
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceChitosan-based enzyme ink for screen-printed bioanodes
Isao Shitanda†
 *ab,
Kanako Oda†a,
Noya Loew*a,
Hikari Watanabea,
Masayuki Itagakiab,
Seiya Tsujimurabc and
Abdelkader Zebdad
*ab,
Kanako Oda†a,
Noya Loew*a,
Hikari Watanabea,
Masayuki Itagakiab,
Seiya Tsujimurabc and
Abdelkader Zebdad
aDepartment of Pure and Applied Chemistry, Faculty of Science and Technology, Tokyo University of Science, 2641 Yamazaki, Noda, Chiba 278-8510, Japan. E-mail: shitanda@rs.tus.ac.jp; noya-loew@rs.tus.ac.jp; Fax: +81-4-7123-9890; Tel: +81-4-7124-1501
bResearch Institute for Science and Technology, Tokyo University of Science, 2641 Yamazaki, Noda, Chiba 278-8510, Japan
cDivision of Material Science, Faculty of Pure and Applied Science, University of Tsukuba, 1-1-1, Tennodai, Tsukuba, Ibaraki 305-5358, Japan
dUGA-Grenoble 1, CNRS, INSERM, TIMC-IMAG UMR 5525, Grenoble 38000, France
First published on 8th June 2021
Abstract
In this study, magnesium oxide (MgO)-templated mesoporous carbon (MgOC) and chitosan cross-linked with genipin (chitosan–genipin) were considered bio-composite inks for screen-printed bioanodes. The fabrication processes were optimized using rheological and structural data, and a bioanode ink containing glucose oxidase (GOx) and 1,2-naphthoquinone (1,2-NQ) was successfully developed. The optimal bioanode-ink contained MgOC pre-treated by washing to achieve a hydrophilic and neutral surface, which helped maintain enzyme activity and resulted in a highly porous electrode structure, which is essential for the accessibility of glucose to GOx. A bioanode fabricated using this ink showed a linear response current dependency up to 8 mM glucose with a sensitivity of 25.83 μA cm−2 mM−1. Combined with a conventional biocathode, an electromotive force of 0.54 V and a maximal power density of 96 μW cm−2 were achieved. These results show that this bio-composite ink can be used to replace the multi-step process of printing with conventional ink followed by drop-casting enzyme and mediator with a one-step printing process.
Introduction
Screen printing is a popular fabrication method of electrodes for biosensor and biofuel cell (BFC) applications.1,2 It offers several advantages, such as fabrication of miniaturized electrodes with versatile designs, as well as the intrinsic adaptability to low-cost, large-scale manufacturing.1–3 While the screen-printing process itself is expected to, at best, maintain the performance of the resulting biosensor or BFC, the design can enhance the performance and/or the usability of the device. In the case of non-invasive devices such as wearable biosensors and BFCs, screen printing allows designing flexible and comfortable devices that can be used to monitor biomarkers in sweat, urine, tears, and saliva and thus provide necessary information for health management and medical diagnoses.4–6 Wang et al. developed tattoo-, textile-, and mouth guard-type biosensors and BFCs that are comfortable to wear on the body.7–11 Their temporary tattoo-type biosensors, for example, could measure up to 100 μM glucose or 20 mM lactate depending on the enzyme used for fabrication.7,8 Our group developed paper-based devices that are inexpensive, lightweight, flexible, and disposable.12–15 For example, we developed a paper-based wearable self-powered glucose biosensor system integrated into a diaper for detecting urine sugar in patients with diabetes.15Generally, screen-printed biosensors and BFCs are fabricated via the printing of wiring, connectors, and base electrode materials on a substrate using conductive inks. Enzymes, mediators, cross-linkers, and protective films are deposited onto the printed electrodes via drop casting. To facilitate mass production, a “one-step deposition” method, in which a bio-composite ink is prepared by mixing enzymes, mediators, and cross-linkers with a conductive material and then printed onto the wiring-equipped substrate, forming the electrode in one step, is favorable.16 In this manner, Galán-Vidal et al. prepared a composite ink containing glucose oxidase and graphite powder dispersed in an epoxy resin, with cyclohexane to adjust the viscosity, and printed a glucose biosensor on a glass fiber circuit board.17 Crouch et al. printed a glucose biosensor on a polyvinyl chloride substrate using an water-based carbon ink containing glucose oxidase and cobalt phthalocyanine.18
Such one-step deposition using a bio-composite ink is one of the quickest and simplest manufacturing techniques for the fabrication of enzyme electrodes. However, to maintain the desired properties of the components, such as enzyme activity, the fabrication of the ink itself requires complicated optimization during its development.16 Possibly because of this need for a complicated optimization, as well as for in-house printing facilities in the developing laboratory, the number of reported bio-composite inks for one-step deposition for fabrication of biosensors and BFCs is limited.16–22 Furthermore, to the best of our knowledge, all printable bio-composite inks have been fabricated using graphite as a conductive material.
