
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceStephapierrines A–H, new tetrahydroprotoberberine and aporphine alkaloids from the tubers of Stephania pierrei Diels and their anti-cholinesterase activities†
Waraluck Chaichompooa,
Pornchai Rojsitthisak *ab,
Wachirachai Pabuprapapc,
Yuttana Siriwattanasathienc,
Pathumwadee Yotmaneec,
Woraphot Haritakund and
Apichart Suksamrarnc
*ab,
Wachirachai Pabuprapapc,
Yuttana Siriwattanasathienc,
Pathumwadee Yotmaneec,
Woraphot Haritakund and
Apichart Suksamrarnc
aDepartment of Food and Pharmaceutical Chemistry, Faculty of Pharmaceutical Sciences, Chulalongkorn University, Bangkok 10330, Thailand. E-mail: pornchai.r@chula.ac.th; Fax: +66-2-254-5195; Tel: +66-2-218-8310
bNatural Products for Aging and Chronic Diseases Research Unit, Chulalongkorn University, Bangkok 10330, Thailand
cDepartment of Chemistry and Center of Excellence for Innovation in Chemistry, Faculty of Science, Ramkhamhaeng University, Bangkok 10240, Thailand
dProgram in Chemical Technology, Faculty of Science and Technology, Suan Dusit University, Bangkok 10700, Thailand
First published on 15th June 2021
Abstract
Eight new alkaloids, which are four new tetrahydroprotoberberine alkaloids, stephapierrines A–D (1–4), and four new aporphine alkaloids, stephapierrines E–H (5–8), together with three new naturally occurring alkaloids (9–11) and thirty-four known alkaloids (12–45) were isolated from the tubers of Stephania pierrei Diels. The structures of the new compounds were elucidated by spectroscopic analysis and physical properties. The structures of the known compounds were characterized by comparison of their spectroscopic data with those previously reported. Compound 42 exhibited the strongest acetylcholinesterase (AChE) inhibitory activity, which was more active than galanthamine, the reference drug. Compound 23 showed the highest butyrylcholinesterase (BuChE) inhibitory activity, which was also more active than galanthamine. Molecular docking studies are in good agreement with the experimental results.
Introduction
Alzheimer's disease (AD) is the major progressive chronic neurodegenerative disease, accounting for an estimated 60–80% of cases of patients suffering from dementia worldwide.1 It is deterioration in cognitive function affecting memory, thinking, learning, judgement, and behavior which is ultimately interfering with the ability of the person to perform daily tasks.2 In 2019, over 50 million people were living with dementia worldwide. This number is expected to increase to 152 million by 2050.3 The impact of AD is not only substantial in economic terms for health care resources and medical services, but also represents the extensive human costs to countries, societies, families, and individuals. Previously reported hypotheses on the progression of AD pathologies included cholinergic dysfunction, amyloid plaques, neurofibrillary tangles, neuroinflammation, and mitochondrial dysfunction, as well as oxidative stress, which are the major hallmarks of AD.4 Cholinergic hypothesis is one of the theories for AD pathology. Acetylcholine (ACh) neurotransmitter used by cholinergic neurons is considered to play a critical role in the peripheral and central nervous systems.5 Acetylcholinesterase (AChE) and butyrylcholinesterase (BuChE) are the biologically important enzymes that hydrolyze ACh.6 In addition, BuChE was associated with AD pathology, such as amyloid-beta (Aβ) plaques.7 It has been reported that acetylcholinesterase inhibitors (AChEIs) decrease the level of AChE in the brains of AD patients, which resists cerebral neurotransmitter acetylcholine metabolism and prolongs ACh action at the synapses.8 Thus, the inhibitions of AChE and BuChE to protect and enhance the ACh levels in patients have been the most attractive therapeutic strategy. US Food and Drug Administration (FDA)-approved medications are palliative and offer only temporary modulate the symptoms by changing the level of neurotransmitters in the brain. Donepezil, rivastigmine, and galanthamine are currently the only three AChEIs available in clinics while memantine is an N-methyl-D-aspartate receptor (NMDAR) antagonist.9 However, these AChEI drugs cannot stop or reverse disease progression and cause several side effects.10 Due to these issues, there is a crucial need to find an effective and safe disease-modifying medication to overcome AD and the research for new medications with potential clinical value seems to be necessary.Plant-derived natural products have long been and will continue to be extremely important as the most promising source of biologically active compounds. A great potential is expected for indigenous plants to be used as a source of new drugs. Among these natural products, alkaloids are considered to be the most promising candidates for the treatment of AD due to their complex nitrogen-containing structures.11 Many alkaloids that are active cholinesterase inhibitors have already been described in various families, for example, the Menispermaceae family.12
The genus Stephania belongs to the Menispermaceae family, a large family of approximately 65 genera and 350 species, distributed in temperate regions of the world. The plants of the genus Stephania are slender climbers with peltate and membranous leaves. The plants of this genus are commonly used in Asian folk medicine to treat a wide range of biological activities including malaria, fever, dysentery, and tuberculosis.13 At least 15 species in the genus Stephania have been found distributed throughout Thailand.14 Stephania pierrei Diels, known in Thai as Sabu-lueat, is a medicinal plant regularly used in traditional remedies and as herbal medicine to treat body oedema, migraine, and heart disease, which are primarily distributed in Cambodia, Thailand, and Vietnam.15 The reported data showed that several types of alkaloids had been isolated from this plant species, including aporphine, tetrahydroprotoberberine, tetrahydrobenzylisoquinoline, and miscellaneous compounds.16,17 However, the anti-cholinesterase activity of this plant has not been reported. From our preliminary investigation on phytochemicals with cholinesterase inhibitory activities from Thai medicinal plants, we found that the active crude extracts of the tubers of S. pierrei showed inhibitory activities on AChE and BuChE. Accordingly, we report herein the isolation, structure elucidation, and absolute configuration assignments of eight new alkaloids (1–8), and three new naturally occurring alkaloids (9–11), together with thirty-four known alkaloids (12–45) from the tuber extracts of S. pierrei. Most of the isolated compounds were also evaluated for their AChE and BuChE inhibitory activities. For further insight into the experimental results, in silico studies were performed.
Results and discussion
The tubers of S. pierrei were extracted successively with n-hexane, EtOAc, and MeOH. Preliminary screening of the hexane, EtOAc and MeOH extracts of this plant revealed significant in vitro cholinesterase inhibitory activities towards AChE with IC50 ranges from 1.01–20.95 ng mL−1 and exhibited BuChE inhibitory activity with the IC50 values of 2.76–17.46 ng mL−1. These active extracts were therefore subjected to chromatographic isolation for the active principles. Based on the spectroscopic analysis and the physical properties, the chemical structures of the isolated compounds were elucidated and characterized as eight previously undescribed alkaloids, which are four new tetrahydroprotoberberine alkaloids stephapierrines A–D (1–4) and four new aporphine alkaloids stephapierrines E–H (5–8). Three new naturally occurring alkaloids, O,N-diacetylasimilobine (9),18 N-acetamidesecocrebanine (10)19 and 2,3-didemethyltetrahydropalmatine (11),20 together with thirty-four known alkaloids (12–45) were identified. The previously described alkaloids were identified as stepholidine (12),21 discretamine (13),22 tetrahydropalmatine (14),23 N-methylstepholidine (15),21 cyclanoline (16),21 N-methyltetrahydropalmatine (17),23 jatrorrhizine (18),24 palmatine (19),25 dehydrocorydaline (20),26 pseudodehydrocorydaline (21),27 roemerine (22),28 (−)-stephanine (23),29 (−)-isolaureline (24),30 crebanine (25),23 dicentrine (26),31 (−)-ushinsunine (27),32 (−)-ayuthianine (28),29 sukhodianine (29),33 (−)-N-fonnylanonaine (30),32 (−)-N-methylasimilobine (31),32 (−)-asimilobine (32),32 (−)-asimilobine-2-O-β-D-glucoside (33),16,34 lanuginosine (34),35 dicentrinone (35),36 oxocrebanine (36),35 8-methoxyuvoriopsine (37),37 dehydroroemerine (38),38 dehydrostephanine (39),39 dehydroisolaureline (40),40 dehydrocrebanine (41),40 dehydrodicentrine (42),41 (−)-crebanine-β-N-oxide (43),42 coclaurine (44)43 and salutaridine (45)44 by physical and spectroscopic data comparisons with those of the literature values (see Fig. 1). The spectroscopic (IR, 1H and 13C NMR and mass) spectra of the new compounds 1–8, and the 1H and 13C NMR spectra, together with the specific optical rotations and mass spectral data of compounds 9–45 are presented in the ESI.†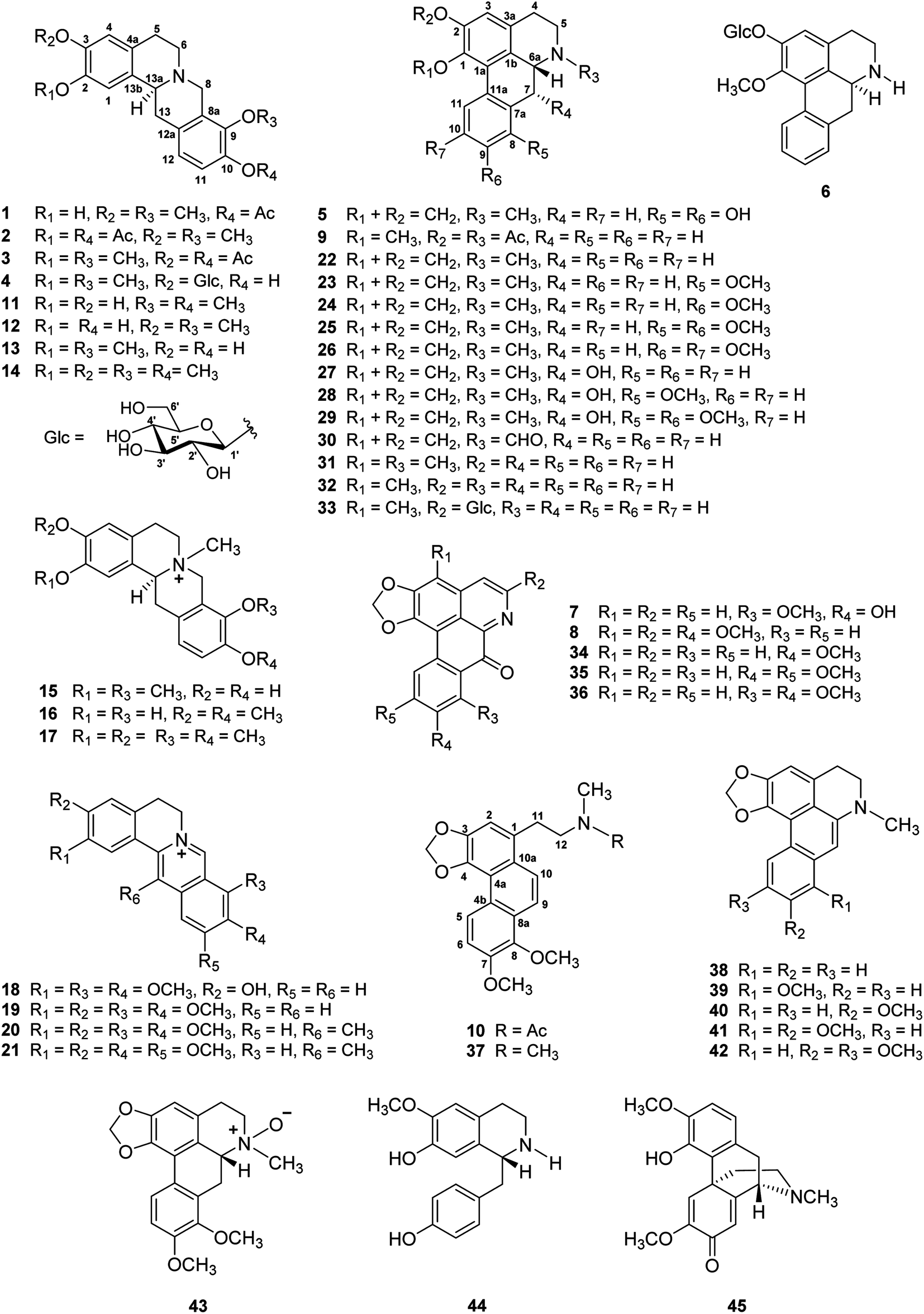 | ||
| Fig. 1 Structures of the isolated compounds 1–45 from the tubers of Stephania pierrei. | ||
Stephapierrine A (1) was assigned the molecular formula C21H23NO5 as deduced from the HR-ESI-TOF-MS at m/z 370.1633 [M + H]+ and NMR data. This alkaloid and all other isolated alkaloids gave positive orange coloration with Dragendorff's reagent. The IR spectrum indicated the presence of a hydroxy group (3380 cm−1), an acetoxy group (1761 cm−1) and aromatic rings (1608 and 1514 cm−1). The 1H NMR data (Table 1) showed two singlet methoxy signals at δH 3.79 and 3.85 (each 3H, s), and an acetoxy methyl proton signal at δH 2.30 (s), together with two ortho-coupled aromatic protons at δH 6.90 and 6.88 (each 1H, d, J = 8.8 Hz, H-11 and H-12) and two aromatic singlet protons at δH 6.78 and 6.58 (1H each, s, H-1 and H-4). The 13C NMR (Table 2) and DEPT spectra showed twenty-one carbon signals which included twelve aromatic carbons between δC 110.6 and 147.7, one carbonyl carbon (δC 169.1), three methyl carbons (δC 20.8, 55.9 and 60.6), four methylene carbons (δC 28.7, 36.0, 51.3 and 53.5), and one methine carbon (δC 58.8). The seventeen-carbon and one-nitrogen skeleton of 1 suggested a tetraoxygenated tetrahydroprotoberberine alkaloid core structure.45,46 This was further confirmed by the presence of two adjacent methylenes as multiplet signals at δH 2.68 and 3.18 (H-5) and δH 2.68 and 3.21 (H-6), methylene signals at δH 3.59 and 4.22 (each d, J = 15.7 Hz, H-8), methylene signals at δH 2.90 (dd, J = 16.4, 12.2 Hz) and 3.28 (dd, J = 16.4, 3.2 Hz) (H-13), together with a partially overlapping signal at δH 3.64 (H-13a). The COSY correlations of these protons are shown in Fig. 2. The structure of 1 was similar to that of stepholidine (12).21 The main difference between the 1H NMR spectrum of 12 and that of 1 was the presence of an acetoxy methyl signal at δH 2.30 (1H, s) which was proven to be at C-10 by HMBC correlations between COCH3 (δH 2.30) and COCH3 (δC 169.1), and C-10 (δC 141.0) (see Fig. 2). The HMBC correlations between the OH signal (δH 5.52) and C-1 (δC 111.2) and C-2 (δC 144.0) allowed the location of this phenolic function at C-2. The location of the methoxy groups at C-3 and C-9 were assigned according to the correlations of the methoxy signals at δH 3.85 and 3.79 with the carbons at δC 145.2 (C-3) and δC 147.7 (C-9), respectively. The structure of compound 1 was confirmed by key HMBC correlations from H-1 to C-3, C-4a and C-13a, H-4 to C-2 and C-5, H-8 to C-6, C-9, C-12a and C-13a, and H-13 to C-13b (see Fig. 2). The structure of compound 1 was further confirmed by the following key NOESY correlations: CH3O-3/H4, H-6/H-5 and H-8, CH3O-9/H-8, and H-13a/H-1 and H-13 (Fig. 3). The absolute configuration of C-13a of this class of alkaloids has been established, for example, by stereoselective asymmetric synthesis.47 The negative sign of specific optical rotation of 1 is consistent with an α-orientation of hydrogen at C-13a. The absolute configuration of 1 was confirmed by the electronic circular dichroism (ECD) spectrum (Fig. S234†), which was similar to those reported for tetrahydroprotoberberine alkaloids,48,49 with negative Cotton effects at 204 nm (Δε −10.69), 243 nm (Δε −50.55) and 287 nm (Δε −6.61). Thus, the absolute configuration of C-13a was established as S-configuration (Fig. 1). Accordingly, compound 1 was identified as 10-O-acetylstepholidine.
