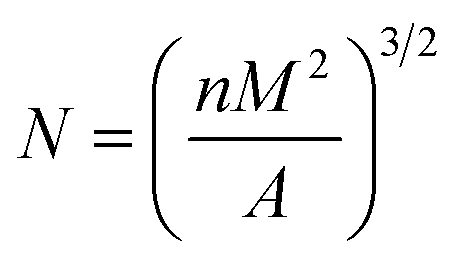 | (1) |
The different crystal phases of PVDF and the content of each phase, as well as the crystal phase conversion during laser sintering and supercritical foaming, were analyzed by FT-IR spectroscopy (Nicolet 6700 Fourier transform infrared Spectrometer from Thermo Scientific, USA). Samples below 2 mm were tested by the ATR method. For the test, the scanning range was 4000–400 cm−1, the scanning was performed 32 times, and the resolution was 4 cm−1. According to the Lambert–Beer law, the relative β-phase content was estimated by the formula (2):33
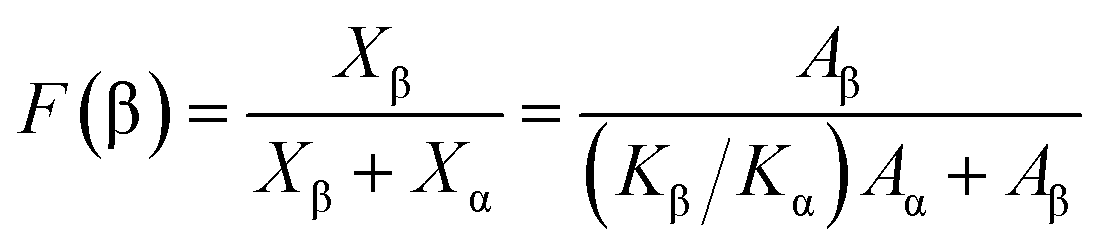 | (2) |
The crystallinity and melting point changes during the sample preparation were measured by the DSC technique (Q20 type from American TA Company). The test process was to load approximately 5–10 mg of the sample into a crucible, increase the temperature from 40 °C to 230 °C at a rate of 10 °C min−1, and then cool down to 40 °C. The crystallinity, Xc, was calculated by eqn (3):34
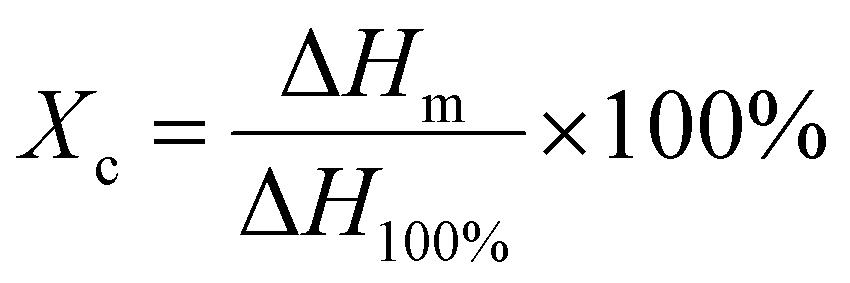 | (3) |
The piezoelectric performance test (NTI AG HS01-37×166) was performed to evenly coat conductive silver glue on both sides of the test wafer. Dried wafers were polarized for 30 min by applying a high voltage of 4 kV in an 80 °C silicone oil bath using a corona poling instrument. After poling, the double-sided conductive aluminum foil was stuck on the front and back of the work-piece connected to the iron plate of the experimental device, and its position was opposite to the impact head. The electric signal generated by a linear motor hitting the sheet at 5 m s−2 was amplified and the output was connected to the computer; finally, the open-circuit voltage and short circuit current of the part were obtained.
Results and discussion
Morphology of the PVDF powder and parts sintered by selective laser
