
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceActive targeting of cancer cells by CD44 binding peptide-functionalized oil core-based nanocapsules†
A. De Capuaab,
A. Palladinoc,
M. Chino d,
C. Attanasio*ac,
A. Lombardid,
R. Vecchione*a and
P. A. Nettiabe
d,
C. Attanasio*ac,
A. Lombardid,
R. Vecchione*a and
P. A. Nettiabe
aCenter for Advanced Biomaterials for Health Care@CRIB, Istituto Italiano di Tecnologia, Largo Barsanti e Matteucci 53, Napoli, 80125, Italy. E-mail: raffaele.vecchione@iit.it
bDepartment of Chemical, Materials & Industrial Production Engineering, University of Naples Federico II, Naples 80125, Italy
cDepartment of Veterinary Medicine and Animal Productions, University of Naples Federico II, Via F. Delpino 1, 80137 Naples, Italy. E-mail: chiara.attanasio@unina.it
dDepartment of Chemical Sciences, University of Naples Federico II, Complesso Universitario Monte S. Angelo, Via Cintia 45, 80126 Naples, Italy
eInterdisciplinary Research Center of Biomaterials, CRIB, University Federico II, P. le Tecchio 80, 80125 Naples, Italy
First published on 13th July 2021
Abstract
Selectivity in tumor targeting is one of the major issues in cancer treatment. Therefore, surface functionalization of drug delivery systems with active moieties, able to selectively target tumors, has become a worldwide-recognized strategy. The CD44 receptor is largely used as a biomarker, being overexpressed in several tumors, and consequently as a target thanks to the identification of the CD44 binding peptide. Here we implemented the CD44 binding peptide logic onto an oil core–polymer multilayer shell, taking into account and optimizing all relevant features of drug delivery systems, such as small size (down to 100 nm), narrow size distribution, drug loading capability, antifouling and biodegradability. Besides promoting active targeting, the oil core-based system enables the delivery of natural and synthetic therapeutic compounds. Biological tests, using curcumin as a bioactive compound and fluorescent tag, demonstrated that CD44 binding peptide-functionalized nanocapsules selectively accumulate and internalize in cancer cells, compared to the control, thanks to ligand–receptor binding.
1. Introduction
During the last decades, the introduction of tumor-targeted therapies has significantly changed the medical treatments of several types of cancer, improving their therapeutic index.1,2 Nanoparticle-based platforms are currently adopted to enhance site-specific accumulation of anticancer drugs in tumor sites, hopefully circumventing the severe side effects that are generally due to the exposure of healthy tissues to the drugs.3–7 These drug delivery systems (DDSs) hold the promise of overcoming current limitations, not only by offering a micro-environment that hosts poorly soluble conventional drugs, but also by actively targeting specific tumor biomarkers. A possible strategy is their decoration with ligands that are specific towards overexpressed receptors. A well-established tumor biomarker is the cluster of differentiation 44 (CD44), a membrane glycoprotein that is involved in several cell–cell interactions, adhesion, and migration events.8 The CD44 receptor binds to diverse extra-cellular matrix (ECM) ligands, mainly hyaluronic acid (HA).9,10 CD44 is found in different types of cells, whereas its isoforms, CD44v3 and CD44v6, are up-regulated in invasive cancer cells (e.g. breast, colon, prostate, and lung cancer cells).11,12 Besides HA, a particular sequence (CD44 binding peptide, CD44BP) has been identified, which binds specifically to CD44v3 and CD44v6 and inhibits tumor cell migration, invasion and angiogenesis via fibroblast growth factor 2 (FGF2) blockade.13–15 CD44BP may thus be identified as a promising candidate for tumor targeting. Furthermore, it inhibits primary B16-F10 tumor growth (melanoma), angiogenesis, lung colonization, and abolishes breast cancer tumor sphere formation, in vitro and in vivo.14,16 Zaiden and co-workers have recently designed a new copolymer, conjugated with CD44BP, able to inhibit tumor growth rate in preclinical models,17 demonstrating that CD44BP may have also interesting therapeutic properties.Oil in water nanoemulsions (O/W NEs) hold many requirements to be excellent DDSs, such as biocompatibility, biodegradability, and the ease of scaling-up. These systems are ideal carriers of hydrophobic drugs by improving their solubility in the blood and, as a consequence, by increasing their bioavailability. They can be strengthened with polymer shells, as previously described;18,19 nevertheless, there are only a few reports on the use of O/W NEs as the core of polymer nanocapsules, due to the difficulty in managing such a delicate template. In order to use O/W NEs as liquid templates for multilayer polymer deposition, it is crucial to finely control size distribution and surface charge, as O/W NE stability can benefit from narrow size distribution and high surface charges. In the last years, we were able to tune the protocol for polymeric multilayer deposition (e.g. alginate, chitosan and hyaluronic acid) around O/W NEs via the layer-by-layer (LbL) technique, even on sizes down to 100 nm.20 Herein, starting from such small O/W NEs, we designed nanocapsules (NC) with the aim to selectively target cancer cells (human primary glioblastoma cell line, U87)21 over healthy ones (human umbilical vein endothelial cells, HUVECs).22 Upon deposition of a bilayer of chitosan and biotinylated hyaluronic acid around the O/W NE core, we were successful in conjugating the nanocarrier surface with a polyethylene glycol (PEG2k) labeled CD44BP via biotin–streptavidin technology (Fig. 1).23–26
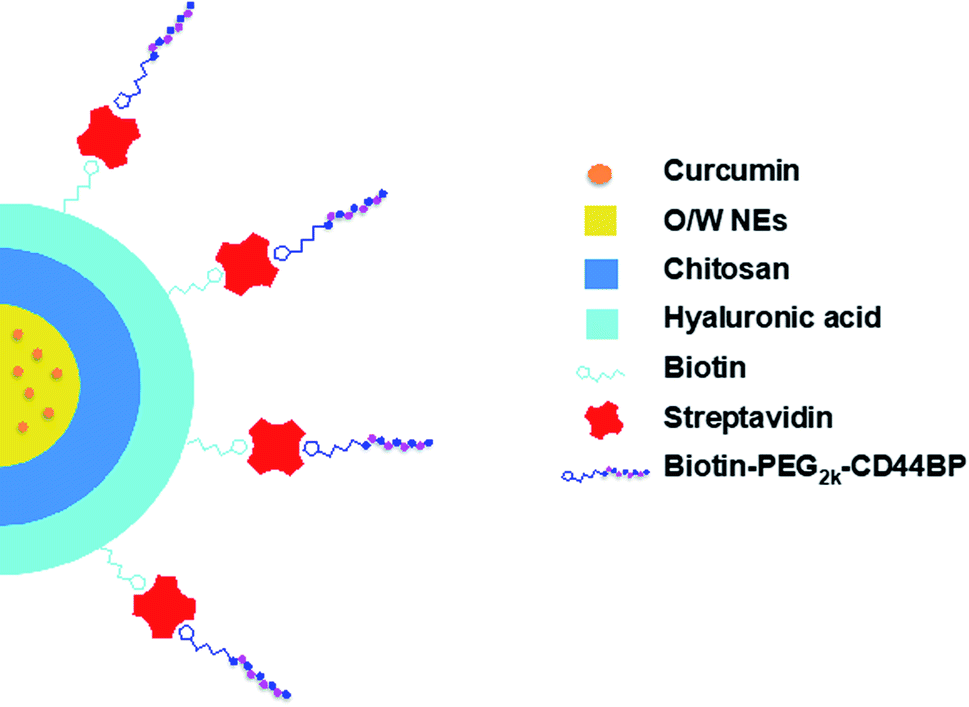 | ||
| Fig. 1 Schematic representation of peptide functionalized NEs. | ||
Poly(ethylene glycol)2k (PEG2k) is known to inhibit NC clearance by eluding the mononuclear phagocyte system (MPS).27 We performed a comprehensive biophysical characterization to find the best conditions for the NC fabrication, as well as to define the NC chemical and morphological features. Finally, we assessed the specificity of the final NC toward tumor cells over-expressing CD44 receptor. To this aim, we loaded the system with curcumin, a natural bioactive compound isolated from the Curcuma longa plant, and compared the anti-cancer activity of the free curcumin against curcumin loaded NC, in presence and in absence of the PEGylated CD44BP coating. We found that the PEGylated CD44BP functionalized NC recognizes tumor sites with higher specificity with respect to un-functionalized NC and free curcumin thanks to CD44BP that recognizes CD44 receptor and to PEG that without CD44BP inhibits cell internalization. Thus, we clearly demonstrate the feasibility of our approach in building a multi-component nanoplatform able to promote a site-specific behaviour.
2. Results
2.1 Hyaluronic acid functionalization and characterization
Prior to deposition onto the nanoemulsion, HA was functionalized with biotin hydrazide exploiting carbodiimide chemistry (Fig. S.1A†).28 The reaction was performed under mild conditions at pH ∼ 6 in order to avoid HA degradation under acidic (pH < 1.6) and basic (pH > 12.6) conditions, as well as to avoid HA gelification.29,30 The reaction outcome and the final functionalization ratio were detected by NMR (see ESI†). The functionalization ratio was found to be 37%, only slightly higher than previous attempts of HA functionalization (33%).292.2 O/W NEs coating and determination of available biotin
Over the last years, LbL-NCs have been largely investigated in our lab. Ultrastable chitosan (CT) coated O/W NEs, from now on indicated as CT–NEs, have been obtained by a previously developed high-pressure homogenization procedure.20,31 Biot–HA was deposited onto the CT–NEs (the resulting system was named Biot–HA–NEs), by controlled mixing of the two components through two syringe pumps (see ESI† for details, Fig. S.2–S.4†). The optimal concentration of Biot–HA was found to be 0.12 wt%, when 0.050 wt% CT was adopted to coat 5.0 wt% of O/W NEs. The optimum ratio between Biot–HA and CT (wt/wt%) was, therefore, 2.4, and kept constant for the next experiments.The exact amount of streptavidin (SAV) able to saturate the Biot–HA over the surface of the CT–NEs was established by isothermal titration calorimetry (ITC) experiments. After Biot–HA deposition, the resulting NC was titrated with a 16 μM SAV solution. The ITC binding curve, obtained by integrating the heat data, had well-behaved sigmoidal trend and it was fitted with an independent and equivalent-sites model (Fig. 2).