| Position | 1b | 2b | 3b | 4c | 11d |
|---|---|---|---|---|---|
| a Assignments were based on 1H–1H COSY, HMQC, HMBC, and NOESY experiments; chemical shifts (δ) are given in ppm.b Recorded in CDCl3.c Recorded in CD3OD.d Recorded in DMSO-d6.e Overlapping signal.f Partially overlapping signal. | |||||
| 1 | 6.78 s | 6.89 s | 6.89 s | 6.92 s | 6.68 s |
| 4 | 6.58 s | 6.68 s | 6.79 s | 6.93 s | 6.63 s |
| 5 | 2.68 m | 2.71 m | 2.64f | 2.69f | 2.54f |
| 3.18 m | 3.14e | 3.08f | 3.07 m | 2.89 m | |
| 6 | 2.68 m | 2.63 m | 2.62f | 2.64f | 2.42f |
| 3.21 m | 3.15e | 3.15f | 3.20 m | 3.08f | |
| 8 | 3.59 d (15.7) | 3.51 d (15.7) | 3.54 d (15.9) | 3.35 d (15.6) | 3.35e |
| 4.22 d (15.7) | 4.17 d (15.7) | 4.18 d (15.9) | 4.19 d (15.6) | 4.02 d (15.7) | |
| 11 | 6.90 d (8.8) | 6.86 d (8.4) | 6.88 d (9.2) | 6.72 d (8.2) | 6.67 d (8.2) |
| 12 | 6.88 d (8.8) | 6.86 d (8.4) | 6.88 d (9.2) | 6.80 d (8.2) | 6.71 d (8.2) |
| 13 | 2.90 dd (16.4, 12.2) | 2.85 dd (16.1, 11.2) | 2.90 dd (16.1, 11.3) | 2.73 dd (16.1, 11.0) | 2.50 dd (15.7, 12.3) |
| 3.28 dd (16.4, 3.2) | 3.23 dd (16.1, 3.4) | 3.27 dd (16.1, 3.5) | 3.41 dd (16.1, 3.5) | 3.14 dd (15.7, 3.4) | |
| 13a | 3.64f | 3.55 dd (11.2, 3.4) | 3.60 dd (11.3, 3.5) | 3.58 dd (11.0, 3.5) | 3.34e |
| 2-OCH3 | 3.82 s | 3.86 s | |||
| 3-OCH3 | 3.85 s | 3.80 s | |||
| 9-OCH3 | 3.79 s | 3.79 s | 3.79 s | 3.80 s | 3.71 s |
| 10-OCH3 | 3.73 s | ||||
| 2-OCOCH3 | 2.29 s | ||||
| 3-OCOCH3 | 2.29 s | ||||
| 10-OCOCH3 | 2.30 s | 2.30 s | 2.31 s | ||
| 1-OH | 8.68 s | ||||
| 2-OH | 5.52 s | 9.06 s | |||
| 1′ | 4.88 d (7.5) | ||||
| 2′ | 3.45 dd (8.7, 7.5) | ||||
| 3′ | 3.48 t (8.7) | ||||
| 4′ | 3.38e | ||||
| 5′ | 3.38e | ||||
| 6′ | 3.68 dd (12.1, 5.2) | ||||
| 3.85 dd (12.1, 1.7) | |||||
| Position | 1b | 2b | 2b | 4c | 11d |
|---|---|---|---|---|---|
| a Assignments were based on 1H–1H COSY, HMQC, HMBC, and NOESY experiments; chemical shifts (δ) are given in ppm.b Recorded in CDCl3.c Recorded in CD3OD.d Recorded in DMSO-d6. | |||||
| 1 | 111.2 | 119.7 | 121.2 | 111.5 | 114.8 |
| 2 | 144.0 | 138.0 | 149.3 | 150.0 | 147.3 |
| 3 | 145.2 | 149.2 | 138.1 | 147.1 | 144.6 |
| 4 | 110.6 | 112.3 | 122.6 | 118.6 | 111.7 |
| 4a | 125.5 | 130.0 | 127.1 | 129.2 | 124.7 |
| 5 | 28.7 | 29.4 | 28.6 | 29.7 | 28.5 |
| 6 | 51.3 | 51.2 | 51.1 | 53.1 | 51.1 |
| 8 | 53.5 | 53.7 | 53.8 | 55.3 | 53.5 |
| 8a | 128.4 | 129.2 | 129.3 | 128.5 | 125.7 |
| 9 | 147.7 | 147.7 | 147.7 | 145.5 | 143.2 |
| 10 | 141.0 | 141.0 | 141.0 | 149.3 | 146.0 |
| 11 | 121.4 | 121.2 | 124.2 | 116.8 | 112.3 |
| 12 | 124.3 | 124.2 | 124.2 | 125.9 | 123.7 |
| 12a | 133.4 | 133.7 | 135.9 | 127.6 | 128.3 |
| 13 | 36.0 | 36.4 | 36.4 | 37.0 | 35.8 |
| 13a | 58.8 | 58.6 | 59.1 | 61.4 | 58.6 |
| 13b | 130.3 | 133.1 | 133.6 | 133.4 | 129.9 |
| 2-OCH3 | 56.0 | 57.5 | |||
| 3-OCH3 | 55.9 | 55.8 | |||
| 9-OCH3 | 60.6 | 60.6 | 60.6 | 60.9 | 59.2 |
| 10-OCH3 | 55.5 | ||||
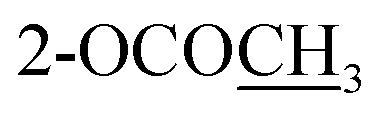 |
20.8 | ||||
 |
169.3 | ||||
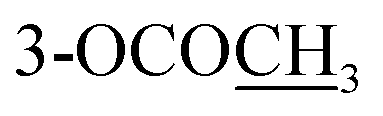 |
20.8 | ||||
 |
169.2 | ||||
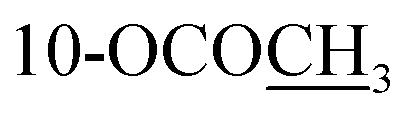 |
20.8 | 20.6 | 20.6 | ||
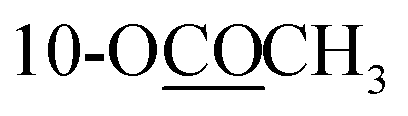 |
169.1 | 169.2 | 169.2 | ||
| 1′ | 103.3 | ||||
| 2′ | 75.4 | ||||
| 3′ | 78.3 | ||||
| 4′ | 71.9 | ||||
| 5′ | 78.7 | ||||
| 6′ | 63.0 | ||||
 | ||
| Fig. 2 1H–1H COSY and HMBC correlations of compounds 1–11. | ||
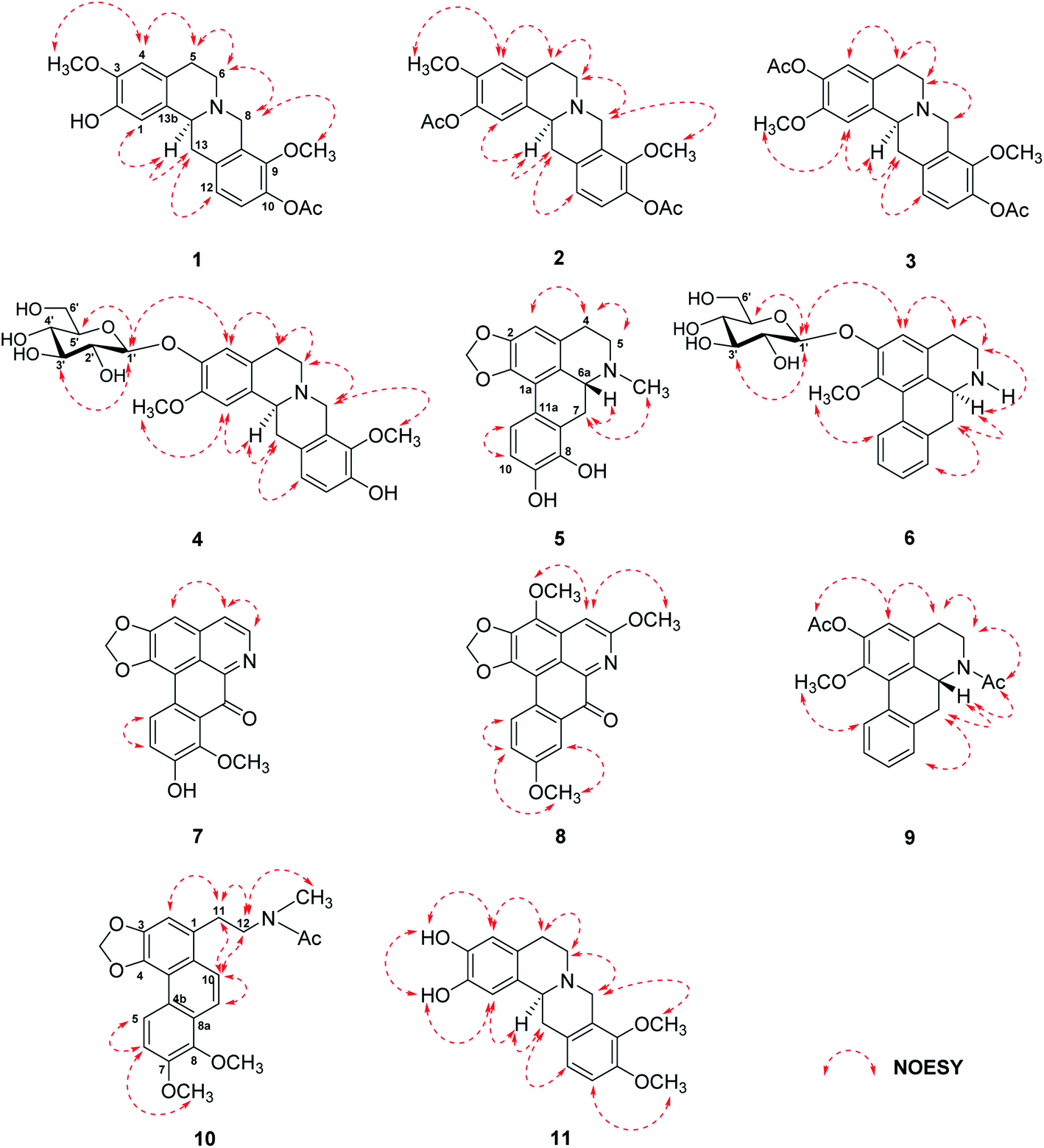 | ||
| Fig. 3 NOESY correlations of compounds 1–11. | ||
The molecular formula of stephapierrine B (2) was determined to be C23H25NO6 on the basis of HR-ESI-TOF-MS (m/z 412.1747 [M + H]+) and NMR data. The IR spectrum showed absorption bands of acetoxy groups (1753 cm−1) and aromatic rings (1623 and 1511 cm−1). The 1H and 13C NMR data (Tables 1 and 2) showed similar spectral features to those of compound 1. The significant difference was the presence of an additional acetoxy group at δH 2.29, and δC 20.8 and 169.3. The HMBC experiments showed correlations between AcO-2 (δH 2.29) and C-2 (δC 138.0), whereas H-1 (δH 6.89) correlated with C-2, C-3 (δC 149.2), C-4a (δC 130.0), C-13a (δC 58.6) and C-13b (δC 133.1) (see Fig. 2). It should be noted that the presence of the acetoxy group at the 2-position resulted in 0.11 and 8.5 ppm down-field shifts of H-1 and C-1 resonances, respectively, as compared with those of compound 1. These observations indicated that the acetoxy group should be located at the 2-position. Compound 2 showed the negative sign of specific optical rotation as that of compound 1, suggesting the same S-configuration or α-orientation of hydrogen at C-13a. This was confirmed by the similar ECD curve of 2 (Fig. S234†) that exhibited similar negative Cotton effects at 206 nm (Δε −14.21), 240 nm (Δε −63.97) and 286 nm (Δε −2.69), respectively. Compound 2 was therefore identified as 2,10-di-O-acetylstepholidine (see Fig. 1).
