
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDesign, synthesis and biological evaluation of 1H-pyrrolo[2,3-b]pyridine derivatives as potent fibroblast growth factor receptor inhibitors†
Xingping Su‡
a,
Zhihao Liu‡a,
Lin Yuea,
Xiuli Wua,
Wei Weia,
Hanyun Quea,
Tinghong Ye a,
Yi Luo*b and
Yiwen Zhang*a
a,
Yi Luo*b and
Yiwen Zhang*a
aSichuan University-University of Oxford Huaxi Joint Centre for Gastrointestinal Cancer, State Key Laboratory of Biotherapy, West China Hospital, Sichuan University, Chengdu, Sichuan 610041, China. E-mail: yiwenzhang@scu.edu.cn
bDepartment of Orthopedics, West China Hospital of Sichuan University, Wai Nan Guo Xue Xiang 37#, 610041, Chengdu, Sichuan, China. E-mail: orthop_luoyi@163.com
First published on 9th June 2021
Abstract
Abnormal activation of FGFR signaling pathway plays an essential role in various types of tumors. Therefore, targeting FGFRs represents an attractive strategy for cancer therapy. Herein, we report a series of 1H-pyrrolo[2,3-b]pyridine derivatives with potent activities against FGFR1, 2, and 3. Among them, compound 4h exhibited potent FGFR inhibitory activity (FGFR1–4 IC50 values of 7, 9, 25 and 712 nM, respectively). In vitro, 4h inhibited breast cancer 4T1 cell proliferation and induced its apoptosis. In addition, 4h also significantly inhibited the migration and invasion of 4T1 cells. Furthermore, 4h with low molecular weight would be an appealing lead compound which was beneficial to the subsequent optimization. In general, this research has been developing a class of 1H-pyrrolo[2,3-b]pyridine derivatives targeting FGFR with development prospects.
1. Introduction
The fibroblast growth factor receptor (FGFR) family has four distinct isoforms (FGFR1–4) found across various tissue types and expressed to different extents under varying conditions.1,2 The FGFR1–4 isoforms mainly consist of highly conserved extracellular ligand-binding domains, a single transmembrane segment, and a cytoplasmic tyrosine kinase domain.3–5 Physiologically, the FGF–FGFR axis is involved in signal transduction pathways that regulate organ development, cell proliferation and migration, angiogenesis, and other processes.6,7 Upon binding to fibroblast growth factors, the receptor undergoes dimerization and autophosphorylation of tyrosine residues in the cytoplasmic tail, resulting in activation of downstream signaling including RAS–MEK–ERK, PLCγ, and PI3K–Akt.8,9Abnormal activation of FGFR signaling pathway due to amplification, fusion or missense mutations in the exon of FGFR family members is associated with the progression and development of several cancers such as breast cancer, lung cancer, prostate cancer, bladder cancer and liver cancer.10–14 Moreover, activation of FGFR-dependent signaling pathways can facilitate cancer initiation, progression, and resistance to cancer therapy. Hence, the FGFR signaling pathway is an important and proven target for cancer therapeutics. Currently, FGFR inhibitors are currently under clinical investigation for the treatment of various cancers such as AZD4547,15 JNJ-42756493 (Erdafitinib),16 CH5183184,17 BGJ-398,18 LY2874455,19 INCB054828 (Pemigatinib)20 and so on, shown in Fig. 1. Recently, FDA has announced approval of Erdafitinib21 and Pemigatinib.22
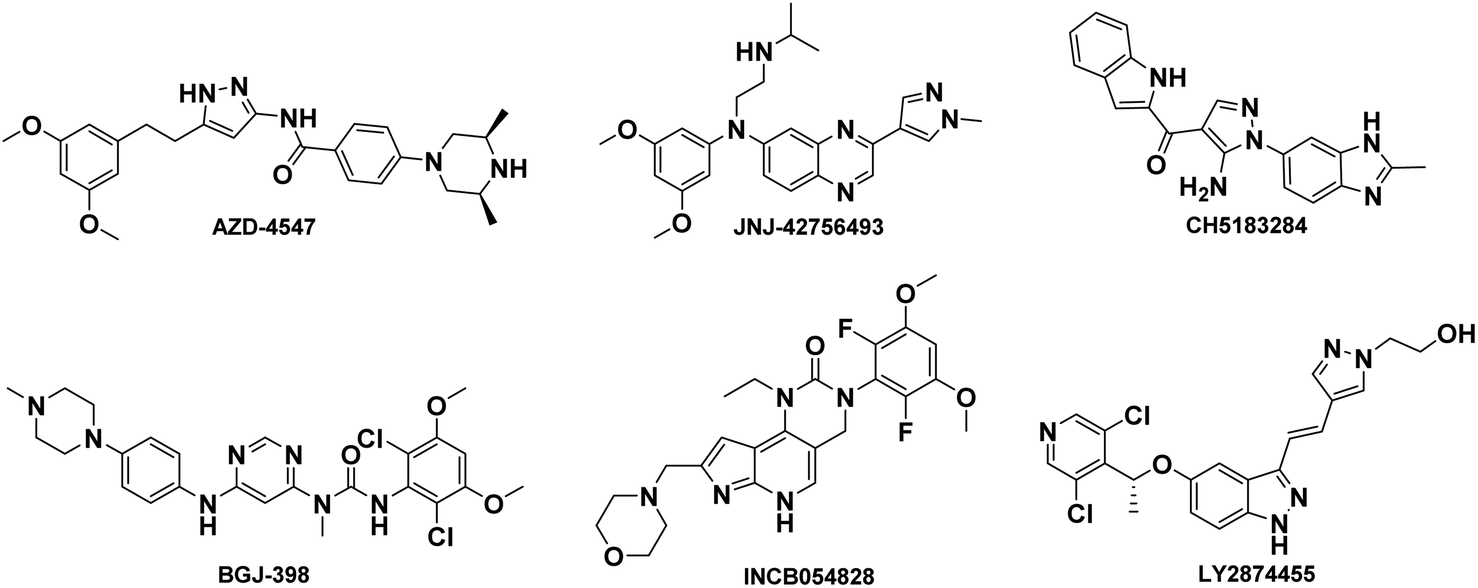 | ||
| Fig. 1 Chemical structure of representative FGFR inhibitors. | ||
During the development of our FGFR inhibitor program, we wish to explore a novel and concise scaffold of selective FGFR inhibitors. Based on previous literature studies, we found that compound 1 has potent FGFR1 inhibitory activity with IC50 value of 1.9 μM (ref. 23) (Fig. 2A). Compared with the existing FGFR inhibitors, compound 1 contains a novel core scaffold 1H-pyrrolo[2,3-b]pyridine with low molecular mass and high ligand efficiency. Jin Q. et al. developed a series of 1H-pyrrolo[2,3-b]pyridine derivatives as FGFR4 inhibitors with potent antiproliferative activity against Hep3B cells.24 Furthermore, 1H-pyrrolo[2,3-b]pyridine also as a new scaffold used in other targets research, such as human neutrophil elastase (HNE).25 Moreover, recent studies have shown that 1H-pyrrolo[2,3-b]pyridine analogues have inhibitory activity against different cancer cell lines.26
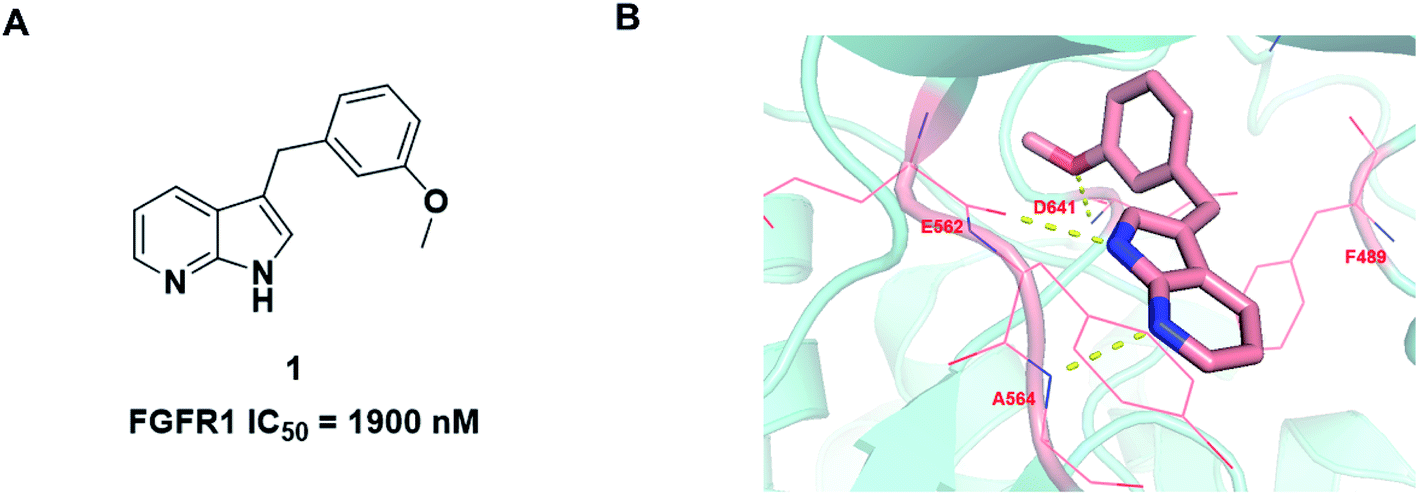 | ||
| Fig. 2 (A) Chemical structure of compound 1. (B) The binding mode of 1 with FGFR1. | ||
We firstly investigated the co-crystallization structure (PDB code 3C4F) to understand interactions between 1 and FGFR1 kinase domain (Fig. 2B). As a hinge binder, 1H-pyrrolo[2,3-b]pyridine ring of 1 could form two hydrogen bonds with the backbone carbonyl of E562 and NH of A564 in the hinge region. The methoxyphenyl motif could potentially occupy hydrophobic pocket in the ATP site and form van der Waals interactions with amino acid residues of the hydrophobic pocket, and its methoxy group could also form a strong hydrogen bond with the NH of D641.