In general, however, printed graphite electrodes are considered to have a small surface area per footprint. To fabricate enzyme electrodes with a small footprint but large surface area, carbon nanomaterials, such as carbon nanotubes and porous carbon materials, are frequently used.23–28 Among the different types of porous carbon materials, magnesium oxide (MgO)-templated mesoporous carbon (MgOC) has been shown to increase the performance of biosensors and BFCs.28–32 MgOC is advantageous because its pore size can be controlled by tuning the template MgO size and it has a high active surface area. Recently, we succeeded in developing a screen-printed lactate biosensor based on modified MgOC.32
Apart from the electrode material, printable inks contain a binder, usually a polymer, and a solvent. The chosen binder should be stable, allow substrate diffusion, and favor enzyme stability. One water-soluble polymer that is becoming popular for the fabrication of enzyme electrodes, although it is usually used as a protective layer or entrapping polymer, is chitosan.33–36 Chitosan is a polysaccharide obtained through the deacetylation of chitin isolated from crustaceans and mushrooms. It is abundant, non-toxic, biodegradable, and biocompatible, and thus, is a promising biopolymer for use in biotechnological applications.36 Functionally, chitosan provides the kind of hydrophobic micro-environment that stabilizes enzyme activity for long periods. Indeed, according to Minteer et al., hydrophobic micelles such as chitosan and Nafion minimize the molecular vibration of an entire enzyme unit, forcing it to stay in place and maintain its native state via repulsive forces between the native hydrophilic exterior of an enzyme and the hydrophobic side chains.37 Zebda et al. demonstrated that the encapsulation of enzymes such as laccase and glucose dehydrogenase significantly increases long-term stability, even under in vivo conditions.38,39
When used as a binder in bio-composite inks, chitosan should prevent enzyme elution while simultaneously allowing the mass transfer of substrates to the enzyme catalytic sites. The permeability of composite materials containing chitosan depends on not only the preparation of chitosan itself but also the interactions of chitosan with the other components of the composite. In other words, the physical and chemical interactions between chitosan and the carbon material determine the porosity of the bio-composite ink and electrodes printed with such an ink. Thus, the porosity can be modified by the addition of a cross-linker, which is often added to improve chitosan stability. As a cross-linker, genipin is of particular interest. Genipin is a natural cross-linking agent obtained through enzymatic hydrolysis of geniposide, which is isolated from Gardenia jasminoides with β-glucosidase. Genipin spontaneously reacts with the amino groups of polymers and proteins and is 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 times less toxic than the commonly used cross-linker glutaraldehyde.40 Chitosan cross-linked with genipin (chitosan–genipin) has been successfully used as a protective layer and entrapment material for enzyme electrodes in biosensors and BFCs.34,41 Conghaile et al. used genipin to cross-link chitosan, glucose oxidase, and a mediator.42 Fernandes et al. used chitosan–genipin as a matrix to immobilize laccase in a carbon paste electrode, with paraffin oil as a binder.43
000 times less toxic than the commonly used cross-linker glutaraldehyde.40 Chitosan cross-linked with genipin (chitosan–genipin) has been successfully used as a protective layer and entrapment material for enzyme electrodes in biosensors and BFCs.34,41 Conghaile et al. used genipin to cross-link chitosan, glucose oxidase, and a mediator.42 Fernandes et al. used chitosan–genipin as a matrix to immobilize laccase in a carbon paste electrode, with paraffin oil as a binder.43
In this study, we developed a bio-composite ink for screen printing a BFC-anode using MgOC as a conductive material and chitosan–genipin as a binder. To the best of our knowledge, this is the first report of a screen-printable bio-composite ink containing mesoporous carbon or chitosan–genipin. Paper-based bioanodes fabricated with this ink were evaluated and compared with the corresponding electrodes fabricated through conventional printing and modified via drop-casting.
Experimental
Materials
MgO-templated carbon CNovel© (MgOC) was obtained from Toyo Tanso (Osaka, Japan). Glucose oxidase (GOx, 100 U mg−1) from Aspergillus niger and chitosan were purchased from Sigma-Aldrich (Tokyo, Japan). Bilirubin oxidase (BOD, BO “Amano” 3) from Myrothecium verrucaria was obtained from Amano Enzyme Inc. (Nagoya, Japan). 1,2-Naphthoquinone (1,2-NQ) was purchased from Kanto Chemical (Tokyo, Japan). 2,2′-Azinobis(3-ethylbenzothiazoline-6-sulfonic acid ammonium salt) (ABTS) was obtained from Tokyo Chemical Industry Co. (Tokyo, Japan). Genipin was purchased from Fujifilm Wako Pure Chemical Co. (Osaka, Japan). Carbon ink (JELCOM CH-10) was obtained from Jujo Chemicals (Tokyo, Japan).All other chemicals were of analytical grade. All aqueous solutions were prepared using ultrapure water.
Rheological characterization
All rheological measurements were performed using a rheometer (MCR 102; Anton Paar, Tokyo, Japan) with parallel plates (pp25, gap 1 mm).To evaluate chitosan–genipin thermal behavior, a sample was prepared by adding genipin to a chitosan solution and stirring for 1 min and analyzed immediately after preparation. A temperature sweep was performed in the range of 25–65 °C increasing at 2 °C min−1. The low vibration frequency (1 Hz) and small deformation (0.01) confirmed that the experimental conditions did not interfere with the gelation process.
To evaluate chitosan–genipin gelation behavior over time, a chitosan–genipin sample was prepared and analyzed at a constant temperature of 60 °C with low vibration frequency (1 Hz) and small deformation (0.01). The gelation point was determined from the intersection of the storage and loss moduli.
To evaluate the coatability of MgOC–chitosan–genipin ink, ink samples were prepared by mixing MgOC into a chitosan–genipin sample before gelation at 1 min after adding genipin (excluding the 1 min stirring step), at 20 min (close to gelation point), at 40 min (after gelation), and at 80 min (well after gelation). Chitosan–genipin was removed from the heat source before the addition of MgOC. The rotational viscosities of the resulting MgOC–chitosan–genipin inks were measured. To simulate screen printing, the shear rate was changed from slow (0.1 s−1 for 60 s) to fast (1000 s−1 for 60 s) and back to slow (0.1 s−1 for 420 s) during the measurement and the change in viscosity during this procedure was recorded.
