
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceThe effect of size, charge state and composition on the binding of propene to yttrium-doped gold clusters†
Júlia Barabás a,
Piero Ferrarib,
Vladimir Kaydashevb,
Jan Vanbuelb,
Ewald Janssens*b and
Tibor Höltzl*cd
a,
Piero Ferrarib,
Vladimir Kaydashevb,
Jan Vanbuelb,
Ewald Janssens*b and
Tibor Höltzl*cd
aDepartment of Inorganic and Analytical Chemistry, Budapest University of Technology and Economics, 1111 Budapest, Hungary
bQuantum Solid-State Physics, Department of Physics and Astronomy, KU Leuven, 3001 Leuven, Belgium. E-mail: ewald.janssens@kuleuven.be
cFurukawa Electric Institute of Technology, 1158 Budapest, Hungary. E-mail: tibor.holtzl@furukawaelectric.com
dMTA-BME Computation Driven Chemistry Research Group, Budapest University of Technology and Economics, 1111 Budapest, Hungary
First published on 1st September 2021
Abstract
The catalytic activity of metal clusters can be easily tuned by their size, charge state, or the introduction of dopant atoms. Here, the dopant-, charge- and size-dependent propene adsorption on gold (Aun+) and yttrium doped gold (Aun−1Y+) clusters (n = 4–20) was investigated using combined gas-phase reaction studies and density functional theory computations. The increased charge transfer between the cluster and propene in the cationic clusters considerably enhances the propene binding on both pure and yttrium-doped species, compared to their neutral cluster counterparts, while yttrium-doping lowers the propene binding strength in a size-dependent way compared to the pure gold clusters. Chemical bonding and energy decomposition analysis indicate that there is no covalent bond between the cluster and propene. The preferred propene binding site on a cluster is indicated by the large lobes of its LUMO, together with the low coordination number of the adsorption site. In small yttrium-doped gold clusters propene can not only bind to the electron-deficient yttrium atom, but also to the partially positively-charged gold atoms. Therefore, by controlling the charge of the clusters, as well as by introducing yttrium dopants, the propene binding strength can be tuned, opening the route for new catalytic applications.
1 Introduction
Transition metal clusters are of interest in catalysis,1–5 not only due to their high activity in different reactions, but also because of their size-,2,6 shape-,7 composition-,8,9 and charge state-dependent6 reactivity. Moreover, gas phase clusters can be useful models of complex, industrially relevant catalysts,10,11 as their reactivity can be investigated with atomic precision and under highly-controlled conditions, while simultaneously they can be studied using accurate quantum chemical methods.1 The obtained insights can assist in the design of novel catalysts with high efficiency and selectivity.In contrast to the noble bulk gold surface, gold nanoparticles and nanoclusters facilitate specific reactions.12,13 Gold clusters,14,15 and other gold-, silver- and copper-based clusters, nanoparticles and surfaces,16–19 were found to be good catalysts for the selective epoxidation of propene via molecular oxygen toward propene oxide, an important industrial precursor material. In particular, experiments on the direct propene oxidation by an oxygen/hydrogen mixture on gold particles deposited on TiO2 opened new possibilities for the polyurethane industry.20 Studies have shown that not only the type of support, but also the size of the nanoparticles are important for the reaction.2 Particles smaller than 2 nm promote propane formation, those with a diameter between 2–5 nm favour direct epoxidation, whereas larger particles combust propene.20 Interestingly, Au particles smaller than 1 nm have been seen to be very reactive towards propene epoxidation.21
The exceptional oxidation activity of nano-gold can be traced back partially to the presence of several low-coordinated reaction sites that favourably activate oxygen,22 as was shown for gold clusters in the gas phase.23–25 The charge state of the gold cluster23–25 and the charge separation between gold and oxygen play an essential role.25 The additional charge transfer due to the co-adsorption of ethylene even leads to O–O bond dissociation.24 Comparing the binding energies of dioxygen on small gold clusters in different charge states (neutral, anionic and cationic), the strongest adsorption was found on the cationic species.26 A computational study also found propene to bind stronger on cationic Aun+ (n = 1–5, 8) clusters, when compared with the neutral and anionic counterparts.27 This is a consequence of the larger electron transfer from the HOMO of propene to the LUMO of the cationic metal cluster. The adsorption of ethylene on charged gold clusters showed that the charge influences not only the binding energy, but also the preference of the π- (where both carbon atoms bind to the same gold atom) or the di–σ (where the carbon atoms bind to two adjacent gold atoms) binding modes.28 These observations reflect the important role of the charge state in the cluster reactivity.