It could be seen from Fig. 2(a) that the shape of PVDF particles was random but close to spherical, and the surface was rough. The measurement results of particle size and distribution are shown in Fig. 2(b). The corresponding values of D50 and D90 were 77.74 and 128.9 μm, indicating that the as-prepared powders were suitable for SLS sintering.35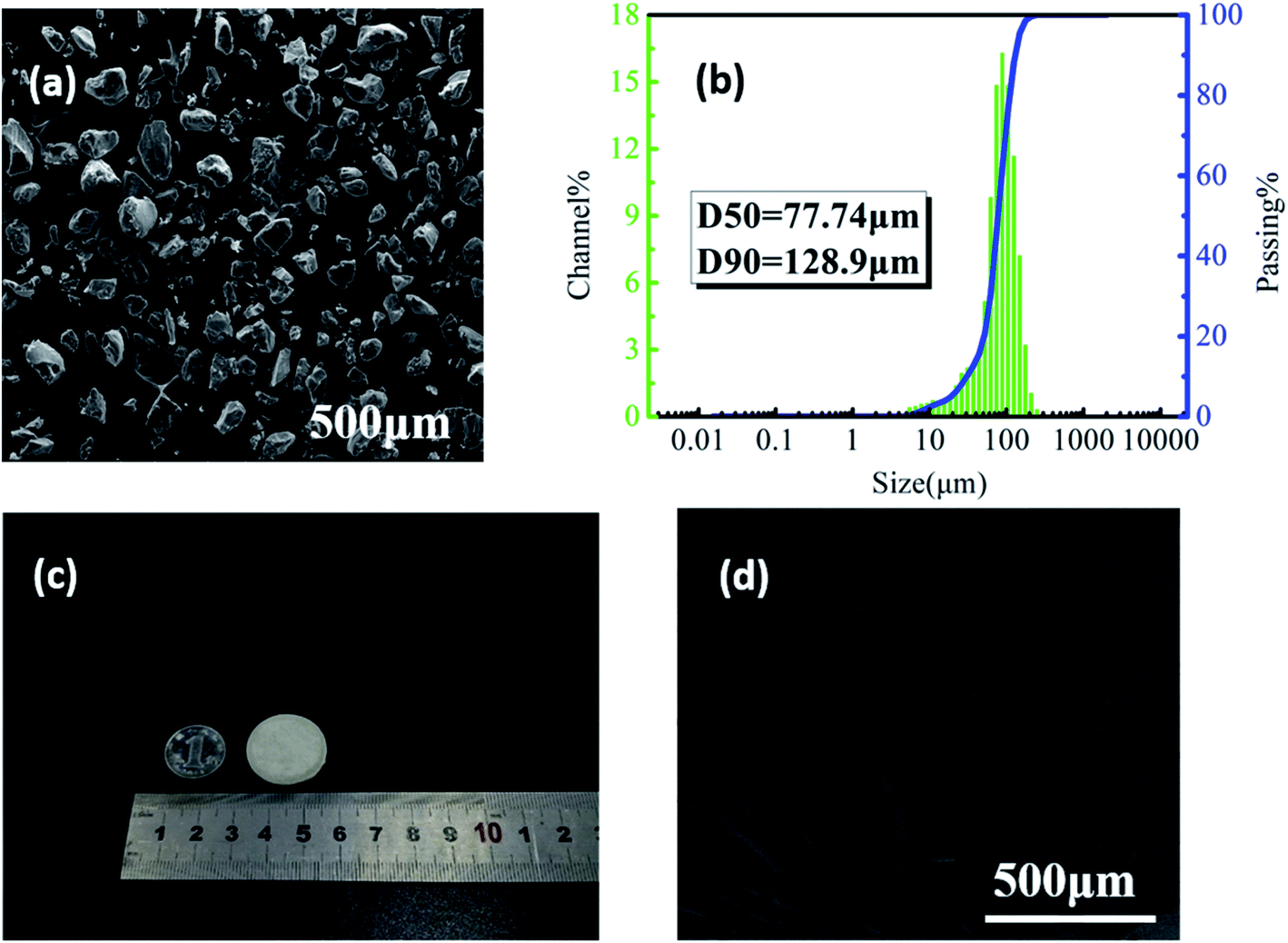 | ||
| Fig. 2 (a) The SEM image of PVDF powder and (b) the particle size distribution; (c) the digital photo and (d) the microstructure of PVDF laser-sintered parts. | ||
Fig. 2(c) shows that the PVDF part printed by SLS with a diameter of 25 mm and a thickness of 2 mm has good dimensional accuracy with no sintering margin or warpage. Some small holes on the surface of the part (Fig. 2(d)) mainly caused by the sintering and incomplete bonding of powder particles can be clearly observed. Studies have shown that under the same external force, the higher the porosity of the piezoelectric material, the more sensitive it is to mechanical changes and a higher piezoelectric output performance.36 In addition, these small holes facilitate the entry of ScCO2 into the sintered part during the subsequent foaming process, making the foaming of the part easier.
Effect of the foaming temperature on the properties of sintered PVDF parts
SEM was used to investigate the foam morphology of PVDF-sintered parts foamed at the temperature range of 160–170 °C, for 2 h at 14 MPa pressure. As the temperature increased, the viscosity of the polymer matrix decreased, and the cell expansion was obvious.37 Therefore, as shown in Fig. 3(a)–(c), the thickness of the cell wall decreased from 40.41 μm (161 °C) to 8.19 μm (165 °C). However, cells collapsed, and the shape was poor at 167 °C, as shown in Fig. 3(d). This is mainly due to the relatively low melt strength of the PVDF matrix during the cell growth stage. In addition, the excessive cell growth led to serious cell aggregation and collapse, forming cells of various sizes and irregular shapes. | ||
| Fig. 3 SEM images of cell structures of the PVDF foam at different foaming temperatures (temperature: (a) 161 °C, (b) 163 °C, (c) 165 °C, and (d) 167 °C). | ||