Pre-treatment of MgOC
To modify the surface characteristics of MgOC, the porous carbon was pre-treated using different methods. Acids are used to dissolve the MgO template, and thus, untreated MgOC (“Normal MgOC”) has an acidic and hydrophilic surface. Baking MgOC at 800 °C in a N2 atmosphere results in the removal of functional groups and the surface becomes hydrophobic and neutral (“Baked MgOC”).44 An intermediate state with a hydrophilic and neutral surface was achieved by washing MgOC with 0.25 M NaOH aq. and pure water (“Washed MgOC”). Treatment with bases introduces basic functional groups and thus neutralizes the acidic surface.45Preparation of the bio-composite ink
To prepare the bio-composite ink, Normal, Baked, or Washed MgOC was first dispersed in a mediator solution and the solvent was removed by filtration. Next, MgOC/mediator was dispersed in an enzyme solution and the solvent was removed again by filtration. Finally, MgOC/mediator/enzyme was mixed with chitosan–genipin, which was prepared by adding genipin to chitosan solution.Fabrication of paper-based BFCs via screen printing
First, Japanese paper (Izumo Tokusengasenshi, Keynote, Japan) was treated with a water repellent (Hajikkusu, Komensu, Japan) and dried at 25 °C for 12 h. Next, five layers of carbon leads were printed with an LS-150TV screen-printer (Newlong Seimitsu Kogyo Co. Ltd., Tokyo, Japan) (Fig. 1a and b). The carbon leads were then dried at 120 °C for 1 d. Finally, the modified electrodes were printed using the bio-composite ink prepared for the bioanode. The size of each electrode was 0.5 cm × 2.0 cm. The bio-composite ink-printed electrodes were dried at 5 °C for 1 d. The BFC was completed with a conventional biocathode prepared according to a previously reported method.15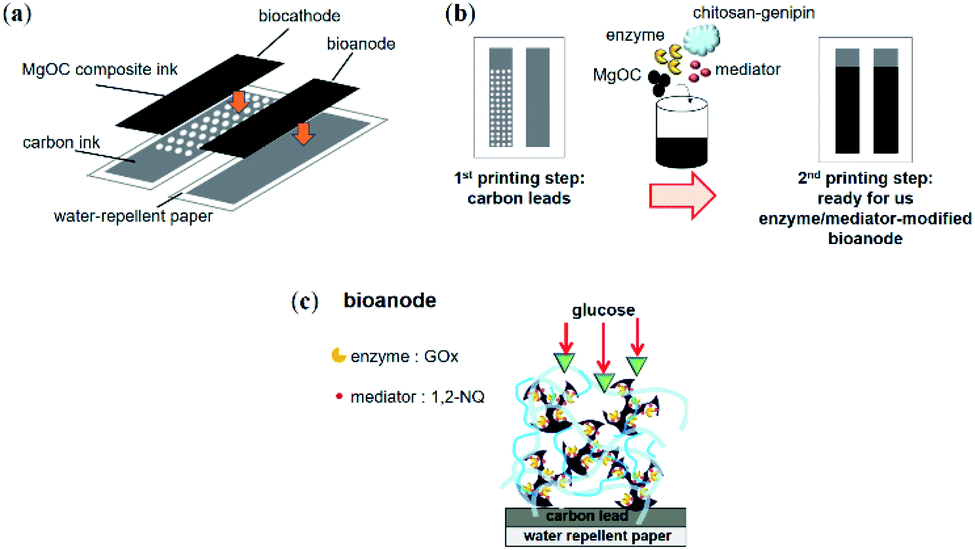 | ||
| Fig. 1 Schematic of a paper-based BFC printed using MgOC bio-composite ink. (a) Design of the BFC with printing layers. (b) Schematic of the printing process conducted using bio-composite ink. (c) Schematic of structure of printed bioanode. | ||
Scanning electron microscopy
The electrode structures were characterized using a scanning electron microscope (SEM, JSM-7600F; JEOL, Tokyo, Japan). For this, the samples were dried under reduced pressure.Electrochemical characterization
Single electrodes were evaluated in a batch cell with a platinum wire used as the counter electrode and Ag/AgCl/saturated KCl as the reference electrode. Glucose was added to the measurement solution before potential application. BFCs were evaluated in an incubator at 36 °C and 70% humidity by dropping 200 μL of 100 mM glucose solution onto the electrodes. All electrodes were used as fabricated. Cyclic voltammetry was performed at a scan rate of 10 mV s−1. Chronoamperometry was performed by applying a potential of 0.2 V. Linear sweep voltammetry was performed at a scan rate of 1 mV s−1. Emstat3 Blue (Palm Sens, Houten, Netherlands) was used as the potentiostat.Results & discussion
Rheological characterization of chitosan–genipin
Depending on the progress of the chitosan–genipin cross-linking reaction, the viscoelastic properties of the resulting gel might change, thus influencing the screen printability of an ink containing chitosan–genipin as a binder. Therefore, the time and temperature of gel formation by covalent cross-links were optimized by measuring the rheological behavior of gelation.To optimize the cross-linking temperature, the change in the storage modulus of chitosan mixed with genipin was observed over increasing temperature (Fig. 2a). The storage modulus showed a sharp increase at around 60 °C, which can be attributed to a higher degree of gel formation caused by an increased rate of the cross-linking reaction at higher temperatures.
