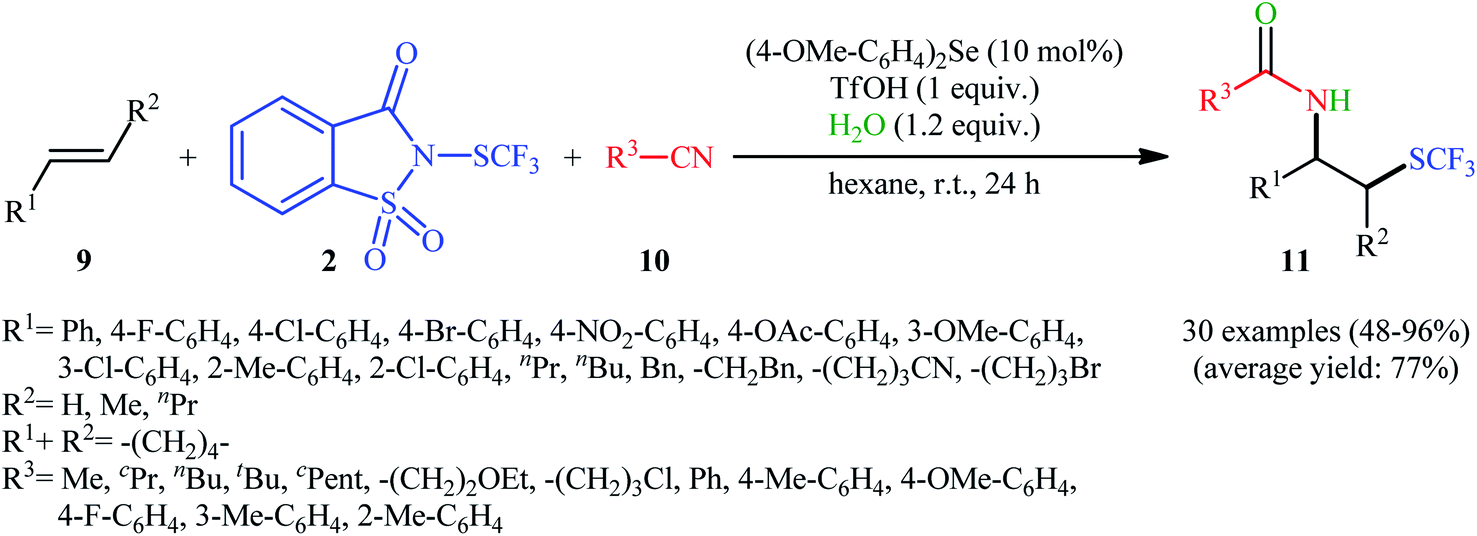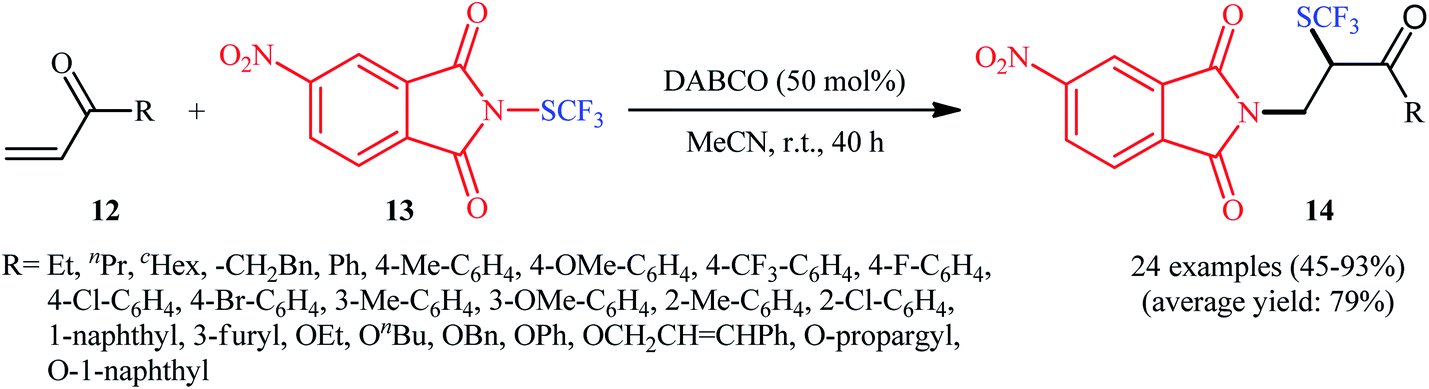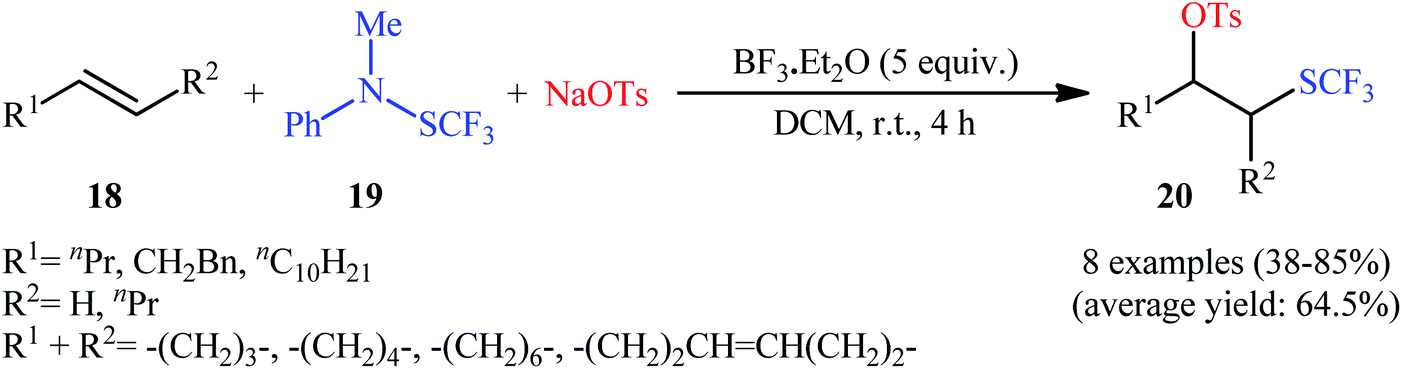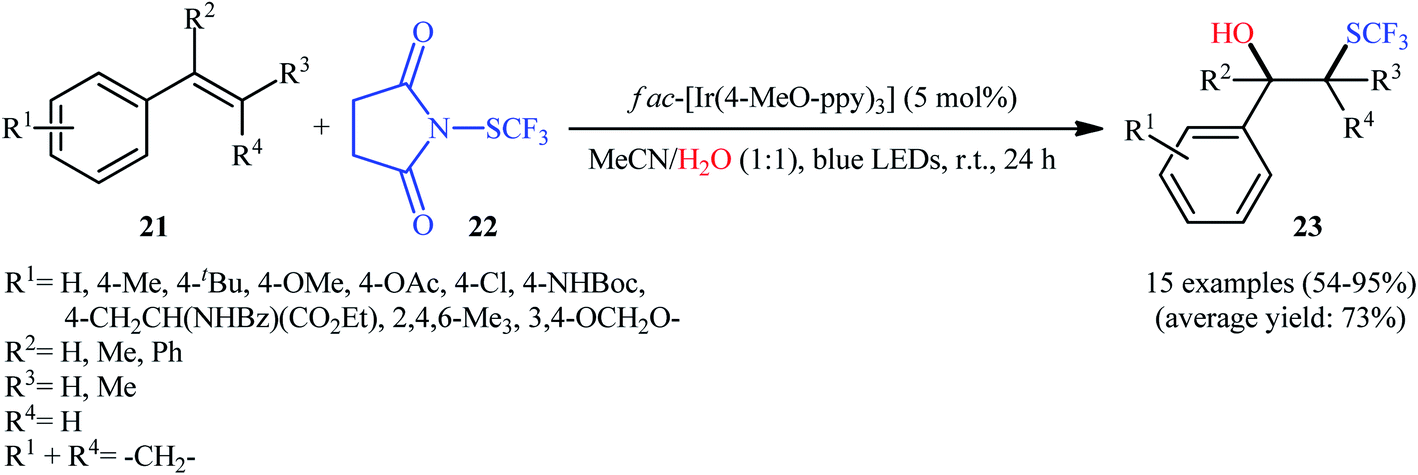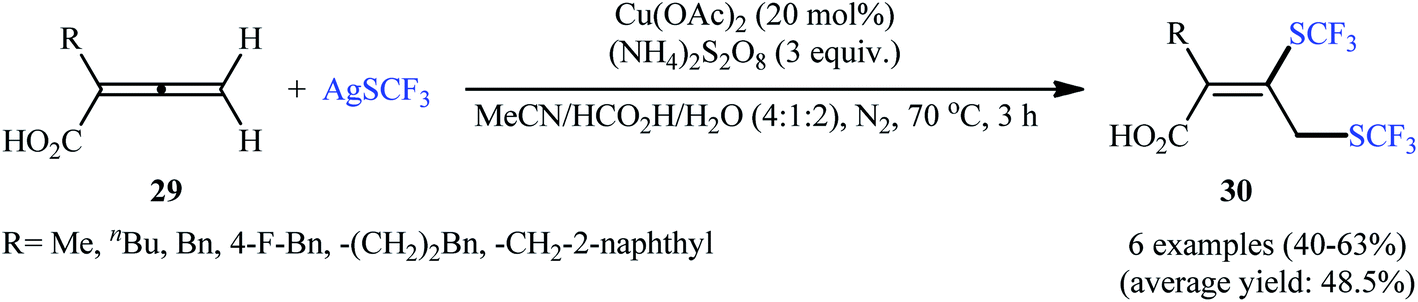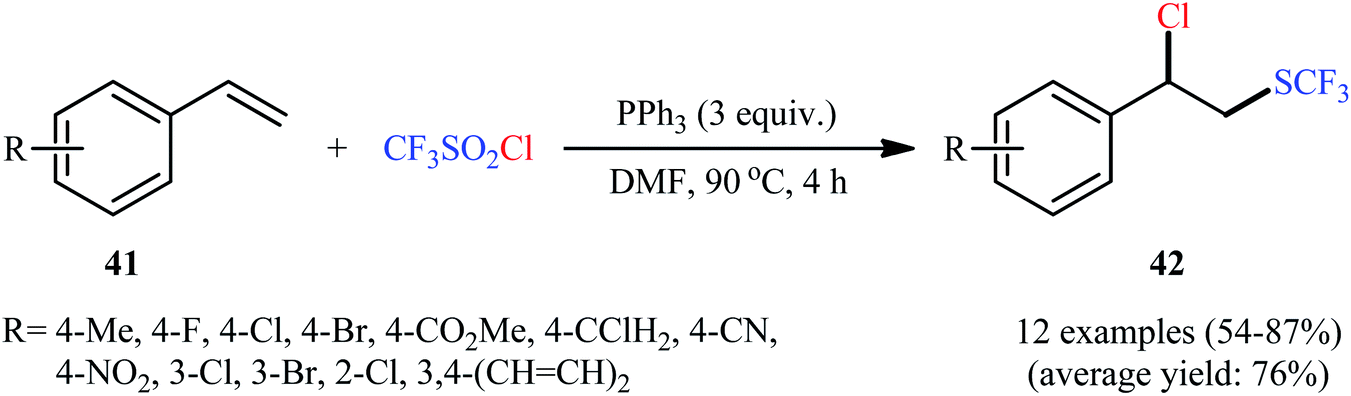DOI:
10.1039/D1RA02606B
(Review Article)
RSC Adv., 2021,
11, 24474-24486
Recent advances in intermolecular 1,2-difunctionalization of alkenes involving trifluoromethylthiolation
Received
2nd April 2021
, Accepted 17th June 2021
First published on 13th July 2021
Abstract
Trifluoromethylthiolative difunctionalization of alkenes, a cheap and abundant feedstock, which installs a trifluoromethylthiol (SCF3) group and another unique functional group across the carbon–carbon double bonds, provides an ideal strategy for the preparation of β-functionalized alkyl trifluoromethyl sulfides and has become a hot topic recently. This review aims to summarize the major progress in this exciting research area, with particular emphasis on the mechanistic aspects of the reaction pathways.