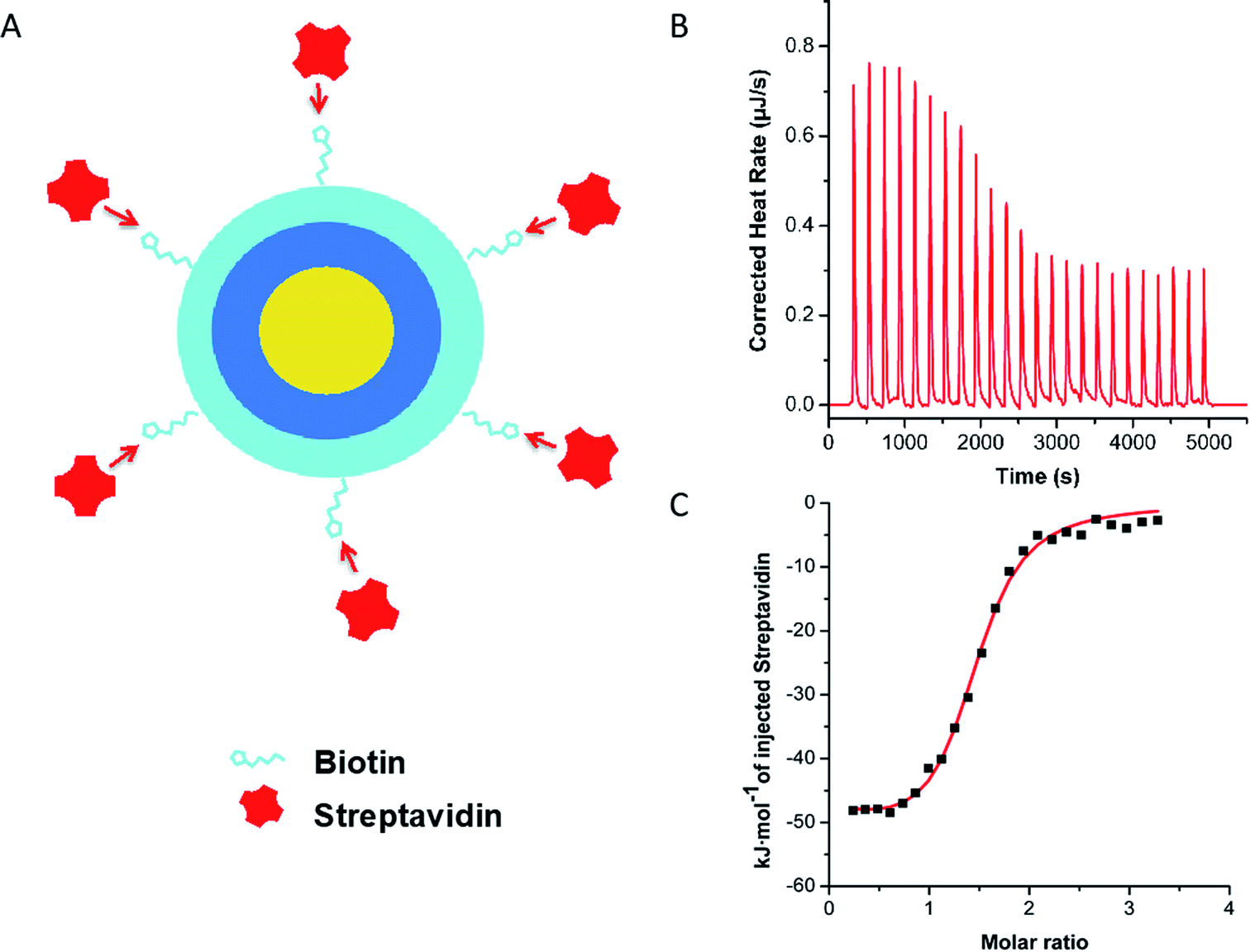 | ||
| Fig. 2 (A) Scheme of ITC experiment. (B) ITC data for titration by stepwise injection of 16 μM SAV solution in NEs, made up of 0.05 wt% Biot–HA-0.02 wt%, CT-2.08 wt%, at 25 °C. (C) Normalized heat of interaction between Biot–HA–CT–NEs and SAV. The solid squares are the experimental data obtained by integrating the raw data and subtracting the heat of ligand dilution into the buffer. | ||
Saturation was reached when SAV matched 2.0 μM concentration. Accordingly, the calculated molar ratio between SAV and Biot–HA (n*) was 1.8 ± 0.50 (averaged over three independent titrations), either when the polymer was dissolved in the bulk solution (see ESI†) or when it was deposited above the CT–NEs. This molar ratio was therefore adopted for the following experiments. As a further test, biotin–streptavidin specific interaction was confirmed by control titration of non-biotinylated HA with SAV (Fig. S.7†).
2.3 Synthesis and purification of PEGylated CD44 binding peptide
CD44BP was chemically synthesized by solid-phase peptide synthesis (SPPS). The synthesized peptide featured a proper spacer of three glycine residues, a nucleophile cysteine residue, and a β-alanine residue at the N-terminal (βA-CGGG-RLVSYNGIIFFLK). The peptide was functionalized at the N-terminal with 5-carboxyfluorescein (5-FAM) as a fluorescent probe for peptide detection (see ESI†). The highly selective thiol–maleimide reaction was chosen for further functionalization, between the cysteine side chain and maleimide–PEG2k–Biot (from now on only PEG) hetero bifunctional linker. The coupling of maleimide–PEG–Biot linker to the peptide was heterogeneously performed on the resin, giving the desired product, 5-FAM–CD44BP–PEG–Biot (Fig. S.17†).The crude product (10% yield) was cleaved from the resin, deprotected, and purified by reverse-phase (RP) flash chromatography (see ESI†). The final purity was ascertained by RP-HPLC to be higher than 95%. Identification was accomplished by MALDI-TOF mass spectrometry (Fig. 3). An intense, singly charged polydisperse gaussian distribution of 44 Da ethylene oxide repeat units was observed with the most abundant ion having a monoisotopic mass of 4704.71 m/z, proving the presence of PEG within the sample. The maleimide–PEG–Biot linker mass spectrum was also registered to assess the mass shift after peptide conjugation (Fig. S.20†). The observed increment of 2271.09 Da perfectly matched the expected monoisotopic mass of 5-FAM–CD44BP (2271.08 Da), clearly demonstrating the occurrence of the desired reaction.
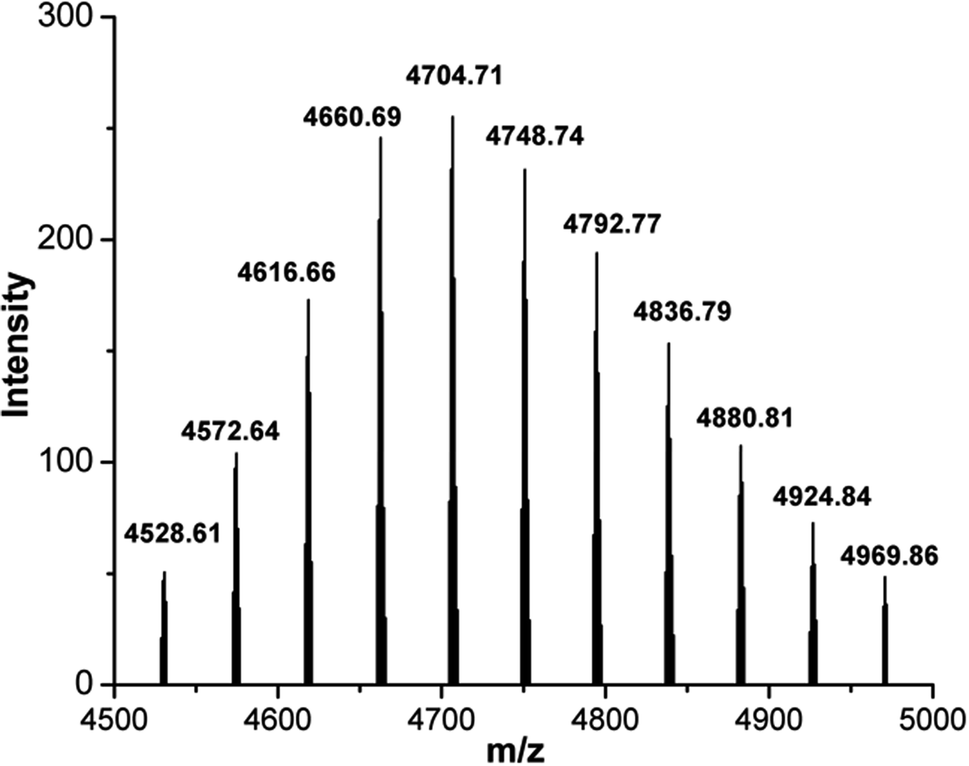 | ||
| Fig. 3 MALDI mass spectra (centroid) of 5-FAM–CD44BP–PEG–Biot peptide. | ||
2.4 Nanocarrier assembly
Once the synthesis and the complete characterization of each component of the proposed NC were carried out, the overall assembly of the multi-component system could be performed. Firstly, SAV was added to the curcumin loaded Biot–HA–NE under sonication at a SAV/biotin ratio in accordance with ITC results. Then, 5-FAM–CD44BP–PEG–Biot was added to the SAV–Biot–HA–NEs under sonication at a 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio, with the SAV taking into account that it is a tetramer can bind up to four biotin molecules.32 The resulting NC is named CD44BP–PEG–NEs. The dynamic light scattering (DLS) analysis confirmed a narrow NC size distribution, according to the polydispersity index (PdI) and Z-potential measurements (Table 1). Additional DLS results about the optimization of polymers deposition by saturation method,33 are reported in ESI.†
:1 ratio, with the SAV taking into account that it is a tetramer can bind up to four biotin molecules.32 The resulting NC is named CD44BP–PEG–NEs. The dynamic light scattering (DLS) analysis confirmed a narrow NC size distribution, according to the polydispersity index (PdI) and Z-potential measurements (Table 1). Additional DLS results about the optimization of polymers deposition by saturation method,33 are reported in ESI.†
| Size (nm) | PdI | Z-Potential (mV) | |
|---|---|---|---|
| NEs | 98 ± 5 | 0.10 ± 0.01 | −31 ± 4 |
| CT–NEs | 111 ± 7 | 0.11 ± 0.05 | +30 ± 10 |
| Biot–HA–NEs | 129 ± 7 | 0.09 ± 0.02 | −35 ± 3 |
| SAV–Biot–HA–NEs | 134 ± 11 | 0.13 ± 0.04 | −29 ± 2 |
| CD44BP–PEG–NEs | 134 ± 10 | 0.11 ± 0.01 | −29 ± 3 |
| PEG–NEs | 134 ± 12 | 0.13 ± 0.04 | −27 ± 1 |
The morphological characterization of curcumin-loaded CD44BP–PEG–NEs was performed by cryo-TEM analysis as reported in Fig. 4. The sample, imaged in its frozen hydrated state, was formed by a well-defined electron-dense core, corresponding to the curcumin loaded in the oil template, which makes difficult the observation of polymer layers around the NEs. The size distribution is in agreement with the DLS analysis. Overall, DLS periodical measurements were carried out to evaluate the dimensional evolution over time. Fig. 4B shows that CD44BP–PEG–NEs maintains its hydrodynamic diameter, and the PdI values remained <0.2 for several weeks indicating that no significant aggregation or dissociation phenomena occurred. Confocal microscopy analysis confirmed the co-localization of SAV and CD44BP–PEG built around the NE (Fig. S.21†).