Based on above analyses, to quest for a novel and concise chemotype of FGFR inhibitors, we kept 1H-pyrrolo[2,3-b]pyridine motif as hinge binder and focused on utilizing structure-based design strategy to design 1H-pyrrolo[2,3-b]pyridine derivatives as potent FGFR inhibitors. Given that the 5-position of 1H-pyrrolo[2,3-b]pyridine is close to G485, a group which could provide hydrogen bond acceptor with suitable size was introduced into the 5-position of 1H-pyrrolo[2,3-b]pyridine ring to form a hydrogen bond with G485 to improve the activity. Meanwhile, the m-methoxyphenyl fragment was altered to various larger substituents to explore the possible interactions within the hydrophobic pocket. In this study, we report the synthesis and biology evaluation of 1H-pyrrolo[2,3-b]pyridine derivatives as potent FGFR inhibitors.
The synthesis of all compounds is shown in Scheme 1. The starting material 5-(trifluoromethyl)-1H-pyrrolo[2,3-b]pyridine was reacted with R-substituted aldehyde at 50 °C to obtain the compounds 3a–3k in 45–60% yield. Subsequently, 3a–3k in the presence of acetonitrile under triethylsilane and trifluoroacetic acid catalysis at reflux to undergo a reduction reaction to furnish 4a–4l in 46–80%.
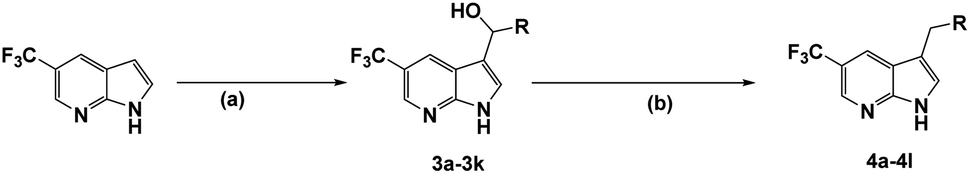 | ||
| Scheme 1 aSynthesis of target compounds. aReagents and conditions: (a) R–CHO, potassium hydroxide, methanol, 50 °C, 5 h, 45–60%; (b) triethylsilane, trifluoroacetic acid, acetonitrile, reflux, 2 h, 46–80%. | ||
All prepared compounds were screened for their inhibitory activity against FGFR1 at the concentration of 0.1 and 1 μM as well as antiproliferative activities against 4T1 (mouse breast cancer cells), MDA-MB-231 and MCF-7 cancer cells at 10 μM. As summarized in Tables 1 and 2, compound 1 with low molecular mass demonstrated FGFR1 potency with an IC50 value of 1900 nM. Based on our analysis above, a suitably sized trifluoromethyl was introduced at the 5-position of the 1H-pyrrolo[2,3-b]pyridine ring (4a) in 1 to form hydrogen bond with G485. Remarkably, the activity against FGFR1 of 4a increased nearly 20-fold compared with compound 1, implying that our strategy was feasible. Then the m-methoxy moiety of 4a was modified to improve FGFR activities. The methoxy group at the 3-position of phenyl ring in 4a replaced with chlorine (4b) decreased FGFR1 potency and cellular activity. Poor activities of 4b may be due to the weak electronegativity of Cl atom in 4b which cannot form strong hydrogen bond with D641. Introduction of 3-trifluoromethyl group at the 3-position of phenyl ring reduced the activities of 4c, which may be result from the inability of trifluoromethyl group to form a hydrogen bond with the NH of D641. The introduction of suitable group at the 3-position of phenyl ring to occupy the hydrophobic pocket improved the FGFR1 activity (4a–e), indicating a suitable size of the group on this position was favorable. Incorporation of a trifluoromethyl (4f) or benzyloxy (4g) at the 4-position of phenyl ring declined both FGFR1 and cellular activities. Then the influence of substituents on 3- and 5-positions of the phenyl ring 4a was investigated. Excitingly, introduction of methoxy groups at the 3- and 5-positions significantly improved against FGFR1 potency (4h) (95% & 0.1 μM) and cellular activity (4T1) (57% & 10 μM). However, multiple substitutions at other positions (4i, 4j, 4k, 4l) exhibited declined FGFR1 and cellular activities. In addition, the presence of a hydroxyl also decreased FGFR1 potency and cellular activity (4a vs. 3a), (4h vs. 3h) which implicated that the hydroxyl group may be too close to its neighboring amino acid F489, resulting in decreased activities.
|
||||||
|---|---|---|---|---|---|---|
| Compound | R | FGFR1 inhibition rate (%) | Inhibition rate 10 μM (%) | |||
| 1 μM | 0.1 μM | 4T1 | MDA-MB-231 | MCF-7 | ||
| a Cell results are given in concentrations of 10 μM after a continuous exposure of 72 h and show means ± SD of two-independent experiments. NS: not significant; NT: not tested. | ||||||
| 1 | — | 55 | 7 | 11 ± 0.02 | 8 ± 2.14 | 16 ± 0.62 |
| 3a | 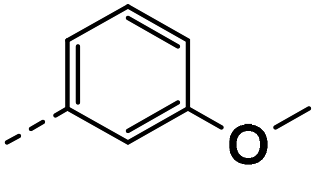 |
78 | 35 | 16 ± 0.01 | 6 ± 1.22 | 33 ± 0.50 |
| 3b | 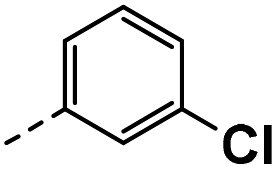 |
25 | 1 | NS | 10 ± 2.88 | 14 ± 3.18 |