1. Introduction
Organofluorine compounds play an exceptionally important role in the field of medicinal chemistry and related areas due to the beneficial effects of the fluorine moiety on the physiochemical and biological properties of molecules.1,2 Intriguingly, up to 25% of the current blockbuster drugs3 and about 40% of contemporary agrochemicals4 contain at least one fluorine atom or a fluorinated functional group in their structures. The trifluoromethylthiol (–SCF3) group is one of the most important fluorine-containing substituents, which is found in many bioactive compounds and commercialized drugs, such as toltrazuril, tiflorex, and fipronil.5 Thus, the introduction of this functionality into organic molecules has gained increasing interest from synthetic organic chemists and significant progress has been achieved in this area over the past few years.6 On the other hand, the direct vicinal difunctionalization of alkenes has recently become a powerful and viable strategy for quickly increasing molecular complexity via concomitant introduction of two functional groups onto the carbon–carbon double bond within a single click.7,8 In this context, some important trifluoromethylthiolative difunctionalization reactions such as carbo-, halo-, amino-, thio-, seleno-, phosphono-, and oxy-trifluoromethylthiolation of alkenes have been developed to afford β-functionalized alkyl trifluoromethyl sulfide derivatives in one-pot with high selectivity (Fig. 1). Despite the significant advances in this exciting research topic over the past few years, no comprehensive and insightful review has appeared in the literature until now. Thus, it is an appropriate time to summarize those achievements. In connection with our recent reviews on organofluorine6f,9 and organosulfur chemistry,10 within this review, we will summarize recent discoveries and accomplishments in the arena of intermolecular trifluoromethylthiolative difunctionalization of alkenes, with the aim of stimulating further research in this hot research area.
 |
| | Fig. 1 Intermolecular trifluoromethylthiolative difunctionalization of alkenes. | |
2. Carbo-trifluoromethylthiolation
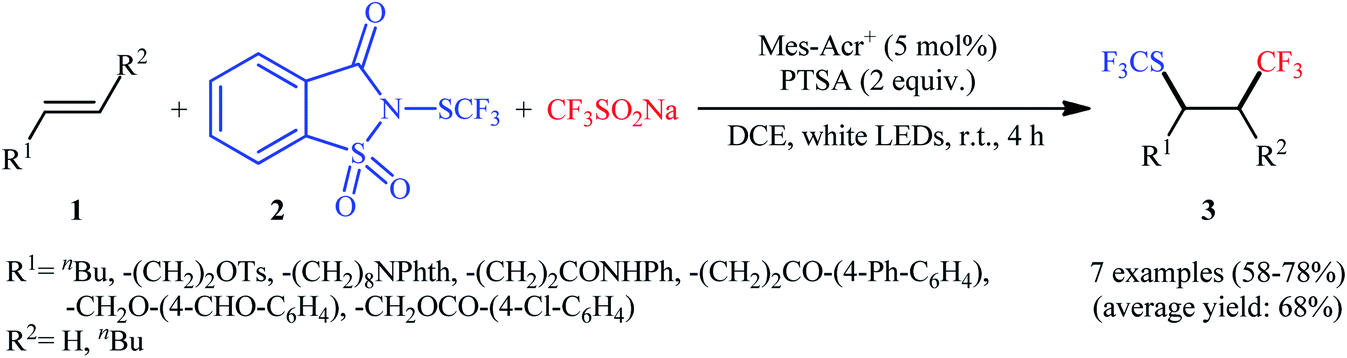
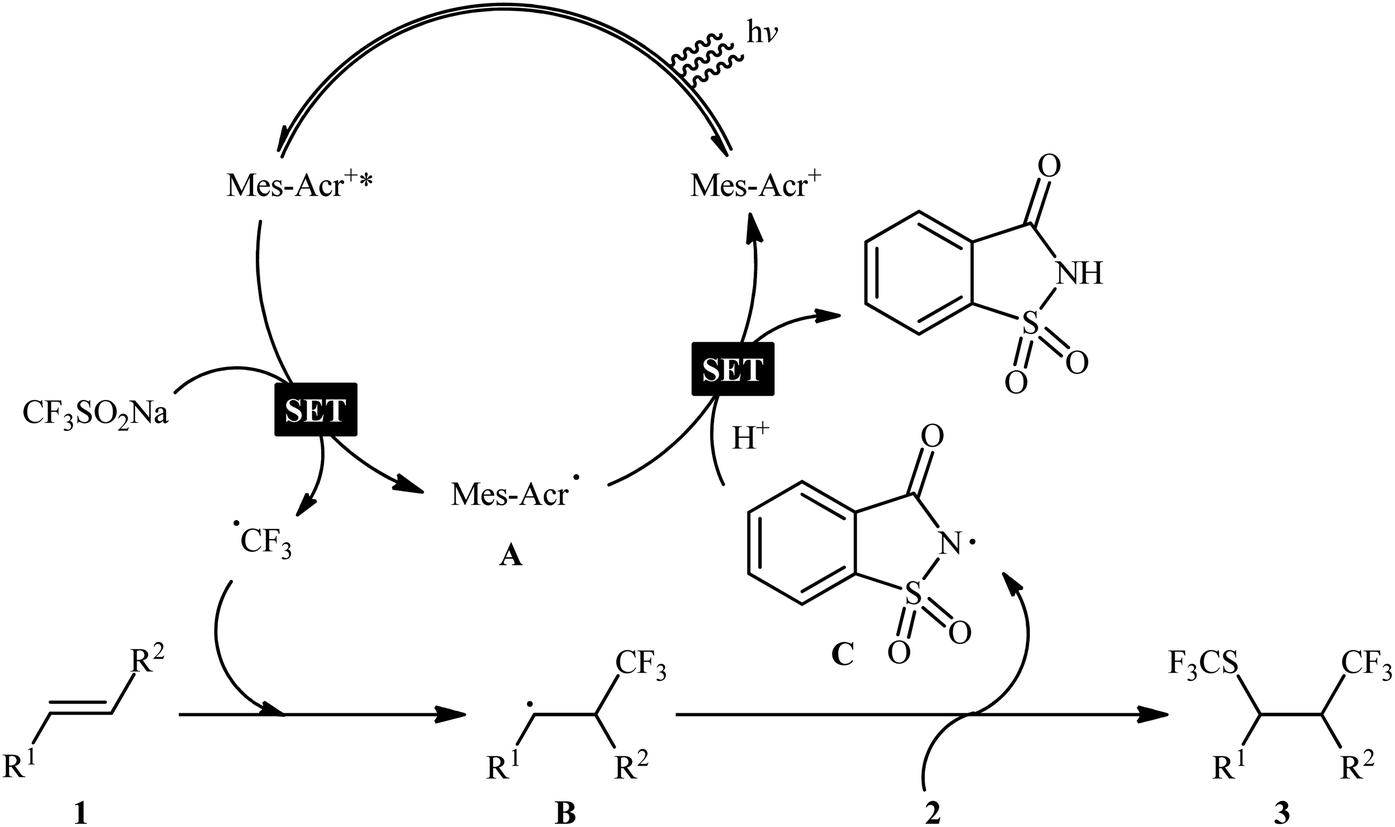
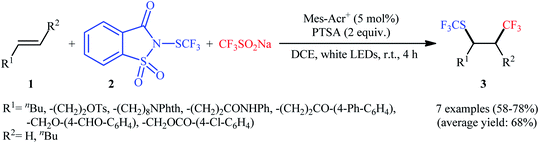
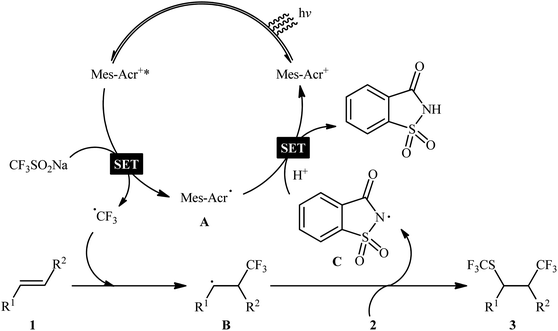
In 2017, Liu and co-workers reported a unique visible-light-driven trifluoromethylthiotrifluoromethylation of alkenes 1 using Langlois reagent (CF3SO2Na) as a solid and easily-handleable source of CF3 and N-trifluoromethylthiosaccharin 2 as a commercially available SCF3 source.11 The reaction was carried out in the presence of 9-mesityl-10-methylacridinium (Mes-Acr+) as an organic photoredox catalyst at room temperature under constant irradiation by a white LED, tolerated various terminal and internal aliphatic alkenes and afforded the corresponding vicinal trifluoromethylthio-trifluoromethylated compounds 3 in good yields and excellent levels of diastereoselectivity, in which the trifluoromethyl group predominantly added to the less sterically hindered end of the double bond (Scheme 1). Interestingly, by simply replacing N-trifluoromethylthiosaccharin reagent with N-chlorophthalimide and N-bromophthalimide, respectively, the optimized reaction conditions could be applied to the vicinal chlorotrifluoromethylation and bromotrifluoromethylation of alkenes. The authors evoked a reaction mechanism initiated by single electron oxidation of CF3SO2Na by the photoexcited Mes-Acr+* to yield a CF3 radical and an acridine radical A. Next, the CF3 radical selectively attacks the less hindered end of alkene 1 to form the β-trifluoromethyl carbon-centered radical B that, after coupling with the in situ generated SCF3 radical, affords the expected products 3. Finally, the nitrogen-centered radical C, which was generated through homolytic N–S bond cleavage of reagent 2, oxidizes the acridine radical by a single electron transfer (SET) process to regenerate the photocatalyst (Scheme 2). In a related investigation, Jiang and Yi along with their colleagues developed an efficient conversion of a diverse range of mono-, di-, and tri-substituted alkenes to the corresponding 1-CF3-2-SCF3-alkanes, using Langlois reagent as both the trifluoromethylating and trifluoromethylthiolating agent under a dual visible light photoredox/transition metal catalytic system.12
 |
| | Scheme 1 Visible-light-driven trifluoromethylthiotrifluoromethylation of alkenes 1 with CF3SO2Na and N-trifluoromethylthiosaccharin 2 reported by Liu. | |
 |
| | Scheme 2 Reaction mechanism for trifluoromethylthiotrifluoromethylation of alkenes 1. | |
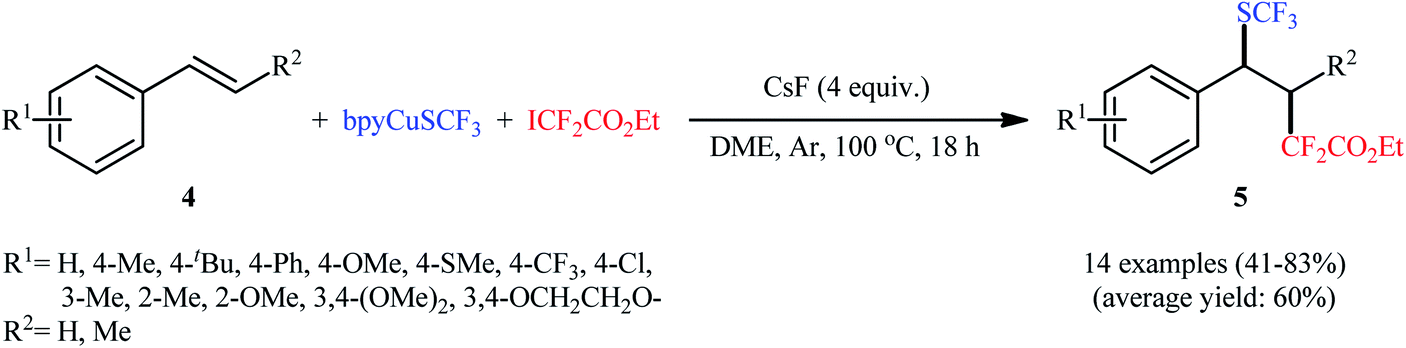
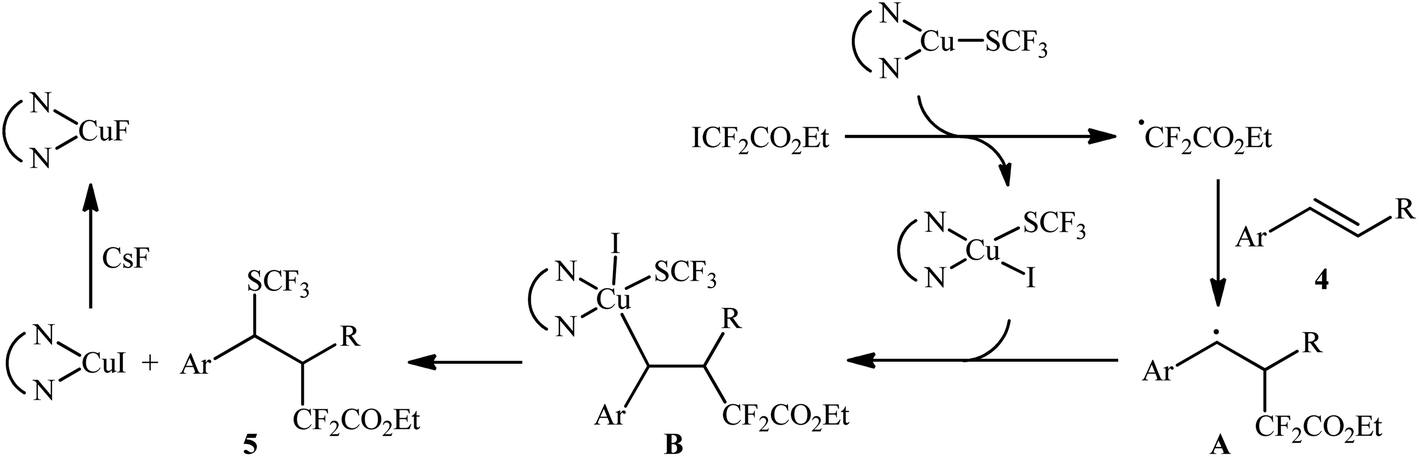
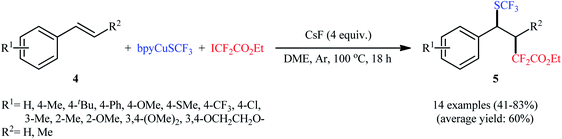
In this context, Liang's research group developed an operationally simple method for the direct trifluoromethylthiolation–difluoroalkylation of alkenes employing (bpy)CuSCF3 (bpy = 2,2′-bipyridine) and ethyl iododifluoroacetate (ICF2CO2Et) as trifluoromethylthiolating and difluoroalkylating reagents, respectively.13 Various styrene derivatives 4 slowly reacted with these reagents to afford the corresponding trifluoromethylthio-difluoroalkylated products 5 in moderate to high yields when running the reaction in the presence of CsF in DME at 100 °C (Scheme 3). Similar to trifluoromethylthiolation–difluoroalkylation, the method could also be used for trifluoromethylselenolation–difluoroalkylation using (bpy)CuSeCF3. However, aliphatic alkenes were not effective in this system. According to the mechanism proposed by the authors, (bpy)CuSCF3 not only serves as a SCF3 source, but also acts as a free-radical initiator of ICF2CO2Et (Scheme 4). Concurrently, a similar alkylative trifluoromethylthiolation of alkenes (aromatic and aliphatic) with NMe4SCN and alkyl halides (bromides, iodides) in the presence of CuI/binap under the condition of visible light was reported by Peters et al.14 A number of primary, secondary, and tertiary alkyl halides were used to establish the general applicability of this methodology. However, the presence of an electron-withdrawing substituent on the substrates is crucial for the success of this reaction.