Stephapierrine C (3) was deduced to have the molecular formula C23H25NO6 by combined analysis of the HR-ESI-TOF-MS m/z 412.1748 [M + H]+ and NMR data. The IR spectrum displayed the absorption bands of acetoxy groups (1758 cm−1) and aromatic rings (1619 and 1512 cm−1). The 1H and 13C NMR data of 3 (Tables 1 and 2) were similar to those of compound 1. The main differences were the presence of an additional acetoxy group (δH 2.29, and δC 20.8 and 169.2) and the methoxy group should be placed at C-2 as was supported by the HMBC correlation between the CH3O-2 and C-2 (δC 149.3) (see Fig. 2). This was confirmed by the NOESY correlation between the CH3O-2 and H-1 (δH 6.89) (see Fig. 3). The location of the acetoxy group at C-3 was also supported by the HMBC correlation of the acetoxy methyl proton (δH 2.29) with C-3 (δC 138.1), together with the correlations of H-4 with C-2, C-3, C-4a (δC 127.1), C-5 (δC 28.6), and C-13b (δC 133.6). Compound 3 exhibited the same negative specific rotation and displayed a similar ECD spectrum as those of compound 1 (Fig. S234†), suggesting the same S-configuration at C-13a. Therefore, compound 3 was established as 3,10-di-O-acetyldiscretamine (see Fig. 1).
The molecular formula of stephapierrine D (4) was determined to be C25H31NO9 from the HR-ESI-TOF-MS (m/z 490.2056 [M + H]+) and NMR data. Its IR data indicated the presence of hydroxy groups (3314 cm−1) and aromatic rings (1611 and 1512 cm−1). The 1H and 13C NMR data of 4 (Tables 1 and 2) were similar to those of discretamine (13), which has also been isolated from S. pierrei in the present work. The significant differences were the presence of an anomeric proton signal at δH 4.88 (1H, d, J = 7.5 Hz, H-1′) along with signals of sugar residue at δH 3.38 (2H, overlapping, H-4′ and H-5′), 3.45 (1H, dd, J = 8.7 and 7.5 Hz, H-2′), 3.48 (1H, t, J = 8.7, H-3′), 3.68 (1H, dd, J = 12.1 and 5.2 Hz, Ha-6′), and 3.85 (1H, dd, J = 12.1 and 1.7 Hz, Hb-6′) in the 1H NMR spectrum, and from the carbon resonances at δC 103.3 (C-1′), 78.29 (C-3′ and C-5′), 75.4 (C-2′), 71.9 (C-4′), and 63.0 (C-6′) in the I3C NMR spectrum. The large coupling constant of the anomeric proton signal together with the remaining characteristic 1H and 13C signals of the sugar residue suggested this sugar moiety to be a β-glucoside.16,50
The anomeric proton signal showed correlation with the anomeric carbon signal at δC 103.3 (C-1′) in the HMQC spectra (Figs. S66–S68†). The sequence of the glucose unit connected to C-3 of the aglycone was deduced from the HMBC correlations of the anomeric H-1′ (δH 4.88) with the aglycone carbon signals at C-3 (δC 147.1), C-3′ (δC 78.3) and C-5′ (δC 78.7), indicating the attachment of β-D-glucose at C-3 (see Fig. 2). In addition, the 1H–1H COSY correlations, H-1′/H-2′, H-2′/H-3′, H-3′/H-4′, H-4′/H-5′, and H-5′/H-6′, were observed (see Fig. 2). Moreover, the NOESY correlations of H-1′ and H-4 (δH 6.93), H-3′ (δH 3.48) and H-5′ (δH 3.38) (Fig. 3) were also key interactions to support this glucoside structure. The β-D-glucosyl nature of this sugar residue was confirmed by enzymatic hydrolysis of 4 with β-glucosidase51 and the hydrolysis products were identified to be discretamine (13)22 and D-glucose by TLC comparison with authentic 13 and D-glucose. Furthermore, the location of the methoxy group at C-2 was supported by the HMBC correlation between the CH3O-2 signal and C-2 (δC 150.0) (see Fig. 2) and this was further confirmed by the NOESY correlation between the CH3O-2 and H-1 (δH 6.92) (see Fig. 3). As the substituent on tetrahydroprotoberberine molecule, including a glucose moiety, does not exert a considerable effect on the optical rotation and ECD spectrum,49,52 the same sign of optical rotation and similar ECD curve of compound 4 (Fig. S234†) when compared with those of compounds 1–3 has led to a conclusion that the absolute configuration at C-13a of 4 is S. On the basis of these findings, compound 4 is the glucoside analogue of discretamine (13).22 Compound 4 was therefore identified as discretamine 3-O-β-D-glucopyranoside (see Fig. 1).
The molecular formula of stephapierrine E (5) was deduced to be C18H18NO4 from HR-ESI-TOF-MS (m/z 312.1224 [M + Na]+) and NMR data. The IR spectrum exhibited the absorption bands of the hydroxy group (3383 cm−1) and aromatic rings (1603 and 1575 cm−1). The 1H NMR spectrum (Table 3) showed the characteristic signals of an aporphine alkaloid as a singlet N-methyl proton at δH 2.64 (3H, s, NCH3), a singlet aromatic proton at δH 6.50 (1H, s, H-3), two ortho-coupled aromatic protons at δH 7.46 (1H, d, J = 8.4 Hz, H-11) and 6.71 (1H, d, J = 8.4 Hz, H-10), one methine proton at δH 3.20 (1H, dd, J = 14.1 and 4.5 Hz, H-6a), one coupled methylene protons at δH 3.09 and 2.64 (2H, overlapping signal, H-5), and another coupled methylene protons at δH 3.14 and 2.68 (2H, overlapping signal, H-4). Additional methylene protons at the 7-position appeared at δH 3.69 (1H, dd, J = 14.5 and 4.5 Hz) and δH 2.19 (1H, dd, J = 14.5 and 14.1 Hz). In addition, the spectrum displayed resonances due to a methylenedioxy proton at δH 6.02 and 5.88 (each 1H, d, J = 1.1 Hz). The 13C NMR and DEPT-135 spectra (Table 4) revealed the presence of 18 signals which included one methylenedioxy carbon, three methylene carbons, four methine carbons, nine quaternary carbons, and one N-methyl carbon. The sixteen-carbon and one-nitrogen skeleton of 5 suggested an aporphine alkaloid core structure.41,53,54 The spectroscopic data for 5 was similar to those of crebanine (25),23 especially the substitution patterns on the molecule. The significant difference is the absence of two methoxy signals in the NMR spectra of 5 (Tables 3 and 4). The structure of compound 5 was further confirmed by HMBC and NOESY experiments (see Fig. 2 and 3). The absolute configuration of compound 5 was deduced by optical rotation experiments. The negative sign of specific optical rotation suggested that the absolute configuration of C-6a was R, or the hydrogen at the C-6a position is in the β-orientation.29 This was supported by the ECD spectrum (Fig. S235†). The structure of 5 was therefore elucidated as di-O-demethylcrebanine.
| Position | 5b | 6b | 7b | 8c | 9b |
|---|---|---|---|---|---|
| a Assignments were based on 1H–1H COSY, HMQC, HMBC, and NOESY experiments; chemical shifts (δ) are given in ppm.b Recorded in CDCl3.c Recorded in CD3OD.d Overlapping signal.e Partially overlapping signal. | |||||
| 3 | 6.50 s | 7.08 s | 7.07 s | 6.91 s | |
| 4 | 2.68d | 2.94 dd (14.9, 2.5) | 7.76 d (5.2) | 7.82 s | 2.73e |
| 3.14d | 3.22e | 2.86e | |||
| 5 | 2.64d | 3.20e | 8.56 d (5.2) | 3.27e | |
| 3.09d | 3.59 brdd (12.6, 5.3) | 4.12 brdd (13.2, 2.0) | |||
| 6a | 3.20 dd (14.1, 4.5) | 4.11 dd (13.9, 4.3) | 4.97 dd (13.8, 3.8) | ||
| 7 | 2.19 dd (14.5, 14.1) | 2.87 dd (13.9, 13.8) | 2.79 t (13.8) | ||
| 3.69 dd (14.5, 4.5) | 3.03 dd (13.8, 4.3) | 2.96 dd (13.8, 3.8) | |||
| 8 | 7.31 dd (7.1, 1.2) | 8.03 s | 7.25–7.32e | ||
| 9 | 7.26 ddd (7.5, 7.1, 1.2) | 7.25–7.32e | |||
| 10 | 6.71 d (8.4) | 7.32 ddd (7.6, 7.5, 1.2) | 7.15 d (8.8) | 7.30 d (9.0) | 7.25–7.32e |
| 11 | 7.46 d (8.4) | 8.33 brd (7.6) | 8.19 d (8.8) | 8.66 d (9.0) | 8.30 brd (7.7) |
| OCH2O | 5.88 d (1.1) | ||||
| 6.02 d (1.1) | 6.28 s | 6.40 s | |||
| 1-OCH3 | 3.72 s | 3.53 s | |||
| 3-OCH3 | 4.03 s | ||||
| 5-OCH3 | 3.82 s | ||||
| 8-OCH3 | 3.92 s | ||||
| 9-OCH3 | 4.03 s | ||||
| 2-OCOCH3 | 2.32 s | ||||
| N–CH3 | 2.64 s | ||||
| N–COCH3 | 2.21 s | ||||
| 1′ | 4.96 d (7.6) | ||||
| 2′ | 3.54 dd (8.9, 7.6) | ||||
| 3′ | 3.48 t (8.9) | ||||
| 4′ | 3.38 dd (8.9, 8.6) | ||||
| 5′ | 3.48 brdd (8.6, 6.0) | ||||
| 6′ | 3.69 dd (12.0, 6.0) | ||||
| 3.92 dd (12.0, 2.1) | |||||
| Position | 5b | 6b | 7b | 8c | 9b |
|---|---|---|---|---|---|
| a Assignments were based on 1H–1H COSY, HMQC, HMBC, and NOESY experiments; chemical shifts (δ) are given in ppm.b Recorded in CDCl3.c Recorded in CD3OD. | |||||
| 1 | 144.0 | 147.9 | 154.3 | 150.6 | 150.0 |
| 1a | 119.0 | 127.1 | 110.4 | 115.3 | 129.8 |
| 1b | 126.5 | 129.0 | 123.3 | 119.9 | 133.8 |
| 2 | 148.9 | 153.1 | 148.3 | 147.7 | 145.4 |
| 3 | 107.6 | 117.6 | 102.7 | 143.7 | 123.8 |
| 3a | 127.5 | 128.5 | 137.9 | 132.6 | 131.9 |
| 4 | 29.7 | 27.7 | 125.8 | 107.9 | 31.4 |
| 5 | 55.0 | 43.4 | 144.9 | 156.6 | 43.5 |
| 6a | 63.9 | 54.9 | 147.1 | 132.6 | 52.7 |
| 7 | 27.5 | 36.1 | 184.1 | 174.7 | 35.0 |
| 7a | 123.3 | 135.7 | 126.3 | 126.9 | 138.4 |
| 8 | 143.5 | 129.6 | 152.0 | 108.1 | 129.9 |
| 9 | 146.6 | 129.6 | 157.9 | 150.8 | 129.7 |
| 10 | 114.5 | 129.1 | 126.3 | 113.4 | 129.5 |
| 11 | 120.7 | 129.9 | 125.9 | 123.0 | 128.7 |
| 11a | 124.6 | 133.2 | 124.9 | 120.3 | 132.7 |
| OCH2O | 102.4 | 104.4 | 102.8 | ||
| 1-OCH3 | 61.7 | 61.3 | |||
| 3-OCH3 | 61.3 | ||||
| 5-OCH3 | 56.2 | ||||
| 8-OCH3 | 61.7 | ||||
| 9-OCH3 | 56.2 | ||||
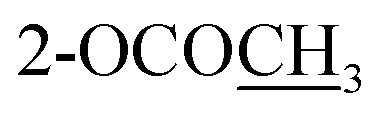 |
22.7 | ||||
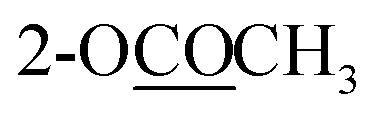 |
171.4 | ||||
| N–CH3 | 44.0 | ||||
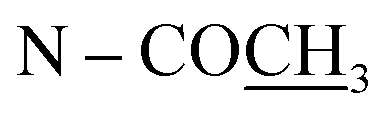 |
21.1 | ||||
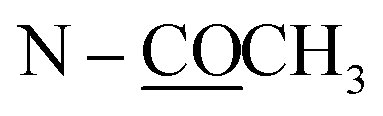 |
172.5 | ||||
| 1′ | 103.0 | ||||
| 2′ | 75.4 | ||||
| 3′ | 78.8 | ||||
| 4′ | 71.9 | ||||
| 5′ | 78.8 | ||||
| 6′ | 63.1 | ||||