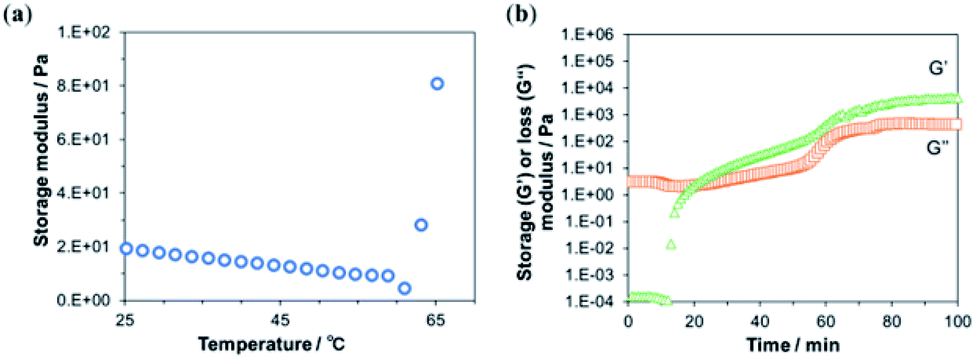 | ||
| Fig. 2 Rheologic characterization of chitosan mixed with genipin. (a) Temperature dependency of the storage modulus. (b) Time dependency of the storage (G′, orange squares) and loss (G′′, green triangles) moduli. | ||
Next, the progress of the cross-linking reaction was further evaluated at 60 °C by measuring the time-dependent changes in the storage and loss moduli (Fig. 2b). At the beginning of the measurement, the loss modulus, which represents the viscous component of the viscoelasticity, was higher than the storage modulus, which represents the elastic component. This indicates a high fluidity of the sample, suggesting that the crosslink density was low immediately after the addition of genipin. After 12 min, the storage modulus increased sharply and intersected with viscosity loss at approximately 20 min (Fig. 2b). This suggests an increase in cross-linking and a transition from solution to gel. The intersection of the storage and loss moduli is called the gel point.
Next, MgOC was mixed with chitosan–genipin at different times in relation to the gel point and the coatability of the resulting ink was evaluated (Fig. 3). For successful screen-printing, the ink should be of medium viscosity and retain this viscosity when shear forces are applied. When MgOC was mixed into chitosan–genipin before gel formation (1 min after mixing chitosan and genipin), the initial viscosity was not recovered after the application of shear forces (Fig. 3a). Immediately after adding the cross-linker genipin, the chitosan–genipin mixture is too fluid, and adding MgOC might hinder further cross-linking. Electrodes printed using ink with this rheological profile tend to blur at the edges, making it difficult to form well-defined patterns.
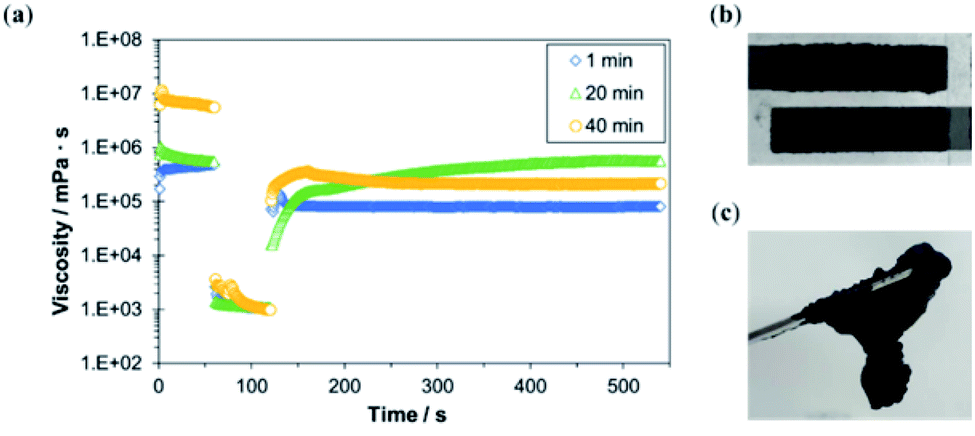 | ||
| Fig. 3 Characterization of MgOC–chitosan–genipin ink mixed at different times in relation to the gel point of chitosan–genipin. (a) Rheologic viscosity measurements following the immediate application of shear forces in the second minute (blue diamonds: 1 min), mixing of ink before the gel point (green triangles: 20 min), mixing of ink at approximately the gel point (yellow circles: 40 min), and mixing of ink after the gel point. (b) Printing test of ink mixed at the approximate gel point (at 20 min). (c) Ink mixed well after the gel point (at 80 min). | ||
When MgOC was mixed into chitosan–genipin at the approximate gel point (20 min after mixing chitosan and genipin), the initial viscosity gradually recovered following the application of shear forces. In this case, sufficient cross-linking had occurred before the addition of MgOC. An ink with this rheological profile should be suitable for screen printing, and a printing test resulted in acceptable electrodes (Fig. 3b).
When MgOC was mixed with chitosan–genipin after the gel point (40 min after mixing chitosan and genipin), the initial viscosity was high, but it did not recover following the application of shear forces (Fig. 3a). The high initial viscosity can be attributed to increased cross-linking before the addition of MgOC. However, the rheological profile suggests that some of these cross-links break when shear forces are applied. The initial viscosity of this ink was too high for screen printing, and it was difficult to press the ink through the stencil. When MgOC was mixed with chitosan–genipin well after the gel point (80 min after mixing chitosan and genipin), the resulting ink was lumpy and not suitable for printing (Fig. 3c). This can be explained by the excessive number of cross-links formed before the addition of MgOC.