 |
| | Scheme 3 Liang's trifluoromethylselenolation–difluoroalkylation of styrenes 4. | |
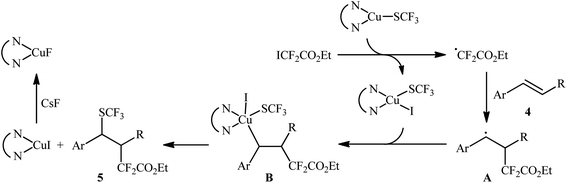 |
| | Scheme 4 The proposed pathway for the formation of trifluoromethylthio-difluoroalkylated products 5. | |
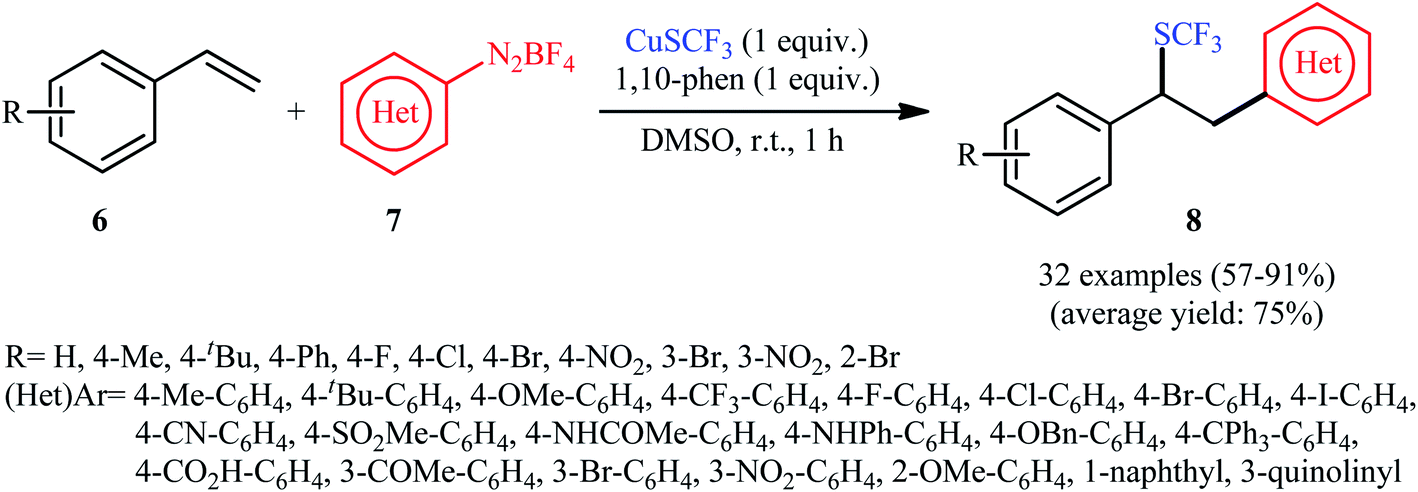
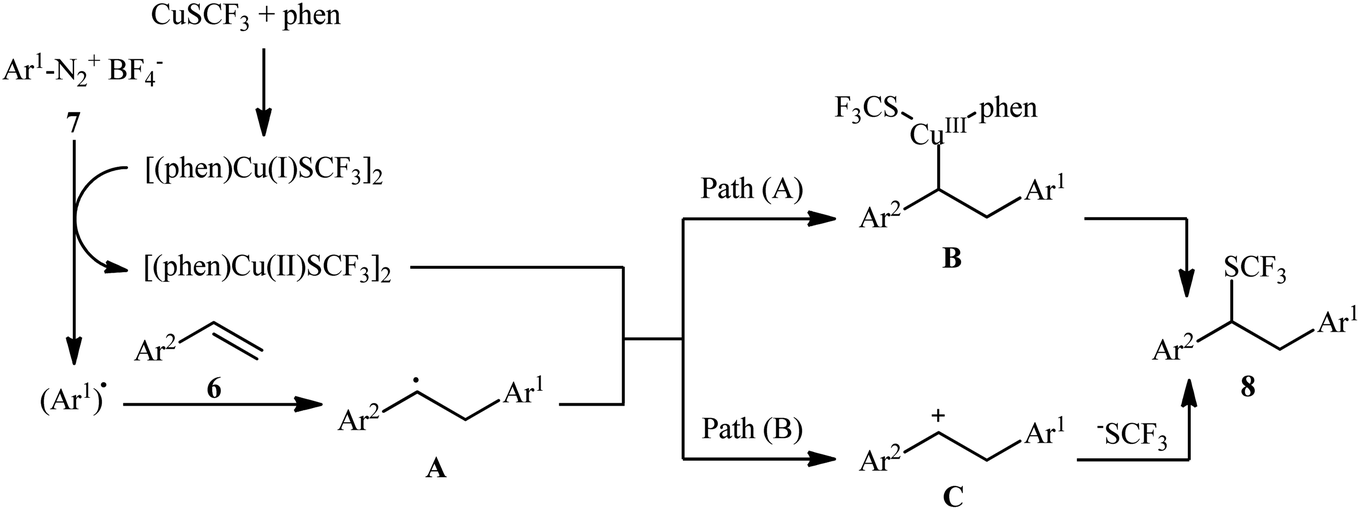
An attractive contribution to this field was reported by Chen et al.,15 who developed a convenient arylative trifluoromethylthiolation of styrenes 6 with arenediazonium salts 7 and copper(I) trifluoromethylthiolate (CuSCF3) to generate valuable (1,2-diarylethyl)(trifluoromethyl)sulfanes 8 (Scheme 5). Both electron-donating and electron-deficient functional groups (e.g., F, Cl, Br, I, OMe, NO2, CN, CO2H, and NHAc) on both substrates were well tolerated by this protocol. Thus, this procedure provides potential opportunities for further manipulation of the products. Heteroaromatic diazonium salts were also compatible substrates in this transformation. However, the method was ineffective for ortho-substituted electron-poor arenediazonium salts. The plausible mechanism proposed by the authors to explain this transformation is illustrated in Scheme 6. Initially, reaction between CuSCF3 and 1,10-phenanthroline (phen) leads to the formation of dimeric complex [(phen)Cu(I)SCF3]2, which after oxidation with arenediazonium salt 7 provides the corresponding copper(II) species and an aryl radical. Subsequent addition of the aryl radical to the styrene 6 results in the formation of benzylic radical A. Next, the reaction of the newly formed radical with [(phen)Cu(II)SCF3]2 affords Cu(III) complex B, which after reductive elimination provides the expected product 8 (Scheme 6, path a). In another possibility, oxidation of benzylic radical A by a Cu(II) cation leads to the formation of cationic intermediate C, which on subsequent attack by a CF3S ion yields the final desired product 3 (Scheme 6, path b).
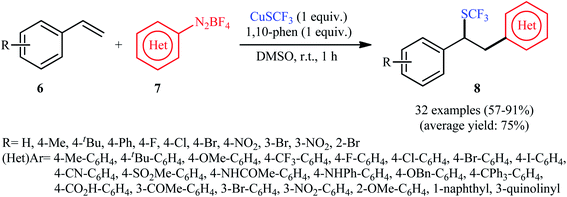 |
| | Scheme 5 Arylative trifluoromethylthiolation of styrenes 6 with arenediazonium salts 7 and CuSCF3. | |
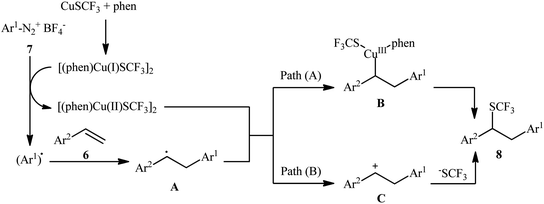 |
| | Scheme 6 Mechanism proposed to explain the formation of (1,2-diarylethyl)(trifluoromethyl)sulfanes 8. | |
3. Amino-trifluoromethylthiolation
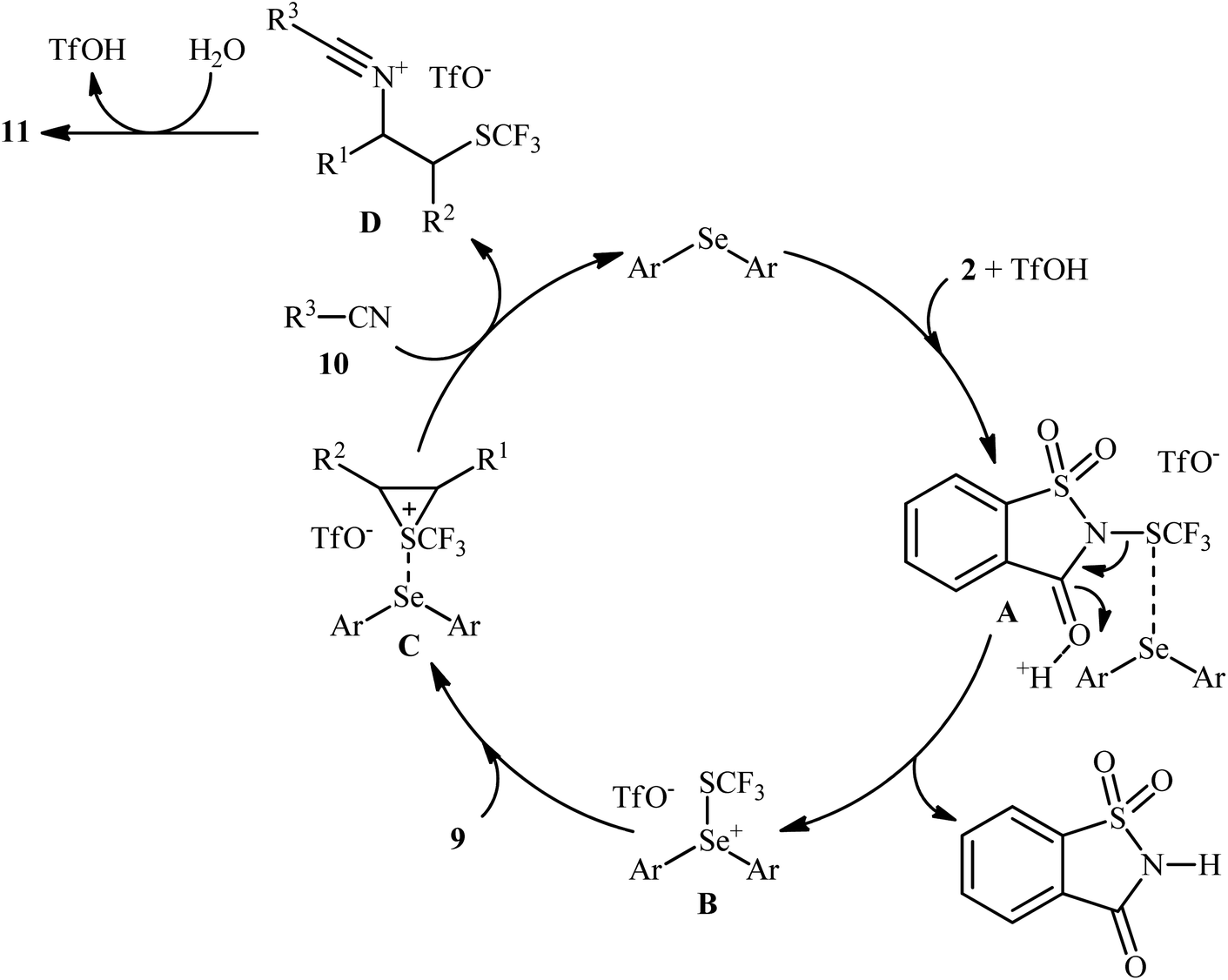
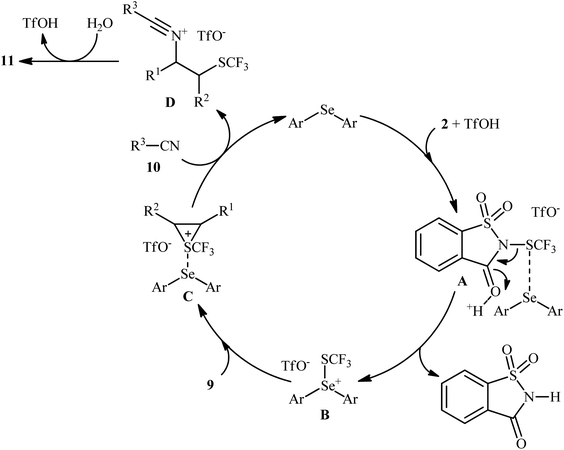
In 2015, Zhao and co-workers reported one of the earliest vicinal trifluoromethylthiolative amination reactions to produce β-SCF3-substituted amides 11 from the readily available alkenes 9, N-SCF3 reagent 2, and nitriles 10 in the presence of water (Scheme 7).16 This transformation was catalyzed by an electron-rich diaryl selenide [bis(4-methoxyphenyl)selane; (4-OMe-C6H4)2Se] and a weak sulfonic acid. The reaction displayed a broad substrate scope, and various important functional groups such as fluoro, chloro, bromo, methoxy, acetoxy, and nitro were tolerated. It is worth noting that, by replacing nitriles with carboxylic acids, various aromatic and aliphatic alkenes could be trifluoromethylthioesterificated in high yields. In this case, no additional reaction solvent was necessary, since the acids played a dual role as the solvent and the substrates. As shown in Scheme 8, a plausible reaction mechanism was suggested by the authors for this difunctionalization reaction. First, the reaction of diaryl selenide with reagent 2 in the presence of TfOH gives selenonium triflate intermediate B and saccharin through transition state A. Later, the highly reactive intermediate B interacts with alkene 9 to form the trifluoromethylthiiranium ion C. A subsequent nucleophilic attack of nitrile 10 on this intermediate C produces the nitrilium ion D through an anti addition involving the opening of the thiiranium intermediate. Finally, hydrolysis of intermediate D affords the expected product 11.