The molecular formula of stephapierrine F (6) was established as C23H27NO7 from the HR-ESI-TOF-MS (m/z 430.1856 [M + H]+) and NMR data. The IR spectrum indicated the presence of hydroxy groups (3311 cm−1) and aromatic rings (1593 cm−1). The 1H NMR data (Table 3) showed the presence of a methoxy signal at δH 3.72, four adjacent aromatic hydrogens δH 7.31 (1H, dd, J = 7.1, 1.2 Hz, H-8), 7.26 (1H, ddd, J = 7.5, 7.1, 1.2 Hz, H-9), 7.32 (1H, ddd, J = 7.6, 7.5, 1.2 Hz, H-10), and 8.33 (1H, brd, J = 7.6 Hz, H-11), which were ascribed to the hydrogens of the unsubstituted D ring of the aporphine alkaloid.54 In addition, the 1H NMR spectrum revealed the presence of a sugar unit with its anomeric proton appearing at δH 4.96 (1H, d, J = 7.6 Hz, H-1′). The remaining sugar proton signals H-2′ to H-6′ are in the region δH 3.38–3.92 exhibiting similar features to those of compound 4. The 13C NMR spectrum (Table 4) showed 23 carbon resonances, of which five methines and one methylene were assigned to the β-glucopyranose moiety, suggested that 6 was an aporphine glucoside. The existence of a β-D-glucosyl moiety was confirmed by enzymatic hydrolysis51 in a similar manner to compound 4. The sugar linkage was determined by the HMBC correlations of H-1′ with C-2 (δC 153.1), C-3′ (δC 78.8) and C-5′ (δC 78.8) (see Fig. 2).50 The HMBC correlation between the methoxy proton at δH 3.72 and a carbon resonance at δC 147.9 (C-1) confirmed that the location of the methoxy group was placed to C-1. Additionally, the structural assignment for compound 6 was supported by the analysis of its NOESY spectrum (see Fig. 3); the H-1′ signal displayed a key interaction with H-3, H-3′ and H-5′. Moreover, H-6a (δH 4.11) displayed key interactions with H-7 (δH 2.87 and 3.03) and H-8 (δH 7.31), whereas CH3O-1 (δH 3.72) showed correlation to H-11 (δH 8.33). The 1H and 13C NMR spectra were consistent with those of (−)-asimilobine-2-O-β-D-glucoside (33).16 However, the sign of specific optical rotation of 6 was opposite to that of 33. The reported specific optical rotation of 33 was −107 (c = 0.1, MeOH).16 It is therefore possible that these two alkaloids are C-6a enantiomers. The ECD of 6 has therefore been determined and it was found that its ECD curve exhibited opposite Cotton effects to those of compounds 5 (see Fig. S235†). This led to a conclusion that the absolute configuration at C-6a of 6 was S. It is normal for the aporphine alkaloids to exist at different chirality at the asymmetric carbon C-6a.57 Based on these data, the structure of compound 6 was established as (+)-asimilobine 2-O-β-D-glucopyranoside.
Stephapierrine G (7) was assigned the molecular formula C18H11NO5 as determined from the HR-ESI-TOF-MS at m/z 344.0523 [M + Na]+ and NMR data. The IR spectrum showed absorption bands of hydroxy groups (3276 cm−1) and a conjugated carbonyl group (1638 cm−1). The 13C NMR spectrum in combination with the DEPT-135 experiment (Table 4) indicated the presence of 18 carbon atoms belonging to eleven quaternary carbons, five methine carbons, one methylene carbon, and one methyl carbon. The 1H NMR spectrum (Table 3) revealed characteristic resonances for protons and carbons at C-4 and C-5 of an aporphine unit as a pair of doublet signals at δH 7.76 and 8.56 (each 1H, d, J = 5.2 Hz), which connected to δC 125.8 and 144.9, respectively. Two singlet signals at δH 7.07 (1H, s) and 6.28 (2H, s) attaching to carbons at δC 102.7, and 104.4, respectively, were assigned to proton signals at C-3 and methylenedioxy protons, respectively. Two doublets at δH 7.15 and 8.19 (each 1H, d, J = 8.8 Hz) were attributed to two ortho-coupled aromatic protons at C-10 and C-11, respectively. In addition, a singlet signal at δH 3.92 (3H, s) was assigned to the methoxy proton at C-8. This assignment was confirmed by the HMBC correlations of the CH3O-8 and H-10 with C-8 (δC 152.0). The correlations of H-11 with C-1a (δC 110.4), C-7a (δC 126.3), C-9 (δC 157.9), and C-11a (δC 124.9) suggested the location of the hydroxy group at C-9 (see Fig. 2). The carbonyl carbon resonance at δC 184.1 together with a high-field resonance of its adjacent carbon (δC 126.3, C-7a) supported that 7 was a 7-oxoaporphine scaffold.58,59 Furthermore, the NOESY correlations of H-3 with H-4 and H-4 with H-5 (Fig. 3) as well as additional key HMBC correlations (Fig. 2) confirmed the structure of compound 7 to be a 7-oxoaporphine analogue. The spectroscopic data suggested that 7 is closely related to oxocrebanine (36),35 which has also been isolated from S. pierrei in the present work. Comparison of the NMR data of compounds 7 and 36 revealed that 7 is 9-de-O-methyloxocrebanine. Thus, compound 7 was concluded to be 1,2-methylenedioxy-8-methoxy-9-hydroxyoxoaporphine.
The sodiated molecular ion of the HR-ESI-TOF-MS of stephapierrine H (8) at m/z 388.0772 and the NMR data have led to the assignment of the molecular formula C20H15NO6. The IR spectrum indicated the presence of a conjugated carbonyl group (1651 cm−1). The 1H and 13C NMR data (Tables 3 and 4) showed two ortho-coupled aromatic protons at δH 7.30 and 8.66 (each 1H, d, J = 9.0 Hz, H-10 and H-11),58 together with two singlet protons at δH 7.82 and 8.03 (each 1H, s, H-4, and H-8). The remaining signals were assignable to three methoxys at δH 3.82 (3H, s, CH3O-5) and 4.03 (6H, s, CH3O-3, and CH3O-9), together with a methylenedioxy signal at δH 6.40 (2H, s). The foregoing 1H NMR data, together with the 13C NMR data (Table 4) suggested 8 to be an oxoaporphine type alkaloid.46,58,59 The HMBC experiments (see Fig. 2) confirmed the structure of compound 8. The structure of 8 was also confirmed by the NOESY correlations (see Fig. 3). On the basis of these data, the structure of compound 9 was established as 1,2-methylenedioxy-3,5,9-trimethoxyoxoaporphine.
Compound 9 was assigned the molecular formula C21H21NO4 as determined from the HR-ESI-TOF-MS (m/z 374.1363 [M + Na]+). The IR spectrum exhibited absorption bands of the acetoxy group (1761 cm−1) and amide group (1636 cm−1). The 1H and 13C NMR spectra (Tables 3 and 4) suggested an aporphine alkaloid core structure.41,53,54 The COSY correlations of these protons are shown in Fig. 2. The 1H NMR data showed the typical resonance of the N-acetyl signal at δH 2.21. The aromatic protons showed four adjacent signals for H-8, H-9 and H-10 in the range δH 7.25–7.32 and at δH 8.30 (1H, brd, J = 7.7 Hz) for H-11, which were attributed to the hydrogens of the unsubstituted D ring of the aporphine alkaloid.54 These observations were confirmed by 1H–1H COSY and HMBC experiments (see Fig. 2). The significant down-field shift of H-11 is the characteristic of this class of alkaloids.55,56 Furthermore, an aromatic singlet signal at δH 6.91 (H-3), a methoxy signal at δH 3.53 and an acetoxy proton signal at δH 2.32 were observed. The 1H and 13C NMR spectra of 9 were in agreement with the structure of O,N-diacetylasimilobine (9),18 which has been synthesized from asimilobine (32).32 This was confirmed by the HMBC experiments (Fig. 2) and the NOESY experiment (Fig. 3). It is noteworthy that the H-11 down-field shift in 9 (δH 8.30) was more pronounced than that of compound 5 (δH 7.46), and this was due to the vicinity of the methylenedioxy group in 5.55,56 The negative sign of specific optical rotation of 9 suggested that the absolute configuration of C-6a was R, or the hydrogen at the C-6a position is in the β-orientation.29 This was supported by the ECD spectrum (Fig. S235†). Based on these data, the structure of compound 9 was identical to (−)-O,N-diacetylasimilobine.18 Compound 9 has been reported in the present work as a naturally occurring alkaloid for the first time.
The molecular formula of compound 10 was deduced to be C22H23NO5 from the HR-ESI-TOF-MS at m/z 382.1258 [M + H]+. The IR spectrum indicated the presence of an acetoxy group (1751 cm−1). The 1H and 13C NMR features of 10 (Table 5) were similar to those of 8-methoxyuvoriopsine (37),37 the significant difference of which was the presence of an N-acetyl group, and the absence of an N-methyl signal. The rest of the 1H and 13C NMR data were consistent with the core structure of both compounds. The structure of 10 was confirmed by HMBC and NOESY analyses (see Fig. 2 and 3, respectively). Compound 10 has been reported as a structurally modified analogue from crebanine (25) and was called N-acetamidesecocrebanine,19 though some of the reported NMR spectroscopic data do not match with our data. This compound has thus been reported in the present work as a naturally occurring alkaloid for the first time.
| Position | 10b | |
|---|---|---|
| δH | δC | |
| a Assignments were based on 1H–1H COSY, HMQC, HMBC, and NOESY experiments; chemical shifts (δ) are given in ppm.b Recorded in CDCl3. | ||
| 1 | 129.8 | |
| 2 | 7.06 s | 110.1 |
| 3 | 145.0 | |
| 4 | 141.9 | |
| 4a | 117.1 | |
| 4b | 127.3 | |
| 5 | 8.80 d (9.2) | 123.7 |
| 6 | 7.28 d (9.2) | 112.7 |
| 7 | 149.9 | |
| 8 | 143.2 | |
| 8a | 123.6 | |
| 9 | 7.92 d (9.6) | 118.6 |
| 10 | 7.81 d (9.6) | 122.9 |
| 10a | 125.1 | |
| 11 | 3.29 m | 31.4 |
| 12 | 3.14 m | 61.9 |
| OCH2O | 6.19 s | 100.9 |
| 7-OCH3 | 4.00 s | 56.3 |
| 8-OCH3 | 3.98 s | 61.2 |
| N–CH3 | 2.81 s | 46.9 |
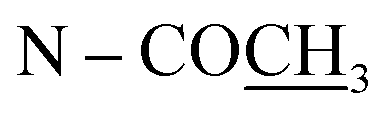 |
2.09 s | 19.5 |
 |
169.7 | |
Compound 11 was assigned the molecular formula C19H21NO4 as deduced from the HR-ESI-TOF-MS at m/z 328.1542 [M + H]+ and NMR data. The IR spectrum revealed the presence of hydroxy groups (3380 cm−1). The 1H and 13C NMR data (Tables 1 and 2) showed similar patterns to those of tetrahydropalmatine (14),23 except that the 1H NMR spectrum of 11 at the aromatic region was less well-defined. The NMR spectra of compound 11 showed only two methoxy signals at δH 3.71 (δC 59.2) and δH 3.73 (δC 55.5). Placement of the two methoxy groups at C-9 and C-10 was based on HMBC correlations of the CH3O-9 proton signal with C-9 (δC 143.2), CH3O-10 signal with C-10 (δC 146.0), H-8 and H-11 with C-9, and H-12 with C-10. This was further confirmed by NOESY correlations between CH3O-9 and H-8, and correlation between CH3O-10 and H-11. In addition, the locations of the hydroxy groups at C-2 and C-3 were deduced from HMBC correlations of H-1 with C-2 (δC 147.3) and C-3 (δC 144.6), and H-4 with C-2 and C-3 (Fig. 2). The positions of the hydroxy groups were confirmed by NOESY correlations (see Fig. 3). Compound 11 was therefore the 2,3-di-O-demethylated analogue of tetrahydropalmatine (14).23 Compound 11 was previously detected as one of the demethylated metabolites of compound 14 from rat's urine by ultra high-performance liquid chromatography–tandem mass spectrometric analysis.20 Thus, we report herein the spectroscopic data of 11, 2,3-didemethyltetrahydropalmatine, which was identified as a new naturally occurring alkaloid in the plant species.