The charge distribution can be tuned by the introduction of a heteroatom, i.e. by chemical doping. For example, a complementary ion-mobility and density functional computational study of silver-doped gold cluster cations showed an inhomogeneous charge distribution, where the positive charge resides almost exclusively on the silver atoms.29 In particular, it was shown that doping both neutral and cationic gold clusters with silver results in a decrease of the reactivity towards CO.30,31 In addition, the bond between propene and gold clusters can be weakened upon exchanging gold with the less electronegative silver in mixed gold-silver clusters.32
As yttrium has an even lower electronegativity than silver, the electron transfer from yttrium to gold is more pronounced. This leads also to decreased CO binding energies of yttrium-doped clusters compared to that of pure gold.33 Y-doping also modifies the geometry of gold clusters, as was shown in combined infrared spectroscopy and density functional theory studies.34–36 Electronically, the trivalent Y dopant alters the electronic shell level structure of pure gold, resulting in closed electronic shells for Au6Y+ and Au16Y+ clusters.37 In contrast, palladium-doping of gold clusters increases the CO adsorption energy, despite the smaller electronegativity of Pd in comparison with Au,9 illustrating that the role of the heteroatom in the adsorption is not trivial. All these studies clearly show that charge state and charge distribution play an important role in the reactivity of metal clusters.
Recently, we investigated the binding of propene on neutral Aun and yttrium doped gold Aun−1Y (n = 5–15) clusters, using a combination of experimental and computational methods.38 The propene binding to gold involves electron donation from the HOMO of the propene to the metal and back-donation to the LUMO of the alkene. Here, we extend our studies to positively charged clusters and address the effect of charge state, cluster size, and yttrium doping on propene adsorption. We use mass spectrometry to study the gas-phase reactivity of the clusters with propene in conjunction with density functional theory (DFT) calculations. The interactions between the clusters and propene are analysed using several computational descriptors, such as the Localized-Orbital Locator (LOL), Bader partial charges, Natural Bond Orbitals (NBOs), Wiberg bond indices and natural charges, as well as energy decomposition (EDA) analysis.
2 Computational methods
The BP86 GGA functional39,40 and the LANL2DZ basis set41–43 were applied to pre-optimize the geometries of the clusters and their propene adducts. Due to the presence of several, low-lying isomers, the accurate determination of the lowest-energy structure of gold cluster cations is challenging.44 We re-optimized structures determined in previous publications for neutral and cationic pure gold clusters.44–48 While the structures of small neutral yttrium doped gold clusters33,34,36,38 were reported earlier, little information is available about the structure of their cationic states;35 therefore, we computed the geometric structures of the low-energy cationic isomers using the Particle Swarm Optimization algorithm of CALYPSO49–51 and investigated the growth patterns using the capping method.52 The obtained low-energy structures were re-optimized using the higher LRC-ωPBE/def2-TZVP level of theory53,54 including the Exchange Dipole Model (XDM) dispersion correction.55,56. (see Tables S1 and S2 in the ESI† for the propene adsorption energies).The method was selected based on CCSD(T)/def2-TZVP benchmark computations57 for Au4+, Au5+, Au6+, Au3Y+, Au4Y+ and Au5Y+ clusters using the MRCC program.58,59 The PBE0, the CAM-B3LYP and the TPSSh functionals were also included for the comparison. It is interesting to note that while the single point energies using the CAM-B3LYP + XDM60 or TPSSh + XDM61 methods accurately described the propene binding to neutral clusters, these were less accurate for the cations.