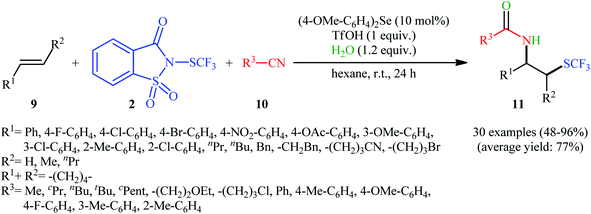 |
| | Scheme 7 Diaryl selenide-catalyzed vicinal amino-trifluoromethylthiolation of alkenes 9 by Shen's reagent 2 and nitriles 10 in the presence of water. | |
 |
| | Scheme 8 Probable mechanism of trifluoromethylthioamination of alkenes 9 by N-SCF3 reagent 2 and nitriles 10. | |
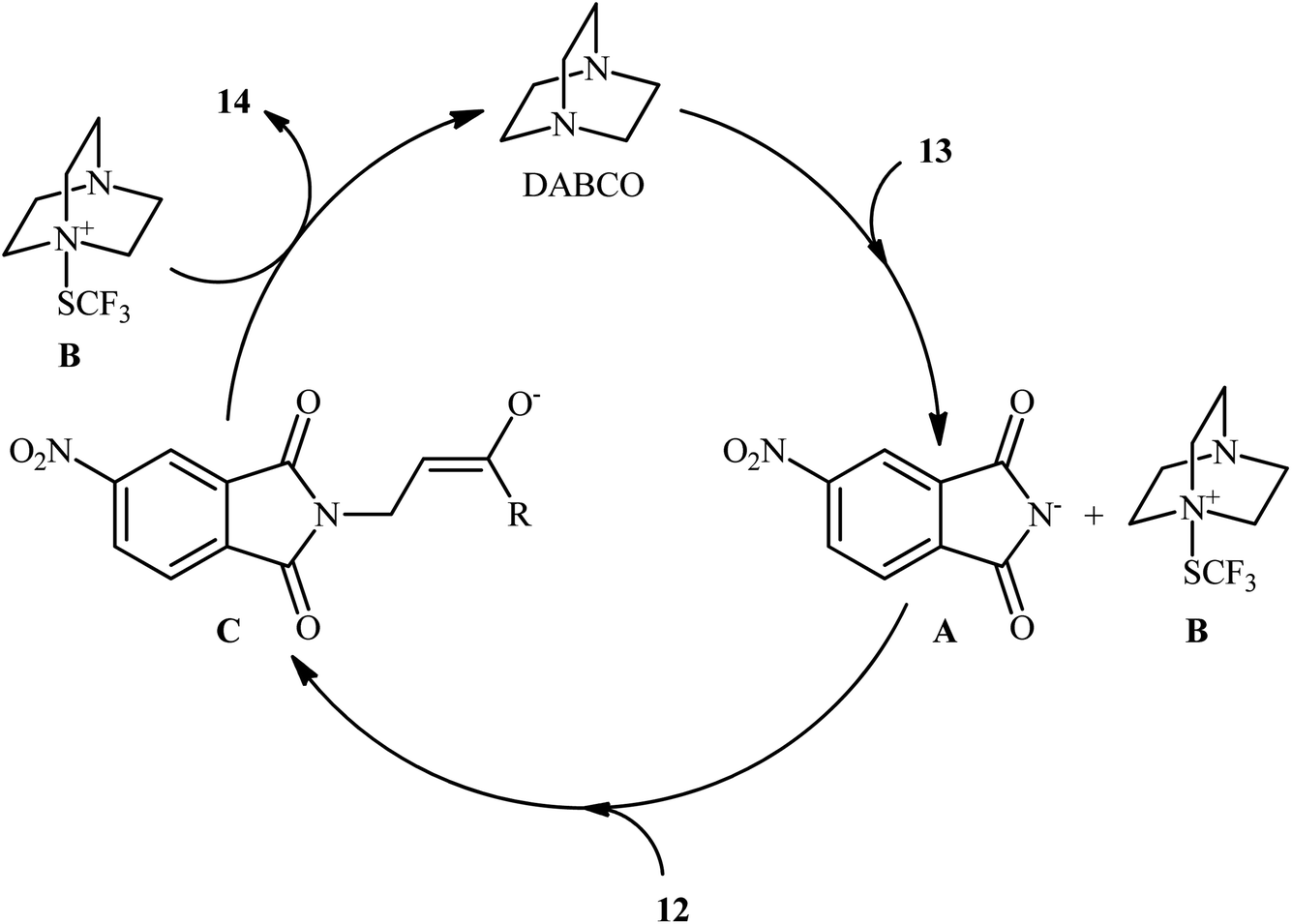
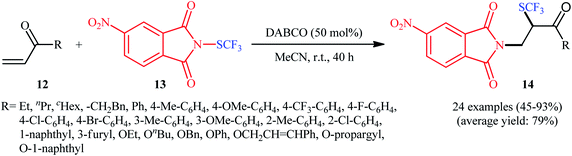
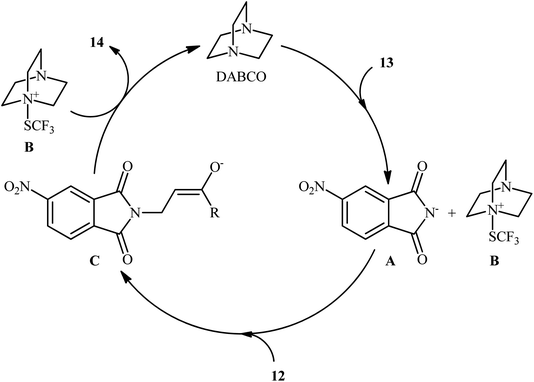
In the same year, the Wang group disclosed a highly atom-economical direct trifluoromethylthioamination of α,β-unsaturated ketones and esters 12 using N-trifluoromethylthio-4-nitrophthalimide 13 as both the nitrogen and SCF3 sources under metal-free and mild conditions (Scheme 9).17 The reaction was conducted in the presence of substoichiometric amounts of DABCO at room temperature under open air, tolerated both aromatic and aliphatic ketones and esters 12, and provided α-trifluoromethylthiolated β-amino carbonyl compounds 14 in moderate to excellent yields. However, acrylonitriles afforded lower yields and α,β-unsaturated amides failed to provide the desired products. It should be mentioned that the synthesized α-SCF3-β-N-phthaloylamino acid esters could be easily converted to the corresponding β-amino acids via hydrogenation with Et3SiH, followed by treatment with NaBH4. Studies showed that when the newly developed reagent 13 was replaced by SCF3-phthalimide, the reaction furnished the corresponding trifluoromethylthioaminated products, albeit with reduced yields. On the contrary, replacing compound 13 by either N-SCF3-aniline or SCF3-succinimide did not give the expected products. Therefore, the authors hypothesized that SCF3-4-nitrophthalimide 13 functions as both the electrophilic SCF3 and nitrogen sources, while SCF3-aniline and SCF3-succinimide are not electrophilic enough to react with alkenes under the identical conditions. The plausible mechanism of this trifluoromethylthioamination is represented in Scheme 10.
 |
| | Scheme 9 DABCO-catalyzed amino-trifluoromethylthiolation of α,β-unsaturated carbonyl compounds 12 with N-trifluoromethylthio-4-nitrophthalimide 13. | |
 |
| | Scheme 10 Proposed mechanism for the reaction in Scheme 9. | |
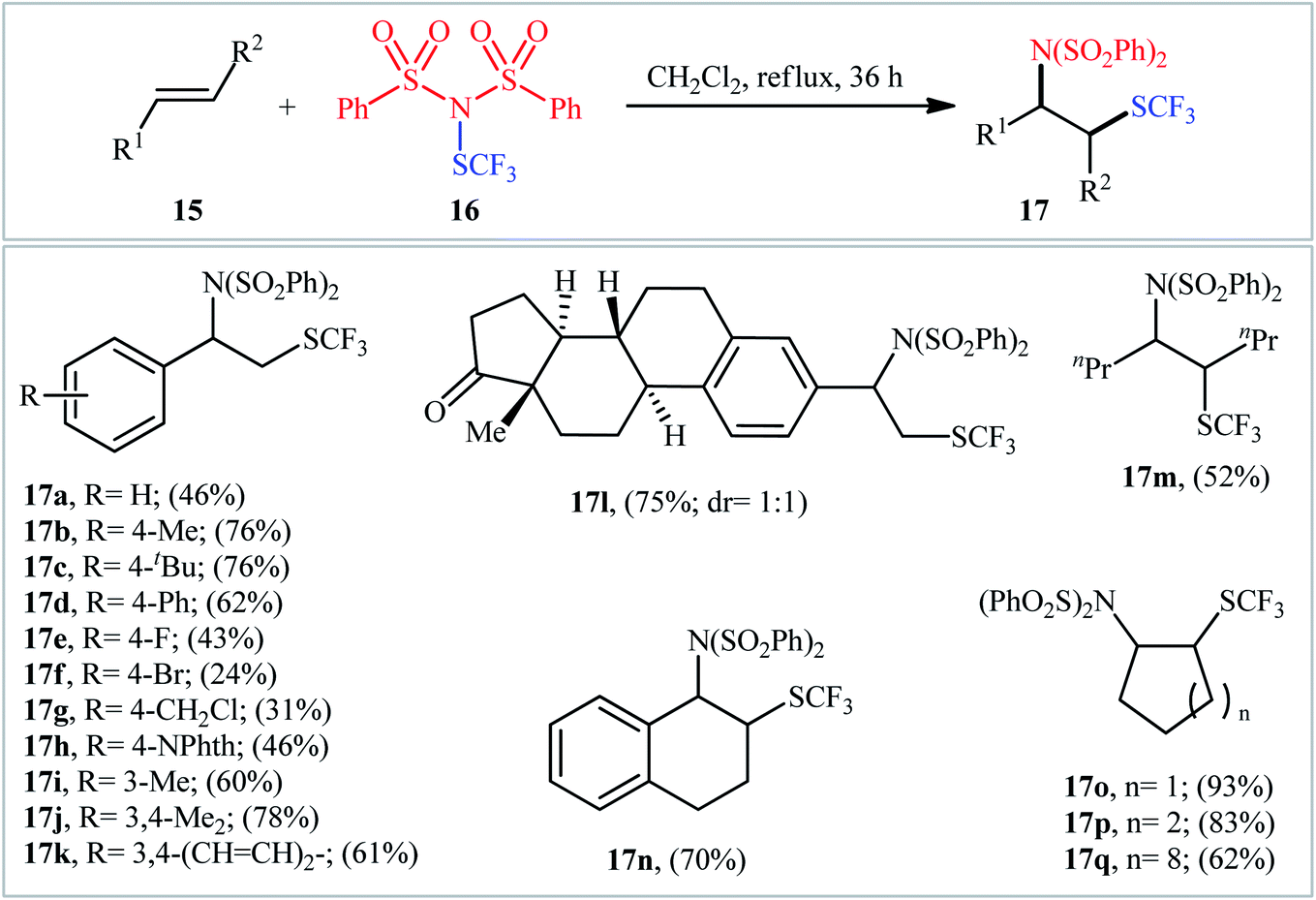
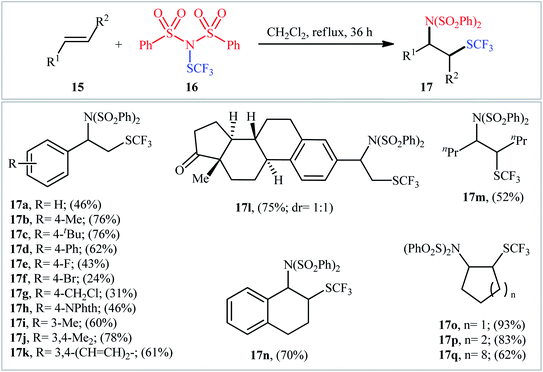
By a similar strategy, Shen and colleagues reported a facile synthesis of β-SCF3-substituted amines 17 from unactivated alkenes 15 and N-trifluoromethylthio-dibenzenesulfonimide 16 in DCM media under catalyst- and additive-free conditions (Scheme 11).18 In this study, 17 aromatic and aliphatic alkenes were amino-trifluoromethylthio difunctionalized with fair to very good yields. Noteworthily, both acyclic and cyclic alkenes were compatible with the reaction conditions. Interestingly, when the reaction was performed in more polar solvent DMF, the corresponding trifluoromethylthiolated alkenes were obtained as the sole products.