Cholinesterase inhibitory activities
Several reports on the cholinesterase inhibitory activities of alkaloids from the Stephania plant species by different groups of researchers have appeared.21,60–63 In the present work, most of the isolated compounds were evaluated for their acetylcholinesterase (AChE) and butyrylcholinesterase (BuChE) inhibitory activities and the results are shown in Table 6. The well-known AChE and BuChE inhibitor, galanthamine, was used as the reference drug. Compound 42 exhibited the highest inhibitory activity against AChE (IC50 1.09 μM) followed by compound 38 (IC50 1.21 μM), with the IC50 values of 1.21 and 1.09 μM, respectively, which were slightly more active than and comparable to galanthamine (IC50 1.21 μM). Compounds 22, 26, 28, 29, 39 and 45 showed high inhibitory activities with the IC50 values of 2.85–8.32 μM, followed by compounds 1, 2, 13, 15, 17, 18, 21, 23–25, 27, 37, 41 and 44 which exhibited moderate AChE inhibitory activity with the IC50 values of 11.33–41.47 μM. Moreover, compounds 3, 14, 19, 30, 32, 34, 35 and 40 showed weak activity with the IC50 values of 71.28–265.82 μM, whereas compounds 11, 12, 33, 36 and 43 were inactive to the test.| Compounds | AChEb | BuChEc |
|---|---|---|
| IC50 (μM) | IC50 (μM) | |
| a Data represent as IC50 values in μM ± S.D. of three independent experiments.b Acetylcholinesterase.c Butyrylcholinesterase.d Inactive at 0.1 mg mL−1.e Galanthamine was used as the reference drug. | ||
| 1 | 41.47 ± 0.77 | Inactived |
| 2 | 15.41 ± 0.54 | Inactived |
| 3 | 149.63 ± 1.33 | 63.12 ± 0.83 |
| 11 | Inactived | Inactived |
| 12 | Inactived | 221.16 ± 1.62 |
| 13 | 26.99 ± 0.58 | 109.33 ± 0.97 |
| 14 | 71.28 ± 1.90 | 65.35 ± 0.80 |
| 15 | 11.33 ± 0.15 | 13.52 ± 0.59 |
| 17 | 21.16 ± 0.40 | 234.34 ± 1.08 |
| 18 | 12.25 ± 0.44 | 31.46 ± 0.20 |
| 19 | 152.59 ± 0.81 | Inactived |
| 21 | 18.31 ± 1.55 | 32.82 ± 0.34 |
| 22 | 8.32 ± 0.12 | 2.85 ± 0.08 |
| 23 | 11.34 ± 0.20 | 2.80 ± 0.07 |
| 24 | 11.94 ± 0.39 | 16.58 ± 0.54 |
| 25 | 17.37 ± 0.22 | 10.51 ± 0.27 |
| 26 | 6.11 ± 0.38 | 26.41 ± 0.43 |
| 27 | 17.63 ± 0.67 | 7.42 ± 0.16 |
| 28 | 6.12 ± 0.63 | 5.87 ± 0.06 |
| 29 | 4.30 ± 0.28 | 22.47 ± 0.10 |
| 30 | 140.15 ± 0.83 | Inactived |
| 32 | 141.47 ± 0.82 | 10.08 ± 0.15 |
| 33 | Inactived | 175.55 ± 1.45 |
| 34 | 73.08 ± 0.33 | 13.60 ± 0.30 |
| 35 | 265.82 ± 0.80 | Inactived |
| 36 | Inactived | Inactived |
| 38 | 1.21 ± 0.09 | 3.34 ± 0.02 |
| 39 | 2.85 ± 0.24 | 3.26 ± 0.05 |
| 40 | 147.18 ± 0.71 | 20.32 ± 0.39 |
| 41 | 32.49 ± 0.52 | 14.11 ± 0.25 |
| 42 | 1.09 ± 0.02 | 5.57 ± 0.15 |
| 43 | Inactived | 150.57 ± 0.54 |
| 44 | 40.86 ± 0.67 | 20.46 ± 0.42 |
| 45 | 7.49 ± 0.66 | 7.83 ± 0.11 |
| Galanthaminee | 1.21 ± 0.11 | 3.59 ± 0.07 |
For the BuChE inhibitory activities, compounds 23 and 22 displayed high inhibitory activity, with the IC50 values of 2.80 and 2.85 μM, respectively, which were approximately 1.3-fold more active than galanthamine (IC50 3.59 μM), followed by compounds 39 and 38, which also showed high activity of 3.26 and 3.34 μM, respectively, which were slightly more active than galanthamine. Compounds 27, 28, 42 and 45 showed high inhibitory activity with the IC50 values in the range 5.57–7.83 μM. Furthermore, compounds 15, 18, 21, 24–26, 29, 32, 34, 40, 41 and 44 exhibited moderate inhibitory activity with the IC50 values of 10.08–22.47 μM, followed by compounds 3, 12–14, 17, 33 and 43 that showed weak anti-BuChE activity with the IC50 values of 63.12–234.34 μM. Compounds 1, 2, 11, 19, 30, 35 and 36 were inactive to the test.
Structure–activity relationship (SAR) studies
Structure–activity relationship (SAR) studies indicated that, for anti-AChE activity, the aporphine alkaloids are the most active group. The protoberberine alkaloids are only moderately active, weakly active, or inactive. SAR discussion mainly focused on the aporphine alkaloids. Among the aporphines bearing the 2,3-methylenedioxy and 6-N-methyl functions, there are three groups of them, based on the structural nature around the C-6a and C-7 positions. For the saturated analogues, the aporphine group, the parent compound 22 exhibited moderate AChE inhibitory activity, with the IC50 of 8.32 μM (see Table S1†). Introduction of a methoxy function on the 8- and 9-positions to give the mono-methoxy analogues 23 and 24 resulted in a decrease in activity (IC50 of 11.34 and 11.94 μM). Introduction of the second methoxy group of 24 to the dimethoxy analogue 25 further reduced AChE inhibitory activity (IC50 17.37 μM). However, introduction of a methoxy group to the 10-position of 24 to the analogue 26 caused a considerable increase in activity (IC50 6.11 μM). The presence of the 10-methoxy function seemed to enhance anti-AChE activity. It should be mentioned that the presence of the 6-N-formyl, instead of the N-methyl, group as well as the absence of the 6-methyl group, resulted in a sharp decrease in activity, as in the case of compounds 30 and 32 that exhibited very weak anti-AChE activity (IC50 140.15 and 141.47 μM). It should also be noted that introduction of oxygen to the amino function of 25 to give the N-oxide analogue 43 resulted in complete loss of AChE inhibitory activity. In the presence of an α-hydroxy group at the 7-position, an increase in inhibitory activity was observed for compounds 28 and 29 (IC50 6.12 and 4.30 μM), except for compound 27 that a decrease in activity was observed. Unfortunately, only three 7-hydroxy analogues of this group were isolated. The most highly active analogues are those with a 6a,7-unsaturated analogues, the dehydroaporphines. Compounds 38, 39 and 42 were strongly active (IC50 1.21, 2.85 and 1.09 μM, respectively). Compounds 38 and 42 were respectively as active as and more active than galanthamine, the reference drug. However, the analogues 40 and 41 showed weak activity (IC50 147.18 and 32.49 μM), suggesting that, while the more rigid dehydroaporphine skeleton contributed to high anti-AChE property, the number and position of the oxygenation patterns highly affected the activity of this group of alkaloids.For the anti-BuChE activity, the aporphine alkaloids are also the most active group. Almost all protoberberine alkaloids are weakly active or inactive. SAR discussion was mainly on the aporphine alkaloids as well as that of the anti-AChE activity. In the aporphine group, compound 23 exhibited high BuChE inhibitory activity, followed by 22 (IC50 of 2.80 and 2.85 μM, respectively (see Table S1†)). The methoxy group on the 8-position seemed to enhance the activity. In contrast, the 9-methoxy group caused a 5.8-fold decrease in the activity of compound 24 (IC50 16.58 μM) when compared with compound 22. While the anti-AChE activity of the 8,9-dimethoxy analogue 25 was 2.8-fold less active than the 9,10-dimethoxy analogue 26, the anti-BuChE activity of 25 was 2.5-fold higher than that of 26 (Table 6). This implied that the oxygen functions at the 8-, 9- and 10-positions differently contributed to cholinesterase inhibitory activities of the aporphine alkaloids. It should also be noted that the presence of the 6-N-formyl group in the analogue 30 caused inactivity in this compound and the absence of the 6-N-methyl group in the analogue 32 did not cause a sharp decrease of anti-BuChE property as that occurred in the anti-AChE case since it exhibited anti-BuChE activity with IC50 of 10.08 μM. It should also be mentioned that, in going from the amino function in 25 to the corresponding N-oxide function in 43, a sharp decrease in BuChE inhibitory activity was observed. In the presence of an α-hydroxy group at the 7-position of the aporphine core structure, in contrast to most of the aporphine group, an approximately 2-fold decrease in inhibitory activity was observed for compounds 27, 28 and 29 (IC50 7.42, 5.87 and 22.47 μM, respectively). For the third group of the aporphine analogues, the dehydroaporphines, a slight decrease in activity was observed for compounds 38, 39, 40 and 41 (IC50 3.34, 3.26, 20.32 and 14.11 μM, respectively). However, the analogue 42 showed a 4.7-fold increase in anti-BuChE activity (IC50 5.57 μM). This implied that the more rigid dehydroaporphine skeleton and the number and position of the methoxy groups contributed to the anti-BuChE activity of this group of alkaloids.
Molecular docking study of the interaction between the potent alkaloids and cholinesterase enzymes
Molecular docking was performed using the AutoDock program to gain insight into the possible interaction of the potent inhibitory activity of the alkaloids with the enzymes AChE (compounds 38 and 42) and BuChE (compounds 22, 23, 38 and 39). The binding energy (ΔG, kcal mol−1) and interaction residues of the alkaloids within the enzyme binding site obtained from the best ranked and the most populated conformation of the docking calculation are summarized in Table 7.| Compound | Residue | Interaction | Distance (Å) | ΔGdocking (Kcal mol−1) | IC50 (μM) |
|---|---|---|---|---|---|
| AChE-inhibitor interaction | |||||
| 38 | F295 | Hydrogen bond | 2.82 | −9.57 | 1.21 |
| W286 | Pi–Pi | 4.02, 4.92, 5.36 | |||
| S293 | Pi–Pi | 3.63 | |||
| F297 | Pi–Pi | 5.16 | |||
| Y341 | Pi–Pi | 4.93 | |||
| 42 | R296 | Hydrogen bond | 1.70 | −8.95 | 1.09 |
| W286 | Pi–Pi | 4.15, 3.71, 5.46, 4.13, 5.59, 5.56 | |||
| Y124 | Pi–Pi | 5.78 | |||
| S293 | Pi–Pi | 4.30 | |||
| Galanthamine | G121 | Pi–σ | 3.76 | −7.91 | 1.21 |
| F338 | Pi–Pi | 5.94 | |||
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) |
|||||
| BuChE-inhibitor interaction | |||||
| 22 | W82 | Pi–Pi | 4.68, 5.40, 5.32, 5.35 | −8.07 | 2.85 |
| Pi–σ | 3.89 | ||||
| T120 | Pi–Pi | 3.36 | |||
| 23 | W82 | Pi–Pi | 4.95, 5.72, 4.38, 5.26 | −8.28 | 2.80 |
| 3.86, 5.26 | |||||
| Pi–σ | 3.42, 3.87 | ||||
| 38 | W82 | Pi–Pi | 4.64, 5.52, 5.21, 5.42 | −8.01 | 3.34 |
| T120 | Pi–σ | 3.37 | |||
| 39 | W82 | Pi–Pi | 5.88, 4.56, 5.31, 5.20 | −7.89 | 3.26 |
| 5.04 | |||||
| Galanthamine | W82 | Pi–σ | 3.83 | −7.41 | 3.59 |
The active site of AChE and BuChE consists of five regions such as the catalytic triad (CT), the anionic site (AS), the acyl pocket (AC), the oxyanion hole (OAH) and the peripheral anionic site (PAS). Thus, the amino acids of S203, E334, H447, W86, Y133, F337, F338, of F295, F297, G121, G122, A204, Y72, D74, Y124, W286 and Y341 are considered as active residues of AChE,64 while W82, Y128, F330, F331, L286, V288, G116, G117, A199, D70 and Y332 are identified as the key residues of BuChE binding gorge.65
As can be seen from Fig. 4, the docking result shows that compounds 38 and 42 were well occupied in the active site of AChE, where compound 38 could form a hydrogen bond with residue F295 and hydrophobic interactions with W286, S293, F297 and Y341. In contrast, compound 42 formed a hydrogen bond with R296 and strong Pi–Pi interactions with W286, Y124 and S293. The binding free energies of compounds 38 and 42 were similarly found to be −9.57 and −8.95 kcal mol−1, respectively, which possessed higher binding affinity with the AChE binding site compared to galanthamine (ΔG of −7.91 kcal mol−1).
 | ||
| Fig. 4 Binding interaction of docking poses of the ligands (yellow) in the vicinity of the target protein AChE (gray). (A) Binding mode of compound 38 and (B) binding mode of compound 42. The residues of the interaction site are shown as ball and stick. The blue and pink dashed lines represent hydrogen bonding and hydrophobic interaction, respectively. | ||
The best conformations of the docked complexes of BuChE and compounds 22, 23, 38 and 39 are shown in Fig. 5. The results showed that these alkaloids mainly interacted with BuChE by forming Pi–Pi interactions with the key residue W82. The free energy of binding for all complexes is similar, being about −8 kcal mol−1. Interestingly, the binding free energies of these alkaloids show good binding affinity with the BuChE binding site compared to galanthamine (−7.41 kcal mol−1).