From these rheological characterizations, it can be concluded that a suitable ink can be prepared by mixing MgOC with chitosan–genipin that has been allowed to gelate for 20 min at 60 °C.
Structural characterization of electrodes printed using MgOC–chitosan–genipin ink
SEM images of MgOC powders after different pre-treatments before mixing with chitosan–genipin showed relatively fine particles of 1–5 μm for Normal and Baked MgOC, and larger particles of ≥5 μm for Washed MgOC (Fig. 4). This result indicated that the washing process led to the aggregation of the MgOC particles. | ||
| Fig. 4 SEM images of (a) Normal, (b) Baked, and (c) Washed MgOC powder following different pre-treatments. | ||
Inks were prepared using the three types of MgOC, and electrodes were printed using the resulting inks. SEM images of these electrodes showed a dense structure for electrodes containing MgOC and Baked MgOC (Fig. 5a and b). The SEM image of the electrode obtained with Washed MgOC showed macropores with an average pore size of 5–10 μm (Fig. 5c).
 | ||
| Fig. 5 SEM images of electrodes printed using MgOC–chitosan–genipin ink prepared with (a) Normal, (b) Baked, or (c) Washed MgOC. | ||
The structures of the electrodes suggested that, while the large secondary particles from Washed MgOC are partially broken down and dispersed in chitosan–genipin, they are not as dispersed as the fine particles of the Normal and Baked MgOC. This could be related to the effect that washing has on MgOC acidity, which could then affect cross-linking. Furthermore, it has been reported that the mechanism of cross-linking chitosan with genipin is pH dependent.46 Indeed, under acidic conditions, genipin acts as a dialdehyde, but its condensation products are considerably more stable than glutaraldehyde and this results in a tight network with shorter cross-links. In contrast, under basic conditions, the reaction is accompanied by genipin self-polymerization, resulting in a loose and flexible network with longer cross-links. Therefore, pH plays an important role in influencing the cross-linking reactions. In addition, the enzyme itself can be cross-linked to chitosan via genipin and the degree of this cross-linking is maximum at pH 3–6 and decreases at near neutral pH.47
As the surface of Normal MgOC is acidic and the surfaces of Baked and Washed MgOC are neutral, this pH dependency might influence cross-linking reaction after the addition of MgOC. In fact, the electrode fabricated with Baked MgOC was slightly more porous than that fabricated with Normal MgOC (Fig. 5a and b). Thus, both surface pH and particle size of the MgOC powder contribute to the structure of the resulting electrode.
To prepare a successful bio-composite ink, the components must be carefully chosen such that the enzyme remains active during fabrication and printing and is substrate accessible within the electrodes. The intended enzyme in this study, GOx, favors neutral over acidic conditions. A possibly flexible chitosan–genipin is also favorable for maintaining enzymes in general. For the accessibility of glucose, hydrophilic conditions and a macroporous structure are favorable (Fig. 1c). Thus, Washed MgOC seemed the most promising candidate to prepare a bioanode ink containing GOx.
Electrochemical characterization of bioanodes printed using bio-composite ink
Ink containing Washed MgOC, GOx, and 1,2-NQ was fabricated, and a bioanode was printed using this ink. The cyclic voltammograms show that, in the presence of glucose, the oxidation current was significantly higher than in the absence of glucose (Fig. 6a). This indicates that the electrode exhibits a high electrocatalytic activity toward glucose oxidation.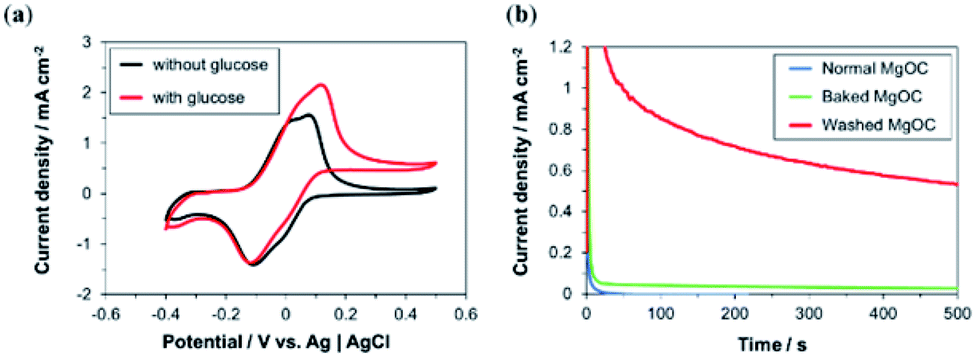 | ||
| Fig. 6 Electrochemical responses of bioanodes printed using bio-composite ink. (a) Cyclic voltammetric response of Washed MgOC-based bioanodes in the (black) absence and (red) presence of 100 mM glucose. (b) Chronoamperometric response of bioanodes based on (blue) Normal, (green) Baked, and (red) Washed MgOC in the presence of 100 mM glucose. | ||
This was confirmed by chronoamperometry measurements (Fig. 6b) of the bioanode delivering a current density of >0.5 mA cm−2 in the presence of glucose (red curve), whereas the current densities delivered by bioanodes based on Normal (blue curve) and Baked MgOC (green curve) were very low. However, a closer observation showed a slightly higher response current for the Baked MgOC-based electrode than the Normal MgOC-based electrode, suggesting that an acidic surface and a dense structure are more detrimental than a hydrophobic surface.