In order to further explore the influence of the foaming temperature on the change of the crystal phase content of PVDF parts, corresponding FT-IR images were obtained and are presented in Fig. 4(a) and (b). The peak at 766 cm−1 is due to the stretching vibration of –CF2 of the α crystal phase, while the vibrational peaks corresponding to the β-phase appear at 840 cm−1. It can be seen from Fig. 4(a) that the peak intensity at 1279 cm−1 and 840 cm−1 gradually enhances as the temperature increases. This change can be further seen by the enlarged image (Fig. 4(b)). The β-phase content of the part increased from 44.2% to 69.5%, as illustrated in Fig. 4(d), indicating that ScCO2 foaming is beneficial in the transformation from the α to β crystal phase. Mechanical stretching has made PVDF molecules extend along the expansion direction of the cell. In addition, the extrusion and friction between the part and the mold wall during the confined space foaming process caused greater stress on PVDF, which further increased the β-phase content.26
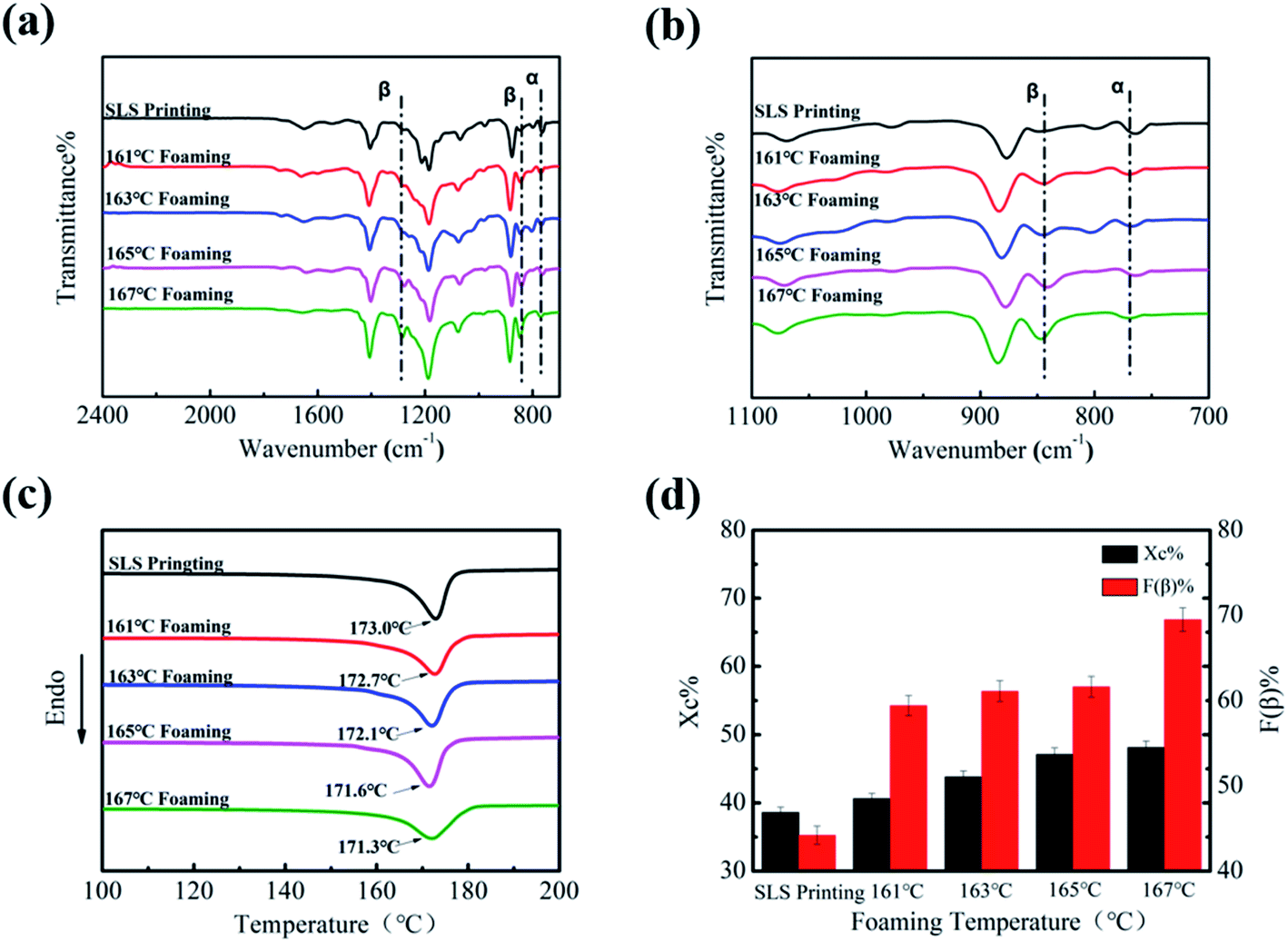 | ||
| Fig. 4 FT-IR of PVDF parts at different foaming temperatures ((a) wavenumber: 2500–1600 cm−1 and (b) wavenumber 1100–700 cm−1), (c) DSC diagram of PVDF parts at different foaming temperatures, and (d) crystallinity and β crystal content of PVDF parts at different foaming temperatures. | ||
DSC was used to express the effect of the foaming temperature on the melting and crystallization behavior of PVDF. As shown in Fig. 4(c), it can be clearly seen that Tm decreased slightly (from 173.0 °C to 171.3 °C) with an increase in the foaming temperature. The main reason for the decrease in Tm is that the plasticizing effect of CO2 weakens the interaction between PVDF molecular chains.38 In addition, the diffusion ability of CO2 increases with the increase in temperature, which promotes the movement of PVDF molecules, so that molecular chains will enter into the crystal lattice more easily.39 Consequently, its crystallinity increased from 38.6% to 48.1% (Fig. 4(d)).