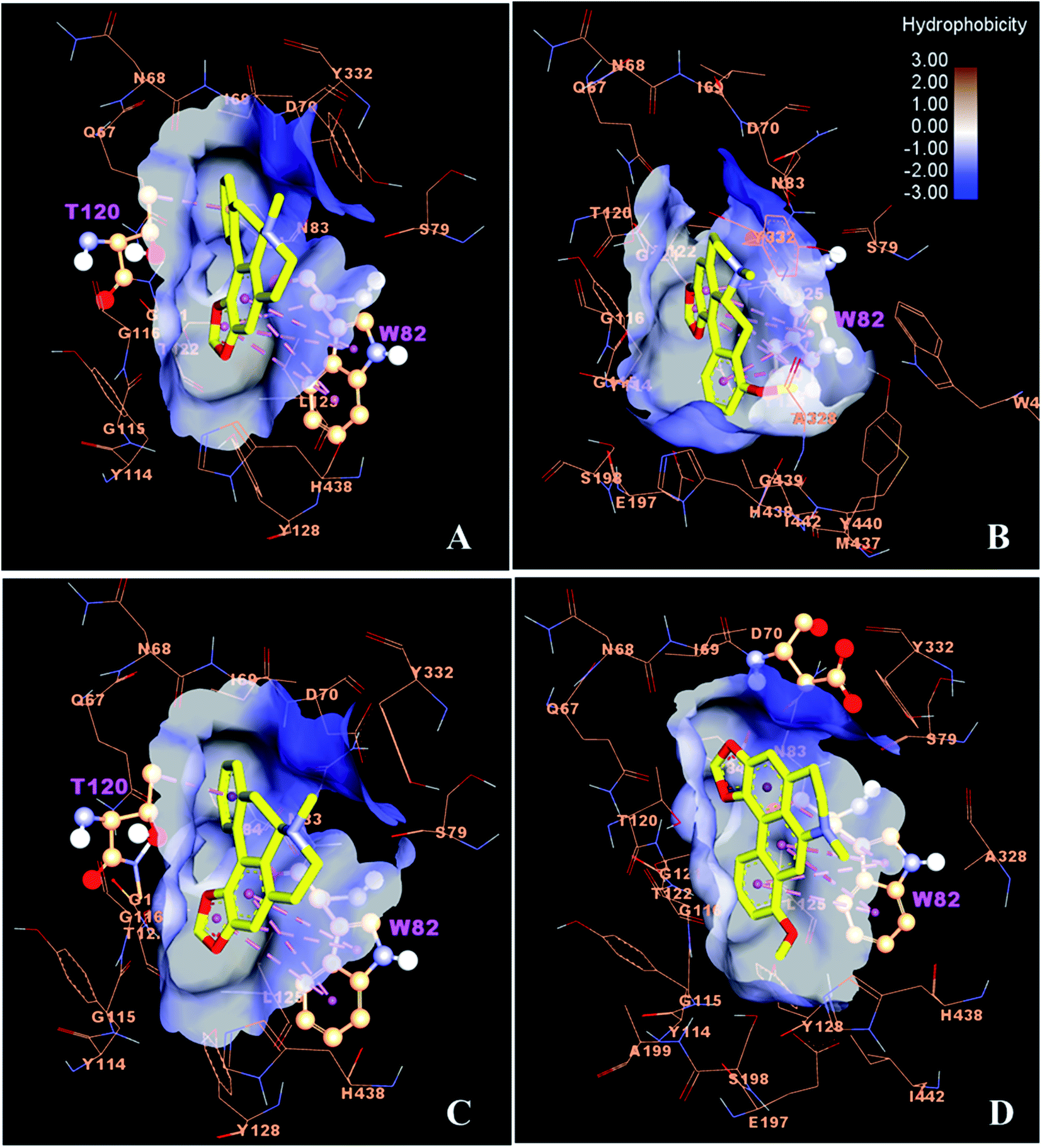 | ||
| Fig. 5 Binding interaction of ligands (yellow) with BuChE enzyme (orange); (A–D) Binding pose of active compounds 22, 23, 38 and 39. The dashed pink bond shows the hydrophobic interaction with the site residue (ball and stick). | ||
The molecular docking results showed that, for the alkaloids isolated from the tubers of S. pierrei, compounds 38 and 42 as well as compounds 22, 23, 38 and 39 exhibited good binding affinity towards AChE and BuChE, respectively, which is in agreement with the experimental IC50 values.
Experimental
General experimental procedures
Optical rotations were measured on a JASCO-1020 polarimeter. Electronic Circular Dichroism (ECD) was recorded on a JASCO J-810 spectropolarimeter. UV spectra were collected on a Shimadzu UV 1800 spectrophotometer. IR spectra were recorded in the ATR mode using a PerkinElmer FT-IR Spectrum 400 spectrophotometer. 1H and 13C NMR spectra were recorded on a Bruker ASCEND 400 FT-NMR spectrometer, operating at 400 MHz (1H) and 100 (13C) MHz. HR-ESI-TOF-MS spectra were measured with a Bruker micrOTOF-QII mass spectrometer. Unless otherwise indicated, column chromatography was carried out using Merck silica gel 60 (particle sizes less than 0.063 mm) and GE Healthcare Sephadex LH-20. For thin-layer chromatography (TLC), Merck pre-coated silica gel 60 F254 plates were used. Spots on TLC were detected under UV light and by spraying with anisaldehyde-H2SO4 reagent followed by heating.Plant material
The tubers of Stephania pierrei were collected from Prachin Buri Province, Thailand in 2019 and the plant species was identified by Assoc. Prof. Nopporn Dumrongsiri, Ramkhamhaeng University. A voucher specimen is deposited at the Faculty of Science, Ramkhamhaeng University, Thailand (Apichart Suksamrarn, No. 101).Extraction and isolation
The fresh tubers of S. pierrei (1.5 kg) were sliced, air-dried, milled, and macerated successively with n-hexane, EtOAc, and MeOH at room temperature. The filtered solution of each extraction was evaporated under reduced pressure at 40–45 °C to give the hexane (2.8 g), ethyl acetate (3.3 g), and methanol (4.5 g) extracts, respectively.The crude hexane extract (2.5 g) was fractionated by column chromatography, using a gradient solvent system of n-hexane, n-hexane–EtOAc, EtOAc, EtOAc–MeOH and MeOH with increasing amounts of the more polar solvent. The eluates were examined by TLC and 7 combined fractions (H1–H7) were obtained. Fraction H1 (280.0 mg) was column chromatographed eluting with a gradient system of n-hexane–CH2Cl2 (10:0.1 to 10:4) to give 5 subfractions (H1.1–H1.5). Subfraction H1.2 (23.0 mg) was rechromatographed over silica gel eluting with n-hexane–CH2Cl2 (10:0.5) to yield compound 38 (5.0 mg). Subfraction H1.3 (200 mg) was subjected to column chromatography eluting with n-hexane–CH2Cl2 (10:1) to afford compounds 39 (15.0 mg), 40 (1.6 mg), 41 (20.0 mg), and 42 (2.4 mg). Subfraction H1.4 (15.0 mg) was rechromatographed over silica gel and eluting with n-hexane–EtOAc (10:0.3) to give compound 30 (1.3 mg). Subfraction H1.5 (18.0 mg) was subjected to column chromatography eluting with n-hexane–CH2Cl2–EtOAc (9:9:0.2) to afford compound 8 (2.2 mg). Fraction H2 (850 mg) was subjected to silica column chromatography eluting with the isocratic condition of CH2Cl2–MeOH (10:0.1) to give 5 subfractions (H2.1–H2.5). Subfraction H2.1 (290 mg) was rechromatographed over silica gel eluting with n-hexane–EtOAc (6:4) to yield compounds 25 (240 mg), and 3 (9.0 mg). Subfraction H2.2, H2.3, and H2.4 were identified to be compounds 22 (7.6 mg), 24 (15.3 mg), and 23 (433.0 mg). Fraction H3 (550.0 mg) was subjected to silica column chromatography eluting with a gradient condition of CH2Cl2–MeOH (10:0.1 to 10:0.3) to give 2 subfractions (H3.1–H3.2). Subfraction H3.1 (350.0 mg) was chromatographed eluting with CH2Cl2–MeOH (10:0.1) to afford compound 2 (50.6 mg) and compound 36 (150.0 mg). Subfraction H3.2 (150.0 mg) was rechromatographed over silica gel eluting with CH2Cl2–MeOH (10:0.2) to yield compound 26 (68.0 mg). Fraction H4 (180.0 mg) was subjected to silica column chromatography eluting with the isocratic condition of CH2Cl2–MeOH (10:0.2) to give 4 subfractions (H4.1–H4.4). Subfraction H4.1 (75.0 mg) was chromatographed on Sephadex LH-20 column eluted with MeOH–CH2Cl2 (7:3) to yield compound 14 (15.0 mg). Subfraction H4.2 was identified to be compound 35 (5.5 mg). Subfraction H4.3 (15.0 mg) was chromatographed eluting with CH2Cl2–MeOH (10:0.3) to give compound 1 (2.5 mg). Subfraction H4.4 was identified to be compound 34 (2.0 mg). Fraction H5 (120.0 mg) was subjected to column chromatography eluting with the isocratic condition of CH2Cl2–MeOH (10:0.4) to give 3 subfractions (H5.1–H5.3). Subfraction H5.1 (25.0 mg) was chromatographed over silica gel eluting with CH2Cl2–MeOH (10:0.2), followed by column chromatography on Sephadex LH-20 eluted with MeOH to yield compound 7 (1.0 mg). Subfraction H5.3 (48.0 mg) was purified on a Sephadex LH-20 column eluted with MeOH to afford compound 17 (15.5 mg). Fraction H6 (80.0 mg) was chromatographed over silica gel and eluted under the isocratic condition of CH2Cl2–MeOH (10:0.8) to yield compounds 21 (12.0 mg), and 20 (2.5 mg). Subfraction H7 (125 mg) was chromatographed on Sephadex LH-20 column eluting with MeOH, followed by silica column chromatography, eluting with gradient solvent system of CH2Cl2–MeOH (10:1) to afford compound 19 (84.0 mg).
The crude EtOAc extract (3.0 g) was fractionated by column chromatography, using a gradient solvent system of EtOAc, EtOAc–MeOH and MeOH with increasing amounts of the more polar solvent. The eluates were examined by TLC and 3 combined fractions (E1–E3) were obtained. Fraction E1 (300.0 mg) was subjected to column chromatography eluting with an isocratic condition of CH2Cl2–MeOH (10:0.2) to give compounds 9 (1.5 mg), 31 (1.0 mg), 10 (1.2 mg), and 32 (3.2 mg). Fraction E2 (450.0 mg) was chromatographed on silica column eluting under isocratic condition of CH2Cl2–MeOH (10:0.5) to provide compounds 11 (3.4 mg), 13 (2.0 mg), and 12 (10.0 mg). Fraction E3 (150.0 mg) was subjected to silica column chromatography eluting under the isocratic condition of CH2Cl2–MeOH (10:1) to give compound 18 (5.0 mg).