| 3c | 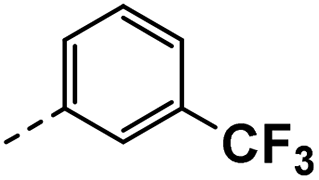 |
22 | 22 | NS | 23 ± 0.63 | 33 ± 0.71 |
| 3d | 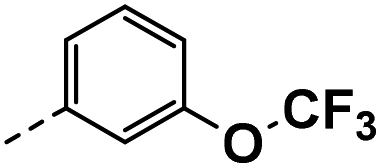 |
22 | −5 | NS | 12 ± 1.50 | 21 ± 0.82 |
| 3e | 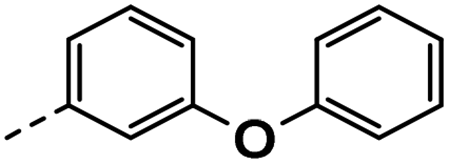 |
34 | 15 | NS | NS | 26 ± 0.88 |
| 3f | 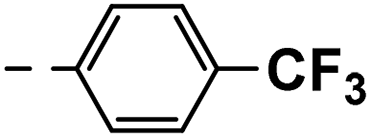 |
25 | 14 | NS | 18 ± 0.70 | 33 ± 1.10 |
| 3g | 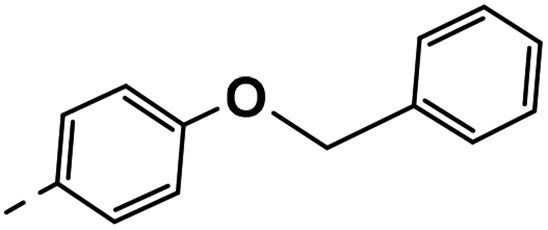 |
25 | −1 | NS | 28 ± 0.23 | 25 ± 1.05 |
| 3h | 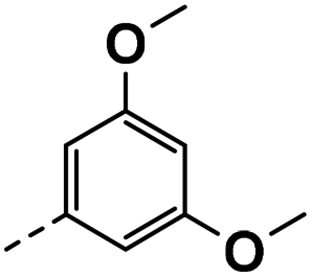 |
89 | 61 | 23 ± 0.01 | 13 ± 3.80 | 33 ± 0.11 |
| 3i | 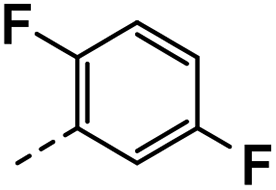 |
61 | 19 | NS | 21 ± 1.87 | 32 ± 1.24 |
| 3j | 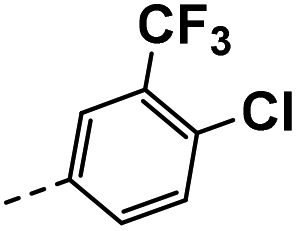 |
17 | 3 | NS | 15 ± 0.97 | 30 ± 1.17 |
| 3k | 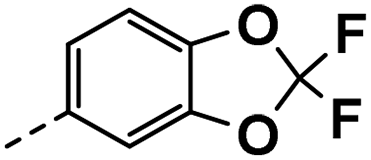 |
11 | 3 | NT | NT | NT |
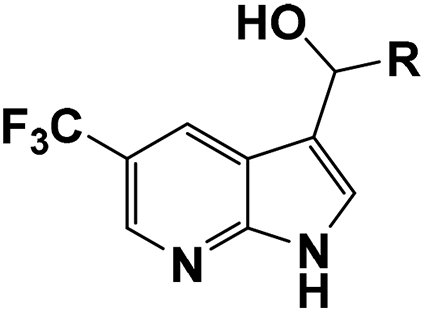
|
||||||
|---|---|---|---|---|---|---|
| Compound | R | FGFR1 inhibition rate (%) | Inhibition rate 10 μM (%) | |||
| 1 μM | 0.1 μM | 4T1 | MDA-MB-231 | MCF-7 | ||
| a Cell results are given in concentrations of 10 μM after a continuous exposure of 72 h and show means ± SD of two-independent experiments. NS: not significant. | ||||||
| 4a |  |
91 | 53 | 18 ± 0.01 | 15 ± 1.16 | 32 ± 0.54 |
| 4b | 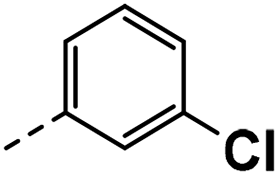 |
56 | 2 | NS | 6.1 ± 0.16 | 29 ± 0.83 |
| 4c | 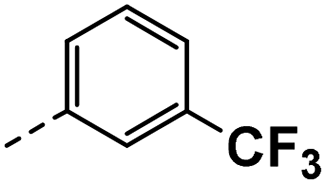 |
19 | 10 | 26 ± 0.04 | 29 ± 0.24 | 51 ± 0.17 |
| 4d | 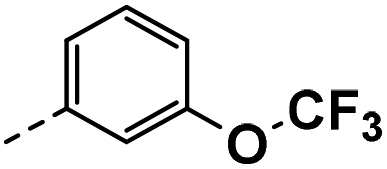 |
21 | 15 | NS | 25 ± 0.36 | 27 ± 3.33 |
| 4e | 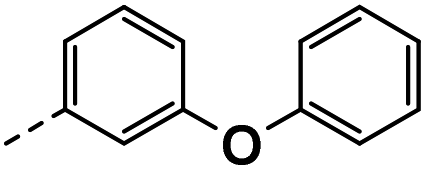 |
64 | 21 | NS | 16 ± 0.54 | 26 ± 1.65 |
| 4f | 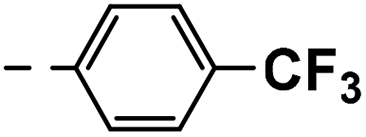 |
21 | 24 | 17 ± 0.02 | 20 ± 2.60 | 44 ± 0.73 |
| 4g | 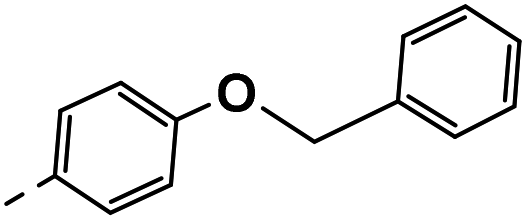 |
29 | −4 | NS | 25 ± 2.44 | 32 ± 0.24 |
| 4h | 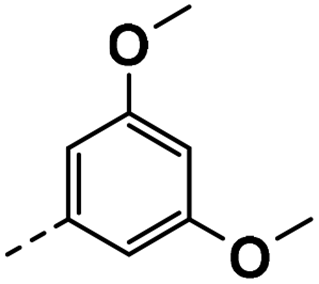 |
91 | 95 | 57 ± 0.01 | 22 ± 0.08 | 33 ± 0.94 |
| 4i | 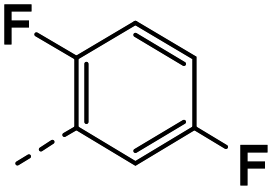 |
78 | 29 | NS | 14 ± 3.18 | 41 ± 0.71 |
| 4j | 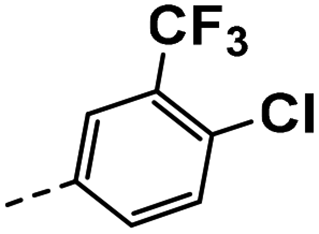 |
16 | 7 | 13 ± 0.02 | 19 ± 0.38 | 46 ± 1.09 |
| 4k | 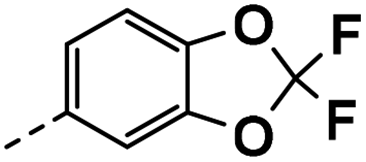 |
40 | 19 | 22 ± 0.03 | 20 ± 0.03 | 46 ± 0.49 |
| 4l | 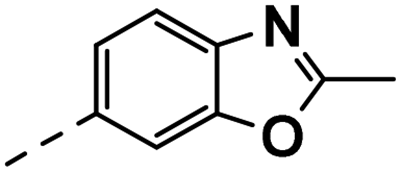 |
76 | 27 | 23 ± 0.01 | 9 ± 0.62 | 22 ± 1.66 |
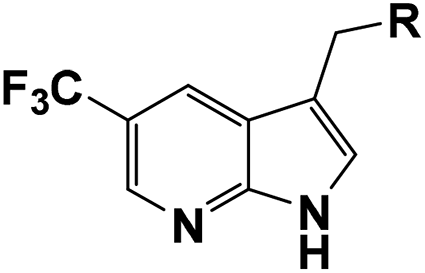
Compound 3h, 4a, 4h and 4l were selected to investigate their inhibitory activities against other FGFR isoforms, including FGFR1, FGFR2, FGFR3 and FGFR4 (Table 3). All these compounds exhibited inhibitory activities to FGFR1–3 in vitro. Among them, 4h showed the best activities that effectively inhibited the activities of FGFR1–4 with IC50 values of 7, 9, 25 and 712 nM, respectively. Based on above results, 4h was chosen for further biological evaluation.
| Compound | IC50 (nM) | |||
|---|---|---|---|---|
| FGFR1 | FGFR2 | FGFR3 | FGFR4 | |
| 3h | 54 | 66 | 320 | >3000 |
| 4a | 83 | 93 | 421 | >3000 |
| 4h | 7 | 9 | 25 | 712 |
| 4l | 266 | 259 | 634 | >3000 |
| AZD-4547 | 0.8 | 1 | 2 | 47 |
To investigate the binding modes of our inhibitors, compound 4h was docked with FGFR1 protein, as showed in Fig. 3. The nucleus 1H-pyrrolo[2,3-b]pyridine could form two hydrogen bonds with the backbone carbonyl of E562 and NH of A564 in the hinge region. In addition, the essential π–π interaction was observed between 3,5-dimethoxyphenyl of 4h and F489. Then, the 3,5-dimethoxyphenyl group could more fully and appropriately occupy the hydrophobic pocket and maintain the formation of hydrogen bonds with D641. As expected, trifluoromethyl substitution at the 5-position of 1H-pyrrolo[2,3-b]pyridine could form a hydrogen bond with G485, which may be a crucial factor in improving the activity of the compound. In addition, the ligand efficiency of 4h (LE = 0.44) has been significantly improved compared with compound 1 (LE = 0.13).
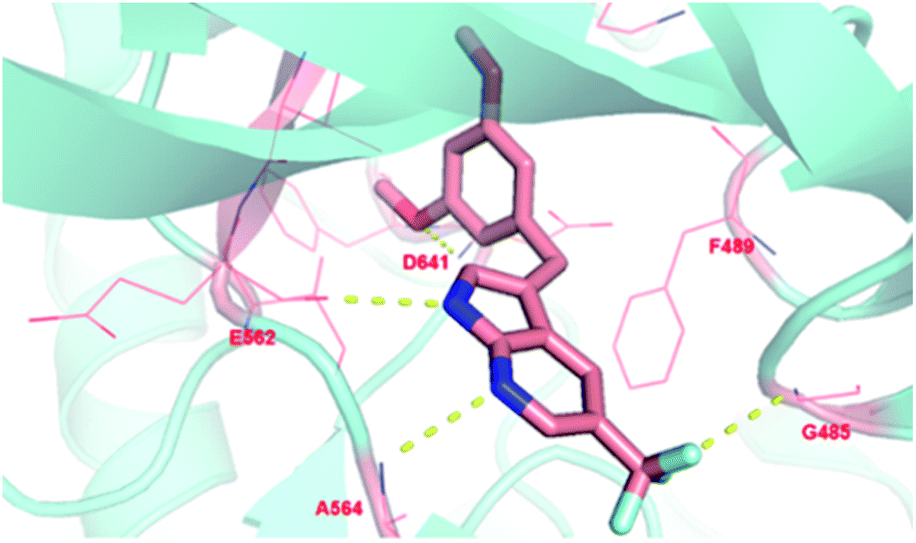 | ||
| Fig. 3 Proposed binding mode of compound 4h to FGFR1 kinase domain. | ||
To further investigate whether 4h could inhibit the proliferation of 4T1 cells, we conducted the colony formation assays. As showed in Fig. 4, the colony formation of 4T1 cells was reduced after treatment with 4h. Moreover, 4h could inhibit the population-dependence growth of 4T1 cells. These results suggest that 4h could inhibit 4T1 cells viability in a concentration-dependent manner.