The open-circuit voltage (Voc) and short-circuit current (Isc) under an impact acceleration of 5 m s−2 were tested. As shown in Fig. 5, the Voc value shows an increasing trend first and then decreases as the foaming temperature increases, and Voc reaches a maximum of 4.5 V at 165 °C. There are two reasons for this phenomenon: first of all, there is friction and stretching inside PVDF during the foaming process, which increases its β-phase content; on the other hand, the 3D porous structure with excellent compression performance can improve the electromechanical coupling ability.40,41
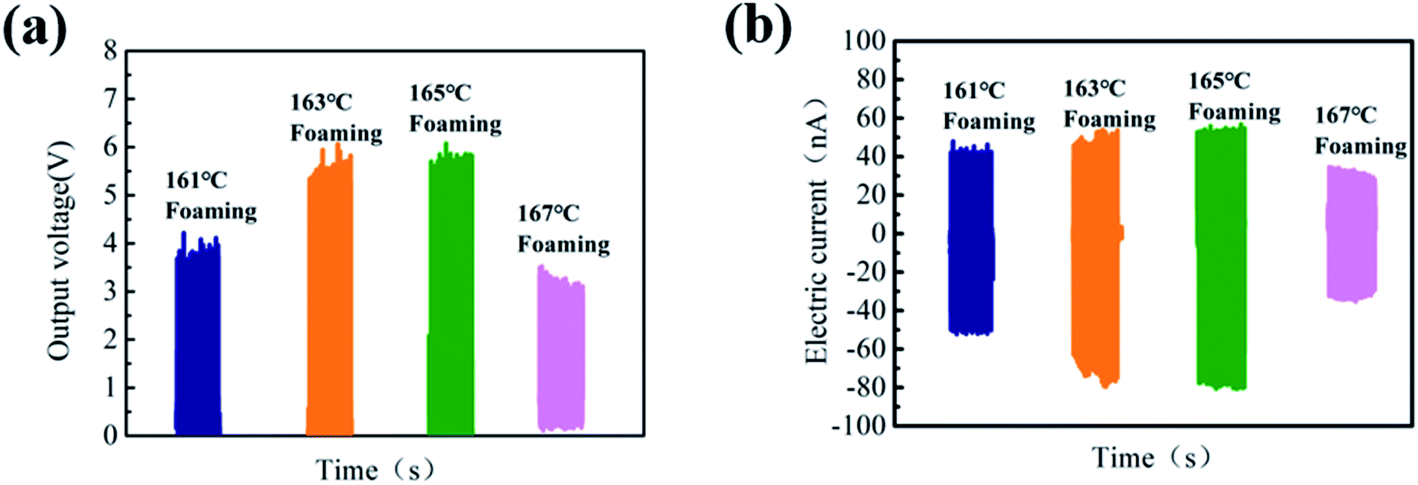 | ||
| Fig. 5 (a) Output voltage of PVDF parts with different foaming temperatures after poling and (b) short-circuit current diagram of PVDF foam parts with different foaming temperatures after poling. | ||
Effect of foaming pressure on the properties of sintered PVDF parts
Herein, the influence of foaming pressure on the performance of PVDF-sintered parts was studied at the optimal foaming temperature of 165 °C. With the increase in pressure from 10 to 16 MPa, SEM images (Fig. 6(a)–(d)) show that the average cell size and cell density changed greatly. It can be clearly seen from Fig. 6(e) that as the pressure increases, the cell diameter gradually decreases from 102 μm at 10 MPa to 65.2 μm at 16 MPa, and the cell density increases from 7.1 × 105 to 2.2 × 106 cells per cm3. This is mainly because at low pressure, the nucleation of the PVDF crystal region is mainly heterogeneous nucleation due to the relatively small plasticizing effect of CO2 and molecular chain activity, resulting in low cell density and large cell size. On the other hand, pressure changes are closely related to the solubility of CO2 in PVDF. The increase in CO2 in the PVDF matrix leads to an increase in nucleation sites, which increases the cell density and reduces the cell size.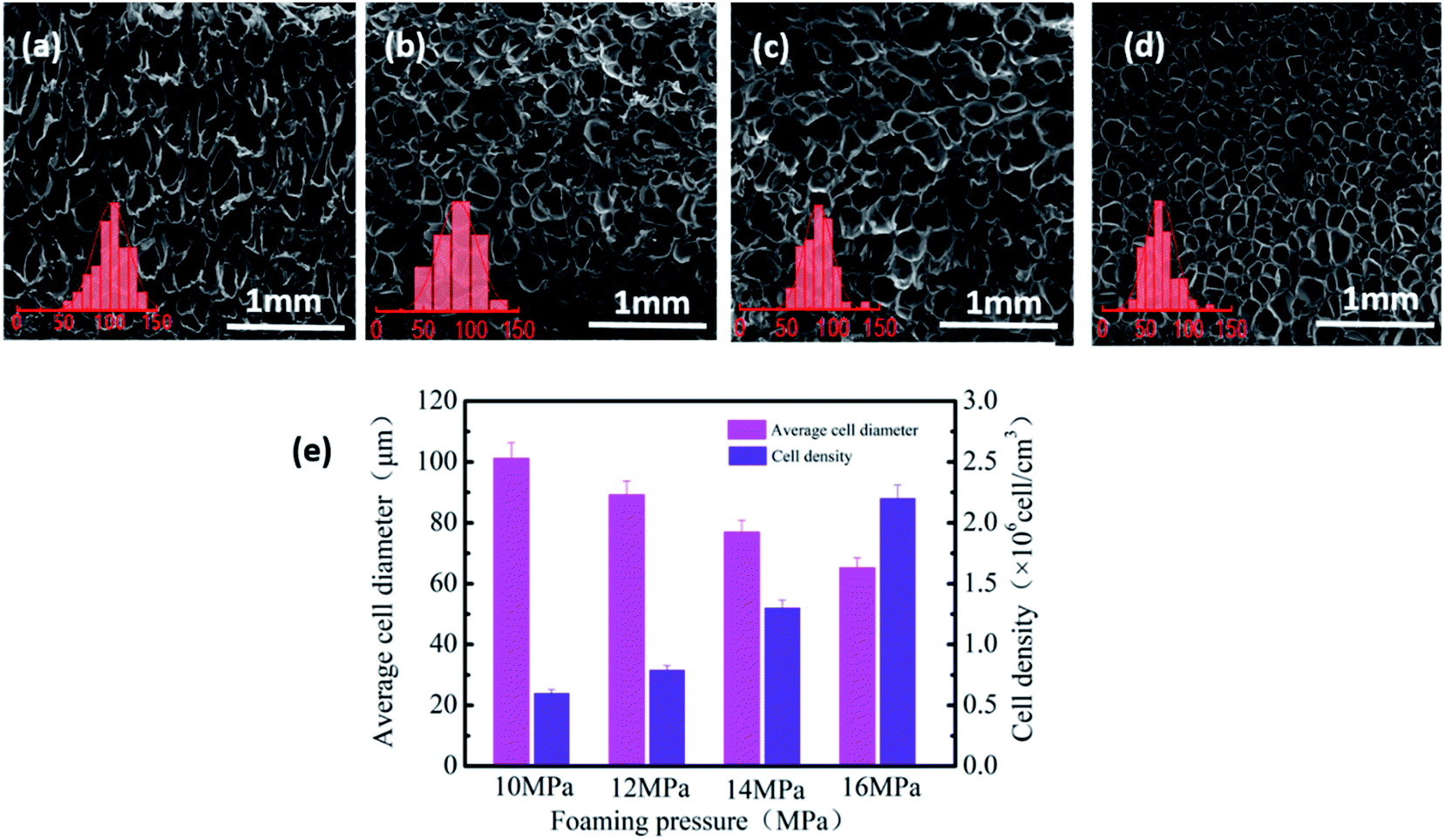 | ||