 |
| | Scheme 11 Shen's synthesis of β-SCF3-substituted amines 17. | |
4. Oxy-trifluoromethylthiolation
The first mention of the direct oxytrifluoromethylthiolation of alkenes can be found in a 2009 paper by Billard et al.,19 who showed that the treatment of aliphatic alkenes 18 with trifluoromethanesulfanylamide 19 and sodium tosylate as the +SCF3 and −OTs sources, respectively, in the presence of 5 equiv. of BF3·Et2O as a mediator in DCM at room temperature afforded the corresponding β-(trifluoromethyl)alkyl tosylates 20 in moderate to high yields with complete regioselectivity (Scheme 12). Other Lewis acids, such as (Bu)2BOTf and TfOSiMe3, were tested and showed lower efficiency than that of BF3·Et2O. Notably, Cu(OTf)2, Yb(OTf)3, La(OTf)3, and In(OTf)3, and Ti(OiPr)4 proved to be completely ineffective. The same difunctionalization reaction was also investigated with alkynes. The reaction gave satisfactory yields, but the regioselectivity was not controlled in the case of disubstituted alkynes since an equal amount of the two regioisomers was obtained. It should be mentioned that the presence of the tosyl group provides the opportunity for further functionalization of the products using convenient SN2 reactions, as exemplified by the synthesis of 2-trifluoromethylsulfanyl cyclohexyl amine from cyclohexene over 3 steps (i.e., oxytrifluoromethylthiolation, azidation, protonation).
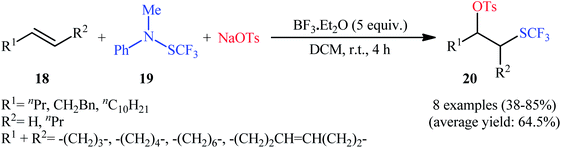 |
| | Scheme 12 Lewis acid-mediated oxytrifluoromethylthiolation of alkenes 18 developed by Billard. | |
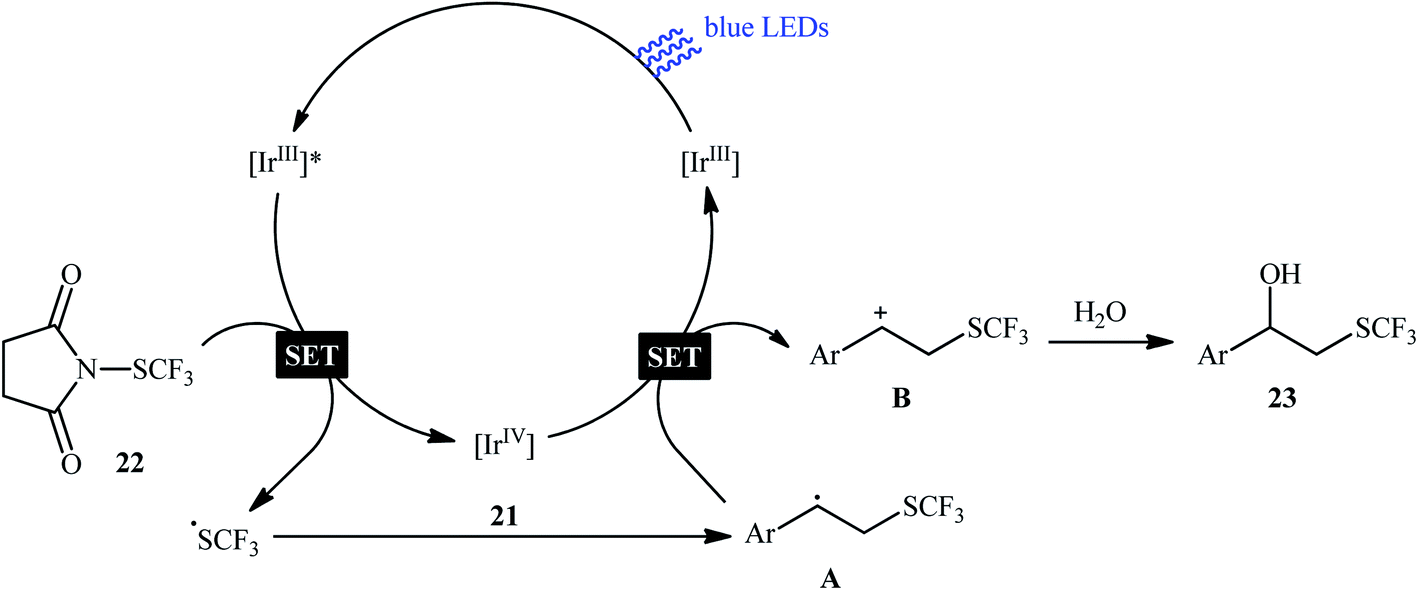
Eight years later, Koike and Akita developed an efficient photoredox-catalyzed regioselective hydroxytrifluoromethylthiolation of alkenes under irradiation of visible light.20 Using 5 mol% of fac-[Ir(4-MeO-ppy)3] (4-MeO-ppy = 2-pyridyl-5-methoxyphenyl) as the catalyst, a library of terminal and internal aromatic alkenes 21 underwent regioselective hydroxytrifluoromethylthiolation with N-trifluoromethylthiosuccinimide 22 and water to afford the corresponding β-trifluoromethylthiolated benzyl alcohols 23 in fair to excellent yields (Scheme 13). Besides water, alcohols (e.g., MeOH, EtOH and iPrOH) also could be utilized as the sources of oxy functional groups. However, the applicability of aliphatic alkenes as starting materials was not investigated in this study. The mechanism of this reaction as proposed by the authors is shown in Scheme 14. Firstly, the ground state photocatalyst IrIII undergoes photoexcitation under irradiation of visible light to produce the excited state [IrIII]*, which after a SET to the electrophilic CF3S reagent 22 leads to the generation of the ˙CF3 radical and the highly oxidized Ir catalyst, IrIV. Thereafter, the ˙CF3 radical reacts with styrene 21 to form the benzyl radical intermediate A, which after oxidization by IrIV provides the carbocationic intermediate B and regenerates photocatalyst IrIII. Finally, nucleophilic attack of water on the carbocation B produces the desired β-SCF3-substituted benzyl alcohol 23. Another independent hydroxytrifluoromethylthiolation method was published by Luo and co-workers using N-CF3S-saccharin reagent and (4-OMe-C6H4)2Se catalyst under conventional conditions.21 However, in this method, the reaction required an oxygen atmosphere. Various styrene derivatives and aliphatic alkenes were tolerated by this protocol. Moreover, similar conditions proved suitable for the intramolecular cyclization of alkenes tethered by carboxylic acid, hydroxy, ester, and sulfamide groups. Subsequently, the same authors extended their methodology to the enantioselective oxytrifluoromethylthiolation of internal alkenes using chiral selenide catalysis.22,23
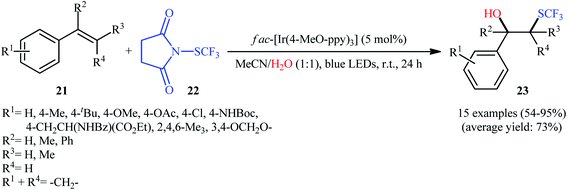 |
| | Scheme 13 Hydroxytrifluoromethylthiolation of alkenes 21 with N-trifluoromethylthiosuccinimide 22 by a visible light-activation system in aqueous acetonitrile. | |
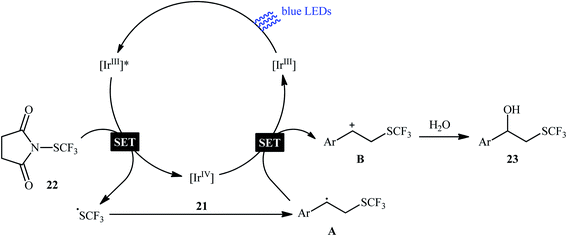 |
| | Scheme 14 Proposed mechanism for the visible light-induced hydroxytrifluoromethylthiolation of alkenes 21 with N-trifluoromethylthiosuccinimide 22 and water. | |
Very recently, the group of Li studied the possibility of synthesizing trifluoromethylthio peroxides through the direct Cu-catalyzed trifluoromethylthiolation–peroxidation of C–C double bonds.24 In this study, a diverse array of β-trifluoromethylthio peroxides 25 were obtained in 31–86% yields with excellent regioselectivities by reaction of various terminal and internal alkenes 24 with AgSCF3 and tert-butyl hydroperoxide (tBuOOH) in refluxing MeCN for 5 h in the presence of a CuSO4/HMPA/K2S2O8 combination as the catalytic system (Scheme 15). The results indicated that the best results were obtained in the case of aromatic and heteroaromatic alkenes. On the contrary, aliphatic alkenes were unfavorable for the reaction and led to lower yields. It is worthwhile to note that the identical condition was also successfully applied for trifluoromethylthiolation–peroxidation of a library of mono- and 1,1-disubstituted allenes. The authors nicely demonstrated the synthetic value of β-trifluoromethylthio peroxide products by the conversion of a peroxide group to hydroxyl and carbonyl moieties. The radical trapping experiments pointed toward a radical reaction mechanism.
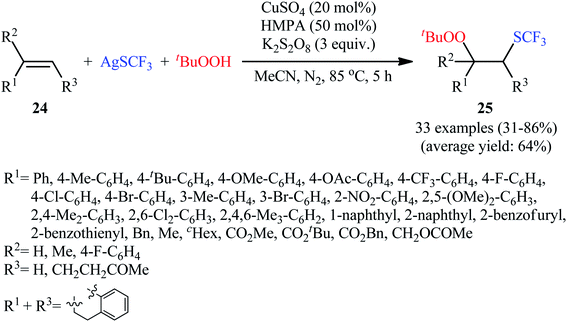 |
| | Scheme 15 Cu-catalyzed trifluoromethylthiolation–peroxidation of alkenes 24 with AgSCF3 and tBuOOH. | |
5. Thio-trifluoromethylthiolation
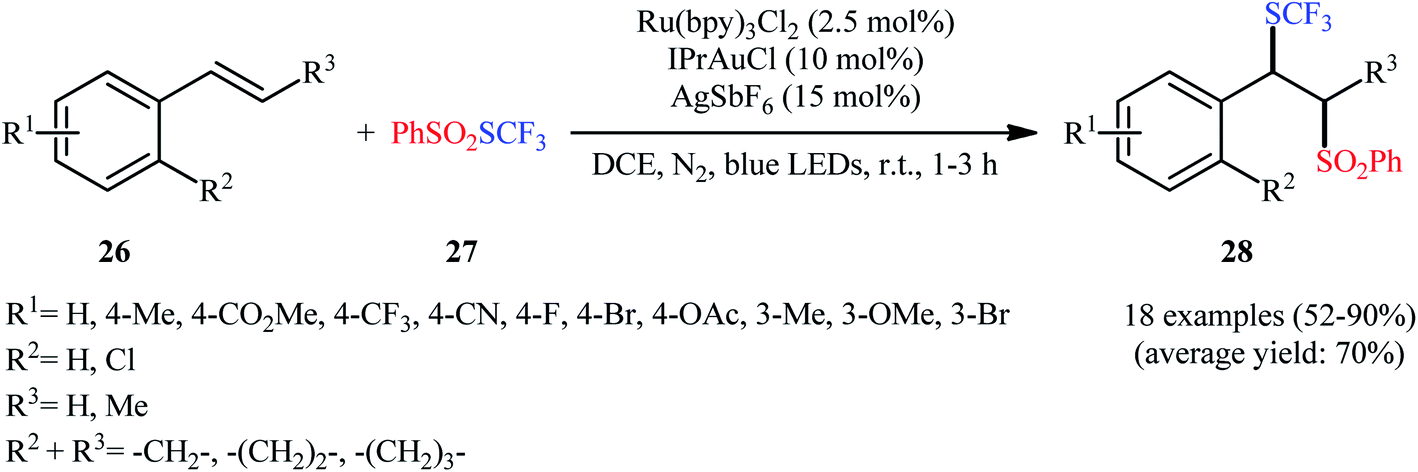
In 2017, Xu et al. for the first time reported the regioselective trifluoromethylthiosulfonylation of alkenes under the action of dual gold and photoredox catalysis.25 They demonstrated that S-(trifluoromethyl)benzenesulfonothioate (PhSO2SCF3; 27) acted as both the trifluoromethylthiolating and sulfonylating agents in the presence of Ru(bpy)3Cl2 as a photocatalyst and IPrAuCl as a cationic gold catalyst under the irradiation of a 100 W blue LED in an inert atmosphere. Additionally, 15 mol% of AgSbF6 was needed as an additive for this transformation. Under the optimized reaction conditions, various styrene derivatives 26 with different substitution patterns and functionalities rapidly underwent this difunctionalization reaction and afforded the corresponding β-SCF3-substituted sulfones 28 in moderate to excellent yields (Scheme 16). The methodology was also applicable for the other related S-alkyl/aryl benzenesulfonothioates like PhSO2SMe, PhSO2SBn, PhSO2SC4H9 and PhSO2SPh. It is worthwhile to note that when the internal alkenes were employed as reaction substrates, very good diastereoselectivities were observed and both trans- and cis alkenes yielded the same diastereomer. Moreover, the authors displayed the synthetic applicability of the prepared β-SCF3-substituted sulfones by high yielding preparation of β-cyano and β-allyl thioethers via the reaction with TMSCN and allylsilane, respectively, in the presence of aluminium chloride. Mechanistically, these reactions are believed to proceed through the sulfur migration substitution pathway.