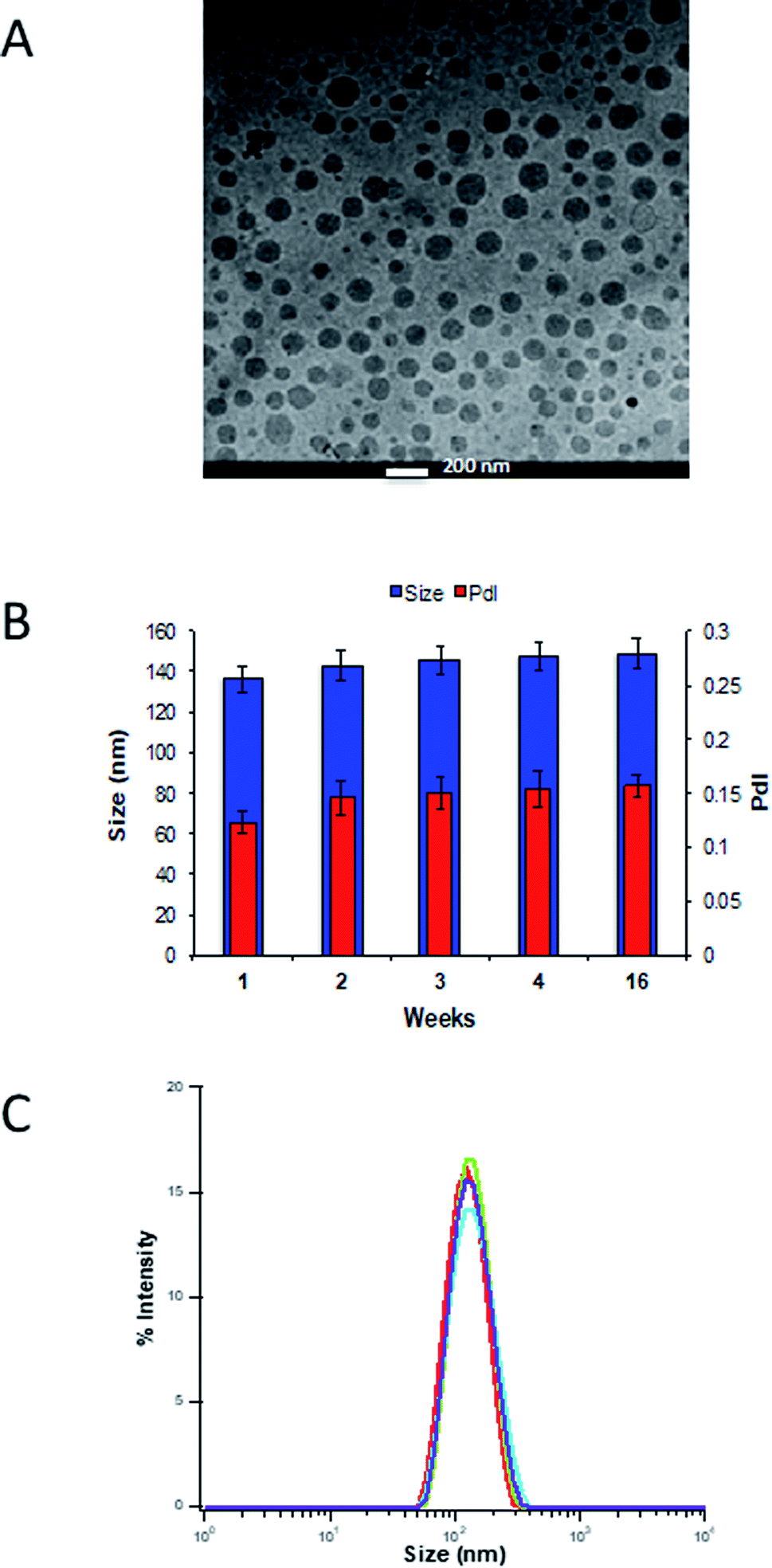 | ||
| Fig. 4 (A) Cryo-TEM projection image for curcumin loaded CD44BP–PEG–NEs. Scale bar is 200 nm. (B) Dimensional behaviour over time of the complete NC measured by DLS analysis. (C) Overlapping of mean hydrodynamic size of each component deposited around NEs. Biot–HA–NEs in red, SAV–Biot–HA–NEs in green, CD44BP–PEG–NEs in light blue and PEG–NEs in violet. | ||
2.5 In vitro selective cytotoxicity analysis
A human primary glioblastoma cell line (U-87), over-expressing CD44 receptor, was chosen as a model to understand the behaviour of CD44BP-functionalized NCs toward cancer cells. Confluent monolayers of U-87 and HUVEC cells were incubated for 4 h with HA-coated O/W NEs, CD44BP–PEG–NEs, and PEG–NEs at a final curcumin concentration of 62.8 μM to check for cell mortality upon exposition to each NC (parameter optimization is reported in ESI, Fig. S.22†). PEG–NEs was used as negative control. In addition, cells were treated with cell medium alone as positive control and with free curcumin as second negative control. Further, HA–NEs acted as second positive control since they included hyaluronic acid, which is the main ligand of CD44 receptor (Fig. 5).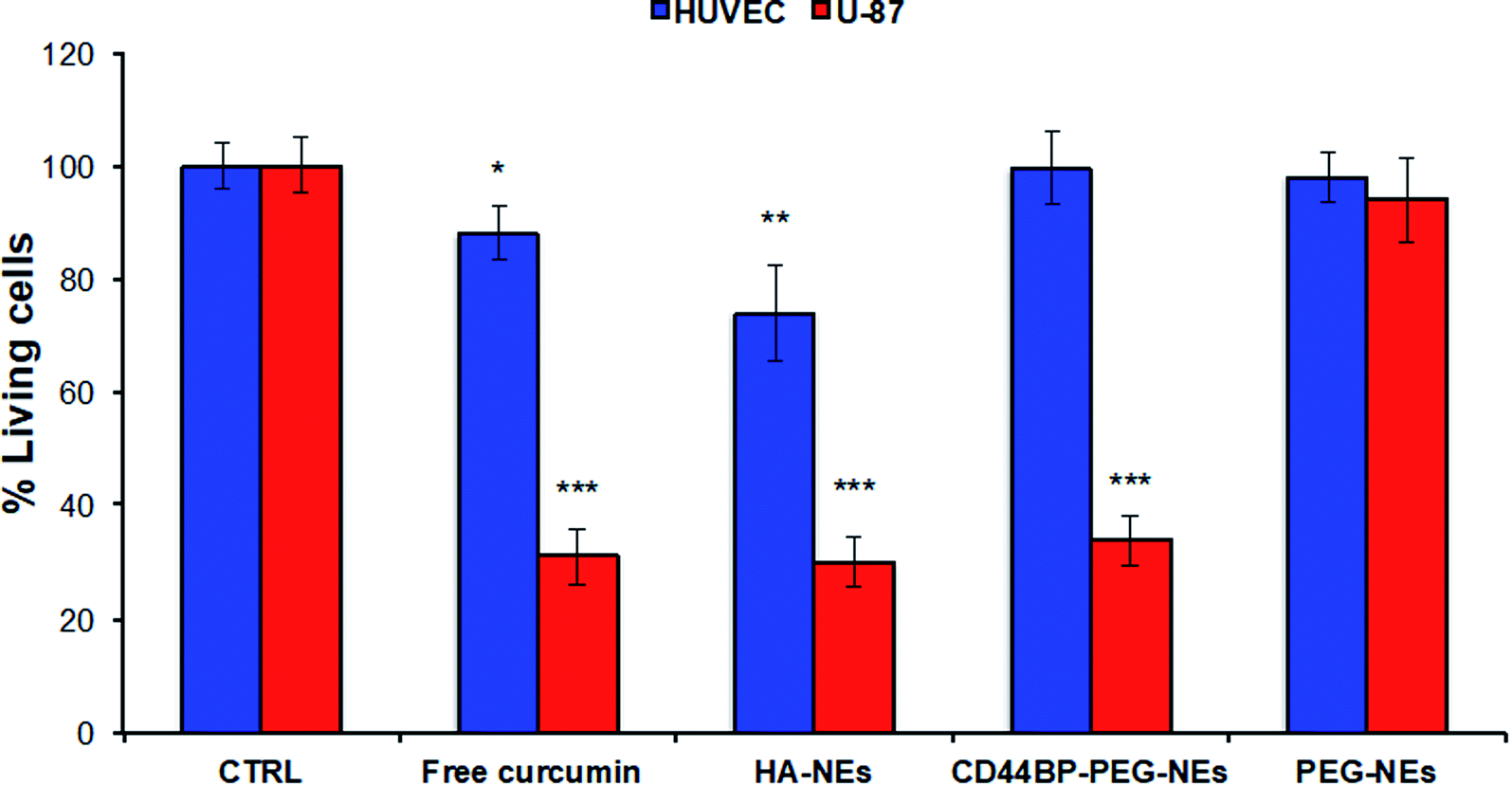 | ||
| Fig. 5 Cytotoxicity assay of curcumin loaded HA–CT–NEs, CD44BP–PEG–NEs, PEG–NEs, and free curcumin. U-87 and HUVEC cells of all groups were treated for 4 h and cell viability was evaluated after 24 h. Data are reported as mean of three independent experiments ± SD (n = 3) and expressed as percentage compared to control group. The asterisk (*) indicates the statistical significance vs. CTRL evaluated by Student's t test considering p < 0.05 (*), p ≤ 0.01 (**), p ≤ 0.001 (***). | ||
Data showed a significant cell mortality of 66 ± 4% of CD44BP–PEG–NEs compared to PEG–NEs in U-87 cells. This is an evident consequence of the peptide capability to enhance NC accumulation on the cell surface, thanks to ligand-receptor recognition, also improving its internalization. Curcumin loaded HA-coated O/W NEs also induced an important cell death in U-87 cells (=70 ± 4%), almost similar to 66 ± 4% observed for CD44BP–PEG–NEs. No cytotoxic effect was observed in HUVEC cells for CD44BP–PEG–NEs, in contrast to the 30% of mortality induced by HA–NEs, thus confirming the specific ligand-receptor interaction, differently from the non-specific behaviour of HA–NEs.