| Fig. 6 SEM images and the cell distribution of PVDF foam under different foaming pressures ((a) 10 MPa, (b) 12 MPa, (c) 14 MPa, and (d) 16 MPa) and (e) the average cell diameter and cell density of PVDF foaming parts. | ||
Fig. 7(a) and (b) shows the FT-IR spectra of PVDF foaming parts at different pressures. As observed in Fig. 7(a) and (b), the peak intensity at 766 cm−1 gradually weakens, while peaks at 1279 and 840 cm−1 gradually increase. This is due to the fact that the expansion and stretching action of the gas after entering the work piece can be enhanced followed by a pressure rise, forming more β phases. Particularly when the pressure drop is large, the PVDF molecular chain has a greater expansion force. The PVDF foam prepared under the conditions of 165 °C and 16 MPa has an uniform-oriented cell morphology. It is worth noting that the high strain caused by the cell orientation growth promotes the TTT conformation of PVDF, which led to an increase in the β-phase content. Therefore, after foaming at 16 MPa, the β-phase content of the PVDF device is as high as 76.2%.
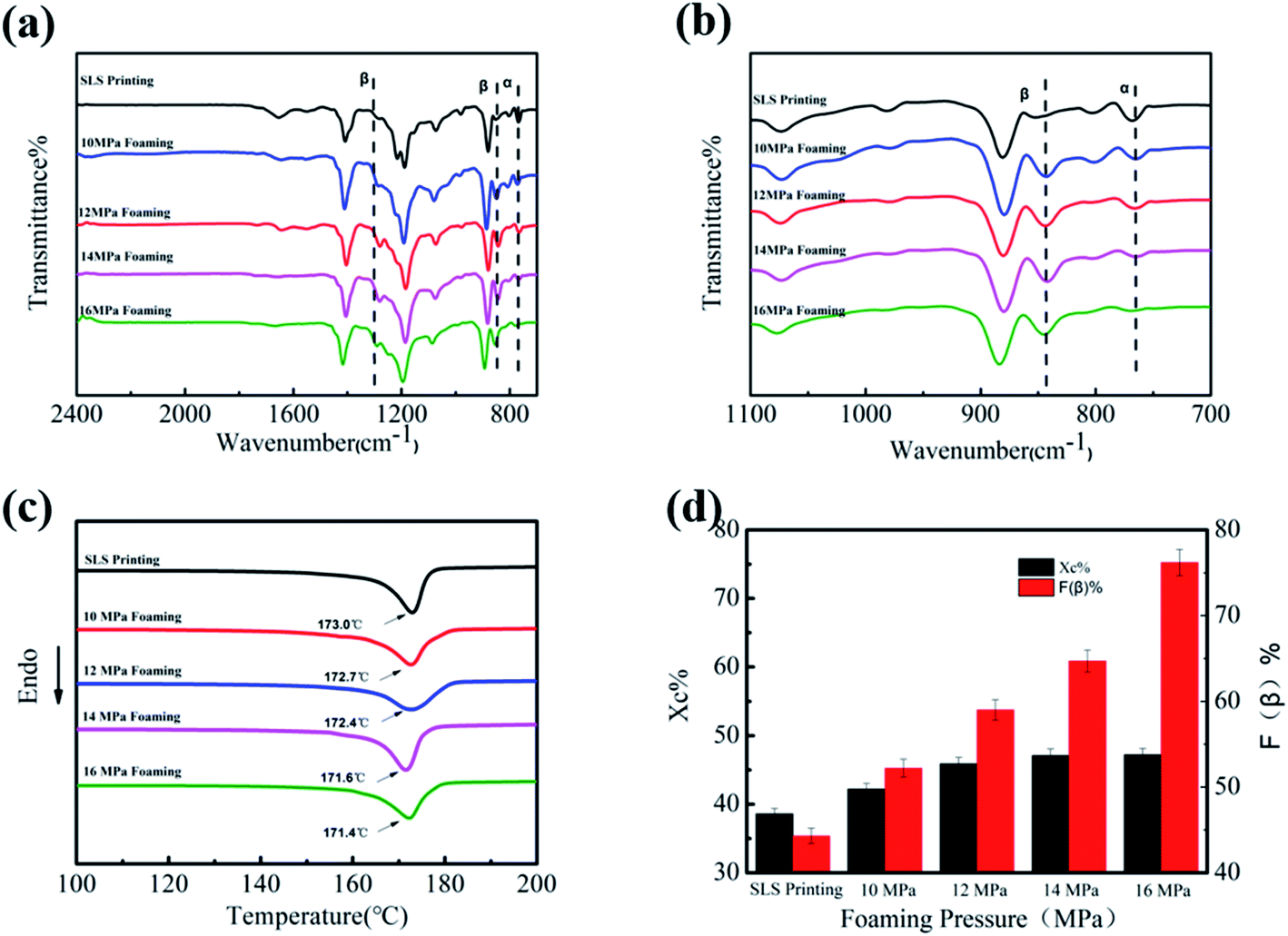 | ||
| Fig. 7 FT-IR of PVDF foaming parts at different foaming pressures ((a) wavenumber: 2500–1600 cm−1 and (b) wavenumber 1100–700 cm−1), (c) the DSC diagram of PVDF foaming parts at different foaming pressures, and (d) crystallinity and β crystal content of PVDF foaming parts at different pressures. | ||
DSC curves show the influence of foaming pressure on the melting and crystallization behavior of products. As shown in Fig. 7(c), the increase in the foaming pressure leads to a slight decrease in Tm (from 173.0 °C to 171.4 °C), and the crystallinity increases from 38.6% to 47.2%. The mobility of the PVDF chain around the crystal nucleus is improved by CO2 due to the plasticizing effect. These results further confirmed that the regularity and arrangement of molecular chains are perfect with the plasticization and subsequent extension of ScCO2.