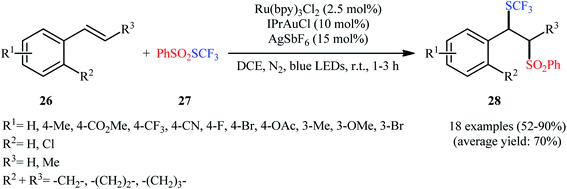 |
| | Scheme 16 Dual gold and visible light-mediated trifluoromethylthiosulfonylation of alkenes 26 with PhSO2SCF3. | |
Concurrently, Qing's research group disclosed an excellent strategy for oxidative bis(trifluoromethylthiolation) of 2-monosubstituted 2,3-allenoic acids 29 employing AgSCF3 as the source of the trifluoromethylthiol group in the presence of a catalytic amount of inexpensive Cu(OAc)2 combined with (NH4)2S2O8 as a stoichiometric oxidant.26 It was found that the solvent had a major impact on the success of this reaction and the highest yield was obtained in MeCN/HCO2H/H2O (4![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1:2). Under the optimized conditions, the reaction exhibited excellent regioselectivity and provided 3,4-bis(trifluoromethylthio) but-2-enoic-acid products 30 in moderate to good yields (Scheme 17). In contrast, when 4-aryl-2,3-allenoic acids were employed under the identical conditions, a different outcome was obtained. In these cases, 4-((trifluoromethyl)thio)furan-2(5H)-ones were selectively generated in modest to excellent yields through radical trifluoromethylthiolation/intramolecular cyclization. While the detailed mechanistic picture for the formation of 3,4-bis(trifluoromethylthio) but-2-enoic-acid products 30 remains unclear, the authors postulated that the reaction may start with the formation of a CF3S radical or CF3SSCF3 via oxidation of AgSCF3 by (NH4)2S2O8 through a AgIISCF3 intermediate. Next, the SCF3 radical adds to the central carbon of allene 29 leading to allylic radical species A and A′. The thermodynamically more favored A is then trapped by a Cu salt to give the SCF3–allyl–Cu complex B. Finally, the trifluoromethylthiolation of this complex B with CF3SSCF3 affords the expected bis-trifluoromethylthiolated products 30 (Scheme 18).
:1:2). Under the optimized conditions, the reaction exhibited excellent regioselectivity and provided 3,4-bis(trifluoromethylthio) but-2-enoic-acid products 30 in moderate to good yields (Scheme 17). In contrast, when 4-aryl-2,3-allenoic acids were employed under the identical conditions, a different outcome was obtained. In these cases, 4-((trifluoromethyl)thio)furan-2(5H)-ones were selectively generated in modest to excellent yields through radical trifluoromethylthiolation/intramolecular cyclization. While the detailed mechanistic picture for the formation of 3,4-bis(trifluoromethylthio) but-2-enoic-acid products 30 remains unclear, the authors postulated that the reaction may start with the formation of a CF3S radical or CF3SSCF3 via oxidation of AgSCF3 by (NH4)2S2O8 through a AgIISCF3 intermediate. Next, the SCF3 radical adds to the central carbon of allene 29 leading to allylic radical species A and A′. The thermodynamically more favored A is then trapped by a Cu salt to give the SCF3–allyl–Cu complex B. Finally, the trifluoromethylthiolation of this complex B with CF3SSCF3 affords the expected bis-trifluoromethylthiolated products 30 (Scheme 18).
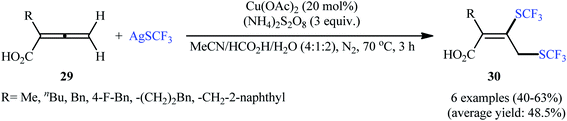 |
| | Scheme 17 Cu-catalyzed bis(trifluoromethylthiolation) of allenes 29 with AgSCF3. | |
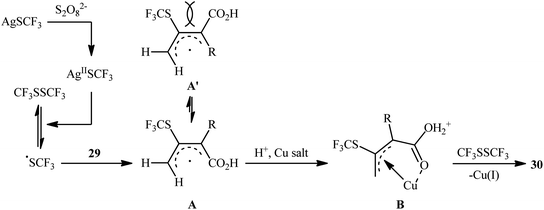 |
| | Scheme 18 Plausible mechanism for the oxidative bis(trifluoromethylthiolation) of 2-monosubstituted 2,3-allenoic acids 29. | |
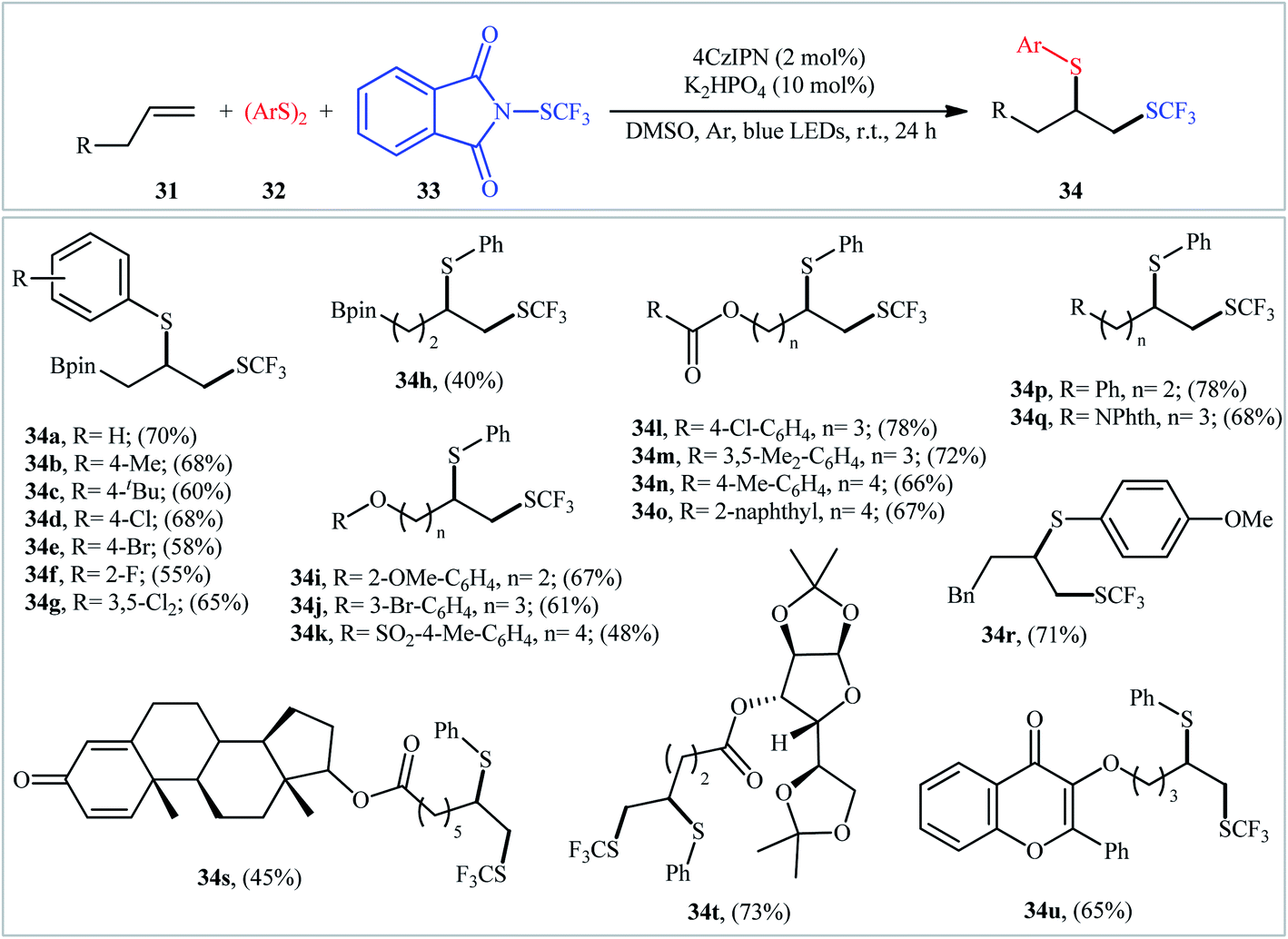
Very recently, Zhang and Ma, along with their co-workers, reported a three-component thio-trifluoromethylthiolation of aliphatic terminal alkenes 31 with aryldisulfides 32 and electrophilic trifluoromethylthiolating reagent 33 through visible light photoredox catalysis.27 This reaction employed 1,2,3,5-tetrakis(carbazol-9-yl)-4,6-dicyanobenzene (4CzIPN) as an organic photocatalyst, K2HPO4 as an additive, and blue LEDs as the light source, leading to the production of aryl(2-((trifluoromethyl)thio)ethyl)sulfane derivatives 34 in moderate to good yields, ranging from 48–78% (Scheme 19). Notably, this methodology was applicable to the difunctionalization of boldenone- and D-glucose-derived terminal alkenes, indicating a potential application in the design and synthesis of biologically active molecules. Unfortunately, the applicability of alkyldisulfides as starting materials was not investigated in this study. Mechanistic elucidation through radical trapping experiments in the presence of 2,2,6,6-tetramethylpiperidin-1-oxyl (TEMPO) indicated a radical pathway for the transformation.