The MeOH extract (4.2 g) was fractionated by column chromatography, using a gradient solvent system of EtOAc, EtOAc–MeOH, MeOH, and 5% water in MeOH with an increasing amount of the more polar solvent. The eluates were examined by TLC and 6 combined fractions (M1–M6) were obtained. Fraction M1 (450.0 mg) was subjected to column chromatography eluting under the isocratic condition of CH2Cl2–MeOH (10:1) to give 3 subfractions (M1.1–M1.3). Subfraction M1.2 (150.0 mg) was rechromatographed eluting with n-hexane–CH2Cl2–MeOH (40:50: 2) to afford compound 27 (30.0 mg). Subfraction M1.3 was identified to be compound 44 (29.1 mg). Fraction M2 (200.0 mg) was chromatographed over silica gel eluting with a gradient solvent system of CH2Cl2–MeOH (10:0.4) to give 4 subfractions (M2.1–M2.4). Subfraction M2.3 was identified to be compound 37 (3.9 mg).
Subfraction M2.4 (120.0 mg) was chromatographed eluting with the isocratic condition of CH2Cl2–MeOH (10:0.5) to yield compound 43 (20.6 mg). Fraction M3 (750.0 mg) was chromatographed on a silica gel column eluting under the isocratic condition of CH2Cl2–MeOH (10:0.5) to give 4 subfractions (M3.1–M3.4). Subfraction M3.2 was identified to be compound 45 (7.4 mg). Subfraction M3.3 (80.0 mg) was subjected to column chromatography eluting with CH2Cl2–MeOH (10:1) to afford compound 33 (9.0 mg). Subfraction M3.4 (30.0 mg) was rechromatographed over silica gel and eluted under the isocratic condition of CH2Cl2–MeOH (100:12) to yield compounds 16 (2.2 mg), and 15 (8.6 mg). Fraction M4 (150.0 mg) was purified by using a Sephadex LH-20 column eluting with MeOH, followed by silica column chromatography eluting with n-hexane–CH2Cl2–MeOH (40:50:10) to provide compound 4 (1.5 mg). Fraction M5 (280.0 mg) was chromatographed over silica gel eluting with an isocratic solvent system of CH2Cl2–EtOAc–MeOH (40:60:10) to give 4 subfractions (M5.1–M5.4). Subfraction M5.1 and M5.2 were identified as compounds 29 (8.8 mg) and 5 (1.3 mg), respectively. Subfraction M5.3 (80.0 mg) was rechromatographed over silica gel and eluted with CH2Cl2–EtOAc–MeOH (20:80:15) to give compound 28 (11.7 mg). Fraction M6 (80.0 mg) was chromatographed on Sephadex LH-20 column eluting with MeOH, followed by silica column chromatography, eluting with an isocratic solvent system of CH2Cl2–EtOAc–MeOH (20:60:20) to afford compound 6 (2.2 mg).
ε): 203 (4.62), 283 (3.51) nm; ECD (MeOH) λmax (Δε) 204 (−10.69), 243 (−50.55), 287 (−6.61) nm; 1H-NMR (CDCl3, 400 MHz), and 13C-NMR (CDCl3, 100 MHz) data: see Tables 1 and 2; ESI-TOF-MS m/z 370.1633 [M + H]+ (calcd for C21H24NO5, 370.1648).ε): 209 (4.93), 276 (3.93) nm; ECD (MeOH) λmax (Δε) 206 (−14.21), 240 (−63.97), 286 (−2.69) nm; 1H-NMR (CDCl3, 400 MHz), and 13C-NMR (CDCl3, 100 MHz) data: see Tables 1 and 2; ESI-TOF-MS m/z 412.1747 [M + H]+ (calcd for C23H26NO6, 412.1754).ε): 204 (5.99), 278 (4.96) nm; ECD (MeOH) λmax (Δε) 206 (−8.98), 240 (−49.83), 286 (−8.12) nm; 1H-NMR (CDCl3, 400 MHz), and 13C-NMR (CDCl3, 100 MHz) data: see Tables 1 and 2; ESI-TOF-MS m/z 412.1748 [M + H]+ (calcd for C23H26NO6, 412.1754).ε): 202 (3.64), 284 (2.70) nm; ECD (MeOH) λmax (Δε) 211 (−11.55), 234 (−41.99), 290 (−4.32) nm; 1H-NMR (CD3OD, 400 MHz), and 13C-NMR (CD3OD, 100 MHz) data: see Tables 1 and 2; ESI-TOF-MS m/z 490.2056 [M + H]+ (calcd for C25H32NO9, 490.2071).ε): 218 (3.79), 283 (3.54) nm; ECD (MeOH) λmax (Δε) 216 (+13.95), 238 (−36.48), 269 (+33.45), 297 (+2.79), 307 (−20.33) nm; 1H-NMR (CD3OD, 400 MHz), and 13C-NMR (CD3OD, 100 MHz) data: see Tables 3 and 4; ESI-TOF-MS m/z 312.1224 [M + Na]+ (calcd for C18H18NNaO4, 312.1230).ε): 210 (3.85), 272 (3.52) nm; ECD (H2O) λmax (Δε) 211 (−34.47), 232 (+69.01), 270 (−9.83), 297 (−0.16), 315 (+7.29) nm; 1H-NMR (CD3OD, 400 MHz), and 13C-NMR (CD3OD, 100 MHz) data: see Tables 3 and 4; ESI-TOF-MS m/z 430.1856 [M + H]+ (calcd for C23H28NO7, 430.1860).ε): 206 (3.80), 247 (3.72), 275 (3.67) nm; 1H-NMR (CD3OD, 400 MHz), and 13C-NMR (CD3OD, 100 MHz) data: see Tables 3 and 4; ESI-TOF-MS m/z 344.0523 [M + Na]+ (calcd for C18H11NNaO5, 344.0529).ε): 219 (3.55), 244 (3.68), 307 (3.25), 319 (3.32) nm; 1H-NMR (CDCl3, 400 MHz), and 13C-NMR (CDCl3, 100 MHz) data: see Tables 3 and 4; ESI-TOF-MS m/z 388.0772 [M + Na]+ (calcd for C20H15NNaO6, 388.0791).Enzymatic hydrolysis of compounds 4 and 6
Compound 4 (0.5 mg) was dissolved in water (0.5 mL) and the mixture was incubated with Sigma β-glucosidase (from almonds, 1.0 mg) at 37–38 °C for 5 h.51 The reaction mixture was extracted with ethyl acetate and the solvent was removed in vacuo. TLC comparison with discretamine (13)22 isolated from S. pierrei in the present work revealed the identity of the aglycone with 13. The aqueous layer was concentrated and analyzed by TLC comparison with authentic D-glucose using CHCl3:MeOH:H2O (3:1.5:0.2).
Compound 6 was similarly hydrolyzed under the same condition and the aglycone and the sugar were similarly analyzed.
Evaluation of AChE and BuChE inhibitory activities
AChE (from Electrophorus electricus), BuChE (from equine serum), galanthamine (as hydrobromide salt), 5,5′-dithiobis(2-nitrobenzoic acid) (DTNB), bovine serum albumin (BSA), acetylthiocholine iodide (ATCI) and S-butyrylthiocholine iodide (BTCI) were purchased from Sigma-Aldrich (St. Louis, Misouri, USA). AChE and BuChE inhibitory activities were conducted by using Ellman's method with slight modification.66 Briefly, the enzyme solutions were prepared at 0.2 unit per mL in 10 mM phosphate buffer solution (PBS, pH 8.0). The assay medium consisted of 20 μL of the enzyme, 140 μL of 10 mM PBS and 20 μL of tested compounds were mixed in a 96-well plate and shaken for 10 min. The reaction was initiated by the addition of 20 μL of a mixture of 5 mM DTNB, 0.1% BSA, and substrate (ATCI or BTCI). The hydrolysis of ASCI was monitored by the yellow 5-thio-2-nitrobenzoate anion formation as a result of the reaction with DTNB and thiocholines, catalyzed by enzymes at a wavelength of 405 nm and the absorbance was measured by using a microplate reader (Sunrise, Switzerland) after 5 min of incubation at room temperature. The percentage of inhibition was calculated by comparing the rate of enzymatic hydrolysis of ASCI for the sample to that of the blank (80% MeOH in buffer). Similarly, BuChE inhibition was performed as described for AChE. All samples were run in triplicate in 96-well microplates and galanthamine was used as a positive control.| Enzyme inhibitory activity assay (%) = [(absorbance of control − absorbance of sample)/ absorbance of control] × 100. |
The IC50 values were determined graphically from inhibition curves (inhibitor concentration vs. percent of inhibition) and each concentration was performed in triplicate.
Molecular docking calculations
The three-dimensional (3D) structures of human AChE in complex with galanthamine and human BuChE in complex with tacrine were obtained from Protein Data Bank with PDB IDs: 4EY667 (resolution: 2.40 Å) and 4BDS65 (resolution: 2.10 Å), respectively. The water molecules were removed and all missing hydrogen atoms were added to this protein using AutoDockTools (ADT).68 The potent alkaloids were structurally sketched using ACDLab69 and optimized at the Hartree-Fock level with a 6-31G basis set using the Gaussian 03 program70 and then converted to mol2 format using GaussView71 for docking study. The Autodock 4.2 program68 was used to examine the binding affinity of the optimized alkaloid structures towards the binding site of AChE and BuChE proteins. Non-polar hydrogens and lone pairs were merged and partial atomic charges were assigned using the Gasteiger method.72 The protein molecule was set rigid throughout the docking, while the ligand compounds were allowed to be flexible by the rotation parameter. The cubical grid box of 80 × 80 × 80 points with a spacing of 0.375 Å was positioned at the active site of the cholinesterase proteins. Galanthamine and tacrine were re-docked into the binding pockets of AChE and BuChE, respectively, to serve as controls. Autogrid4 was used to attain a rigid grid maps. Then, autodock4 was used with Lamarckian genetic algorithms and by default of the protocol to gain the 200 independent docking runs. After the run, the docked conformation with the lowest binding energy in the most populated cluster of each compound was selected for detailed analysis and further studies. Visualization of the docking results was performed using Acceryls Discovery Studio 2.5 (Acceryls, Inc., San Diego, CA, USA).
Conclusions
In summary, eight new alkaloids (1–8), three new naturally occurring alkaloids (9–11), together with thirty-four known alkaloids (12–45), have been isolated from Stephania pierrei tubers. Most of the isolated compounds were evaluated for acetylcholinesterase (AChE) and butyrylcholinesterase (BuChE) inhibitory activities. Compound 42 exhibited the most potent AChE inhibitory effect, followed by compound 38, which were respectively slightly more active than and comparable to galanthamine. Moreover, compound 23 exhibited the highest BuChE inhibitory activity, which was 1.3-fold more active than galanthamine, followed by compounds 22, 38 and 39 which also showed high inhibitory activity. Molecular docking studies are well in agreement with these experimental results. The alkaloids 42 and 23 may respectively be regarded as lead compounds for anti-AChE and anti-BuChE drug development.Author contributions
Waraluck Chaichompoo: methodology, investigation, data curation, formal analysis, visualization, and writing – original draft. Pornchai Rojsitthisak: conceptualization, resources, supervision, funding acquisition, project administration, and writing – review & editing. Wachirachai Pabuprapap: investigation, data curation, formal analysis, visualization, and writing – review & editing. Yuttana Siriwattanasathien: investigation, validation, data curation, and formal analysis. Pathumwadee Yotmanee: investigation, data curation, formal analysis, software, and writing – review & editing. Woraphot Haritakun: investigation, and data curation. Apichart Suksamrarn: conceptualization, resources, supervision, funding acquisition, project administration, and writing – review & editing.Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
The authors express gratitude to the Thailand Science Research and Innovation (TSRI) Fund (CU_FRB640001_01_33_3), the Ratchadaphiseksomphot Endowment Fund for the Natural Products for Ageing and Chronic Diseases, Chulalongkorn University (GRU 6404733002-1) (P. R.) and The Thailand Research Fund (DBG6180030) (A. S. and P. R.). This research is supported by Ratchadapisek Somphot Fund for Postdoctoral Fellowship, Chulalongkorn University (W. C.). Support from the Center of Excellence for Innovation in Chemistry, Ministry of Higher Education, Science, Research and Innovation is gratefully acknowledged.Notes and references
- Alzheimer's Association, Alzheimers. Dement., 2020, 16, 391–460 CrossRef PubMed.
- T. McLaughlin, H. Feldman, H. Fillit, M. Sano, F. Schmitt, P. Aisen, C. Leibman, L. Mucha, J. M. Ryan, S. D. Sullivan, D. E. Spackman, P. J. Neumann, J. Cohen and Y. Stern, Alzheimers. Dement., 2010, 6, 482–493 CrossRef PubMed.
- Alzheimer's Disease International, World Alzheimer Report 2019: Attitudes to dementia, Alzheimer's Disease International, London, 2019 Search PubMed.
- Y. Ju, H. Chakravarty and K. Y. Tam, ACS Chem. Neurosci., 2020, 11, 3346–3357 CrossRef CAS PubMed.
- H. A. Hassan, A. E. Allam, D. H. Abu-Baih, M. F. A. Mohamed, U. R. Abdelmohsen, K. Shimizu, S. Y. Desoukey, A. M. Hayallah, M. A. Elrehany, K. M. Mohamedi and M. S. Kamel, RSC Adv., 2020, 10, 36920–36929 RSC.