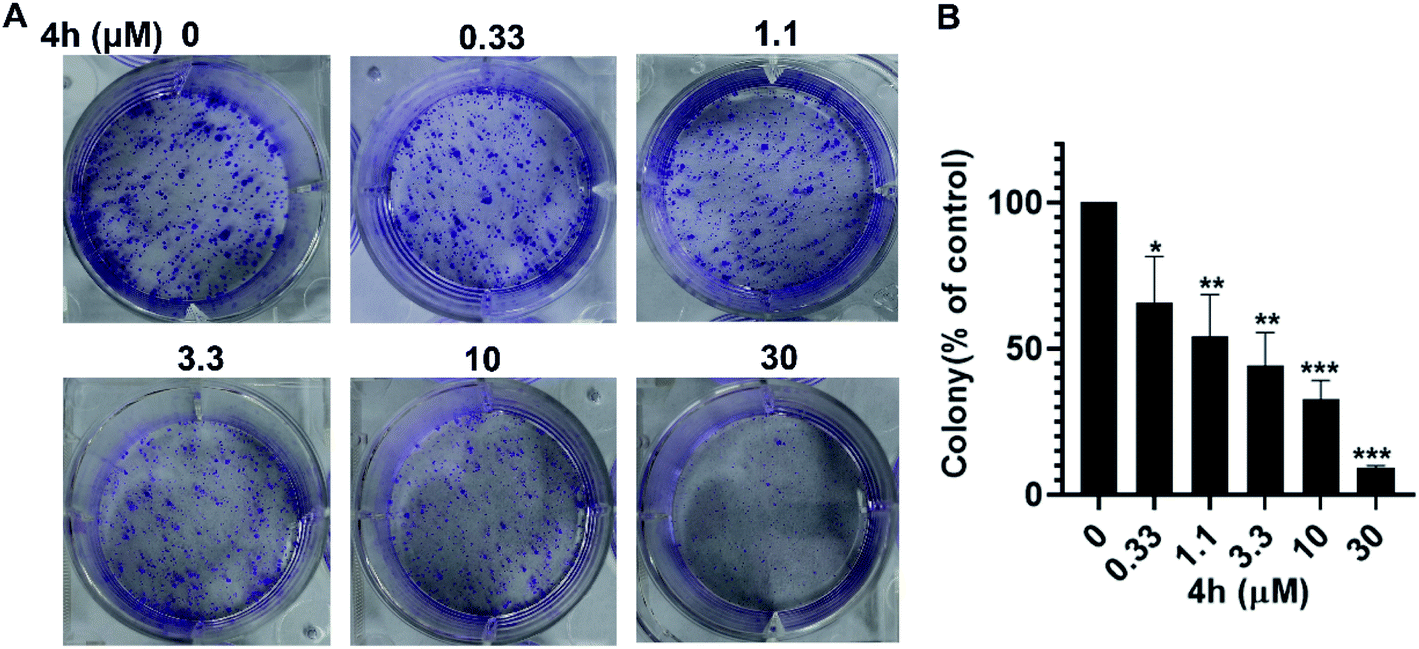 | ||
| Fig. 4 4h inhibits the proliferation of 4T1 cells. (A) The effects of 4h on colony formation in 4T1 cell for 8–12 days. (B) The statistical results of colony formation assays were presented as surviving colonies. Data are expressed as mean ± SD. From two-independent experiments (*p < 0.05, **p < 0.01 and ***p < 0.001). | ||
To quantify whether the anti-survival activity of 4h in 4T1 cells was related to apoptosis. We analyzed the level of apoptosis by FCM using the Annexin V–FITC/PI double labelling technique. As Fig. 5A indicated, the 4h could induce 4T1 cells apoptosis compared with vehicle after treatment with 4h for 24 h. Then we performed western blot analysis to further characterize 4h-induced apoptosis (Fig. 5B). The expression level of anti-apoptotic protein Bcl2 was decreased, whereas the proapoptotic protein cleaved caspase-3 was increased in a dose-dependent manner after 4h treatment in 4T1 cells. These data suggest that 4h could induce 4T1 cell apoptosis.
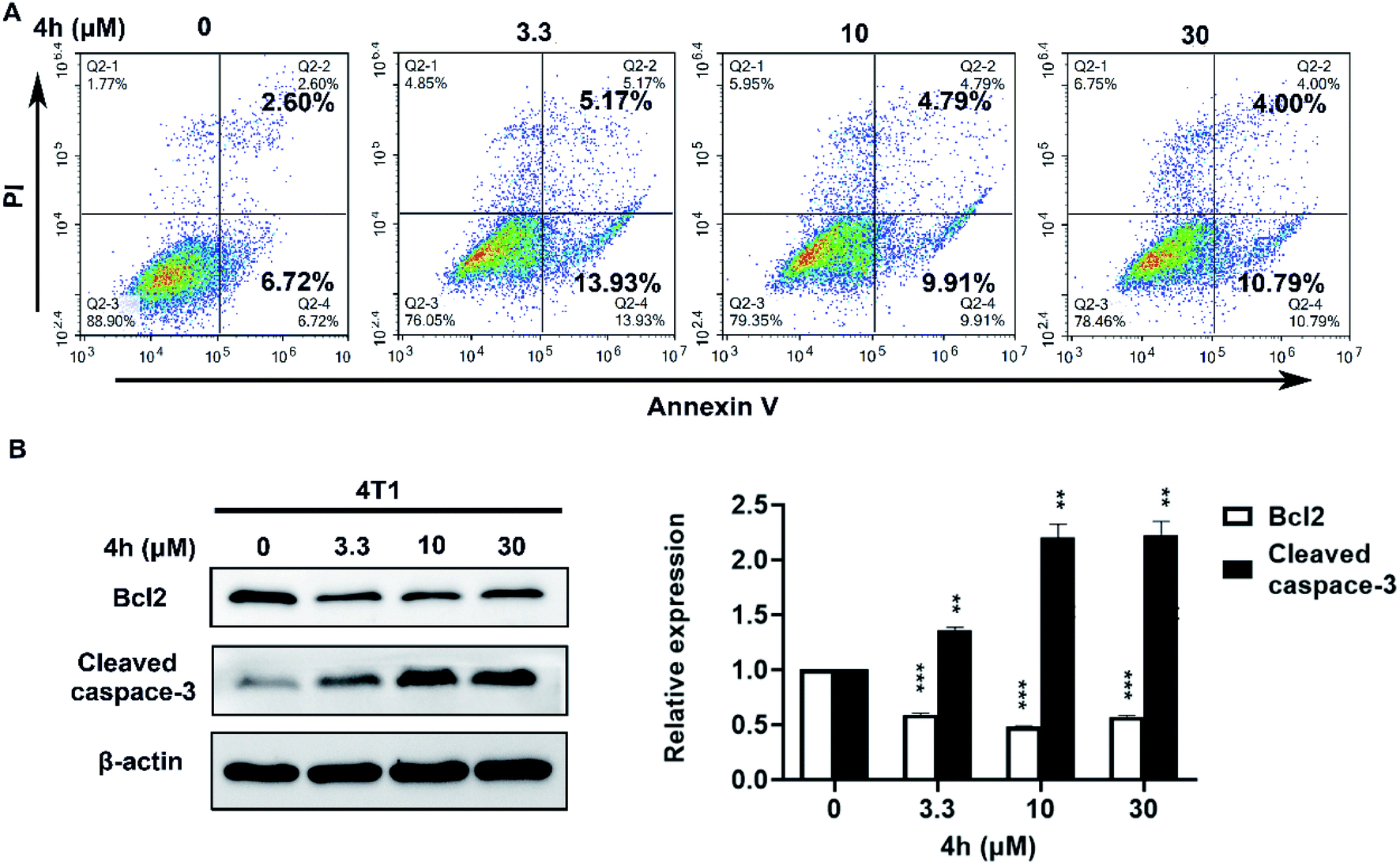 | ||
| Fig. 5 4h induces 4T1 cell apoptosis. (A) 4T1 cells were exposed to 4h at indicated doses for 24 h. Annexin V/PI double labeling technology detects the level of cell apoptosis, and the flow cytometer detects the level of cell apoptosis. (B) Western blot analyses of 4T1 cells treated with different concentrations of 4h for 24 h to evaluate protein expression of Bcl2 and cleaved caspase-3. The protein expression was quantified by densitometry analysis using Image-Pro Plus. Data are expressed as mean ± SD from two-independent experiments (**p < 0.01, ***p < 0.001, compared to control). | ||
Next, we used FCM to detect the change of Δψm after staining with the fluorescent dye Rh123. As showed in Fig. 6A, treatment with 4h for 24 h resulted in a loss of Δψm in 4T1 cells. Furthermore, we examined the ROS level by FCM using the DCFH-DA indicator (Fig. 6B). The results showed that the level of ROS in 4T1 cells increased after treatment with 4h for 24 h. These data indicated that 4h was able to induce apoptosis of 4T1 cell, and it might be via the mitochondrial apoptosis pathway.