The initial cluster–propene adduct geometries were systematically generated using our program in di–σ and π binding modes and optimized subsequently using DFT calculations. The barrierless nature of propene adsorption was confirmed by placing the propene 5 Å from the atom to which it was bound in the adduct and optimizing this structure. The adsorption energy is defined as the difference of the adduct energy and the sum of the cluster and the propene energies. The calculated adsorption energy was corrected with the zero-point vibrational energy, defined as ΔZPE = ZPE(Aun−1Y+–C3H6) − ZPE(Aun−1Y+) − ZPE(C3H6). The basis set superposition error (BSSE) was compensated using the counterpoise method.62,63 The DFT computations were carried out using the Q-Chem 5.2 and 5.3 quantum chemical software packages.64 The chemical bonding was analysed by the LOL,65 computed using the D-Grid code,66,67 Bader theory68 using the Multiwfn program,69 and NBO analysis, Wiberg bond indices, and natural charges,70,71 employing the NBO code incorporated in Q-Chem. EDA67 was carried out as implemented in Q-Chem 5.2. The orbital–orbital interactions were analysed using the ALMO-EDA method and the Complementary Occupied-Virtual Pairs (COVPs).67 Molecules were visualized by the PyMol program.72 The isosurfaces of the Kohn–Sham orbitals and COVPs were visualized using IQMol.73
3 Experimental methods
The experimental approach followed here is similar to that in ref. 38. In brief, pure Aun+ and doped Aun−1Y+ clusters were produced by laser ablation, in a source described in detail elsewhere.74 Two independent YAG lasers ablate almost simultaneously Au and Y targets, creating a plasma that is cooled down by the inclusion of He carrier gas (backing pressure of 8 bar). The mixture of Au–Y–He then expands supersonically into vacuum, through a conical nozzle, creating a molecular beam composed of Aun+ and Aun−1Y+ clusters. Other charge states are also present, but only cationic clusters are analysed. For these experiments, the source was kept at room temperature. After collimation via a 1 mm skimmer, the molecular beam enters a low-pressure collision cell,75 where the clusters interact with C3H6 gas at a controlled pressure (PC3H6). The size dependent intensities of cluster–C3H6 complexes are analysed by reflectron time-of-flight mass