The results of Voc and Isc under the impact acceleration of 5 m s−2 are shown in Fig. 8. It is likely to cause greater mechanical deformation due to smaller cell size and larger cell density and is more conducive to the collection of electric charges, which greatly improves the piezoelectric performance. Thus, the highest output voltage can go up to 8 V when the foaming pressure is 16 MPa.
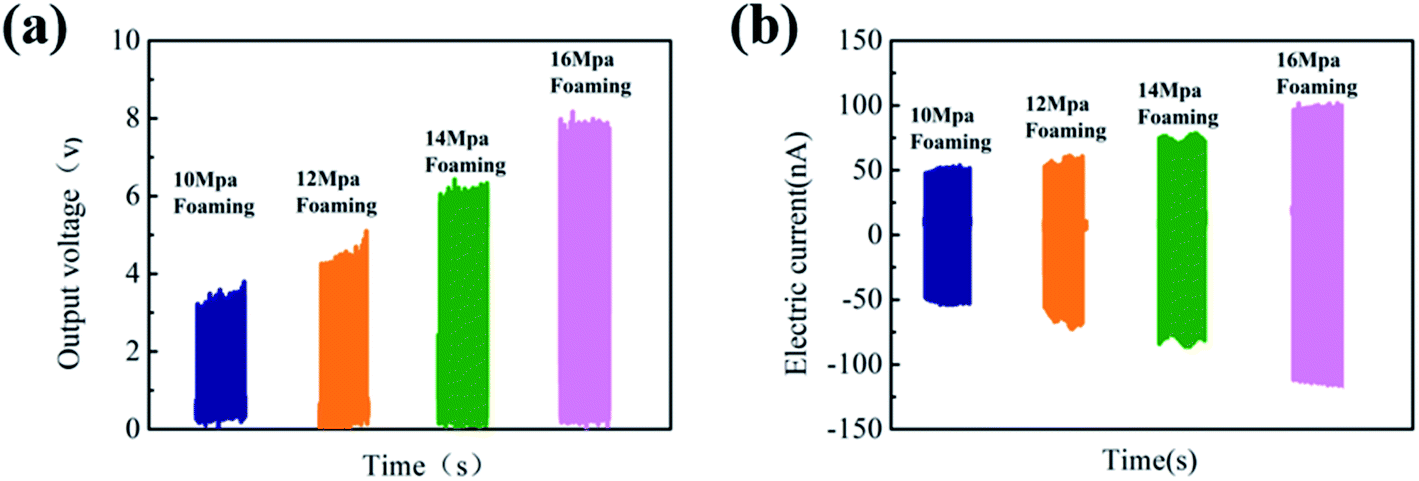 | ||
| Fig. 8 (a) Output voltage of PVDF parts with different foaming pressures after poling and (b) short-circuit current diagram of PVDF foam parts with different foaming pressures after poling. | ||
The energy harvesting and charging capabilities of 3D porous parts after foaming under optimal conditions were also studied. Fig. 9(a) presents the regularly arranged cell structure, which increased the β-phase content and mechanical deformation, leading to an enhanced electromechanical coupling ability of the printed PVDF part. The Voc of the 3D porous part could reach about 8 V, as shown in Fig. 9(b), and can light up 4 commercially available LEDs without converting to direct current. Furthermore, the ability of experimental products to supply power to energy storage devices (batteries, capacitors, etc.) was tested. As shown in Fig. 9(c), the 4.7 μF 50 V capacitor could be successfully charged to 3.51 V in 275 s by designing a bridge rectifier to convert alternating current to direct current.