- M. M. Mesulam, A. Guillozet, P. Shaw, A. Levey, E. G. Duysen and O. Lockridge, Neuroscience, 2002, 110, 627–639 CrossRef CAS PubMed.
- J. Gabriel, M. R. Almeida, M. H. Ribeiro, J. Duraes, M. Tabuas-Pereira, A. C. Pinheiro, R. Pascoal, I. Santana and I. Baldeiras, Neurosci. Lett., 2017, 641, 101–106 CrossRef PubMed.
- D. G. Wilkinson, P. T. Francis, E. Schwam and J. Payne-Parrish, Drugs Aging, 2004, 21, 453–478 CrossRef CAS PubMed.
- F. Zemek, L. Drtinova, E. Nepovimova, V. Sepsova, J. Korabecny, J. Klimes and K. Kuca, Expert Opin. Drug Saf., 2014, 13, 759–774 CAS.
- P. Patil, A. Thakur, A. Sharma and S. J. S. Flora, Drug Dev. Res., 2020, 81, 165–183 CrossRef CAS PubMed.
- D. M. Pereira, F. Ferreres, J. M. A. Oliveira, L. Gaspar, J. Faria, P. Valentão, M. Sottomayor and P. B. Andrade, Phytomedicine, 2010, 17, 646–652 CrossRef CAS PubMed.
- E. L. Konrath, C. S. Passos, L. C. Klein-Júnior and A. T. Henriques, J. Pharm. Pharmacol., 2013, 65, 1701–1725 CrossRef CAS PubMed.
- D. K. Semwal, R. Badoni, R. Semwal, S. K. Kothiyal, G. J. P. Singh and U. Rawat, J. Ethnopharmacol., 2010, 132, 369–383 CrossRef CAS PubMed.
- T. Smitinand, Thai Plant Names, Rev. Edit. Office of the Forest Herbarium, Department of Natural Park, Wildlife and Plant Conservation, Bangkok, 2014 Search PubMed.
- C. Dary, S. Hul, S. Kim and F. Jabbour, Edinb. J. Bot., 2015, 72, 423–428 CrossRef.
- K. Likhitwitayawuid, C. K. Angerhofer, H. Chai, J. M. Pezzuto and G. A. Cordell, J. Nat. Prod., 1993, 56, 1468–1478 CrossRef CAS PubMed.
- J. Maliwong, P. Sahakitpichan, N. Chimnoi, S. Ruchirawat and T. Kanchanapoom, Phytochem. Lett., 2021, 43, 140–144 CrossRef.
- M. Tomita and M. Kozuka, Yakugaku Zasshi, 1965, 85, 77–82 CrossRef CAS.
- H. Wang, X. Cheng, S. Kong, Z. Yang, H. Wang, Q. Huang, J. Li, C. Chen and Y. Ma, Molecules, 2016, 21, 1555 CrossRef PubMed.
- W. Xiao, X. Zhuang, G. Shen, Y. Zhong, M. Yuan and H. Li, J. Sep. Sci., 2014, 37, 696–703 CrossRef CAS PubMed.
- K. Ingkaninan, P. Phengpa, S. Yuenyongsawad and N. Khorana, J. Pharm. Pharmacol., 2006, 58, 695–700 CrossRef CAS PubMed.
- J. R. G. S. Almeida, J. T. Lima, H. R. Oliveira, M. R. Oliveira, P. R. M. Meira, A. S. S. C. Lucio, J. M. Barbosa Filho and L. J. Quintans Junior, Nat. Prod. Res., 2011, 25, 1908–1915 CrossRef CAS PubMed.
- S. Nantapap, C. Loetchutinat, P. Meepowpan, N. Nuntasaen and W. Pompimon, Am. J. Appl. Sci., 2010, 7, 1057–1065 CrossRef CAS.
- S. Singh, T. D. Singh, V. P. Singh and V. B. Pandey, Pharm. Biol., 2010, 48, 158–160 CrossRef CAS PubMed.
- Y. D. Min, M. C. Yang, K. H. Lee, K. R. Kim, S. U. Choi and K. R. Lee, Arch. Pharmacal Res., 2006, 29, 757–761 CrossRef CAS PubMed.
- S. Tong and J. Yan, J. Liq. Chromatogr. Relat. Technol., 2005, 28, 2979–2989 CrossRef CAS.
- T. M. Hung, M. K. Na, N. T. Dat, T. M. Ngoc, U. J. Youn, H. J. Kim, B. S. Min, J. P. Lee and K. H. Bae, J. Ethnopharmacol., 2008, 119, 74–80 CrossRef CAS PubMed.
- J. I. Kunitomo, Y. Okamoto, E. Yuge and Y. Nagai, Yakugaku Zasshi, 1969, 89, 1691–1695 CrossRef CAS PubMed.
- J. T. Blanchfield, D. P. A. Sands, C. H. L. Kennard, K. A. Byriel and W. Kitching, Phytochemistry, 2003, 63, 711–720 CrossRef CAS.
- F. Roblot, R. Hocquemiller and A. Cave, J. Nat. Prod., 1983, 46, 862–873 CrossRef CAS.
- W. G. Ma, Y. Fukushi, S. Tahara and T. Osawa, Fitoterapia, 2000, 71, 527–534 CrossRef CAS PubMed.
- C. Y. Chen, F. R. Chang and Y. C. Wu, J. Chin. Chem. Soc., 1997, 44, 313–319 CrossRef CAS.
- B. Tantisewie and K. Pharadai, J. Nat. Prod., 1982, 45, 355–357 CrossRef.
- C. Dary, S. S. Bun, G. Herbett, F. Mabrouki, H. Bun, S. Kim, F. Jabbour, S. Hul, B. Baghdikian and E. Ollivier, Nat. Prod. Res., 2017, 31, 802–809 CrossRef CAS PubMed.
- E. M. K. Wijeratne, Y. Hatanaka, T. Kikuchi, Y. Tezuka and A. A. L. Gunatilaka, Phytochemistry, 1996, 42, 1703–1706 CrossRef.
- B. N. Zhou, R. K. Johnson, M. R. Mattern, X. Wang, S. M. Hecht, H. T. Beck, A. Ortiz and D. G. I. Kingston, J. Nat. Prod., 2000, 63, 217–221 CrossRef CAS PubMed.
- M. Leboeuf and A. Cave, Phytochemistry, 1972, 11, 2833–2840 CrossRef CAS.
- P. H. Guinaudeau, M. Leboeuf, M. Debray, A. Cave and R. R. Paris, Planta Med., 1975, 27, 304–318 CrossRef PubMed.
- J. I. Kunitomo, M. Oshikata and M. Akasu, Yakugaku Zasshi, 1981, 101, 951–955 CrossRef CAS PubMed.
- X. Wang, J. Qi, P. Zhao, C. Liu, M. Yang, H. Wang, Y. Y. Hu, Z. Liao and X. Xia, Chem. Nat. Compd., 2020, 56, 572–575 CrossRef CAS.
- M. P. Cava, Y. Watanabe, K. Bessho and M. J. Mitchell, Tetrahedron Lett., 1968, 20, 2437–2442 CrossRef CAS PubMed.
- D. H. Le, K. Nishimura and T. Tanahashi, Nat. Prod. Commun., 2016, 11, 949–952 CrossRef PubMed.
- E. V. Costa, M. F. C. Sampaio, M. J. Salvador, A. Nepele and A. Barison, Quim. Nova, 2015, 38, 769–776 CAS.
- F. Bracher, W. J. Eisenreich, J. Muhlbacher, M. Dreyer and G. Bringmann, J. Org. Chem., 2004, 69, 8602–8608 CrossRef CAS PubMed.
- H. Corrodi and E. Hardegger, Helv. Chim. Acta, 1956, 39, 889–897 CrossRef CAS.
- R. Hocquemiller, C. Debitus, F. Roblot and A. Cave, J. Nat. Prod., 1984, 47, 353–362 CrossRef CAS.
- S. G. Pyne and B. Dikic, J. Org. Chem., 1990, 55, 1932–1936 CrossRef CAS.
- B. Ringdahl, R. P. K. Chan, J. C. Craig and R. H. F. Manske, J. Nat. Prod., 1981, 44, 75–79 CrossRef CAS.
- S. M. Hu, S. X. Xu, X. S. Yao, C. C. Cui, Y. Tezuka and T. Kikuchi, Chem. Pharm. Bull., 1993, 41, 1866–1868 CrossRef CAS PubMed.
- P. Suebsakwong, W. Chulrik, W. Chunglok, J. X. Li, Z. J. Yao and A. Suksamrarn, RSC Adv., 2020, 10, 10461–10470 RSC.
- L. T. Byrne, J. M. Sasse, B. W. Skelton, A. Suksamrarn and A. H. White, Aust. J. Chem., 1987, 40, 785–794 CrossRef CAS.
- H. X. Ge, J. Zhang, C. Kai, J. H. Liu and B. Y. Yu, Appl. Microbiol. Biotechnol., 2012, 93, 2357–2364 CrossRef CAS PubMed.
- L. M. Jackman, J. C. Trewella, J. L. Moniot, M. Shamma, R. L. Stephens, E. Wenkert, M. Leboeuf and A. Cavé, J. Nat. Prod., 1979, 42, 437–449 CrossRef CAS.
- M. Shamma, The isoquinoline alkaloids, Academic Press, New York, 1972 Search PubMed.
- M. Shamma and J. L. Moniot, Experientia, 1975, 32, 282–283 CrossRef.
- Y. W. Ge, S. Zhu, M. Y. Shang, X. Y. Zang, X. Wang, Y. J. Bai, L. Li, K. Komatsu and S. Q. Cai, Phytochemistry, 2013, 86, 201–207 CrossRef CAS PubMed.
- B. Ringdahl, R. P. K. Chan, J. C. Craig, M. P. Cava and M. Shamma, J. Nat. Prod., 1981, 44, 80–85 CrossRef CAS.
- F. N. Samita, L. P. Sandjo, I. O. Ndiege, A. Hassanali and W. Lwande, Beilstein J. Org. Chem., 2013, 9, 447–452 CrossRef CAS PubMed.
- W. Silprakob, N. Sukhamsri, C. Kuhakarn, S. Hongthong, S. Jariyawat, K. Suksen, R. Akkarawongsapat, J. Limthongkul, N. Nantasaen and V. Reutrakul, Nat. Prod. Commun., 2018, 13, 1471–1474 CrossRef.
- T. M. Hung, N. H. Dang, J. C. Kim, H. S. Jang, S. W. Ryoo, J. H. Lee, J. S. Choi, K. H. Bae and B. S. Min, Planta Med., 2010, 76, 1762–1764 CrossRef CAS PubMed.
- J. W. Dong, L. Cai, Y. S. Fang, H. Xiao, Z. J. Li and Z. T. Ding, Fitoterapia, 2015, 104, 102–107 CrossRef CAS PubMed.
- X. P. Kong, H. Q. Ren, E. Y. L. Liu, K. W. Leung, S. C. Guo, R. Duan, T. T. X. Dong and K. W. K. Tsim, Molecules, 2020, 25, 5914 CrossRef CAS PubMed.
- L. Cai, J. W. Dong, L. X. Zhao, H. Zhou, Y. Xing, Y. Li, Z. J. Li, W. H. Duan, X. J. Li and Z. T. Ding, Process Biochem., 2016, 51, 933–940 CrossRef CAS.
- J. Cheung, M. J. Rudolph, F. Burshteyn, M. S. Cassidy, E. N. Gary, J. Love, M. C. Franklin and J. J. Height, J. Med. Chem., 2012, 55, 10282–10286 CrossRef CAS PubMed.
- F. Nachon, E. Carletti, C. Ronco, M. Trovaslet, Y. Nicolet, L. Jean and P. Y. Renard, Biochem. J., 2013, 453, 393–399 CrossRef CAS PubMed.
- G. L. Ellman, K. D. Courtney, V. Andres and R. M. Featherstone, Biochem. Pharmacol., 1961, 7, 88–95 CrossRef CAS PubMed.
- J. Cheung, M. J. Rudolph, F. Burshteyn, M. S. Cassidy, E. N. Gary, J. Love, M. C. Franklin and J. J. Height, J. Med. Chem., 2012, 55, 10282–10286 CrossRef CAS PubMed.
- G. M. Morris, R. Huey, W. Lindstrom, M. F. Sanner, R. K. Belew, D. S. Goodsell and A. J. Olson, J. Comput. Chem., 2009, 16, 2785–2791 CrossRef PubMed.
- Acdlabs.com. ACD/ChemSketch for Academic and Personal Use :ACD/Labs.com, 2015. [online] available at: http://www.acdlabs.com/resources/freeware/chemsketch/[accessed 2 May 2015] Search PubMed.
- M. J. Frisch, et al., Gaussian 03, Revision C.02, Gaussian, Inc., Wallingford CT, 2004 Search PubMed.
- R. Dennington, T. Keith and J. Millam. GaussView, Semichem Inc., Shawnee Mission KS., 2009 Search PubMed.
- J. Gasteiger and M. Marsili, Tetrahedron, 1980, 36, 3219–3228 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1ra03276c |
| This journal is © The Royal Society of Chemistry 2021 |