spectrometry. In the absence of a buffer gas and considering the small heat capacity of the clusters, the cluster–C3H6 complexes are heated up by the C3H6 adsorption energy and the alkene may desorb on the time scale of the experiment. Under these conditions, the fraction of the clusters that form C3H6 complexes, as recorded by the mass spectrometer, contains information about the cluster–C3H6 bond strength.764 Results
4.1. Lowest energy structures of Aun+, Aun+–C3H6, Aun−1Y+ and Aun−1Y+–C3H6
The geometries of the putative lowest-energy gold and yttrium-doped gold cluster cations are depicted in Fig. 1. The structures of the neutral lowest-energy clusters and other low-energy cationic isomers together with their XYZ coordinates are available in the ESI.† While the 2D to 3D transition occurs at n = 11 for neutral gold clusters5,77 this transition takes place at n = 8 for the cations, in agreement with earlier studies.44–46,48 Both the lowest energy 2D and 3D isomers are used as Au8+ precursor for the propene adsorption, as they differ by only 0.03 eV in energy. Only for n = 6 and 11, the neutral and cationic clusters have the same geometric structures.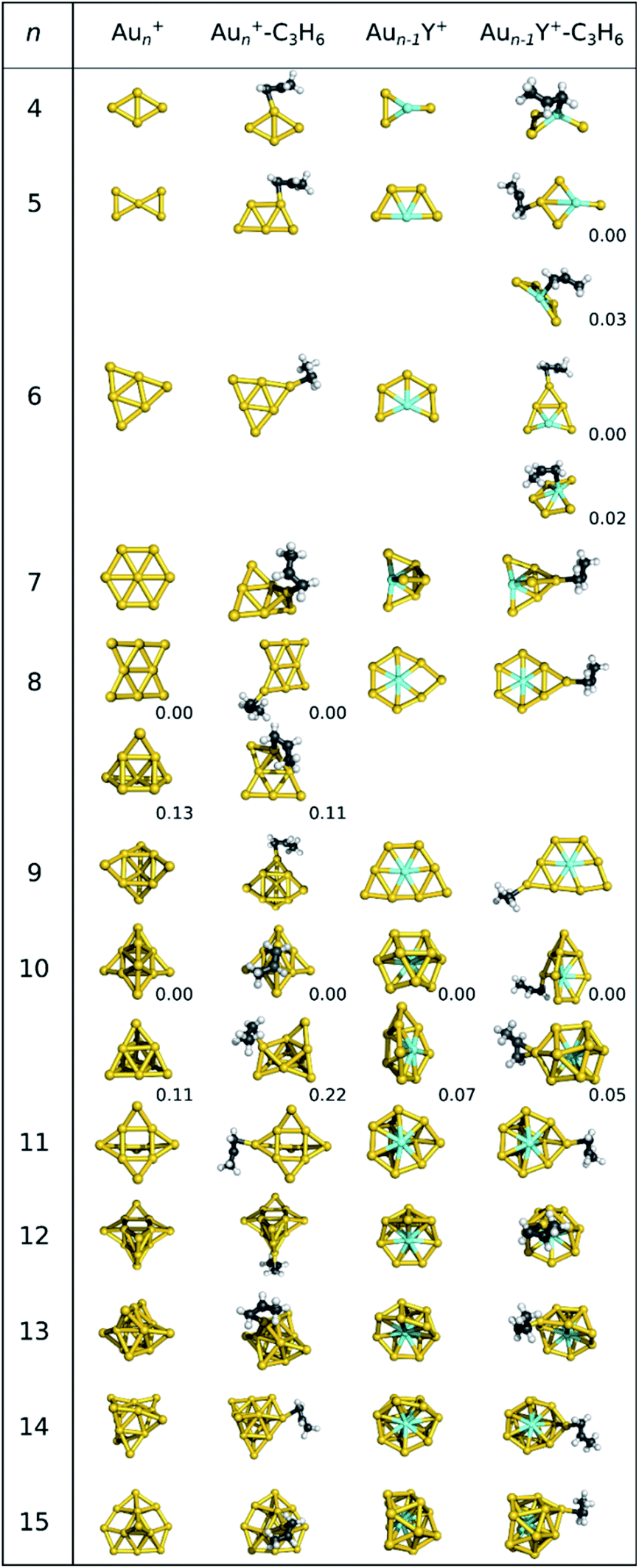 | ||
| Fig. 1 Optimized lowest-energy structures of Aun+ and Aun−1Y+ (n = 5–15) as well as their C3H6 adducts at the LRC-ωPBE+XDM/def2-TZVP level of theory. For Au4Y+ and Au5Y+, two low-lying isomers are shown, since the propene adsorbs favourably on both the yttrium and a gold atom. For Au8+ and Au10+ the energy differences between the two isomers are very small, thus both are shown here as well. In case multiple isomers are shown, relative energies in eV are given. Other isomers with Erel < 0.1 eV are presented in the ESI (Fig. S2†). | ||
Propene binds to the gold atom with the lowest coordination number in Aun+, where one of the low-lying unoccupied orbitals of the bare cluster has a large lobe27,38 (see orbitals in ESI†). In the case of n = 5, 8, 9 and 13, several energetically low-lying isomers of cationic gold cluster propene adducts were found (geometries are available in the ESI†). The geometries of most cationic gold clusters, except Au5+, Au7+ and Au8+, do not significantly change upon propene adsorption. Interestingly, propene binding reverses the relative stabilities of the 2D and 3D geometries for n = 7 and 8.