2.6 Cellular uptake analysis
A cellular uptake assay by confocal microscopy was performed to corroborate cytotoxicity results and understand if the observed mortality could be attributed to curcumin. Based on curcumin intrinsic fluorescence properties, we used it as a probe for NCs detection. Confluent monolayers of U-87 and HUVEC cells were incubated for 4 h with curcumin-loaded HA–NEs, CD44BP–PEG–NEs and PEG–NEs, in the same experimental conditions of cytotoxicity assays as previously described. Also in this case, the control (CTRL) group consisted in cells treated with cell medium alone. Fig. 6 shows confocal microscopy images of U-87 cell monolayers after NC uptake. Curcumin appeared in red, while cell cytoplasm and nuclei were displayed in green and blue respectively. Images are representative of at least three independent experiments, in which ten images were examined for each treatment. Fig. 7 shows the confocal microscopic images of HUVEC cells treated with curcumin loaded HA–NEs and CD44BP–PEG–NEs. Fig. 8 reports the plot of curcumin mean fluorescence intensity normalized for the cell number both in the case of U-87 and HUVEC. Interestingly, HA–NEs resulted in completely non-specific accumulation for the two cell lines. In contrast, clearly improved uptake was detected for CD44BP–PEG–NEs, which is therefore specific for CD44 over-expressing tumor cells (U-87). The specificity of our system is also confirmed by the very weak signal found in HUVEC cells (Fig. 8).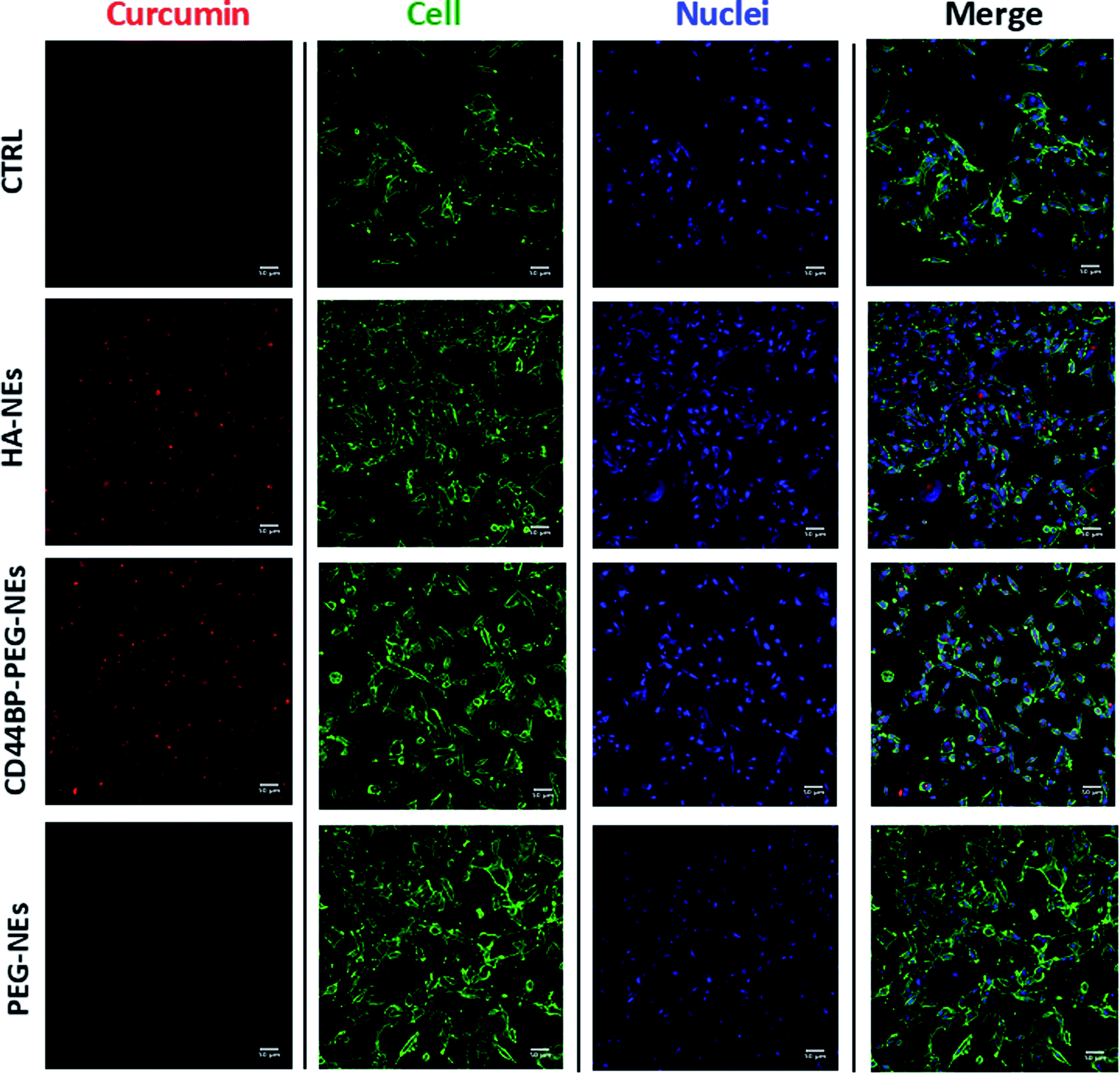 | ||
| Fig. 6 Confocal images of U-87 cells. Untreated, curcumin-loaded HA–NEs, CD44BP–PEG–NEs and PEG–NEs interactions with a confluent monolayer of U-87. Nuclei (blue) and cellular membranes (green) were stained with DAPI and WGA 555 respectively, while curcumin, and therefore the extent of its uptake, is displayed in red. Scale bar is 50 μm. | ||
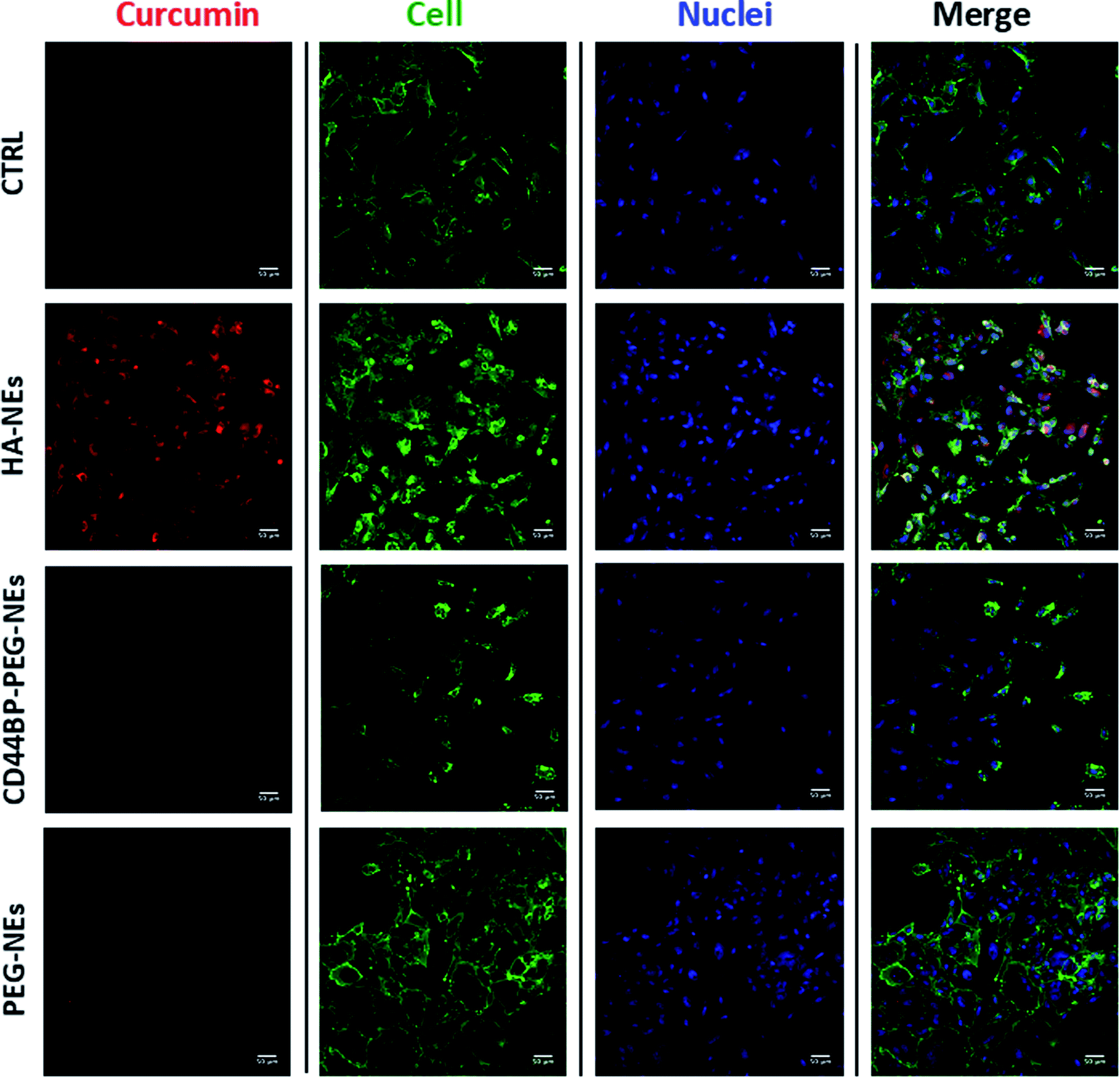 | ||
| Fig. 7 Confocal images of HUVEC cells. Untreated, curcumin loaded HA–CT–O/W NEs, CD44BP–PEG–O/W NEs and PEG–O/W NEs interactions with a confluent monolayer of HUVEC. Nuclei (blue) and cellular membranes (green) were stained with DAPI and WGA 555 respectively, while curcumin, and therefore the extent of its uptake, is displayed in red. Scale bar is 50 μm. | ||
 | ||
| Fig. 8 (A) Plot of mean fluorescence intensity of curcumin normalized to cell number. U-87 and HUVEC cells were treated with curcumin loaded in HA–NEs, CD44BP–PEG–NEs and PEG–NEs. Data are reported as mean ± SD (n = 3). (B) Confocal images of U-87 and HUVEC cells. Untreated, curcumin loaded HA–NEs, CD44BP–PEG–NEs and PEG–NEs interactions with a confluent monolayer of U-87 and HUVEC. Nuclei (blue) and cellular membranes (green) were stained with DAPI and WGA 555 respectively, while curcumin, and therefore the extent of its uptake, is displayed in red. Scale bar is 50 μm. | ||
3. Discussion
The strategy for the implementation of a targeting motif onto an O/W nanocarrier here proposed was based on the binding affinity between streptavidin and biotin, one of the strongest non-covalent assemblies currently known. Biotin tag enabled us to build up NCs by means of a step-by-step strategy, needed to avoid troublesome post-purification steps, which are hardly compatible with liquid systems, like NEs. Given its in situ and non-destructive nature, ITC is an excellent technique to study the interaction between nanoparticles and biomolecules.34 The titration of Biot–HA–NEs with SAV shows an enthalpically driven process, counterbalanced by an unfavourable entropic contribution to Gibbs energy, due to the formation of new interactions between biotin and SAV upon binding. As expected, the unfavourable entropic contribution can be a consequence of the loss of conformational degrees of freedom in both the interacting molecules. By comparing these results to those obtained by titrating free Biot–HA polymer (Fig. S.6 and Table S.5†), the value of ΔG is essentially the same, and nonetheless both enthalpic and entropic contributions were found significantly higher in absolute value. This may be explained by a more favourable biotin exposition outside the carrier, or by a more intrinsically ordered conformation of the HA polymer when deposited around the NC. Nevertheless, we found that LbL technique does not affect biotin–SAV stoichiometry, as provided by n* value that was found essentially the same either when the polymer was dissolved in the bulk solution or when it was deposited above the CT–NEs. Therefore, we have clear indication that LbL technique has not been bearing on availability of biotin to bind SAV. A realistic idea of the number of biotin molecules that are involved in the binding can be calculated as