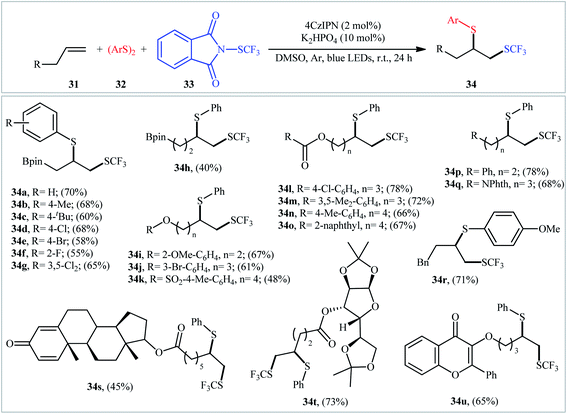 |
| | Scheme 19 Visible light-induced thio-trifluoromethylthiolation of unactivated terminal alkenes 31 with aryldisulfides 32 and Phth-SCF3 reagent 33. | |
6. Seleno-trifluoromethylthiolation
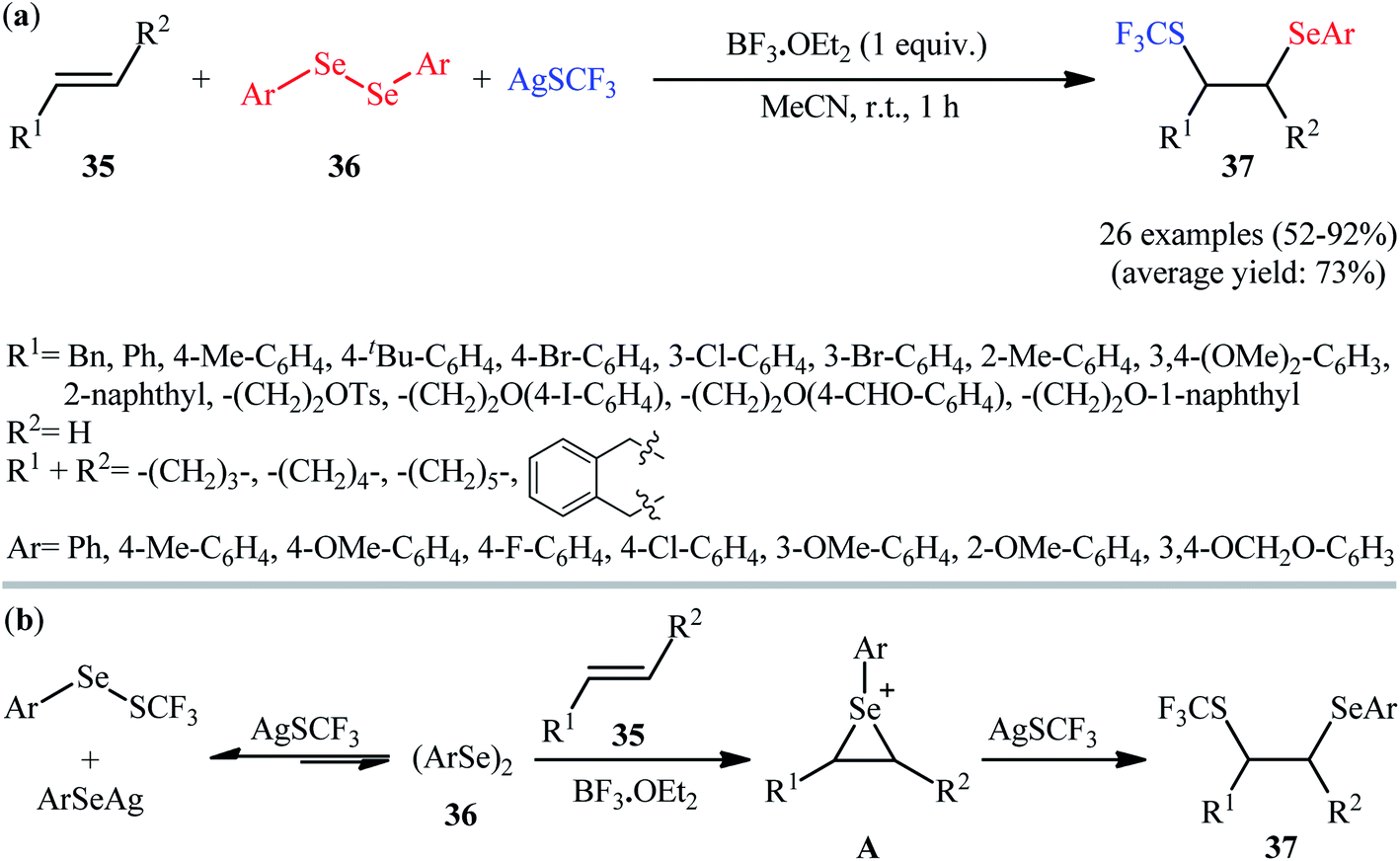
In 2019, Anbarasan and Saravanan devised an efficient method for the direct selenotrifluoromethylthiolation of alkenes 35 with diaryl diselenides 36 and AgSCF3 at room temperature by means of BF3·OEt2 as an activator.28 18 alkenes (aromatic and aliphatic) and 8 diaryl diselenides (electron-rich and electron-poor) were used as substrates in this study and 26 1,2-dichalcogenated products 37 were synthesized in moderate to excellent yields within 1 h (Scheme 20a). Interestingly, the prepared difunctionalized products could be easily converted to the corresponding vinyl trifluoromethyl thioethers when treated with m-CPBA in DCM at room temperature. The mechanistic pathway proposed by the authors to explain this difunctionalization reaction is based on the formation of an episelenonium intermediate, followed by its regioselective ring opening with AgSCF3 (Scheme 20b).
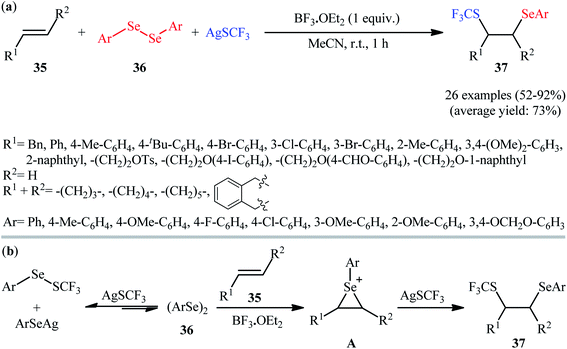 |
| | Scheme 20 (a) Metal-free selenotrifluoromethylthiolation of alkenes 35 with diaryl diselenides 36 and AgSCF3. (b) Proposed mechanism for the formation of 1,2-dichalcogenated products 37. | |
7. Phosphono-trifluoromethylthiolation
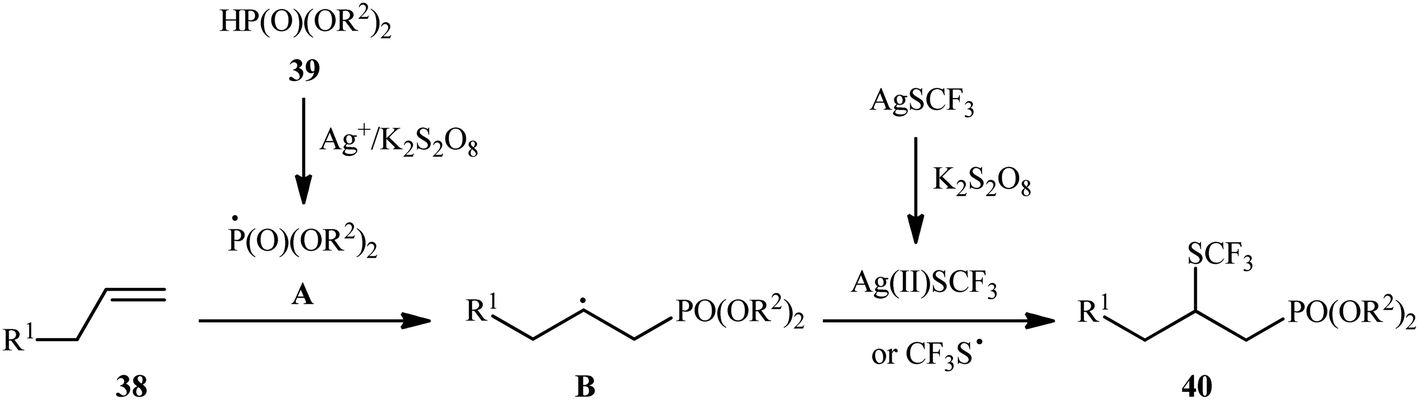
In 2018, Liu and co-workers illustrated an interesting regioselective oxidative phosphonotrifluoromethylthiolation of unactivated alkenes with P(O)–H compounds and AgSCF3 in which 4 equiv. of inexpensive K2S2O8 were employed as an oxidant.29 A variety of aliphatic alkenes 38 and dialkyl phosphonates 39 were well tolerated in the reaction process and gave the β-trifluoromethylthiol phosphonates 40 in 38–76% yields (Scheme 21). However, both aromatic alkenes and diaryl phosphonates were incompatible in the reaction and the applicability of other hydrogen phosphoryl compounds (H-phosphinates and secondary phosphine oxides) as substrates was not investigated in this study. On the basis of experimental observations, the authors suggested a plausible mechanism for this reaction as depicted in Scheme 22. First, the oxidation of the phosphonate 39 with Ag+/K2S2O8 into its corresponding phosphonyl radical A takes place. Thus, its addition with alkene 38 gives the alkyl radical B, which undergoes coupling with a CF3S radical or Ag(II)SCF3 (produced by the oxidation of AgSCF3 with K2S2O8) releasing the required product 40.
 |
| | Scheme 21 Phosphonotrifluoromethylthiolation of aliphatic alkenes 40 with dialkyl phosphonates 41 and AgSCF3. | |
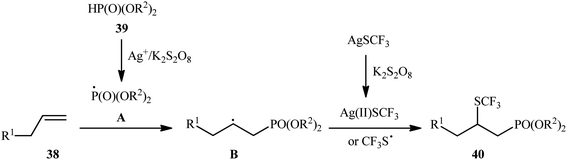 |
| | Scheme 22 Plausible mechanism for the reaction in Scheme 21. | |
8. Chloro-trifluoromethylthiolation
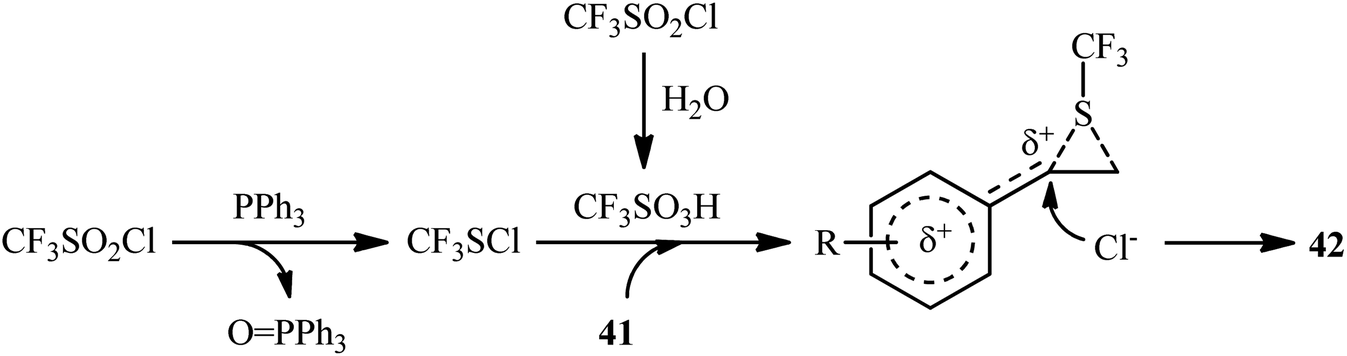
In 2018, Yi–Zhang's group studied the direct chlorotrifluoromethylthiolation of unactivated C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C double bonds with triflyl chloride (CF3SO2Cl) as both the chlorine and SCF3 sources under metal-free conditions.30 Thus, in the presence of over-stoichiometric amounts of PPh3 as a low-cost reductant in DMF under an air atmosphere, chlorotrifluoromethylthiolation of various styrenes 41 having both electron-withdrawing and electron-releasing groups with CF3SO2Cl furnished the corresponding (2-chloro-2-arylethyl)(trifluoromethyl)sulfanes 42 in moderate to high yields and excellent Markovnikov-selectivity (Scheme 23). The reaction was also amenable to the chlorotrifluoromethylthiolation of terminal aliphatic alkenes, while anti-Markovnikov adducts were the major regioisomers. However, internal alkenes did not respond to the reaction under standard conditions. Interestingly, this chlorotrifluoromethylthiolation chemistry was also successfully extended to terminal alkynes. Similar to trifluoromethylthiolation, the method could be used for difluoromethylthiolation using CF2HSO2Cl as a reagent. The plausible mechanism of this difunctionalization reaction is represented in Scheme 24.
C double bonds with triflyl chloride (CF3SO2Cl) as both the chlorine and SCF3 sources under metal-free conditions.30 Thus, in the presence of over-stoichiometric amounts of PPh3 as a low-cost reductant in DMF under an air atmosphere, chlorotrifluoromethylthiolation of various styrenes 41 having both electron-withdrawing and electron-releasing groups with CF3SO2Cl furnished the corresponding (2-chloro-2-arylethyl)(trifluoromethyl)sulfanes 42 in moderate to high yields and excellent Markovnikov-selectivity (Scheme 23). The reaction was also amenable to the chlorotrifluoromethylthiolation of terminal aliphatic alkenes, while anti-Markovnikov adducts were the major regioisomers. However, internal alkenes did not respond to the reaction under standard conditions. Interestingly, this chlorotrifluoromethylthiolation chemistry was also successfully extended to terminal alkynes. Similar to trifluoromethylthiolation, the method could be used for difluoromethylthiolation using CF2HSO2Cl as a reagent. The plausible mechanism of this difunctionalization reaction is represented in Scheme 24.
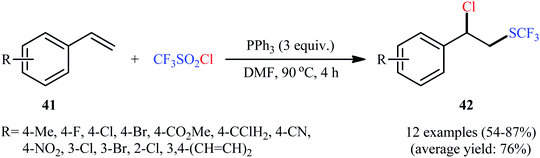 |
| | Scheme 23 Metal-free chlorotrifluoromethylthiolation of styrenes 41 with CF3SO2Cl. | |
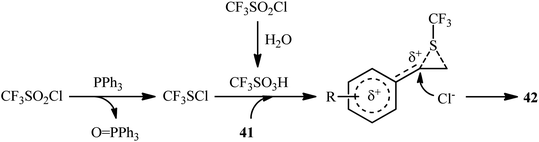 |
| | Scheme 24 Proposed mechanistic pathways for the reaction in Scheme 23. | |
Concurrently, Yang and co-workers reported that various terminal and internal alkenes could regio-selectively undergo chlorotrifluoromethylthiolation with Phth-SCF3 and SOCl2 mediated by Fe2O3, furnishing the corresponding trifluoromethylthiolated chlorides in moderate to almost quantitative yields.31 It is worth noting that quaternary carbon centers could be readily built up using internal alkene substrates with a >20:1 diastereomeric ratio under the reaction conditions.