The slow decrease in current shown in Fig. 6b can be attributed to the charging of the electrical double layer. The large physical area of the electrode (1 cm2), the very large specific surface area due to the macroporous and mesoporous structures of the electrode, as well as the high substrate concentration used in the measurement are known causes for a large double-layer capacity. Leakage of loosely bound mediator or enzyme is unlikely to be the cause for this decrease for two reasons. The first reason is in the fabrication process of the bio-composite ink. The stepwise binding of mediator and enzyme to MgOC with the removal of solvent by filtration should not leave many loosely bound mediator and enzyme molecules on MgOC. Furthermore, the subsequent addition of the binder should trap the mediator and enzyme in the mesopores of MgOC. Second, previous studies have shown that water-insoluble mediators such as 1,2-NQ and various enzymes are immobilized stably in MgOC-electrodes.32,48
Next, the response of the bioanode printed using Washed MgOC-based bio-composite ink was investigated in the presence of various concentrations of glucose (Fig. 7). The response current increased linearly up to at least 8 mM glucose with a sensitivity of 25.83 μA cm−2 mM−1. This dynamic range and sensitivity suggest that the bioanode could, for example, be used as a urine glucose sensor. The normal range for glucose in urine is 0–0.8 mM, and diabetes is suspected at glucose concentrations of ≥3 mM.
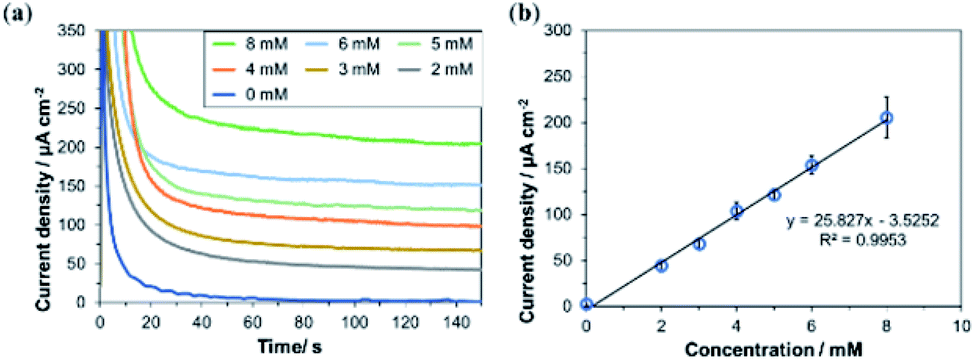 | ||
| Fig. 7 Concentration dependency of bioanodes printed using Washed MgOC-based bio-composite ink. (a) Chronoamperometric response in the presence of various concentrations of glucose. (b) Current density vs. glucose concentration (n = 3, confidence interval 90%, linear regression). | ||
Characterization of the bio-composite-ink-bioanode-based BFCs
Finally, we investigated the performances of the bioelectrode fabricated with the bio-composite ink as a bioanode for wearable BFCs. The BFCs were prepared by combining a bioanode printed using the bio-composite ink and a conventional biocathode with printed MgOC and BOD immobilized in the mesopores of MgOC by drop-casting.15An electromotive force of 0.54 V and a maximum output density of 96 μW cm−2 were obtained (Fig. 8). A current of at least 30 μA cm−2 was achieved at 0.35 V with the 1 cm2 per electrode cell, which is a sufficient power source for ultra-low-power wireless transmission devices without the need for a power booster circuit.
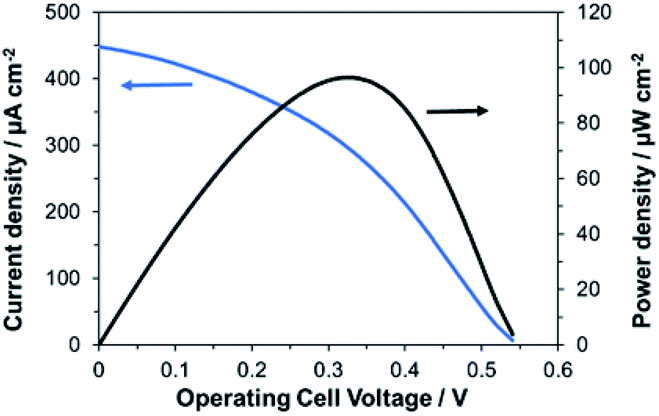 | ||
| Fig. 8 Current and power density curves of BFCs using the bio-composite-ink-bioanode and a drop-casted biocathode in the presence of 100 mM glucose. | ||
Conclusions
In this study, a new bio-composite ink for screen-printing bioanodes for paper-based BFCs was developed successfully. The ink contained GOx, 1,2-NQ, MgOC, and chitosan–genipin, and could be used for one-step deposition of bioanodes. Pre-treating the MgOC by washing to achieve a neutral hydrophilic surface and the highly porous structure of the resulting electrode was essential for the accessibility of glucose to the enzyme. The hydrophilic and neutral conditions were also helpful in maintaining enzyme activity. A paper-based bioanode fabricated via one-step deposition using this bio-composite ink showed a linear current dependency response to at least 8 mM glucose with a sensitivity of 25.83 μA cm−2 mM−1. Combined with a biocathode fabricated via drop casting, an electromotive force of 0.54 V and a maximal power density of 96 μW cm−2 were achieved. These results show that this bio-composite ink can be used to replace the multi-step process of printing bioanodes with conventional ink followed by drop-casting the enzyme and mediator with a one-step printing process. In the future, a corresponding biocathode ink will complete the simple fabrication process for the BFCs.Author contributions
Isao Shitanda: conceptualization, writing – original draft preparation, supervision. Kanako Oda: investigation, writing – original draft preparation. Noya Loew: writing – review & editing. Hikari Watanabe: supervision. Masayuki Itagaki: supervision. Seiya Tsujimura: conceptualization, writing – review & editing. Abdelkader Zebda: conceptualization, writing – review & editing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was partially supported by JST-ASTEP Grant Number JPMJTS1513, JSPS Grant Number 17H02162 and Private University Research Branding Project (2017–2019) from the Ministry of Education, Culture, Sports, Science and Technology, and the Tokyo University of Science Grant for President's Research Promotion. The authors also thank the Auvergne Rhones Alpes program “Cooperation International” for its financial support.Notes and references
- K. Yamanaka, M. C. Vestergaard and E. Tamiya, Sensors, 2016, 16, 1761, DOI:10.3390/s16101761.