follows. According to the NMR data, biotinylation of HA has a fractional yield of 37%, meaning that 37% of carboxylic groups on the total amount of HA monomers was actually functionalized with biotin. This correspond to ∼244 biotin molecules linked for each Biot–HA, by averaging the HA polymer molecular weight. Conversely, ITC data shows that the amount of biotin molecules (conjugated to HA) available to bind the SAV was <1% (∼approximately 2 streptavidin over 244 available biotins), for both the free Biot–HA and the assembled NC. A possible explanation for such a difference derives from the large repertoire of conformations that HA could assume. Therefore, numerous biotin molecules could be hidden inside the network of the polymer chains.The successive functionalization step was performed with the aim to provide the NC with an active recognition module. To this end, CD44BP was preventively PEGylated, not only to increase NC shelf life, but also to enhance exposition on the carrier. Solid-phase synthesis significantly simplifies the purification of unreacted maleimide–PEG–biotin from PEGylated peptide, which differs only by 2000 Da. The completely functionalized NCs were further characterized by DLS. As reported in Table 1, an increment of size and Z-potential inversion occurred passing from NEs to CT–NEs and Biot–HA–NEs because chitosan forms a positive layer around the oil core and Biot–HA forms a negative core. The next steps of conjugation did not change significantly the nanoparticle size, in agreement with the low degree of bound biotin and the small dimensions of SAV (∼4–7 nm) with respect to the NC overall size.35,36
One of the strengths of O/W NE, which is the core of our NCs, is the possibility to carry a huge amount of hydrophobic compounds, such as curcumin, which can be, therefore, loaded at mM concentrations.
Curcumin has been shown to suppress multiple signalling pathways and inhibit cell proliferation, invasion, metastasis, and angiogenesis, by controlling several genes involved in cell death.37 However, curcumin bioactivity is limited by its poor water solubility, reported to be 11 ng mL−1 (0.03 μM) in aqueous buffer (pH 5).38 This feature makes the preparation of aqueous solution of curcumin for intravenous use extremely hard.39,40 Chao Cheng et al. reported that the inhibitory effects of free curcumin (dissolved in DMSO) in U-87 cell line are mostly evident when the free molecule is used at a concentration of 40 μM (this concentration was reached by adding DMSO).41 It has also been shown, by intravenous administration (i.v.) in rabbits, that an aqueous nanosuspension of 25.6 μM curcumin (CUR-NS) displays a significantly different pharmacokinetic compared to the free molecule (dissolved in dimethyl acetamide), with a mean residence time which is 11.2-fold longer.42
In this context, with our CD44BP–PEG–NEs (2.8 wt% of oil), featured by a curcumin concentration of 312 μM, we expect an increment of curcumin bioavailability for i.v. administration. We based this hypothesis not only on the increase of curcumin concentration, but also on its increased half-life along with the targeting motif we used to functionalize our NC for active targeting. NEs protect curcumin from hydrolysis and degradation, thus keeping intact its anti-cancer activity.43 In addition, NEs were functionalized with a PEGylated peptide, which not only accumulates to cancer site thanks to peptide-receptor recognition, as previously stated, but also it improves the half-life of NCs thanks to the anti-fouling properties of PEG chains.
Overall, the results of cytotoxicity assays clearly highlight that peptide functionalized NCs are able to target cancer cells in a specific and selective manner respect to HA–NEs. However, as described before, the concentration of loaded curcumin in our NCs is so high that may induce a slight toxicity. This effect justifies the detectable toxicity we observed for both free curcumin and curcumin loaded HA–NEs in the case of HUVEC cells.44,45 As expected, for both cell lines, PEG–NEs has demonstrated to be totally safe, confirming the complete shielding provided by PEG chains and also the validity of the here proposed approach.
Moreover, cellular uptake assay showed that HA was unable to be highly specific for tumor cells under our experimental conditions, while it did not affect healthy ones, although HA is largely used as ligand for active targeting. On the opposite, our peptide-functionalized NCs not only displayed tumor-selectivity, but also accumulated into U-87 51.5% more than HA-coated O/W NEs. In addition, PEG–NEs were not detected in both the cell lines, highlighting that the ligand-receptor interaction is responsible for NCs internalization.