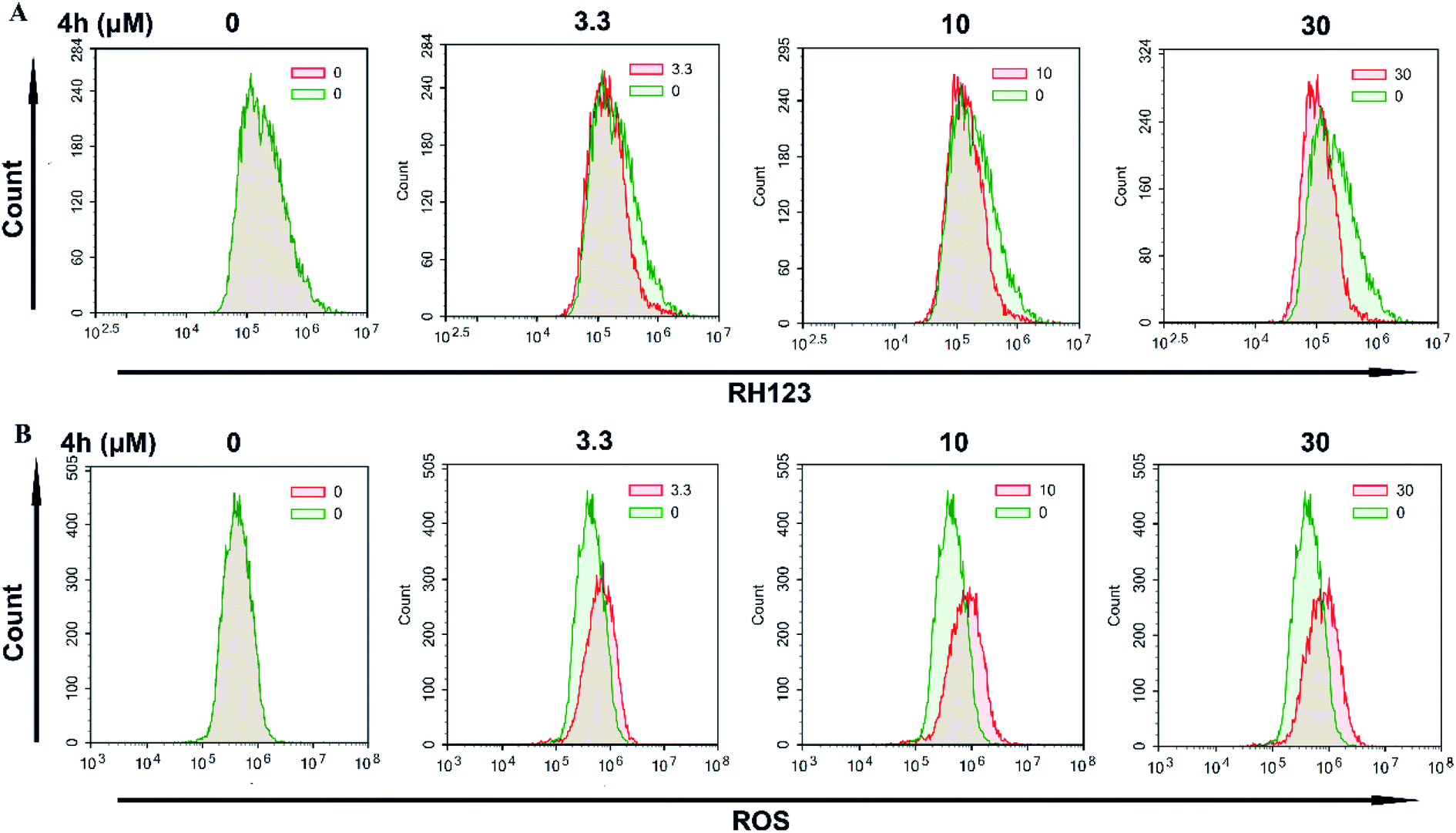 | ||
| Fig. 6 Changes in mitochondrial membrane potential and accumulation of ROS. (A) 4T1 cells were treated with different concentrations of 4h for 24 h. (B) 4T1 cells were treated with 4h at indicated doses for 24 h, and then ROS levels were analyzed with a flow cytometer. | ||
Furthermore, we evaluated the effect of compound 4h on the migration and invasion abilities of 4T1 cells through the transwell chamber assay. As showed in Fig. 7, 4h significantly reduced the migration and invasion abilities of 4T1 cells after treatment of 4h for 24 h. Compared to the control group, 4T1 cells migration was inhibited by 36.1%, 77.3%, and 93.8% following treatment with 3.3, 10 and 30 μM 4h, respectively (Fig. 7A). Similarly, 4T1 cells also exhibited significantly decreased invasion in the presence of 4h compared to control group (Fig. 7B). Furthermore, we detected the expression level of several key proteins by western blot. After 4h interfered with 4T1 cells for 24 h, the expression of MMP9 decreased with the increase of 4h concentration, while the expression of TIMP2 gradually increased (Fig. 7C). The results implied that 4h could enhance inhibition of migration and invasion of 4T1 cells which associated with down-regulation of MMP9 and up-regulation of TIMP2.
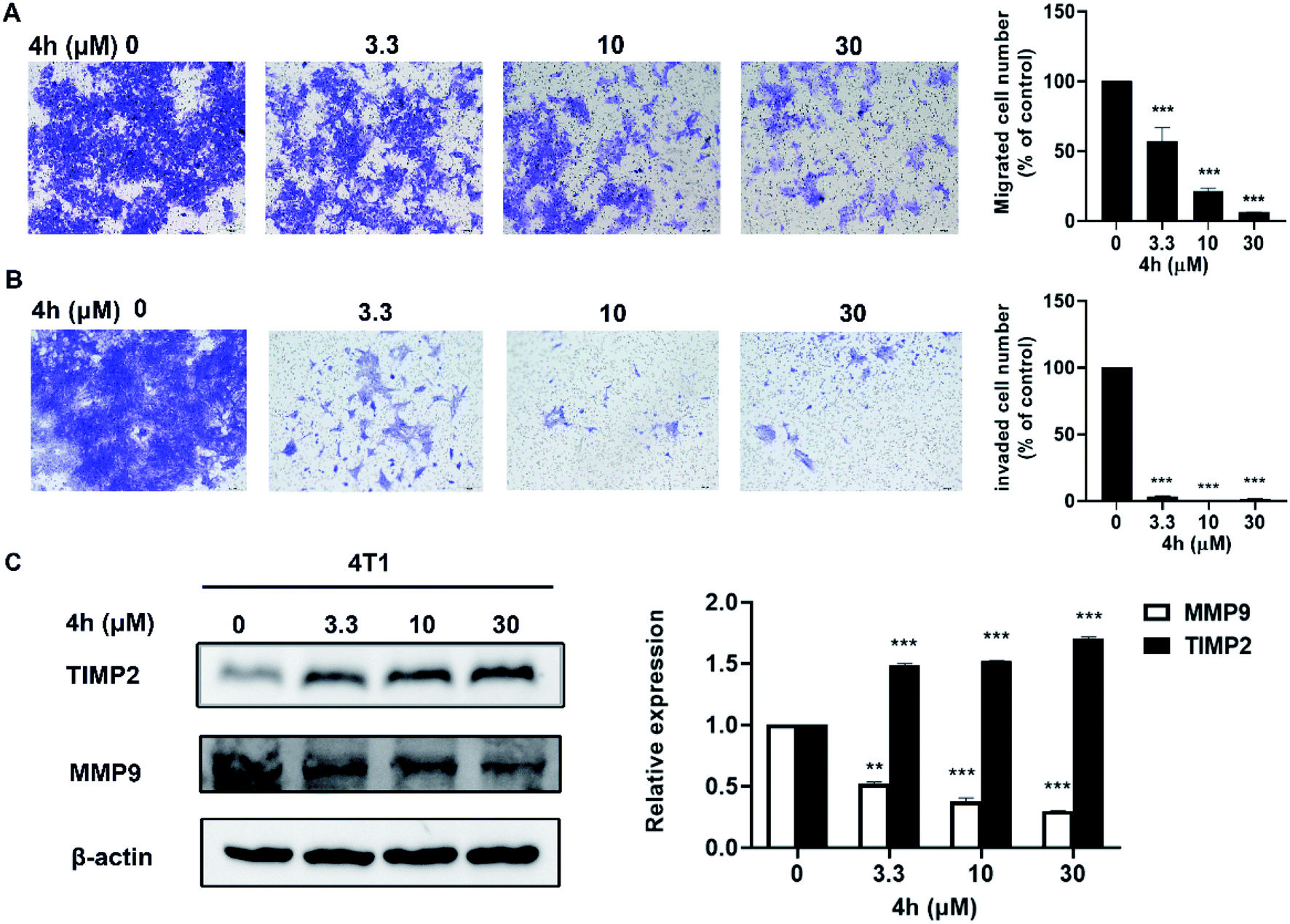 | ||
| Fig. 7 Inhibition of migratory and invasive in 4T1 cells by 4h treated. (A) 4T1 cells were seeded in the top chamber of transwell with serum-free medium, without Matrigel in the upper chamber, treated with vehicle or various concentrations of 4h. After about 24 h, migrated cells were fixed, stained, photographed (10×) and quantified. (B) 4T1 cell were treated with different concentrations of 4h and allowed to invade through Matrigel for 24 h. Invaded cell numbers were counted (10×). (C) Western blot analyses of 4T1 cells treated (24 h) with different concentrations of 4h were used to evaluate protein expression of MMP9 and TIMP2. β-Actin served as loading control. Data are expressed as mean ± SD from two-independent experiments (**p < 0.01, ***p < 0.001, compared to control). | ||
In conclusion, we discovered a series of 1H-pyrrolo[2,3-b]pyridine derivatives as potent FGFR inhibitors. Structure optimization of 1 led to the identification of 4h, which had pan-FGFR inhibitory activities against FGFR1–4 (IC50 values of 7, 9, 25 and 712 nM, respectively) and nearly 300-fold higher FGFR1 activity than compound 1, showing a highly ligand efficiency (ligand efficiency increased from 0.13 to 0.44). Accordingly, we selected compound 4h for further biological activity evaluation, and the results showed that 4h could inhibit proliferation, induce apoptosis, and significantly inhibit the migration and invasion of 4T1 cells. These data indicated that low molecular weight 4h would be a promising lead compound with a large optimization space for further drug development.