- Z. Taleat, A. Khoshroo and M. Mazloum-Ardakani, Microchim. Acta, 2014, 181, 865–891, DOI:10.1007/s00604-014-1181-1.
- A. Othman, A. Karimi and S. Andreescu, J. Mater. Chem. B, 2016, 4, 7178–7203, 10.1039/C6TB02009G.
- J. R. Windmiller and J. Wang, Electroanalysis, 2013, 25, 29–46, DOI:10.1002/elan.201200349.
- A. J. Bandodkar, I. Jeerapan and J. Wang, ACS Sens., 2016, 1, 464–482, DOI:10.1021/acssensors.6b00250.
- C. Gonzalez-Solino and M. D. Lorenzo, Biosensors, 2018, 8, 11–28, DOI:10.3390/bios8010011.
- A. J. Bandodkar, W. Jia, C. Yardımcı, X. Wang, J. Ramirez and J. Wang, Anal. Chem., 2015, 87, 394–398, DOI:10.1021/ac504300n.
- W. Jia, A. J. Bandodkar, G. Valdés-Ramírez, J. R. Windmiller, Z. Yang, J. Ramírez, G. Chan and J. Wang, Anal. Chem., 2013, 85, 6553–6560, DOI:10.1021/ac401573r.
- W. Jia, G. Valdés-Ramírez, A. J. Bandodkar, J. R. Windmiller and J. Wang, Angew. Chem., Int. Ed., 2013, 52, 7233–7236, DOI:10.1002/anie.201302922.
- I. Jeerapan, J. R. Sempionatto, A. Pavinatto, J. M. You and J. Wang, J. Mater. Chem. A, 2016, 4, 18342–18353, 10.1039/C6TA08358G.
- J. Kim, S. Imani, W. R. de Araujo, J. Warchall, G. Valdés-Ramírez, T. R. L. C. Paixão, P. P. Mercier and J. Wang, Biosens. Bioelectron., 2015, 74, 1061–1068, DOI:10.1016/j.bios.2015.07.039.
- I. Shitanda, T. Yamaguchi, Y. Hoshi and M. Itagaki, Chem. Lett., 2013, 42, 1369–1370, DOI:10.1246/cl.130681.
- I. Shitanda, S. Kato, Y. Hoshi, M. Itagaki and S. Tsujimura, Chem. Commun., 2013, 49, 11110–11112, 10.1039/C3CC46644B.
- I. Shitanda, M. Momiyama, N. Watanabe, T. Tanaka, S. Tsujimura, Y. Hoshi and M. Itagaki, ChemElectroChem, 2017, 4, 2460–2463, 10.1039/C3CC46644B.
- I. Shitanda, Y. Fujimura, S. Nohara, Y. Hoshi, M. Itagaki and S. Tsujimura, J. Electrochem. Soc., 2019, 166, B1063, DOI:10.1149/2.1501912jes.
- M. Albareda-Sirvent, A. Merkoçi and S. Alegret, Sens. Actuators, B, 2000, 69, 153–163, DOI:10.1016/S0925-4005(00)00536-0.
- C. A. Galán-Vidal, J. Muñoz, C. Domínguez and S. Alegret, Sens. Actuators, B, 1998, 52, 257–263, DOI:10.1016/S0925-4005(98)00276-7.
- E. Crouch, D. C. Cowell, S. Hoskins, R. W. Pittson and J. P. Hart, Anal. Biochem., 2005, 347, 17–23, DOI:10.1016/j.ab.2005.08.011.
- C. A. Galán-Vidal, J. Muñoz, C. Domínguez and S. Alegret, Sens. Actuators, B, 1997, 45, 55–62, DOI:10.1016/S0925-4005(97)00270-0.
- I. Rohm, M. Genrich, W. Collier and U. Bilitewski, Analyst, 1996, 121, 877–881, 10.1039/AN9962100877.
- R. Nagata, K. Yokoyama, S. A. Clark and I. Karube, Biosens. Bioelectron., 1995, 10, 261–267, DOI:10.1016/0956-5663(95)96845-P.
- G. Hughes, R. M. Pemberton, P. Nicholas and J. P. Hart, Electroanalysis, 2018, 30, 1616–1620, DOI:10.1002/elan.201800104.
- A. Sanati, M. Jalali, K. Raeissi, F. Karimzadeh, M. Kharaziha, S. S. Mahshid and S. Mahshid, Microchim. Acta, 2019, 186, 773, DOI:10.1007/s00604-019-3854-2.
- F. Otero and E. Magner, Sensors, 2020, 20, 3561, DOI:10.3390/s20123561.
- I. Shitanda, S. Nohara, Y. Hoshi, M. Itagaki and S. Tsujimura, J. Power Sources, 2017, 360, 516–519, DOI:10.1016/j.jpowsour.2017.06.043.