4. Conclusion
In this work, we propose a surface functionalization of oil core polymer NCs, featuring a bioactive peptide on their surface, namely CD44BP, which recognizes CD44 receptor, for active targeting. Indeed, we demonstrated that functionalized NCs could treat cancer cells in a specific and efficient way, thanks to CD44BP–receptor binding. As an example, oil-based NC was loaded with curcumin as anticancer compound. Noteworthy, our approach holds broad application, because the hydrophobic nature of oil allows loading different synthetic anticancer drugs, such as, paclitaxel. Moreover, we introduced biotin moieties on the HA polymer, which constitutes the external layer of the oil-based carrier. Thanks to high binding affinity with biotin, SAV was used as linker to conjugate the synthetized CD44BP peptide to the entire system, while keeping nanocarrier stability. Nevertheless, our system may be considered fully modular, meaning that each module may be easily exchanged and tailored according to the specific requirements of the final target. In doing that, an easy additive approach was used for NCs assembly, exploiting ITC analysis to evaluate the right amount of the components to be added step by step, avoiding successive purification steps, thus, simplifying the overall preparation procedure. A well-established performance comparison between curcumin loaded CD44BP–PEG–NEs and HA–NEs was performed. U-87 and HUVEC cell lines were chosen as model of cancer site and healthy one respectively. Our results clearly demonstrate not only the ability of peptide-functionalized NCs to be tumor-selective, but also to accumulate in CD44 over-expressing cells 51.5% more than HA–NEs does. Although HA was largely used as ligand for active targeting, we show its inability to be specific for tumor cells. Our results are particularly encouraging and in vivo performance of our newly designed NCs against cancer cells specifically expressing CD44 will be soon performed. In the future, other targets will be explored by opportune choice of the targeting moiety.5. Experimental section
5.1 Materials
Soybean oil and surfactant Lipoid E80 (egg lecithin powder 80–85% enriched with phosphatidylcholine and 7–9.5% content in phosphatidylethanolamine) were purchased from Lipoid GmbH and used without further purification. For preparation of all nanoemulsions and solutions, Millipore Milli-Q water was used. Chitosan (CT, LMW 90–150 kDa, DDA 84% determined via 1H-NMR). 1-Ethyl-3-(3-dimethylaminopropyl)carbodiimide (EDC), N-hydroxysuccinimide (NHS), biotin hydrazide, curcumin (from Curcuma longa Turmeric, powder), streptavidin (SAV), 1-hydroxybenzotriazole hydrate (HOBt), N,N′-diisopropylcarbodiimide (DIC), N,N-diisopropylethylamine (DIEA), trifluoroacetic acid (TFA), dimethyl sulfoxide (DMSO), dichloromethane (DCM), anhydrous N,N-dimethyl-formamide (DMF), N-methyl-2-pyrrolidone (NMP), 1,2-ethanedithiol (EDT), triisopropylsilane (TIS) piperidine, acetone, diethyl ether, dimethyl sulfoxide (DMSO) and deuterium oxide (D2O) were purchased from Sigma Aldrich. Hyaluronic acid 250 kDa and biotin–PEG2k–maleimide were purchased from Creative PEGWorks. Atto 655-SAV was purchased from Atto-Tec. Dialysis membranes were purchased from Spectrum Laboratories Inc. Hyaluronic acid 250 kDa and biotin–PEG2k–maleimide were purchased from Creative PEGWorks. N-α-Fmoc amino acids, HCTU and 5-carboxy fluorescein were purchased from NovaBiochem.5.2 Particle size and Z-potential measurements
Nanoemulsion-based nanocarriers were characterized at each step of preparation by measuring size and polydispersity index (PdI), using Zetasizer Nano ZS device (Malvern Instruments) with a 4 mW He-Ne ion laser at the wavelength of 633 nm and a photodiode detector at an angle of 173°. All the samples were diluted to a droplet concentration of 0.025 wt% by using acetic acid 20 mM at pH 4 for monolayer, and Milli-Q water for emulsions and bilayer suspensions. The calculation of the particle size distribution was performed using a default refractive index ratio (1.59) and 5 runs for each measurement (1 run lasting 100 s), at least 3 times for each sample. A particle electrophoresis instrument (Zetasizer zs nano series ZEN 3600, Malvern Instruments Ltd., Malvern, U.K.) was used for the Z-potential determinations. Samples were diluted as for the particle size analysis. Setting 50 runs for each measurement carried out the Z-potential analysis. Samples were collected into polystyrene cuvettes and measured three times and the results presented are the averages of these measurements. Experiments were carried out at 25 °C. Zetasizer software (Malvern Instruments) was used to obtain the data. Cumulative analysis was used to give the Z-average value, hydrodynamic diameter, polydispersity index and the intensity size distribution graphs.5.3 Preparation of biotinylated hyaluronic acid
The biotin–hyaluronic acid (Biot–HA) conjugate was prepared as follows: 0.52 mmol of NHS and 0.52 mmol of EDC were added to a 1% (w/v) solution of HA (10 mL solution, 0.26 mmol HA monomer) in water, pH 6.0, under stirring at room temperature for 60 min to facilitate a homogeneous dispersion of reagents in the reaction solution. Then an amount of 0.052 mmol of biotin hydrazide, dissolved in DMSO, was added to the reaction mixture, overnight. The resulting polymer was dialyzed against Milli-Q water using a 12–14 kDa dialysis membrane. The water was changed twice a day for 3 days, and then the polymer was lyophilized.285.4 Nuclear magnetic resonance spectroscopy (NMR)
Biot–HA was characterized by Nuclear Magnetic Resonance (NMR). NMR spectra were recorded using an Agilent 600 MHz (14 T) spectrometer equipped with a DD2 console and a One NMR HX probe. Biot–HA (1 mg) was dissolved in 600 μL of 90/10 H2O/D2O solution.1H 1D spectra were recorded at 300 K using 1024 scans to obtain a good signal-to-noise ratio. Water signal was reduced using a PRESAT pulse sequence. Spectra were transformed and analyzed using VNMRJ 4.0 software.
5.5 Oil-in-water nanoemulsion
Firstly, a 20 wt% oil in water pre-emulsion was prepared. 5.8 g of Lipoid E 80 (egg lecithin powder 80–85% enriched with phosphatidyl choline and 7–9.5% content in phosphatidyl ethanolamine) were dissolved in 24 mL of soybean oil (density at 20 °C of 0.922 g mL−1) at 60 °C using the immersion sonicator (Ultrasonic Processor VCX500 Sonic and Materials), performing runs of 10 s for 1 min at 10% of sonication amplitude (microtip screwed). Then, 100 mg of curcumin were dissolved in the oil phase for 15 min at 60 °C during pre-emulsion preparation. Subsequently, the oil phase was added to the aqueous phase (Milli-Q water), and mixed using the immersion sonicator with runs of 10 s for 8 min at 70% of amplitude (a pulse-on and a pulse-off respectively of 10 s). The pre-emulsion was finally homogenized for 3 single cycles and 200 steps at a pressure of 2000 bar by a high-pressure homogenizer (110P series microfluidizer) to obtain the final nanoemulsion.5.6 Polymers deposition above oil in water nanoemulsion
Firstly, a layer of CT was deposited around the oil template at a final concentration of oil and chitosan of 10 wt% and 0.1 wt%, respectively. 0.1 M acetic acid solution of CT (0.125 wt%) was prepared with a final pH = 4.20 wt% oil nanoemulsion was added quickly to the CT solution under vigorous stirring and kept under stirring for 15 min to allow uniform CT deposition. The nanoemulsion with the first positive layer of CT was passed through a high-pressure valve homogenizer at 700 bars for 100 continuous steps. The next hyaluronic acid layer was prepared by aid of two syringe pumps (HARVARD APPARATUS 11 PLUS) and an ultrasonic bath (FALC INSTRUMENTS). Starting from the secondary nanoemulsion 10 wt% oil–0.1 wt% CT, a negative charged polymer layer was deposited by mixing 1:1 (v:v) of a 0.24 wt% aqueous solution of biotinylated hyaluronic acid with the secondary nanoemulsion suspension. The two liquid phases were injected at the same flow rate (0.4 mL min−1) through two polymicro flexible fused silica capillaries (inner diameter of 200 μm) interfaced at their extremities (Molex). Each drop was then collected inside a glass tube immersed in the ultrasonic bath at room temperature, 59 kHz and 100% power for 15 min.
5.7 Peptide synthesis and purification
Targeting peptide was synthesized manually in a modified version, with a spacer of three glycine residues and a cysteine residue at the N-terminal (H-CGGG-RLVSYNGIIFFLK-NH2). Protocols involving the use of 9-fluorenylmethoxycarbonyl (Fmoc) chemistry were used. The peptide scale synthesis was 0.1 mmol. It was assembled on an H-Rink-amide ChemMatrix® resin with a substitution level of 0.4 mmol g−1, using solid-phase synthetic strategy.Once loaded the resin in the reaction vessel, it was swelled three times for 3 min with DMF and three times with NMP for 3 min.
The following protected amino acids were used to synthesize the peptide:
Fmoc-Lys(Boc)-OH; Fmoc-Leu-OH; Fmoc-Phe-OH; Fmoc-Ile-OH; Fmoc-Gly-OH; Fmoc-Asn(Trt)-OH; Fmoc-Tyr(tBu)-OH; Fmoc-Ser(tBu)-OH; Fmoc-Val-OH; Fmoc-Arg(Pbf)-OH; Fmoc-Cys(Mmt)-OH; Fmoc-Ala-OH.
Fmoc group was removed at the beginning of cycle with a 20% piperidine solution in DMF. After deprotection, the resin was washed with DMF to remove the residual piperidine. The peptide resin was then ready for coupling. The carboxyl group of each Fmoc-amino acid was activated by addition of 7.5 equivalents of HCTU 0.48 M. Single coupling was conducted for each amino acid by addition of 15 equivalents of N,N′-diisopropylethylamine. The pre-activated Fmoc-amino acid reacted with the free amino-terminal group of the growing peptide chain on the resin using DMF as the reaction solvent. The resin was tested for the presence of unreacted amines using the Kaiser reagent. If the test was positive, the coupling reaction was repeated. Capping cycle was introduced after each coupling step to prevent deletion by-products using Ac2O/HOBt/DIEA solution in DMF. Deprotection, coupling and capping steps were repeated with each subsequent amino acid, until the chain assembly was completed. When the coupling was complete, the resin was washed with DMF. The peptide was cleaved from the resin by treating with 94% TFA/2.5% EDT/2.5% water/1% TIS for 2 h. The mixture was then concentrated under reduced pressure and transferred to glass centrifugal tubes for compound precipitation using ice-cold diethyl ether, which was performed repeatedly.
5.8 5-Carboxyfluorescein–peptide conjugation
The peptide was conjugated at the N-term with 5-carboxyfluorescein (5-FAM). Another non-α-amino acid (β-alanine) was introduced at the N-term before the coupling reaction with 5-FAM to avoid an Edman-type elimination reaction.46 β-Alanine conjugation at the N-term was performed as described before. The coupling reaction of 5-carboxyfluorescein was conducted on the resin with DIC/HOBt/DIEA (1:1:2) 0.1 M using DMF as solvent, overnight under nitrogen flow. At completion of the synthesis, the resin was washed several times with DMF, NMP, DCM, isopropanol and methanol, and finally dried.