2. Experimental section
2.1. Chemistry
All reagents and chemicals used in this study do not require further purification and are commercially available. TLC was performed on 0.20 mm silica gel 60 F254 plates (Qingdao Ocean Chemical Factory, Shandong, China). Visualization of spots on TLC plates were done by UV light and I2. 1H (400 MHz) and 13C (101 MHz) NMR spectra were recorded on Bruker Avance 400 spectrometer (Bruker Company, Germany) with CDCl3, DMSO-d6 or CD3OD as solvent and TMS as an internal standard. All chemical shift values were reported in units of δ (ppm). The following abbreviations were used to indicate the peak multiplicity: s = singlet; d = doublet; t = triplet; q = quart; m = multiplet; high-resolution mass data (MS) were obtained by Q-TOF Premier mass spectrometer (Micromass, Manchester, UK).3b: 1H NMR (400 MHz, DMSO-d6) δ 12.04 (s, 1H), 8.53 (d, J = 2.2 Hz, 1H), 8.18 (d, J = 2.2 Hz, 1H), 7.55–7.42 (m, 3H), 7.42–7.34 (m, 2H), 6.04 (d, J = 4.6 Hz, 1H), 5.96 (d, J = 4.6 Hz, 1H). 13C NMR (101 MHz, DMSO-d6) δ 150.52, 144.45, 139.79, 139.75, 131.73, 128.51, 128.47, 126.21, 125.55, 125.51, 119.74, 117.88–116.60 (m), 117.27, 68.15. HRMS m/z (ESI) calcd for C15H11N2OF3Cl [M + H]+: 327.0507, found: 327.0513.
3c: 1H NMR (400 MHz, DMSO-d6) δ 12.07 (s, 1H), 8.53 (d, J = 2.4 Hz, 1H), 8.19 (d, J = 2.2 Hz, 1H), 7.87 (s, 1H), 7.74 (d, J = 7.4 Hz, 1H), 7.63–7.53 (m, 2H), 7.50 (d, J = 1.8 Hz, 1H), 6.15 (d, J = 4.6 Hz, 1H), 6.10 (d, J = 4.6 Hz, 1H); 13C NMR (101 MHz, DMSO-d6) δ 150.51, 146.94, 139.80, 130.80, 129.55 (d, J = 19.5 Hz), 130.17–128.56 (m), 126.93, 126.35, 125.46 (d, J = 3.9 Hz), 124.01 (d, J = 4.0 Hz), 122.94 (d, J = 3.9 Hz), 119.44, 117.26 (q, J = 31.3 Hz), 117.18, 68.16. HRMS m/z (ESI) calcd for C16H11N2OF6 [M + H]+: 361.0770, found: 361.0770.
3d: 1H NMR (400 MHz, chloroform-d) δ 9.83 (s, 1H), 8.57 (d, J = 2.1 Hz, 1H), 8.16 (d, J = 2.0 Hz, 1H), 7.47–7.36 (m, 3H), 7.24–7.15 (m, 2H), 6.20–6.15 (m, 1H); 13C NMR (101 MHz, chloroform-d) δ 150.10, 149.53, 145.00, 140.68, 130.04, 126.17, 125.91, 125.00, 124.73, 123.22, 120.37, 118.98, 118.67, 117.46, 69.63, 29.71. HRMS m/z (ESI) calcd for C16H11N2O2F6 [M + H]+: 377.0719, found: 377.0684.
3e: 1H NMR (400 MHz, DMSO-d6) δ 12.10–11.96 (m, 1H), 8.53 (d, J = 2.1 Hz, 1H), 8.10 (d, J = 2.2 Hz, 1H), 7.48 (d, J = 2.4 Hz, 1H), 7.35 (m, J = 7.9, 6.2, 3.4 Hz, 3H), 7.25–7.18 (m, 1H), 7.17–7.08 (m, 2H), 7.01–6.93 (m, 2H), 6.89 (m, J = 8.1, 2.6, 1.1 Hz, 1H), 6.01 (d, J = 4.6 Hz, 1H), 5.91 (d, J = 4.6 Hz, 1H). 13C NMR (101 MHz, DMSO-d6) δ 157.21, 156.94, 150.52, 147.86, 139.72, 139.68, 130.39, 130.16, 126.98, 126.27, 125.52 (d, J = 3.8 Hz), 124.28, 123.79, 121.94, 119.79, 118.88, 117.81–116.51 (m), 68.44. HRMS m/z (ESI) calcd for C21H16N2O2F3 [M + H]+: 385.1158, found: 385.1156.
3f: 1H NMR (400 MHz, DMSO-d6) δ 12.08 (s, 1H), 8.53 (d, J = 2.2 Hz, 1H), 8.20 (d, J = 2.3 Hz, 1H), 7.70 (s, 4H), 7.50 (d, J = 2.4 Hz, 1H), 6.23–6.03 (m, 2H). 13C NMR (101 MHz, DMSO-d6) δ 162.75, 150.49, 150.15, 139.81, 128.08, 127.77, 127.32, 126.95, 126.40, 126.16, 125.47, 124.25, 123.46, 119.38, 117.27 (q, J = 31.5 Hz), 68.26. HRMS m/z (ESI) calcd for C16H11N2OF6 [M + H]+: 361.0770, found: 361.0767.
3g: 1H NMR (400 MHz, methanol-d4) δ 8.45 (d, J = 2.1 Hz, 1H), 8.13 (d, J = 2.3 Hz, 1H), 7.40–7.32 (m, 2H), 7.33–7.21 (m, 6H), 7.10 (t, J = 2.1 Hz, 1H), 7.06 (dt, J = 8.1, 1.0 Hz, 1H), 7.02–6.87 (m, 2H), 6.05 (s, 1H), 5.07 (s, 2H). 13C NMR (101 MHz, DMSO-d6) δ 158.08, 149.06, 144.43, 138.33, 136.45, 128.21, 127.19, 126.56, 126.24, 124.94, 124.86, 118.24, 118.03, 117.29–116.69 (m), 113.08, 112.04, 68.73, 68.49. HRMS m/z (ESI) calcd for C22H18N2O2F3 [M + H]+: 399.1315, found: 399.1305.
3h: 1H NMR (400 MHz, DMSO-d6) δ 12.04 (s, 1H), 8.53 (d, J = 2.2 Hz, 1H), 8.18 (d, J = 2.2 Hz, 1H), 7.55–7.42 (m, 3H), 7.42–7.34 (m, 2H), 6.04 (d, J = 4.6 Hz, 1H), 5.96 (d, J = 4.6 Hz, 1H). 13C NMR (101 MHz, DMSO-d6) δ 150.52, 144.45, 139.79, 139.75, 131.73, 128.93, 128.73, 128.51, 128.47, 126.21, 125.55, 125.51, 119.74, 117.34, 117.27, 117.03, 68.15. HRMS m/z (ESI) calcd for C17H16N2O3F3 [M + H]+: 353.1108, found: 353.1108.
3i: 1H NMR (400 MHz, DMSO-d6) δ 12.10 (s, 1H), 8.55 (d, J = 2.2 Hz, 1H), 8.28 (d, J = 2.2 Hz, 1H), 7.49 (dd, J = 6.2, 3.1 Hz, 1H), 7.39 (d, J = 2.5 Hz, 1H), 7.26–7.07 (m, 2H), 6.24 (d, J = 4.9 Hz, 1H), 6.11 (dd, J = 5.0, 1.8 Hz, 1H). 13C NMR (101 MHz, DMSO-d6) δ 159.96, 157.59 (d, J = 2.0 Hz), 156.70 (d, J = 2.2 Hz), 154.30, 150.37, 139.87 (d, J = 3.8 Hz), 134.43 (dd, J = 16.1, 7.0 Hz), 126.42, 125.28 (d, J = 4.0 Hz), 124.27, 118.07, 117.30, 115.64 (d, J = 24.3 Hz), 114.52 (d, J = 20.2 Hz), 62.54. HRMS m/z (ESI) calcd for C15H10N2OF5 [M + H]+: 329.0708, found: 329.2583.
3j: 1H NMR (400 MHz, DMSO-d6) δ 12.10 (s, 1H), 8.55 (d, J = 2.2 Hz, 1H), 8.25 (d, J = 2.2 Hz, 1H), 7.98 (d, J = 2.0 Hz, 1H), 7.75 (dd, J = 8.3, 2.1 Hz, 1H), 7.72–7.63 (m, 1H), 7.50 (d, J = 2.4 Hz, 1H), 6.26–6.09 (m, 2H). 13C NMR (101 MHz, DMSO-d6) δ 150.51, 145.57, 139.93, 139.89, 132.31, 131.93, 129.37, 129.35, 126.53, 125.75, 125.70, 125.48, 125.44, 119.06, 118.09–116.25 (m), 67.57. HRMS m/z (ESI) calcd for C16H10N2OF6Cl [M + H]+: 395.0380, found: 395.0367.
3k: 1H NMR (400 MHz, DMSO-d6) δ 12.06 (s, 1H), 8.53 (d, J = 2.1 Hz, 1H), 8.24 (d, J = 2.1 Hz, 1H), 7.56–7.41 (m, 2H), 7.39–7.27 (m, 2H), 6.05 (d, J = 10.5 Hz, 2H). 13C NMR (101 MHz, chloroform-d) δ 150.14, 144.08, 143.37, 140.46, 139.05, 131.67, 129.13, 126.24, 125.06, 123.22, 121.63, 118.75, 117.58, 109.29, 108.00, 69.88. HRMS m/z (ESI) calcd for C16H10N2O3F5 [M + H]+: 373.0606, found: 373.0583.
4b: 1H NMR (400 MHz, DMSO-d6) δ 12.00 (s, 1H), 8.52 (d, J = 2.1 Hz, 1H), 8.24 (d, J = 2.2 Hz, 1H), 7.53 (d, J = 2.4 Hz, 1H), 7.33 (s, 4H), 4.13–4.09 (m, 2H). 13C NMR (101 MHz, DMSO-d6) δ 150.36, 140.50, 139.69, 139.65, 130.99, 130.72, 128.72, 126.95, 124.76, 124.72, 121.65, 118.60, 117.17 (q, J = 31.3 Hz), 114.30, 30.24. HRMS m/z (ESI) calcd for C15H11N2F3Cl [M + H]+: 311.0557, found: 311.0593.