Multiple low-lying isomers of Aun−1Y+ exist for several cluster sizes, making it difficult to determine their accurate stability order. Therefore, we considered all isomers with Erel < 0.15 eV for building the cluster-propene adducts (see the ESI† for other isomers and the Cartesian coordinates). Au6Y+ is the smallest cluster with a 3D structure in its most stable geometry, but surprisingly Au7Y+ and Au8Y+ again exhibit 2D geometries. These 2D clusters are even larger than in the case of the neutral yttrium doped gold clusters, which are 3D from Au8Y+ onward. For n = 8 and 14, the geometries of the most stable bare cationic clusters agree with those of the neutral clusters.38 Propene prefers to bind to the yttrium atom in Au3Y+, while gold atom bound adducts are more stable for Au4Y+ and Au5Y+. This is remarkable, as our study on neutral Aun−1Y pointed out that donation from propene to the unoccupied d atomic orbitals of yttrium is an important stabilizing factor.38
It is known that metal clusters can activate and even break C–H bonds.8,78,79 We computed the dissociation reaction paths (Fig. S6 in the ESI†) for Au5Y+, Au6Y+ and Au6+ clusters. Isomers with dissociated propene are higher in energy than those with intact propene and they are separated by large energy barriers. Therefore, dissociation is unlikely, and the population of structures with intact propene is considerably higher than those with the dissociated molecule. Hence, we further discuss only adducts with intact propene.
4.2. Propene adsorption energies
The adsorption energies of propene on Aun+ and Aun−1Y+ (n = 4–15) are plotted in Fig. 2, in comparison with those of the neutral adducts, recomputed from ref. 38. The cations systematically have higher propene binding energies.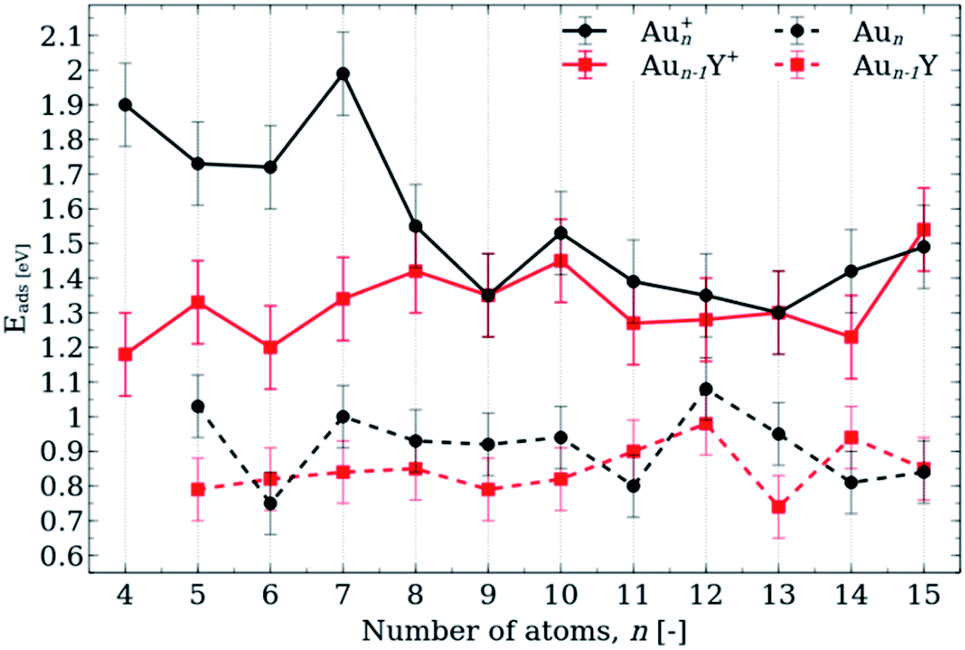 | ||
| Fig. 2 Propene adsorption energies, Eads, on cationic (solid line) and neutral (dashed line) Aun+,0 (black) and Aun−1Y+,0 (red) clusters (n = 4–15) calculated using the LRC-ωPBE+XDM/def2-TZVP method. Error bars for DFT calculations were estimated using CCSD(T)/def2-TZVP benchmarks (Table S1†). The energies corresponding to the neutral species are recomputed from ref. 38. | ||
The propene binding energies of Aun+ clusters show a pronounced size dependence with values in the range of 1.3–2 eV, in contrast with the relatively small variations in the propene binding energy with size for the Aun−1Y+ clusters (range of 1.1–1.6 eV). For the Aun+ clusters, Au7+ has the largest adsorption energy of 1.99 eV, while among the Aun−1Y+ series, the propene binding is strongest for Au14Y+ (Eads ∼ 1.54 eV).