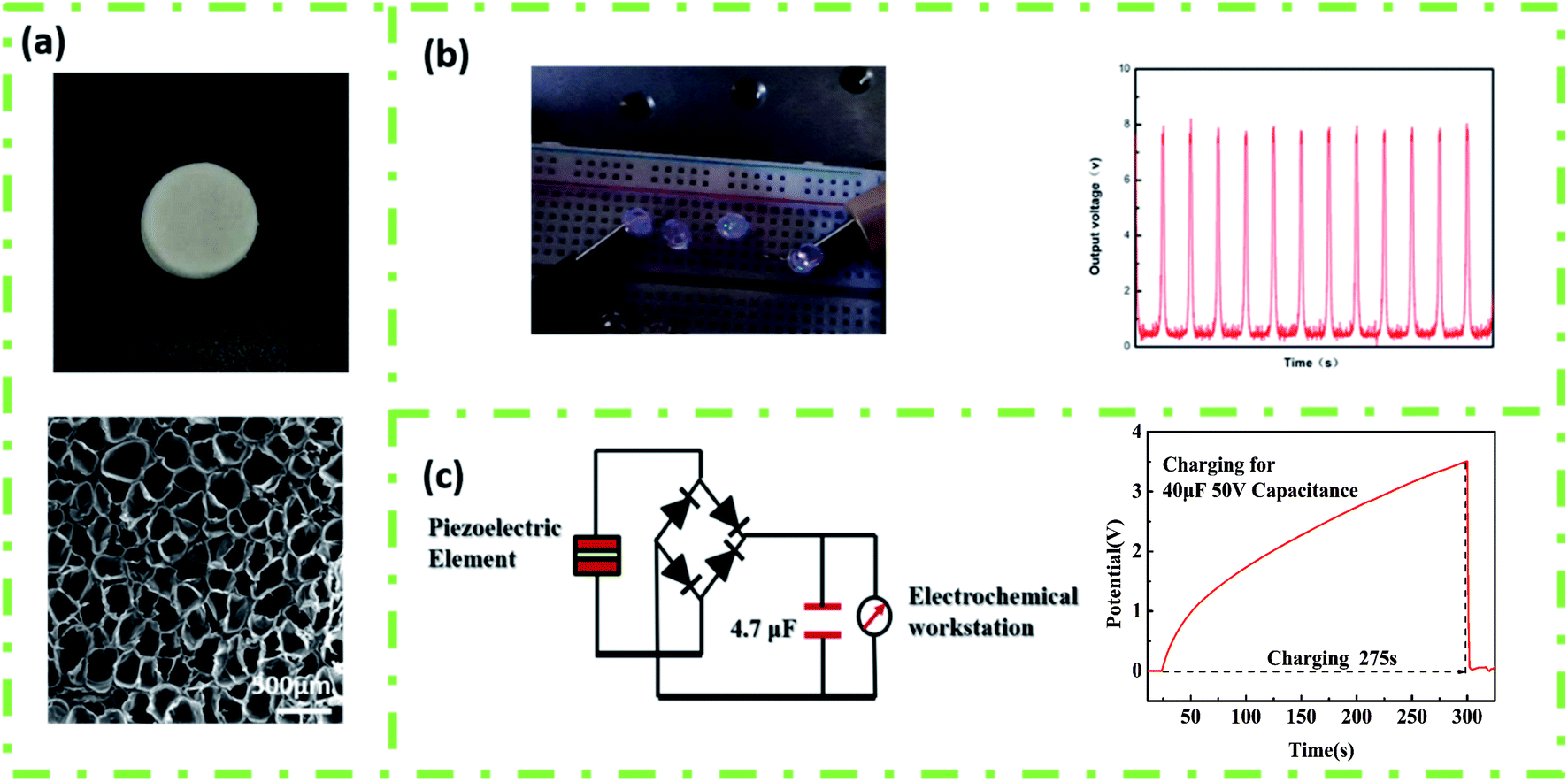 | ||
| Fig. 9 (a) The macroscopic and internal hole structure of the piezoelectric element, (b) linear motor (acceleration 5 m s−2, lighting up 4 LEDs), and (c) 4.7 μF 50 V capacitor, charging for 275 s. | ||
Conclusion
Selective laser sintering and supercritical carbon dioxide foaming were coupled to fabricate three-dimensional porous piezoelectric devices. In particular, the performance was further optimized by exploring the foaming temperature and pressure. Piezoelectric parts obtained by the above method can not only obtain a higher β-crystallite content but also reduce the compression modulus of PVDF. Furthermore, the open-circuit voltage of the obtained 3D PVDF device can be as high as 8 V, which could light up 4 LED lights and charge 4.7 μF 50 V capacitor to 3.51 V in 275 s. 3D printing combined with the supercritical carbon dioxide foaming technology proposed in this article has strategic prospects and valuable potential in the preparation of three-dimensional porous piezoelectric devices for improving the piezoelectric energy storage performance.Conflicts of interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.Acknowledgements
This work is supported by National Natural Science Foundation of China (51933007 and 51720105012).References
- D. Mandal, S. Yoon and K. J. Kim, Macromol. Rapid Commun., 2011, 32, 831–837 CrossRef CAS PubMed.
- H. Wu, Y. A. Huang, F. Xu, Y. Q. Duan and Z. P. Yin, Adv. Mater., 2016, 28, 9881–9919 CrossRef CAS PubMed.
- X. T. Yuan, X. Y. Gao, J. K. Yang, X. Y. Shen, Z. M. Li, S. J. You, Z. H. Wang and S. X. Dong, Energy Environ. Sci., 2020, 13, 152–161 RSC.
- K. Kim, W. Zhu, X. Qu, C. Aaronson, W. R. McCall, S. C. Chen and D. J. Sirbuly, ACS Nano, 2014, 8, 9799–9806 CrossRef CAS PubMed.
- G. Zhu, Z. Zeng, L. Zhang and X. Yan, Comput. Mater. Sci., 2008, 44, 224–229 CrossRef CAS.
- J. Jiang, S. J. Tu, R. F. Fu, J. J. Li, F. Hu, B. Yan, Y. C. Gu and S. Chen, ACS Appl. Mater. Interfaces, 2020, 12, 33989–33998 CrossRef CAS PubMed.
- G. H. Tabhane and S. M. Giripunje, Polym. Test., 2020, 88, 106564 CrossRef CAS.
- Y. Li, X. Wen, M. Nie and Q. Wang, ACS Omega, 2017, 2, 7230–7238 CrossRef CAS PubMed.
- J. I. Scheinbeim, B. A. Newman and K. D. Pae, Bull. Am. Phys. Soc., 1979, 24, 379 Search PubMed.