4c: 1H NMR (400 MHz, DMSO-d6) δ 12.02 (s, 1H), 8.53 (d, J = 2.1 Hz, 1H), 8.30 (d, J = 2.1 Hz, 1H), 7.72 (s, 1H), 7.68–7.57 (m, 2H), 7.57–7.46 (m, 2H), 4.23 (s, 2H). 13C NMR (101 MHz, DMSO-d6) δ 150.35, 143.04, 139.69, 133.08, 129.85, 129.69, 129.38, 127.07, 125.37 (d, J = 3.9 Hz), 124.84 (d, J = 3.7 Hz), 123.16 (d, J = 4.1 Hz), 118.56, 117.80–116.16 (m), 114.00, 30.58. HRMS m/z (ESI) calcd for C16H11N2F6 [M + H]+: 345.0821, found: 345.0833.
4d: 1H NMR (400 MHz, DMSO-d6) δ 12.01 (s, 1H), 8.52 (d, J = 2.1 Hz, 1H), 8.25 (d, J = 2.1 Hz, 1H), 7.59 (d, J = 2.4 Hz, 1H), 7.48–7.29 (m, 3H), 7.17 (d, J = 8.1 Hz, 1H), 4.18 (s, 2H). 13C NMR (101 MHz, DMSO-d6) δ 150.37, 148.90, 144.39, 139.65, 130.66, 128.08, 126.97, 124.84, 121.36, 118.88, 118.54, 117.21 (q, J = 31.5 Hz), 113.95, 55.34, 30.62. HRMS m/z (ESI) calcd for C16H11N2OF6 [M + H]+: 361.0770, found: 361.0784.
4e: 1H NMR (400 MHz, DMSO-d6) δ 12.06–11.90 (m, 1H), 8.52 (d, J = 2.1 Hz, 1H), 8.19 (d, J = 2.1 Hz, 1H), 7.54 (d, J = 2.3 Hz, 1H), 7.38–7.25 (m, 3H), 7.14–7.06 (m, 2H), 7.03–6.89 (m, 3H), 6.81 (dd, J = 8.2, 2.4 Hz, 1H), 4.11 (s, 2H). 13C NMR (101 MHz, DMSO-d6) δ 157.17, 157.02, 150.36, 143.77, 139.63, 130.39, 127.04, 126.88, 124.74, 124.18, 123.72, 119.27, 118.78, 118.63, 117.13 (q, J = 31.3 Hz), 116.71, 114.33, 30.85. HRMS m/z (ESI) calcd for C21H16N2OF3 [M + H]+: 369.1209, found: 369.1253.
4f: 1H NMR (400 MHz, DMSO-d6) δ 12.06 (s, 1H), 8.54 (d, J = 2.1 Hz, 1H), 8.36 (d, J = 2.1 Hz, 1H), 7.86 (s, 1H), 7.61 (dd, J = 6.8, 1.9 Hz, 3H), 4.22 (s, 2H). 13C NMR (101 MHz, DMSO-d6) δ 150.33, 146.47, 139.74, 129.64, 127.36, 127.16, 127.04, 126.21, 125.65, 124.75, 124.33, 118.61, 117.24 (d, J = 31.3 Hz), 113.78, 30.67. HRMS m/z (ESI) calcd for C16H11N2F6 [M + H]+: 345.0821, found: 345.0839.
4g: 1H NMR (400 MHz, DMSO-d6) δ 11.96 (s, 1H), 8.52 (d, J = 2.2 Hz, 1H), 8.24 (d, J = 2.1 Hz, 1H), 7.51 (d, J = 2.2 Hz, 1H), 7.42–7.28 (m, 4H), 7.19 (t, J = 7.9 Hz, 1H), 7.03–6.94 (m, 1H), 6.89 (d, J = 7.6 Hz, 1H), 6.83 (s, 1H), 5.05 (s, 2H), 4.07 (s, 2H). 13C NMR (101 MHz, DMSO-d6) δ 158.88, 150.37, 143.05, 139.56, 137.56, 129.83, 128.80, 128.08, 126.81, 124.76, 121.40, 118.71, 117.09 (q, J = 31.3 Hz), 115.62, 114.56, 112.63, 69.54, 31.02. HRMS m/z (ESI) calcd for C22H18N2OF3 [M + H]+: 383.1366, found: 383.1354.
4h: 1H NMR (400 MHz, DMSO-d6) δ 11.96 (s, 1H), 8.52 (d, J = 2.1 Hz, 1H), 8.29 (d, J = 2.2 Hz, 1H), 7.53 (d, J = 2.3 Hz, 1H), 6.49 (d, J = 2.3 Hz, 2H), 6.31 (t, J = 2.3 Hz, 1H), 4.03 (s, 2H), 3.69 (s, 6H). 13C NMR (101 MHz, DMSO-d6) δ 160.90, 150.35, 143.78, 139.56 (d, J = 4.1 Hz), 126.79, 124.87, 124.83, 118.70, 116.92 (q, J = 31.5 Hz), 114.52, 107.12, 98.11, 55.50, 31.27. HRMS m/z (ESI) calcd for C17H16N2O2F3 [M + H]+: 337.1158, found: 337.1147.
4i: 1H NMR (400 MHz, DMSO-d6) δ 12.05 (s, 1H), 8.54 (d, J = 2.1 Hz, 1H), 8.34 (d, J = 2.1 Hz, 1H), 7.51 (d, J = 2.3 Hz, 1H), 7.23 (td, J = 9.2, 4.7 Hz, 2H), 7.08 (m, J = 12.2, 8.1, 3.6 Hz, 1H), 4.13 (s, 2H). 13C NMR (101 MHz, DMSO-d6) δ 159.76, 158.11, 157.37 (d, J = 2.1 Hz), 155.74, 150.21, 139.74, 130.30 (dd, J = 18.6, 7.8 Hz), 127.19, 124.67, 118.46, 114.99, 114.84, 112.55, 49.06, 24.23. HRMS m/z (ESI) calcd for C15H10N2F5 [M + H]+: 313.0759, found: 313.0752.
4j: 1H NMR (400 MHz, DMSO-d6) δ 12.06 (s, 1H), 8.54 (d, J = 2.1 Hz, 1H), 8.36 (d, J = 2.1 Hz, 1H), 7.86 (s, 1H), 7.61 (dd, J = 6.8, 1.9 Hz, 3H), 4.22 (s, 2H). 13C NMR (101 MHz, DMSO-d6) δ 150.33, 141.83, 139.80, 134.58, 132.06, 128.60, 128.13, 127.25, 127.04, 124.85, 122.02, 118.49, 117.29 (q, J = 31.7 Hz), 113.62, 29.92. HRMS m/z (ESI) calcd for C16H10N2F6Cl [M + H]+: 379.0430, found: 379.0435.
4k: 1H NMR (400 MHz, DMSO-d6) δ 12.01 (s, 1H), 8.52 (d, J = 2.1 Hz, 1H), 8.31 (d, J = 2.1 Hz, 1H), 7.55 (d, J = 2.4 Hz, 1H), 7.38 (d, J = 1.8 Hz, 1H), 7.30 (d, J = 8.3 Hz, 1H), 7.18 (dd, J = 8.2, 1.7 Hz, 1H), 4.13 (s, 2H). 13C NMR (101 MHz, DMSO-d6) δ 150.31, 143.24, 141.48, 139.70, 139.68 (d, J = 4.1 Hz), 138.55, 131.61, 126.98, 124.77 (d, J = 4.0 Hz), 124.53, 118.51, 117.22 (q, J = 31.7 Hz), 114.37, 110.63, 110.17, 30.60. HRMS m/z (ESI) calcd for C16H10N2O2F5 [M + H]+: 357.0657, found: 357.0675.
4l: 1H NMR (400 MHz, DMSO-d6) δ 12.02 (s, 1H), 8.53 (d, J = 2.1 Hz, 1H), 8.30 (d, J = 2.1 Hz, 1H), 7.72 (s, 1H), 7.68–7.57 (m, 2H), 7.57–7.46 (m, 2H), 4.23 (s, 2H). 13C NMR (101 MHz, DMSO-d6) δ 169.35, 164.01, 151.06, 150.37, 148.37, 139.81, 139.60, 138.52, 126.86, 125.20, 124.77 (d, J = 4.4 Hz), 119.08, 117.11 (d, J = 31.6 Hz), 114.90, 110.52, 31.03, 14.52. HRMS m/z (ESI) calcd for C17H13N3OF3 [M + H]+: 332.1005, found: 332.1001.
2.2. Biological
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :5, 60 μL per well, BD Biosciences). After Matrigel polymerization, the lower compartment of the chambers was filled with 600 μL medium with 10% FBS and 5 × 104 cells in 100 μL serum-free medium were placed in the upper part of each transwell and treated with different concentrations of 4h. After incubation for 24 h, cells on the upper side of the filter were removed. Cells located on the underside of the filter were fixed with methanol and stained with 0.5% crystal violet. Next, migrated cells were counted and photographed under a light microscope. The results were expressed as the percentage inhibition rate of migration compared with the untreated group.