Finally, it should be noted that transition metal complexes play important roles in intermolecular 1,2-difunctionalization of alkenes and the synthesis of organic compounds.32–40
9. Conclusion
The direct vicinal difunctionalization of unsaturated hydrocarbons represents one of the most powerful and fascinating tactics for increasing molecular complexity by forging two chemical bonds across an unsaturated carbon–carbon bond in a single reaction. In this family of reactions, intermolecular trifluoromethylthiolative 1,2-difunctionalization of alkenes has attracted a lot of attention from researchers in recent years due to the potential biological activities of the resulting β-functionalized alkyl trifluoromethyl sulfides. As shown in this review, over the last few years, a handful of attractive carbo- and hetero-trifluoromethylthiolation reactions of alkenes were developed that allow for efficient, clean, and selective synthesis of various β-functionalized alkyl trifluoromethyl sulfides. Noteworthily, the majority of these reactions were performed at room temperature and under transition-metal-free conditions. Challenges that remain to be faced in the future include: (i) development of chiral catalytic systems for enantioselective difunctionalization of trifluoromethylthiolations; (ii) extension of hetero-trifluoromethylthiolation reactions to other heteroatoms such as silica- and bora-trifluoromethylthiolations; and (iii) further investigation of the scope and limitations of some reactions, such as phosphono- and seleno-trifluoromethylthiolations.
Conflicts of interest
There are no conflicts to declare.
References
-
(a) E. P. Gillis, K. J. Eastman, M. D. Hill, D. J. Donnelly and N. A. Meanwell, J. Med. Chem., 2015, 58, 8315–8359 CrossRef CAS PubMed;
(b) M. Inoue, Y. Sumii and N. Shibata, ACS Omega, 2020, 5, 10633–10640 CrossRef CAS.
-
(a) M. M. Alauddin, Am. J. Nucl. Med. Mol. Imaging, 2012, 2, 55–76 CAS;
(b) F. John, O. Muzik, S. Mittal and C. Juhász, Mol. Imaging Biol., 2020, 22, 805–819 CrossRef CAS.
- Y. Zhou, J. Wang, Z. Gu, S. Wang, W. Zhu, J. L. Aceña, V. A. Soloshonok, K. Izawa and H. Liu, Chem. Rev., 2016, 116, 422–518 CrossRef CAS PubMed.
- V. V. Grushin, Acc. Chem. Res., 2010, 43, 160–171 CrossRef CAS.
- X.-H. Xu, K. Matsuzaki and N. Shibata, Chem. Rev., 2015, 115, 731–764 CrossRef CAS PubMed.
-
(a) F. Toulgoat, S. Alazet and T. Billard, Eur. J. Org. Chem., 2014, 2415–2428 CrossRef CAS;
(b) J.-H. Lin, Y.-L. Ji and J.-C. Xiao, Curr. Org. Chem., 2015, 19, 1541–1553 CrossRef CAS;
(c) H. Chachignon and D. Cahard, Chin. J. Chem., 2016, 34, 445–454 CrossRef CAS;
(d) H. Zheng, Y. Huang and Z. Weng, Tetrahedron Lett., 2016, 57, 1397–1409 CrossRef CAS;
(e) A.-L. Barthelemy, E. Magnier and G. Dagousset, Synthesis, 2018, 50, 4765–4776 CrossRef CAS;
(f) M. Hamzehloo, A. Hosseinian, S. Ebrahimiasl, A. Monfared and E. Vessally, J. Fluorine Chem., 2019, 224, 52–60 CrossRef CAS.
-
(a) J. Lin, R. J. Song, M. Hu and J. H. Li, Recent advances in the intermolecular oxidative difunctionalization of alkenes, Chem. Rec., 2019, 19, 440–451 CrossRef CAS PubMed;
(b) J. B. Peng, Adv. Synth. Catal., 2020, 362, 3059–3080 CrossRef CAS;
(c) Y. C. Wu, Y. T. Xiao, Y. Z. Yang, R. J. Song and J. H. Li, ChemCatChem, 2020, 12, 5312–5329 CrossRef CAS;
(d) A. Whyte, A. Torelli, B. Mirabi, A. Zhang and M. Lautens, ACS Catal., 2020, 10, 11578–11622 CrossRef CAS;
(e) H.-Y. Tu, S. Zhu, F.-L. Qing and L. Chu, Synthesis, 2020, 52, 1346–1356 CrossRef CAS;
(f) Z.-L. Li, G.-C. Fang, Q.-S. Gu and X.-Y. Liu, Chem. Soc. Rev., 2020, 49, 32–48 RSC;
(g) S. Yang, Y. Chen and Z. Ding, Org. Biomol. Chem., 2020, 18, 6983–7001 RSC;
(h) N. Yue and F. R. Sheykhahmad, J. Fluorine Chem., 2020, 109629 CrossRef CAS.
- A. Bakhtiary, M. R. P. Heravi, A. Hassanpour, I. Amini and E. Vessally, RSC Adv., 2021, 11, 470–483 RSC.
-
(a) A. Monfared, S. Ebrahimiasl, M. Babazadeh, S. Arshadi and E. Vessally, J. Fluorine Chem., 2019, 220, 24–34 CrossRef CAS;
(b) A. Hosseinian, Y. J. Sadeghi, S. Ebrahimiasl, A. Monfared and E. Vessally, J. Sulfur Chem., 2019, 40, 565–585 CrossRef CAS;
(c) C. Yang, A. Hassanpour, K. Ghorbanpour, S. Abdolmohammadi and E. Vessally, RSC Adv., 2019, 9, 27625–27639 RSC;
(d) N. H. Jabarullah, K. Jermsittiparsert, P. A. Melnikov, A. Maseleno, A. Hosseinian and E. Vessally, J. Sulfur Chem., 2020, 41, 96–115 CrossRef CAS.
-
(a) E. Vessally, K. Didehban, R. Mohammadi, A. Hosseinian and M. Babazadeh, J. Sulfur Chem., 2018, 39, 332–349 CrossRef CAS;
(b) E. Vessally, R. Mohammadi, A. Hosseinian, K. Didehban and L. Edjlali, J. Sulfur Chem., 2018, 39, 443–463 CrossRef CAS;
(c) F. A. H. Nasab, L. Z. Fekri, A. Monfared, A. Hosseinian and E. Vessally, RSC Adv., 2018, 8, 18456–18469 RSC;
(d) A. Hosseinian, L. Zare Fekri, A. Monfared, E. Vessally and M. Nikpassand, J. Sulfur Chem., 2018, 39, 674–698 CrossRef CAS;
(e) A. Hosseinian, S. Ahmadi, F. A. H. Nasab, R. Mohammadi and E. Vessally, Top. Curr. Chem., 2018, 376, 1–32 CrossRef;
(f) A. Hosseinian, S. Arshadi, S. Sarhandi, A. Monfared and E. Vessally, J. Sulfur Chem., 2019, 40, 289–311 CrossRef CAS;
(g) N. H. Jabarullah, K. Jermsittiparsert, P. A. Melnikov, A. Maseleno, A. Hosseinian and E. Vessally, J. Sulfur Chem., 2020, 41, 96–115 CrossRef CAS;
(h) X. Lu, Q. Yi, X. Pan, P. Wang and E. Vessally, J. Sulfur Chem., 2020, 41, 210–228 CrossRef CAS;
(i) S. Sarhandi, M. Daghagheleh, M. Vali, R. Moghadami and E. Vessally, Chem. Rev. Lett., 2018, 1, 9–15 Search PubMed;
(j) M. R. J. Sarvestani, N. Mert, P. Charehjou and E. Vessally, J. Chem. Lett., 2020, 1, 93–102 Search PubMed;
(k) M. Daghagheleh, M. Vali, Z. Rahmani, S. Sarhandi and E. Vessally, Chem. Rev. Lett., 2018, 1, 23–30 Search PubMed;
(l) L. Sreerama, E. Vessally and F. Behmagham, J. Chem. Lett., 2020, 1, 9–18 Search PubMed;
(m) S. Shahidi, P. Farajzadeh, P. Ojaghloo, A. Karbakhshzadeh and A. Hosseinian, Chem. Rev. Lett., 2018, 1, 37–44 Search PubMed.
- J. Fang, Z. K. Wang, S. W. Wu, W. G. Shen, G. Z. Ao and F. Liu, Chem. Commun., 2017, 53, 7638–7641 RSC.
- S. Liang, J. Wei, L. Jiang, J. Liu, Y. Mumtaz and W. Li, CCS Chem., 2021, 3, 265–273 CrossRef.
- B. S. Zhang, L. Y. Gao, Z. Zhang, Y. H. Wen and Y. M. Liang, Chem. Commun., 2018, 54, 1185–1188 RSC.
- J. He, C. Chen, G. C. Fu and J. C. Peters, ACS Catal., 2018, 8, 11741–11748 CrossRef CAS PubMed.
- Z. Xiao, Y. Liu, L. Zheng, C. Liu, Y. Guo and Q. Y. Chen, J. Org. Chem., 2018, 83, 5836–5843 CrossRef CAS PubMed.
- J. Luo, Z. Zhu, Y. Liu and X. Zhao, Org. Lett., 2015, 17, 3620–3623 CrossRef CAS PubMed.
- Q. Xiao, Q. He, J. Li and J. Wang, Org. Lett., 2015, 17, 6090–6093 CrossRef CAS PubMed.
- P. Zhang, M. Li, X. S. Xue, C. Xu, Q. Zhao, Y. Liu, H. Wang, Y. Guo, L. Lu and Q. Shen, J. Org. Chem., 2016, 81, 7486–7509 CrossRef CAS.
- A. Ferry, T. Billard, B. R. Langlois and E. Bacqué, Angew. Chem., Int. Ed., 2009, 121, 8703–8707 CrossRef.
- Y. Li, T. Koike and M. Akita, Asian J. Org. Chem., 2017, 6, 445–448 CrossRef CAS.
- Z. Zhu, J. Luo and X. Zhao, Org. Lett., 2017, 19, 4940–4943 CrossRef CAS PubMed.
- X. Liu, Y. Liang, J. Ji, J. Luo and X. Zhao, J. Am. Chem. Soc., 2018, 140, 4782–4786 CrossRef CAS.
- J. Xu, Y. Zhang, T. Qin and X. Zhao, Org. Lett., 2018, 20, 6384–6388 CrossRef CAS.
- Y. Chen, Y. Ma, L. Li, M. Cui and Z. Li, Org. Chem. Front., 2020, 7, 1837–1844 RSC.
- H. Li, C. Shan, C. H. Tung and Z. Xu, Chem. Sci., 2017, 8, 2610–2615 RSC.
- S. Pan, Y. Huang, X. H. Xu and F. L. Qing, Org. Lett., 2017, 19, 4624–4627 CrossRef CAS PubMed.
- X. Li, Q. Zhang, W. Zhang, J. Ma, Y. Wang and Y. Pan, Beilstein J. Org. Chem., 2021, 17, 551–557 CrossRef CAS PubMed.
- P. Saravanan and P. Anbarasan, Chem. Commun., 2019, 55, 4639–4642 RSC.
- Z. Xiao, Y. Liu, L. Zheng, X. Zhou, Y. Xie, C. Liu, Y. Guo and Q. Y. Chen, Tetrahedron, 2018, 74, 6213–6219 CrossRef CAS.
- L. Jiang, T. Ding, W. B. Yi, X. Zeng and W. Zhang, Org. Lett., 2018, 20, 2236–2240 CrossRef CAS PubMed.
- Y. Jia, H. Qin, N. Wang, Z. X. Jiang and Z. Yang, J. Org. Chem., 2018, 83, 2808–2817 CrossRef CAS.
- H. Zhang, et al., The Role of Superoxide Radicals, ACS Omega, 2020, 5, 18007–18012 CrossRef CAS.
- X. Pan, J. Wei, M. Zou, J. Chen, R. Qu and Z. Wang, Water Res., 2021, 194, 116916 CrossRef CAS PubMed.
- M. Zou, et al., Sci. Total Environ., 2021, 771, 144743 CrossRef CAS PubMed.
- W. Cao, et al., Environ. Sci. Pollut. Res., 2021, 28, 31301–31311 CrossRef PubMed.
- Z. Liu, A. Ebadi, M. Toughani, N. Mert and E. Vessally, RSC Adv., 2020, 10, 37299–37313 RSC.
- S. Ahmadi, A. Hosseinian, P. D. Kheirollahi Nezhad, A. Monfared and E. Vessally, Iran. J. Chem. Chem. Eng., 2019, 38, 1–19 CAS.
- E. Vessally, S. Mohammadi, M. Abdoli, A. Hosseinian and P. Ojaghloo, Iran. J. Chem. Chem. Eng., 2020, 39, 11–19 CAS.
- W. Xu, D. Guo, A. G. Ebadi, M. Toughani and E. Vessally, Transition-metal catalyzed carboxylation of organoboron compounds with CO2, J. CO2 Util., 2021, 45, 101403 CrossRef CAS.
- A. Hassanpour, M. R. P. Heravi, A. Ebadi, A. Hosseinian and E. Vessally, J. Fluorine Chem., 2021, 245, 109762 CrossRef CAS.
|
| This journal is © The Royal Society of Chemistry 2021 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence b
b