4.3. Experimental results
The calculated size-dependent propene adsorption energies on Aun+ and Aun−1Y+ clusters are complemented with mass spectrometric results of collision-cell experiments. Fig. 3a presents a representative mass spectrum of the clusters exposed to propene gas in the reaction cell (PC3H6 = 0.2 Pa). Next to the Aun+, secondary peaks, corresponding to the Aun+–C3H6 complexes, are seen. Such secondary peaks are hardly visible for the doped Aun−1Y+ clusters.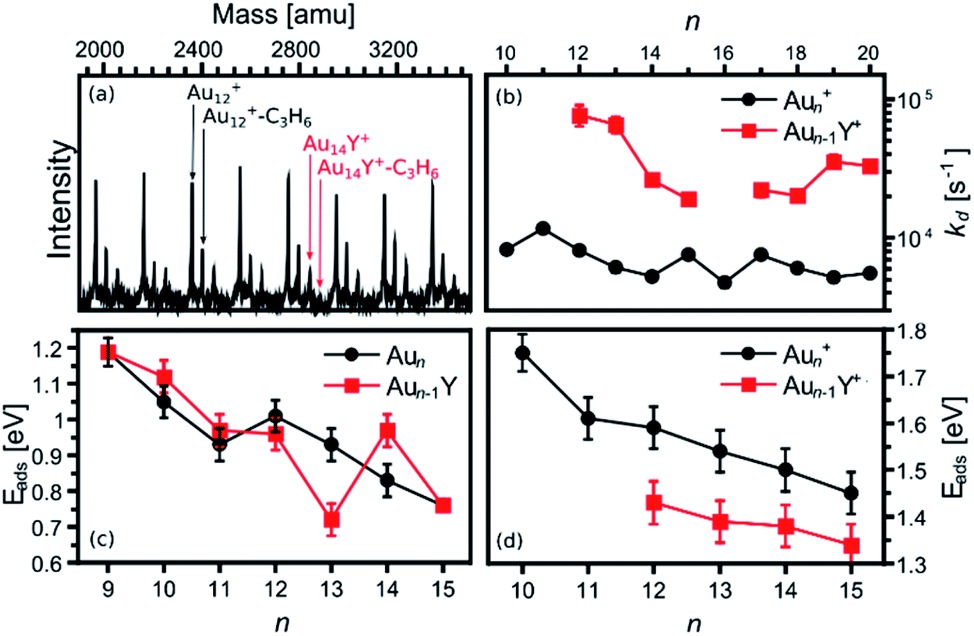 | ||
| Fig. 3 (a) Representative example of a mass spectrum of Aun+ and Aun−1Y+ clusters, measured under the exposure of propene (PC3H6 = 0.2 Pa). (b) Desorption rates of propene from Aun+–C3H6 and Aun−1Y+–C3H6. The missing points in the figure correspond to clusters for which no propene adduct was observed in the mass spectra. (c) and (d) present the adsorption energies extracted from RRKM simulations, using as input the experimental dissociation rates for neutrals (from ref. 38) and cations, respectively. Error bars on the RRKM modelled values have been estimated by scaling all vibration frequencies by a factor 0.9 and a factor 1.1. | ||
Quantification of the Y doping effect on the propene adsorption is achieved by recording mass spectra as a function of PC3H6. As detailed in ref. 38, this allows obtaining desorption rates of propene (kd), which depend on the cluster–propene adsorption energies. The extracted desorption rates are presented in Fig. 3b. As expected from the low Aun−1Y+–C3H6 intensities (Fig. 3a), all kd rates are higher for the Y doped than for the pure cationic gold clusters, indicating a reduction of the propene adsorption energies upon Y doping.