5.9 Biotin–PEG2k–maleimide CD44BP conjugation
Biotin–PEG2k–maleimide (from now on PEG) was linked to the peptide thanks to covalent bond between maleimide and the lateral chain of cysteine. Firstly, Mmt protecting group of cysteine was selectively removed whilst the peptide remained attached to the solid phase. It was performed by several wash cycles using 1% trifluoroacetic acid (TFA), 5% triisopropylsilane (TIS) in DCM. The cleavage is repeated at least 10 times until no more yellow color, due to Mmt release, can be detected. The thiol free group of cysteine was conjugated to the maleimide group of biotin–PEG–maleimide (1:5) directly on resin, using DMF as solvent and 10 molar excess of DIEA, overnight under nitrogen flow. At completion of the synthesis, the resin was washed several times with DMF, NMP, DCM, isopropanol and methanol to remove unreacted linker. Biotin–PEG–maleimide-peptide was cleaved from the resin by treating with 94% TFA/2.5% EDT/2.5% water/1% TIS for 2 h, precipitated in ice-cold diethyl ether and lyophilized.
5.10 MALDI-TOF analysis of PEGylated peptides
PEGylated peptide was characterized by matrix-assisted laser desorption/ionization mass spectrometry coupled to two time of flight detectors (MALDI-TOF-TOF). The sample was prepared with a final concentration of ∼2 pmol μL−1 in the matrix by mixing the peptide with a solution of 60% of α-cyano-4-hydroxycinnamic acid (CHCA) and 40% of 5-dihydroxybenzoic acid (DHB).The two matrix solutions were prepared as follows:
1. 20 mg mL−1 of CHCA in a solution of H2O 5% formic acid in ACN (30/70 v/v)
2. 20 mg mL−1 of DHB in a solution of H2O 0.1% TFA in ACN (30/70 v/v)
Approximately, 0.25 μL of the sample was deposited on the MALDI plate, after a layer deposition of a saturated solution of CHCA in acetone, and allowed to dry prior to analysis. The mass spectra were recorded on an AB SCIEX TOF/TOF 5800 instrument operated in the reflector positive mode. MALDI-TOF MS analyses were conducted at a laser intensity of 4287 units and laser pulse rate of 400 Hz with a set mass range of 1000 to 6000 Da. A continuous stage motion set in a random pattern at 600 μm s−1 was used for sampling. Calibration was performed using Cal mix 5 from AB SCIEX as calibrants, which contained des-Arg1-bradykinin, angiotensin I, Glu1-fibrinopeptide B, adrenocorticotropic hormone ACTH (1–17 clip), ACTH (18–39 clip) and ACTH (7–38 clip) resulting in a mass accuracy of 50 ppm. Each spectrum represents the sum of 2040 laser pulses from randomly chosen spots per sample position. Raw data were analyzed using TOF/TOF Series Explorer software provided by the manufacturer and are reported as monoisotopic masses.
5.11 Nanocarrier assembly
The SAV solution was prepared by dissolving 1 mg in 1 mL of Milli-Q water (16.6 μM). It was added to the Biot–HA 0.12 wt%–CT 0.05 wt%–NEs 5 wt% oil, under sonication for 15 min and T = 20 °C, at a final concentration of 5.69 μM. In the same way the compound CD44BP–PEG–biotin was added under sonication for 15 min and T = 20 °C to the SAV–Biot–HA–NEs at a molar ratio 2:1 between CD44BP–PEG–biotin and SAV. The final concentrations were 3.2 μM and 6.4 μM for SAV and CD44BP, respectively, while the final oil weight percentage was 2.78 wt%. The NCs were characterized at each step of preparation measuring the size and Z-potential by dynamic light scattering.
5.12 Isothermal titration calorimetry analysis
Isothermal titration calorimetry (ITC) experiments were performed using a Nano ITC Low Volume from TA Instruments (USA) with a cell volume of 170 μL. Titration experiments were carried out at 25 °C. In the case of biotin hydrazide–SAV titration, the sample cell was filled with a solution of SAV 6 μM in PBS 10 mM, and the reference cell was filled with Milli-Q water for all the experiments. 25 injections of 2 μL each of biotin hydrazide 100 μM in PBS 10 mM were added to SAV solution every 200 s at a continuous stirring rate of 200 rpm. The interaction heat for each injection was calculated after correction for the heat coming from biotin hydrazide dilution that acts as blank. In the case of SAV–HA–biotin titration, the sample cell was filled with a solution of 0.05 wt% Biot–HA. It was titrated by 25 injections of 2 μL each of a solution of SAV 16 μM. For SAV–(Biot–HA–NEs) titration, the sample cell was filled with the 0.05 wt%-Biot–HA–0.02 wt%-CT–2.08 wt%-NEs solution, and the reference cell was filled with Milli-Q water. 25 injections of 2 μL each of a solution of SAV 16 μM were added to the sample cell, every 200 s at a continuous stirring rate of 200 rpm. Control experiments were performed between the solution of SAV 16 μM and the HA 0.05 wt% solution. The interaction heat for each injection was calculated after correction for the heat coming from SAV dilution that acts as blank. The binding constant (Ka), enthalpy change (ΔH°) and stoichiometry (n = binding sites) of the interaction process were obtained by fitting the binding isotherm to the equivalent and independent binding sites model, by using the NanoAnalyze software, version 2.4.1 (TA Instruments). The remaining thermodynamic parameters of the interaction were calculated using the relationships:| ΔG° = −RTlnKa |
| ΔG° = ΔH° − TΔS° |
5.13 Cryo-TEM characterization
For the preparation of the frozen-hydrated sample the plunge freezing method was performed. Briefly a drop of 3 μL of the samples were put on a previously glow-discharged 200 mesh holey carbon grids (Ted Pella, USA); after that, the grid was inserted in the chamber of a FEI Vitrobot Mark IV (FEI company, the Netherlands) at 4 °C and 90% of humidity. The sample droplet was blotted with filter paper for 1 s, (blot force 1, drain time 0.5 s) and then the grid was plunged into the liquid propane. The grid was then stored in liquid nitrogen in a grid box until it was, finally, transferred to a cryo-specimen 626 holder (Gatan, Inc., USA) and loaded into the cryo-transmission electron microscope for imaging. To obtain the image of the nanoparticles it was used a Tecnai G2 20, a cryo-tomo transmission electron microscope (FEI Company, the Netherland) equipped with LaB6 emitter (acceleration voltage of 200 kV) and recorded at with a 2k × 2k CCD-Eagle 2HS camera. The frozen-hydrated sample is radiation-sensitive material so to avoid any sample damage the observation was carried out in Low Dose Mode.5.14 Cell culture
U-87 human primary glioblastoma cells were grown in DMEM (10% FBS, 1% L-Glu, 1% penicillin streptomycin). Human umbilical vein endothelial cells (HUVECs) from Thermo Fisher Scientific were grown in Medium 200 supplemented with LSGS kit (Life Technologies). Cell culture was always performed at 37 °C in 5% CO2 and 95% relative humidity (RH).5.15 Cytotoxicity analysis
Cell viability was quantified by PrestoBlue assay (Invitrogen), non-treated cells were used as control. Briefly, 1 × 104 U-87 cells were seeded in a 96-well plate and incubated for different timepoints (30 min, 1 h, 2 h, and 4 h) with curcumin loaded CD44BP–PEG–NEs and free curcumin, diluted 1:5 in medium at a final curcumin concentration of 62.8 μM. Control experiments were carried out by treating cells with curcumin loaded CD44BP–PEG–NEs, HA–NEs and PEG–NEs for 4 h at the same experimental conditions described before. PrestoBlue assay was performed after 24 h according to the manufacturer's procedure. Fluorescence of PrestoBlue reagent solution (excitation 535 nm) was read at 615 nm by using a spectrofluorometer (Wallac 1420 Victor2, Perkin-Elmer, USA). All experiments were performed in triplicate.
5.16 Uptake of curcumin loaded NCs
U-87 human primary glioblastoma cells and HUVECs were grown in their respective medium as described before. After seeding, cells (2 × 105) were left 2 h to allow attachment. Then, they were incubated and treated with curcumin loaded CD44BP–NEs, HA–NEs and PEG–NEs, for 4 h in cell specific medium at 37 °C. Cells were then washed twice with PBS and fixed for 20 min in 4% PFA. Nuclei and cell membranes were labelled by DRAQ5 (excitation 633 nm) and WGA 555, respectively, while for curcumin the values set were 488 nm for excitation and 500–530 nm for emission. Fluorescence intensity was analysed by Zeiss LSM 710 confocal microscope. Images were reconstructed by ImageJ.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors want to thank Dr Fabio Formiggini for his support in confocal imaging and Dr Valentina Mollo for her support during cryo-EM experiments.References
- J. Yoo, C. Park, G. Yi, D. Lee and H. Koo, Active Targeting Strategies Using Biological Ligands for Nanoparticle Drug Delivery Systems, Cancers, 2019, 11(5), 640 CrossRef CAS.
- R. Bazak, M. Houri, S. El Achy, S. Kamel and T. Refaat, Cancer active targeting by nanoparticles: a comprehensive review of literature, J. Cancer Res. Clin. Oncol., 2015, 141, 769–784 CrossRef CAS.
- O. M. Koo, I. Rubinstein and H. Onyuksel, Role of nanotechnology in targeted drug delivery and imaging: a concise review, Nanomedicine, 2005, 1, 193–212 CrossRef CAS PubMed.
- J. K. Patra, et al., Nano based drug delivery systems: recent developments and future prospects, J. Nanobiotechnol., 2018, 16, 71 CrossRef.