:5, 60 μL per well, BD Biosciences). After Matrigel polymerization, the lower compartment of the chambers was filled with 600 μL medium with 10% FBS and 5 × 104 cells in 100 μL serum-free medium were placed in the upper part of each transwell and treated with different concentrations of 4h. After incubation for 24 h, cells on the upper side of the filter were removed. Cells located on the underside of the filter were fixed with methanol and stained with 0.5% crystal violet. Next, migrated cells were counted and photographed under a light microscope. The results were expressed as the percentage inhibition rate of migration compared with the untreated group.Free energy of ligand binding:
|
ΔG = −RTlnKd
| (1) |
Binding energy per atom (ligand efficiency):
| ΔLE = ΔG/n (non-hydrogen atoms) | (2) |
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by Post-Doctor Research Project, West China Hospital, Sichuan University, China (No. 2018HXBH009) and the Fundamental Research Funds for the Central Universities (No. 2020SCU12020, the Postdoctoral Foundation of Sichuan University).Notes and references
- M. Kostas, E. M. Haugsten, Y. Zhen, V. Sorensen, P. Szybowska, E. Fiorito, S. Lorenz, N. Jones, G. A. de Souza, A. Wiedlocha and J. Wesche, Mol. Cell. Proteomics, 2018, 17, 850–870 CrossRef CAS PubMed.
- M. Wei, X. Peng, L. Xing, Y. Dai, R. Huang, M. Geng, A. Zhang, J. Ai and Z. Song, Eur. J. Med. Chem., 2018, 154, 9–28 CrossRef CAS PubMed.
- Y. K. Chae, K. Ranganath, P. S. Hammerman, C. Vaklavas, N. Mohindra, A. Kalyan, M. Matsangou, R. Costa, B. Carneiro, V. M. Villaflor, M. Cristofanilli and F. J. Giles, Oncotarget, 2017, 8, 16052–16074 CrossRef PubMed.
- G. C. Ghedini, R. Ronca, M. Presta and A. Giacomini, Expert Rev. Anticancer Ther., 2018, 18, 861–872 CrossRef CAS PubMed.
- A. De Luca, R. Esposito Abate, A. M. Rachiglio, M. R. Maiello, C. Esposito, C. Schettino, F. Izzo, G. Nasti and N. Normanno, Int. J. Mol. Sci., 2020, 21(18), 6856 CrossRef CAS PubMed.
- R. Porta, R. Borea, A. Coelho, S. Khan, A. Araujo, P. Reclusa, T. Franchina, N. Van Der Steen, P. Van Dam, J. Ferri, R. Sirera, A. Naing, D. Hong and C. Rolfo, Crit. Rev. Oncol. Hematol., 2017, 113, 256–267 CrossRef PubMed.
- F. T. Liu, N. G. Li, Y. M. Zhang, W. C. Xie, S. P. Yang, T. Lu and Z. H. Shi, Eur. J. Med. Chem., 2020, 186, 111884 CrossRef CAS PubMed.
- M. Katoh and H. Nakagama, Med. Res. Rev., 2014, 34, 280–300 CrossRef CAS PubMed.
- A. Giacomini, P. Chiodelli, S. Matarazzo, M. Rusnati, M. Presta and R. Ronca, Pharmacol. Res., 2016, 107, 172–185 CrossRef CAS PubMed.
- W. Yan, X. Wang, Y. Dai, B. Zhao, X. Yang, J. Fan, Y. Gao, F. Meng, Y. Wang, C. Luo, J. Ai, M. Geng and W. Duan, J. Med. Chem., 2016, 59, 6690–6708 CrossRef CAS PubMed.
- I. S. Babina and N. C. Turner, Nat. Rev. Cancer, 2017, 17, 318–332 CrossRef CAS PubMed.
- L. Balek, I. Gudernova, I. Vesela, M. Hampl, V. Oralova, M. Kunova Bosakova, M. Varecha, P. Nemec, T. Hall, G. Abbadessa, N. Hatch, M. Buchtova and P. Krejci, Bone, 2017, 105, 57–66 CrossRef CAS PubMed.
- Y. Wang, Y. Dai, X. Wu, F. Li, B. Liu, C. Li, Q. Liu, Y. Zhou, B. Wang, M. Zhu, R. Cui, X. Tan, Z. Xiong, J. Liu, M. Tan, Y. Xu, M. Geng, H. Jiang, H. Liu, J. Ai and M. Zheng, J. Med. Chem., 2019, 62, 7473–7488 CrossRef CAS PubMed.
- Y. Zhou, C. Wu, G. Lu, Z. Hu, Q. Chen and X. Du, J. Cancer, 2020, 11, 2000–2007 CrossRef CAS PubMed.
- P. R. Gavine, L. Mooney, E. Kilgour, A. P. Thomas, K. Al-Kadhimi, S. Beck, C. Rooney, T. Coleman, D. Baker, M. J. Mellor, A. N. Brooks and T. Klinowska, Cancer Res., 2012, 72, 2045–2056 CrossRef CAS PubMed.
- T. Nishina, S. Takahashi, R. Iwasawa, H. Noguchi, M. Aoki and T. Doi, Invest. New Drugs, 2018, 36, 424–434 CrossRef CAS PubMed.
- Y. Nakanishi, N. Akiyama, T. Tsukaguchi, T. Fujii, K. Sakata, H. Sase, T. Isobe, K. Morikami, H. Shindoh, T. Mio, H. Ebiike, N. Taka, Y. Aoki and N. Ishii, Mol. Cancer Ther., 2014, 13, 2547–2558 CrossRef CAS PubMed.
- V. Guagnano, P. Furet, C. Spanka, V. Bordas, M. Le Douget, C. Stamm, J. Brueggen, M. R. Jensen, C. Schnell, H. Schmid, M. Wartmann, J. Berghausen, P. Drueckes, A. Zimmerlin, D. Bussiere, J. Murray and D. Graus Porta, J. Med. Chem., 2011, 54, 7066–7083 CrossRef CAS PubMed.
- G. Zhao, W. Y. Li, D. Chen, J. R. Henry, H. Y. Li, Z. Chen, M. Zia-Ebrahimi, L. Bloem, Y. Zhai, K. Huss, S. B. Peng and D. J. McCann, Mol. Cancer Ther., 2011, 10, 2200–2210 CrossRef CAS PubMed.
- P. C. C. Liu, H. Koblish, L. Wu, K. Bowman, S. Diamond, D. DiMatteo, Y. Zhang, M. Hansbury, M. Rupar, X. Wen, P. Collier, P. Feldman, R. Klabe, K. A. Burke, M. Soloviev, C. Gardiner, X. He, A. Volgina, M. Covington, B. Ruggeri, R. Wynn, T. C. Burn, P. Scherle, S. Yeleswaram, W. Yao, R. Huber and G. Hollis, PLoS One, 2020, 15, e0231877 CrossRef CAS PubMed.
- The website of Erdafitinib approved by FDA, https://www.fda.gov/drugs/new-drugs-fda-cders-new-molecular-entities-and-new-therapeutic-biological-products/novel-drug-approvals-2019.
- The website of Pemigatinib approved by FDA, https://www.fda.gov/drugs/new-drugs-fda-cders-new-molecular-entities-and-new-therapeutic-biological-products/novel-drug-approvals-2020.
- J. Tsai, J. T. Lee, W. Wang, J. Zhang, H. Cho, S. Mamo, R. Bremer, S. Gillette, J. Kong, N. K. Haass, K. Sproesser, L. Li, K. S. Smalley, D. Fong, Y. L. Zhu, A. Marimuthu, H. Nguyen, B. Lam, J. Liu, I. Cheung, J. Rice, Y. Suzuki, C. Luu, C. Settachatgul, R. Shellooe, J. Cantwell, S. H. Kim, J. Schlessinger, K. Y. Zhang, B. L. West, B. Powell, G. Habets, C. Zhang, P. N. Ibrahim, P. Hirth, D. R. Artis, M. Herlyn and G. Bollag, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 3041–3046 CrossRef CAS PubMed.
- Q. Jin, D. Zhang, M. Gao, C. Jiang and J. Zhang, Bioorg. Med. Chem., 2021, 29, 115862 CrossRef CAS PubMed.
- L. Crocetti, M. P. Giovannoni, I. A. Schepetkin, M. T. Quinn, A. I. Khlebnikov, N. Cantini, G. Guerrini, A. Iacovone, E. Teodori and C. Vergelli, Bioorg. Med. Chem., 2018, 26, 5583–5595 CrossRef CAS PubMed.
- S. Narva, S. Chitti, B. R. Bala, M. Alvala, N. Jain and V. G. Kondapalli, Eur. J. Med. Chem., 2016, 114, 220–231 CrossRef CAS PubMed.
- T. Ye, X. Wei, T. Yin, Y. Xia, D. Li, B. Shao, X. Song, S. He, M. Luo, X. Gao, Z. He, C. Luo, Y. Xiong, N. Wang, J. Zeng, L. Zhao, G. Shen, Y. Xie, L. Yu and Y. Wei, Breast Cancer Res. Treat., 2014, 143, 435–446 CrossRef CAS PubMed.
- Y. Li, C. Gan, Y. Zhang, Y. Yu, C. Fan, Y. Deng, Q. Zhang, X. Yu, Y. Zhang, L. Wang, F. He, Y. Xie, T. Ye and W. Yin, Front. Pharmacol., 2019, 10, 1195 CrossRef CAS PubMed.
- C. Gan, Y. Li, Y. Yu, X. Yu, H. Liu, Q. Zhang, W. Yin, L. Yu and T. Ye, Bioorg. Med. Chem., 2019, 27, 115089 CrossRef CAS PubMed.
- A. L. Hopkins, C. R. Groom and A. Alex, Drug Discovery Today, 2004, 9, 430–431 CrossRef PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1ra02660g |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2021 |