Using the extracted desorption rates of propene, adsorption energies can be estimated by conducting a RRKM analysis (see ref. 38 and the ESI† for details). This analysis was performed here for the cationic species, and can be compared with results obtained in ref. 38 for the neutral clusters. Both adsorption energies are presented in panels (c) and (d) of Fig. 3. In general, it is observed that the adsorption energies are higher for the cationic clusters, and that in several cases, Y-doping reduces these energies. This latter observation is particularly true for the cationic clusters, with reduced adsorption energies upon Y doping for all the investigated cluster sizes. For the neutral species, however, the effect of doping is more complex, with either enhancement or reduction in Eads depending on cluster size.
The reduction in Eads upon Y-doping is in general well reproduced for the cationic clusters by the DFT calculations. While the size-to-size energies are not perfectly reproduced, the calculations clearly show a lower adsorption energy of propene on the doped clusters (within the combined errors of the experiment and the calculations). For the neutral species, the particular features at n = 13 and 14 are well reproduced by theory. Below n = 11, the RRKM analysis shows slightly higher adsorption energies on the doped clusters, which are not seen in the DFT calculations. In both cases, however, the changes in Eads induced by doping are small, below the uncertainty of the analysis.
4.4. Charge distribution and chemical bonding analysis
The charge distribution is an important quantity for the propene adsorption, since propene adsorbs for all Aun+ clusters, except for n = 6, 7 and 12, to the gold atom with the highest positive charge. It is interesting to note that the positive charge in Aun+ is unevenly distributed over the constituting atoms. In Au11+, Au13+ and Au15+, a few atoms are negatively charged (Table S3†).Yttrium adopts a large positive partial charge (>1 e) in the smallest Aun−1Y+ (n = 4–7) clusters, while its charge gradually decreases with the cluster size (Table S4†). Remarkably, the Y dopant becomes even negatively charged as the cluster size increases, despite the electronegativity of Au being larger than that of Y. As Y is an early transition metal, d electrons become itinerant in the host gold cluster and combine with the valence s atomic orbitals of Au. Thus, formally Au can donate electrons to the unoccupied d orbitals of Y. Consequently, the valence d orbital population of Y formally increases with the increase of the number of the adjacent Au atoms.
In the case of Aun−1Y+ clusters, propene may either adsorb to the Y or to a Au atom.38 Small neutral clusters (n = 5–8) prefer Y binding. This is due to the favoured propene π electron donation to the unoccupied d atomic orbitals of Y.38 Au and Y binding sites compete at n = 9. At larger cluster sizes, propene prefers to bind to a Au atom, as the Y d electron population increases with the cluster size, hindering the electron donation from propene π orbitals. Contrary to the neutrals, propene can bind to both Y and a Au atom of the cationic Au4Y+ and Au5Y+ clusters, as can be seen in Fig. 1. Au-bound propene is also possible in the case of Au3Y+, however, it is 0.18 eV higher in energy than the Y-bound isomer.
The natural charges show that Au atoms with a significantly high positive partial charge can compete with Y as the preferred binding site for propene (Fig. 4). All Au atoms of Au3Y+ are negatively charged. Consequently, propene binding to gold is not favoured for this cluster. In Au4Y+ and Au5Y+ there are positively charged gold atoms, leading to stable gold atom-bound propene adducts. Most of the gold atoms of the small neutral Aun−1Y (n = 5–9) clusters (see Table S7 in the ESI† for the charges and Fig. S8† for the electrostatic potentials) are negatively charged and therefore, Au binding is generally not favoured. Two exceptions are the slightly positively charged Au atoms in Au6Y+ and in Au8Y+. In these cases, Au atom-bound propene isomers are relatively stable, but higher in energy than the Y-bound isomer. For Au6Y+, there is a 0.2 eV energy difference between the Y and the Au atom bound isomers, while for Au8Y+ this energy difference is only 0.06 eV.