- B. Alamusi, J. Xue, L. Wu, N. Hu, J. Qiu, C. Chang, S. Atobe, H. Fukunaga, T. Watanabe, Y. Liu, H. Ning, J. Li, Y. Li and Y. Zhao, Nanoscale, 2012, 4, 7250–7255 RSC.
- J. Lv, J. Chen and P. S. Lee, SusMat, 2021, 2, 1–18 Search PubMed.
- S. Biswas, B. Dutta and S. Bhattacharya, Eur. Polym. J., 2017, 86, 1–16 CrossRef CAS.
- C. Zhang, Y. Li, W. Kang, X. Liu and Q. Wang, SusMat, 2021, 1, 127–147 CrossRef.
- Y. Zhang, C. R. Bowen and S. Deville, Soft Matter, 2019, 15, 825–832 RSC.
- H. Q. Liang, Q. Y. Wu, L. S. Wan, X. J. Huang and Z. K. Xu, J. Membr. Sci., 2014, 465, 56–67 CrossRef CAS.
- C. A. Petroff, T. F. Bina and G. R. Hutchison, ACS Appl. Energy Mater., 2019, 2, 6484–6489 CrossRef CAS.
- W. Q. Qian, K. Zhao, D. Zhang, C. R. Bowen, Y. H. Wang and Y. Yang, ACS Appl. Mater. Interfaces, 2019, 11, 27862–27869 CrossRef CAS PubMed.
- Y. C. Sun, D. Terakita, A. C. Tseng and H. E. Naguib, Smart Mater. Struct., 2015, 24, 085034 CrossRef.
- S. Bonyadi and T. S. Chung, J. Membr. Sci., 2009, 331, 66–74 CrossRef CAS.
- S. Cha, S. M. Kim, H. Kim, J. Ku, J. I. Sohn, Y. J. Park, B. G. Song, M. H. Jung, E. K. Lee, B. L. Choi, J. J. Park, Z. L. Wang, J. M. Kim and K. Kim, Nano Lett., 2011, 11, 5142–5147 CrossRef CAS PubMed.
- Z. Zhang, C. Yao, Y. Yu, Z. Hong, M. Zhi and X. Wang, Adv. Funct. Mater., 2016, 26, 6760–6765 CrossRef CAS PubMed.
- F. Qi, N. Chen and Q. Wang, Mater. Des., 2018, 143, 72–80 CrossRef CAS.
- Z. Li, Z. Wang, X. Gan, D. Fu, G. Fei and H. Xia, Macromol. Mater. Eng., 2017, 302, 1700211 CrossRef.
- W. R. McCall, K. Kim, C. Heath, G. La Pierre and D. J. Sirbuly, ACS Appl. Mater. Interfaces, 2014, 6, 19504–19509 CrossRef CAS PubMed.
- Y. Jin, N. Chen, Y. Li and Q. Wang, RSC Adv., 2020, 10, 20405–20413 RSC.
- Y. Xiang and H. Lin, Polym. Sci., Ser. A, 2018, 60, 342–349 CrossRef CAS.
- D. Feng, L. Li and Q. Wang, RSC Adv., 2019, 9, 4072–4081 RSC.
- S. Doroudiani, C. B. Park and M. T. Kortschot, Polym. Eng. Sci., 1996, 36, 2645–2662 CrossRef CAS.
- J. E. Lee and S. N. Leung, CrystEngComm, 2018, 20, 4080–4089 RSC.
- Y. Xiang, L. Xue, J. Shen, H. Lin and F. Liu, J. Appl. Polym. Sci., 2014, 131, 51273211 CrossRef.
- H. Zhang, G. Zhang, Q. Gao, M. Tang, Z. Ma, J. Qin, M. Wang and J.-K. Kim, Chem. Eng. J., 2020, 379, 122304 CrossRef.
- S. Siripurapu, Y. J. Gay, J. R. Royer, J. M. DeSimone, R. J. Spontak and S. A. Khan, Polymer, 2002, 43, 5511–5520 CrossRef CAS.
- S. H. Kim, J. W. Ha, S. G. Lee, E. H. Sohn, I. J. Park, H. S. Kang and G. R. Yi, Langmuir, 2019, 35, 8816–8822 CrossRef CAS PubMed.
- N. Soin, D. Boyer, K. Prashanthi, S. Sharma, A. A. Narasimulu, J. Luo, T. H. Shah, E. Siores and T. Thundat, Chem. Commun., 2015, 51, 8257–8260 RSC.
- Z. Li, Y. Jia and S. Bai, RSC Adv., 2018, 8, 2880–2886 RSC.
- H. Chung and S. Das, Mater. Sci. Eng., A, 2006, 437, 226–234 CrossRef.
- S. Cha, S. M. Kim, H. Kim, J. Ku, J. I. Sohn, Y. J. Park, B. G. Song, M. H. Jung, E. K. Lee, B. L. Choi, J. J. Park, Z. L. Wang, J. M. Kim and K. Kim, Nano Lett., 2011, 11, 5142–5147 CrossRef CAS PubMed.
- M. Tang and T.-C. Wang, J. Taiwan Inst. Chem. Eng., 2017, 73, 146–153 CrossRef CAS.
- D. Xu, H. Zhang, L. Pu and L. Li, Compos. Sci. Technol., 2020, 192, 108108 CrossRef CAS.
- J. E. Lee, Y. Guo, R. E. Lee and S. N. Leung, RSC Adv., 2017, 7, 48712–48722 RSC.
- J. W. Zhong, Q. Z. Zhong, X. N. Zang, N. Wu, W. B. Li, Y. Chu and L. W. Lin, Nano Energy, 2017, 37, 268–274 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2021 |