- J. Liu, R. Zhang and Z. P. Xu, Nanoparticle-Based Nanomedicines to Promote Cancer Immunotherapy: Recent Advances and Future Directions, Small, 2019, 15, 1900262 CrossRef PubMed.
- R. H. Fang, A. V. Kroll and L. Zhang, Nanoparticle-Based Manipulation of Antigen-Presenting Cells for Cancer Immunotherapy, Small, 2015, 11, 5483–5496 CrossRef CAS.
- E. Calzoni, et al., Biocompatible Polymer Nanoparticles for Drug Delivery Applications in Cancer and Neurodegenerative Disorder Therapies, J. Funct. Biomater., 2019, 10, 4 CrossRef CAS.
- Y. Yan, X. Zuo and D. Wei, Concise Review: Emerging Role of CD44 in Cancer Stem Cells: A Promising Biomarker and Therapeutic Target, Stem Cells Transl. Med., 2015, 4, 1033–1043 CrossRef CAS.
- L. Cortes-Dericks and R. A. Schmid, CD44 and its ligand hyaluronan as potential biomarkers in malignant pleural mesothelioma: evidence and perspectives, Respir. Res., 2017, 18, 1–12 CrossRef.
- J. Lesley, R. Hyman, N. English, J. B. Catterall and G. A. Turner, CD44 in inflammation and metastasis, Glycoconjugate J., 1997, 14, 611–622 CrossRef CAS PubMed.
- S. Jothy, CD44 and its partners in metastasis, Clin. Exp. Metastasis, 2003, 20, 195–201 CrossRef CAS PubMed.
- L. A. Hefler, et al., The prognostic value of immunohistochemically detected CD44v3 and CD44v6 expression in patients with surgically staged vulvar carcinoma, Cancer, 2002, 94, 125–130 CrossRef.
- M. Makino, et al., Identification of cell binding sites in the laminin α5-chain G domain, Exp. Cell Res., 2002, 277, 95–106 CrossRef CAS PubMed.
- S. Hibino, et al., Identification of an active site on the laminin α5 chain globular domain that binds to CD44 and inhibits malignancy, Cancer Res., 2004, 64, 4810–4816 CrossRef CAS PubMed.
- S. Hibino, et al., Laminin α5 chain metastasis- and angiogenesis-inhibiting peptide blocks fibroblast growth factor 2 activity by binding to the heparan sulfate chains of CD44, Cancer Res., 2005, 65, 10494–10501 CrossRef CAS PubMed.
- X. Yang, S. K. Sarvestani, S. Moeinzadeh, X. He and E. Jabbari, Effect of CD44 Binding Peptide Conjugated to an Engineered Inert Matrix on Maintenance of Breast Cancer Stem Cells and Tumorsphere Formation, PLoS One, 2013, 8, 1–15 CrossRef.
- M. Zaiden, et al., CD44-Targeted Polymer–Paclitaxel Conjugates to Control the Spread and Growth of Metastatic Tumors, Mol. Pharm., 2018, 15, 3690–3699 CrossRef CAS.
- J. Szafraniec-Szczęsny, M. Janik-Hazuka, J. Odrobińska and S. Zapotoczny, Polymer capsules with hydrophobic liquid cores as functional nanocarriers, Polymers, 2020, 12, 1–25 CrossRef.
- S. Deng, M. R. Gigliobianco, R. Censi and P. Di Martino, Polymeric nanocapsules as nanotechnological alternative for drug delivery system: current status, challenges and opportunities, Nanomaterials, 2020, 10, 847 CrossRef CAS PubMed.
- R. Vecchione, et al., Ultrastable Liquid–Liquid Interface as Viable Route for Controlled Deposition of Biodegradable Polymer Nanocapsules, Small, 2016, 12, 3005–3013 CrossRef CAS PubMed.
- S. Lim, et al., Glioblastoma-secreted soluble CD44 activates tau pathology in the brain, Exp. Mol. Med., 2018, 50, 5 Search PubMed.
- J. Dai, et al., Osteopontin induces angiogenesis through activation of PI3K/AKT and ERK1/2 in endothelial cells, Oncogene, 2009, 28, 3412–3422 CrossRef CAS PubMed.
- C. M. Dundas, D. Demonte and S. Park, Streptavidin–biotin technology: improvements and innovations in chemical and biological applications, Appl. Microbiol. Biotechnol., 2013, 97, 9343–9353 CrossRef CAS PubMed.
- E. S. Shibu, et al., Singlet-Oxygen-Sensitizing Near-Infrared-Fluorescent Multimodal Nanoparticles, Angew. Chem., Int. Ed., 2013, 52, 10559–10563 CrossRef CAS PubMed.
- Z. Chai, et al., A facile approach to functionalizing cell membrane-coated nanoparticles with neurotoxin-derived peptide for brain-targeted drug delivery, J. Controlled Release, 2017, 264, 102–111 CrossRef CAS PubMed.
- T. Fotticchia, et al., Enhanced Drug Delivery into Cell Cytosol via Glycoprotein H-Derived Peptide Conjugated Nanoemulsions, ACS Nano, 2017, 11, 9802–9813 CrossRef CAS PubMed.
- R. Juliano, Biological Barriers to Nanocarrier-Mediated Delivery of Therapeutic and Imaging Agents, in Nanobiotechnology II: More Concepts and Applications, John Wiley and Sons, 2007, pp. 261–284 Search PubMed.
- B. Polyak, S. Geresh and R. S. Marks, Synthesis and characterization of a biotin–alginate conjugate and its application in a biosensor construction, Biomacromolecules, 2004, 5, 389–396 CrossRef CAS PubMed.
- T. Pouyani and G. D. Prestwich, Biotinylated Hyaluronic Acid: A New Tool for Probing Hyaluronate–Receptor Interactions, Bioconjugate Chem., 1994, 5, 370–372 CrossRef CAS PubMed.
- I. Gatej, M. Popa and M. Rinaudo, Role of the pH on Hyaluronan Behavior in Aqueous Solution, Biomacromolecules, 2005, 6(1), 61–67 CrossRef CAS PubMed.
- R. Vecchione, et al., Tunable stability of monodisperse secondary O/W nano-emulsions, Nanoscale, 2014, 6, 9300–9307 RSC.
- C. M. Dundas, D. Demonte and S. Park, Streptavidin–biotin technology: improvements and innovations in chemical and biological applications, Appl. Microbiol. Biotechnol., 2013, 97, 9343–9353 CrossRef CAS PubMed.
- S. Mun, E. A. Decker and D. J. McClements, Effect of molecular weight and degree of deacetylation of chitosan on the formation of oil-in-water emulsions stabilized by surfactant–chitosan membranes, J. Colloid Interface Sci., 2006, 296, 581–590 CrossRef CAS PubMed.
- I. Fotticchia, et al., Energetics of ligand–receptor binding affinity on endothelial cells: an in vitro model, Colloids Surf., B, 2016, 144, 250–256 CrossRef CAS.
- S. D. Perrault and W. C. W. Chan, In vivo assembly of nanoparticle components to improve targeted cancer imaging, Proc. Natl. Acad. Sci. U. S. A., 2010, 107(25), 11194–11199 CrossRef CAS PubMed.
- H. P. Erickson, Size and Shape of Protein Molecules at the Nanometer Level Determined by Sedimentation, Gel Filtration, and Electron Microscopy, Biol. Proced. Online, 2009, 11, 32 CrossRef CAS.
- J. Ravindran, S. Prasad and B. B. Aggarwal, Curcumin and Cancer Cells: How Many Ways Can Curry Kill Tumor Cells Selectively?, AAPS J., 2009, 11(3), 495–510 CrossRef CAS PubMed.
- H. H. Tønnesen, M. Másson and T. Loftsson, Studies of curcumin and curcuminoids. XXVII. Cyclodextrin complexation: Solubility, chemical and photochemical stability, Int. J. Pharm., 2002, 244, 127–135 CrossRef.
- K. Sigfridsson, A. J. Lundqvist and M. Strimfors, Particle size reduction for improvement of oral absorption of the poorly soluble drug UG558 in rats during early development, Drug Dev. Ind. Pharm., 2009, 35, 1479–1486 CrossRef CAS PubMed.
- Y. Gao, et al., Preparation, characterization, pharmacokinetics, and tissue distribution of curcumin nanosuspension with TPGS as stabilizer, Drug Dev. Ind. Pharm., 2010, 36, 1225–1234 CrossRef CAS.
- C. Cheng, et al., Curcumin induces G2/M arrest and triggers apoptosis via FoxO1 signaling in U87 human glioma cells, Mol. Med. Rep., 2016, 13, 3763–3770 CrossRef CAS PubMed.
- Y. Gao, et al., Preparation, characterization, pharmacokinetics, and tissue distribution of curcumin nanosuspension with TPGS as stabilizer, Drug Dev. Ind. Pharm., 2010, 36, 1225–1234 CrossRef CAS PubMed.
- R. Vecchione, et al., Curcumin bioavailability from oil in water nano-emulsions: In vitro and in vivo study on the dimensional, compositional and interactional dependence, J. Controlled Release, 2016, 233, 88–100 CrossRef CAS.
- H. J. Koo, et al., Introduction of Methyl Groups at C2 and C6 Positions Enhances the Antiangiogenesis Activity of Curcumin, Sci. Rep., 2015, 5, 14205 CrossRef CAS.
- B. Soltani, N. Bodaghabadi, G. Mahpour, N. Ghaemi and M. Sadeghizadeh, Nanoformulation of curcumin protects HUVEC endothelial cells against ionizing radiation and suppresses their adhesion to monocytes: potential in prevention of radiation-induced atherosclerosis, Biotechnol. Lett., 2016, 38, 2081–2088 CrossRef CAS.
- G. H. Bird, W. C. Crannell and L. D. Walensky, Chemical synthesis of hydrocarbon-stapled peptides for protein interaction research and therapeutic targeting, Curr. Protoc. Chem. Biol., 2011, 3, 99–117 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1ra03322k |
| This journal is © The Royal Society of Chemistry 2021 |