- X. Chen, L. Yin, J. Lv, A. J. Gross, M. Le, N. G. Gutierrez, Y. Li, I. Jeerapan, F. Giroud, A. Berezovska, R. K. O'Reilly, S. Xu, S. Cosnier and J. Wang, Adv. Funct. Mater., 2019, 29, 1905785, DOI:10.1002/adfm.201905785.
- N. Marković, F. Conzuelo, J. Szczesny, M. B. González García, D. Hernández Santos, A. Ruff and W. Schuhmann, Electroanalysis, 2019, 31, 217–221, DOI:10.1002/elan.201800462.
- S. Tsujimura, K. Murata and W. Akatsuka, J. Am. Chem. Soc., 2014, 136, 14432–14437, DOI:10.1021/ja5053736.
- I. Shitanda, K. Takamatsu, A. Niiyama, T. Mikawa, Y. Hoshi, M. Itagaki and S. Tsujimura, J. Power Sources, 2019, 436, 226844, DOI:10.1016/j.jpowsour.2019.226844.
- I. Shitanda, T. Kato, R. Suzuki, T. Aikawa, Y. Hoshi, M. Itagaki and S. Tsujimura, Bull. Chem. Soc. Jpn., 2020, 93, 32–36, DOI:10.1246/bcsj.20190212.
- A. Niiyama, K. Murata, Y. Shigemori, A. Zebda and S. Tsujimura, J. Power Sources, 2019, 427, 49–55, DOI:10.1016/j.jpowsour.2019.04.064.
- I. Shitanda, M. Mitsumoto, N. Loew, Y. Yoshihara, H. Watanabe, T. Mikawa, S. Tsujimura, M. Itagaki and M. Motosuke, Electrochim. Acta, 2021, 368, 137620, DOI:10.1016/j.electacta.2020.137620.
- A. B. Engel, Y. Holade, D. Cornu, K. Servat, T. W. Napporn, K. B. Kokoh and S. Tingry, J. Electrochem. Soc., 2016, 164, G29, DOI:10.1149/2.0571702jes.
- S. El Ichi, A. Zebda, J. P. Alcaraz, A. Laaroussi, F. Boucher, J. Boutonnat, N. Reverdy-Bruas, D. Chaussy, M. Belgacem, P. Cinquin and D. Martin, Energy Environ. Sci., 2015, 8, 1017–1026, 10.1039/C4EE03430A.
- Y. Zhang, Y. Li, W. Wu, Y. Jiang and B. Hu, Biosens. Bioelectron., 2014, 60, 271–276, DOI:10.1016/j.bios.2014.04.035.
- H. Yi, L. Q. Wu, W. E. Bentley, R. Ghodssi, G. W. Rubloff, J. N. Culver and G. F. Payne, Biomacromolecules, 2005, 6, 2881–2894, DOI:10.1021/bm050410l.
- S. Besic and S. D. Minteer, Micellar Polymer Encapsulation of Enzymes, in Enzyme Stabilization and Immobilization: Methods and Protocols, ed. S. D. Minteer, Springer, New York, NY, 2017, pp. 93–108, DOI:10.1007/978-1-4939-6499-4_8.
- S. El Ichi, A. Zebda, A. Laaroussi, N. Reverdy-Bruas, D. Chaussy, M. N. Belgacem, P. K. Cinquin and D. Martin, Chem. Commun., 2014, 50, 14535–14538, 10.1039/C4CC04862H.
- A. B. Tahar, A. Szymczyk, S. Tingry, P. Vadgama, M. Zelsmanne, S. Tsujumura, P. Cinquin, D. Martin and A. Zebda, J. Electroanal. Chem., 2019, 847, 113069, DOI:10.1016/j.jelechem.2019.04.029.
- L. Xu, Y. A. Huang, Q. J. Zhu and C. Ye, Int. J. Mol. Sci., 2015, 16, 18328–18347, DOI:10.3390/ijms160818328.
- K. Hyun, S. Kang, J. Kim and Y. Kwon, ACS Appl. Mater. Interfaces, 2020, 12, 23635–23643, DOI:10.1021/acsami.0c05564.
- P. Ó. Conghaile, R. Kumar, M. L. Ferrer and D. Leech, Electrochem. Commun., 2020, 113, 106703, DOI:10.1016/j.elecom.2020.106703.
- S. C. Fernandes, D. M. P. de Oliveira Santos and I. C. Vieira, Electroanalysis, 2013, 25, 557–566, DOI:10.1002/elan.201200564.
- W. Shen, Z. Li and Y. Liu, Recent Pat. Chem. Eng., 2007, 1, 27–40, DOI:10.2174/2211334710801010027.
- A. Rehman, M. Park and S.-J. Park, Coatings, 2019, 9, 103, DOI:10.3390/coatings9020103.
- E. E. E. Flores, F. D. Cardoso, L. B. Siqueira, N. C. Ricardi, T. H. Costa, R. C. Rodrigues, M. P. Klein and P. F. Hertz, Process Biochem., 2019, 84, 73–80, DOI:10.1016/j.procbio.2019.06.001.
- M. P. Klein, C. R. Hackenhaar, A. S. G. Lorenzoni, R. C. Rodrigues, T. M. H. Costa, J. L. Ninow and P. F. Hertz, Carbohydr. Polym., 2016, 137, 184–190, DOI:10.1016/j.carbpol.2015.10.069.
- I. Shitanda, H. Inoue, Y. Yoshihata, N. Loew and M. Itagaki, Biosens. Bioelectron., 2021, 178, 113014, DOI:10.1016/j.bios.2021.113014.
Footnote |
| † These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2021 |