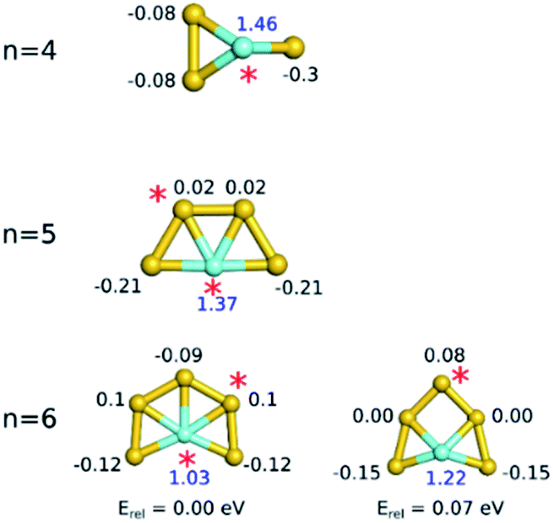 | ||
| Fig. 4 Natural charges (in units of electron charge) of the atoms in the small Aun−1Y+ clusters (blue: yttrium, black: gold). Red asterisks indicate the propene binding sites. | ||
While propene prefers to bind to the most positively charged Au atoms in the larger Aun−1Y+ clusters, there is no one-to-one correspondence between the increased charge and the adsorption energy. This indicates that not only the charge of the metal atoms, but also other factors influence the adsorption. The large lobes of the LUMOs show the preferred propene binding sites (see the ESI† for details). These suggest the importance of the electron donation from propene to the cluster. Following propene adsorption on a gold atom (both for doped and pure clusters), the natural charge of that atom increases, while that of atoms in its vicinity decreases slightly. If the propene adsorbs on the yttrium dopant atom, the charge of the yttrium decreases (Table S5†).
The analysis of the LOLs, bond critical points (Bader analysis) and the Wiberg bond indices demonstrates that there is no covalent bond between the cationic cluster and propene. Similarly, the NBO analysis suggests a donation and back donation mechanism for the adsorption. Details are available in the ESI.† Thus, non-covalent interactions and charge transfer are responsible for the propene binding. The EDA assists in the characterization of the non-covalent interaction between cluster and propene and quantifies the importance of the different terms. It decomposes the adsorption energy as a sum of charge transfer (between the cluster and propene), frozen (due to Pauli-repulsion), polarization (of the electrons in the propene and in the cluster), relaxation (due to the geometry change of the cluster and the propene molecule in the adduct compared to the isolated particles), dispersion energy, and zero-point vibrational energy terms:38
| Eadsorption = Echarge transfer − Efrozen + Epolarization − Erelaxation + Edispersion+ ΔEZPE. | (1) |
The decomposition of the propene adsorption energies on Aun+ and Aun−1Y+ is shown in Fig. 5. The charge transfer and the frozen terms have the largest contributions to the total energy, and these terms follow similar size-dependences in the propene adducts of the pure and doped cluster cations. These terms have opposite signs in eqn (1), however, the charge transfer term is larger and thus, the attractive interaction dominates. As for the neutral clusters,38 the charge transfer energies in cationic cluster-propene adducts follow similar trends to that of the frozen term, although the charge transfer is considerably more important in the cations. This clearly shows the role of the charge transfer in the propene binding and explains the increased propene adsorption energies in the cationic clusters as compared to the neutrals.
The polarization term exhibits a more pronounced size dependence in the Aun+–propene adducts than in the Y-doped species. This is in line with the highly size-dependent propene adsorption energies in Aun+. It is interesting to note that the relaxation terms are considerably larger in the cationic clusters than in the previously investigated neutral species,38 in line with the propene adsorption induced geometry distortions observed for several cluster sizes. The dispersion energies and the ΔZPEs have only small contributions and are weakly size-dependent. Thus, the charge transfer, the frozen, the polarization and the relaxation terms determine the adsorption energies in both Aun+ and Aun−1Y+ clusters.
It is important to note that only the most stable isomers are analysed in Fig. 5, thus for Au4Y+ and Au5Y+ we present only the gold-atom-bound isomers. Generally, both the charge transfer and the frozen terms are lower in the yttrium-bound propene complexes than in the gold-bound isomers. Nevertheless, the adsorption energy shows smaller size-dependence than in the case of the pure gold clusters, as the different terms compensate each other.
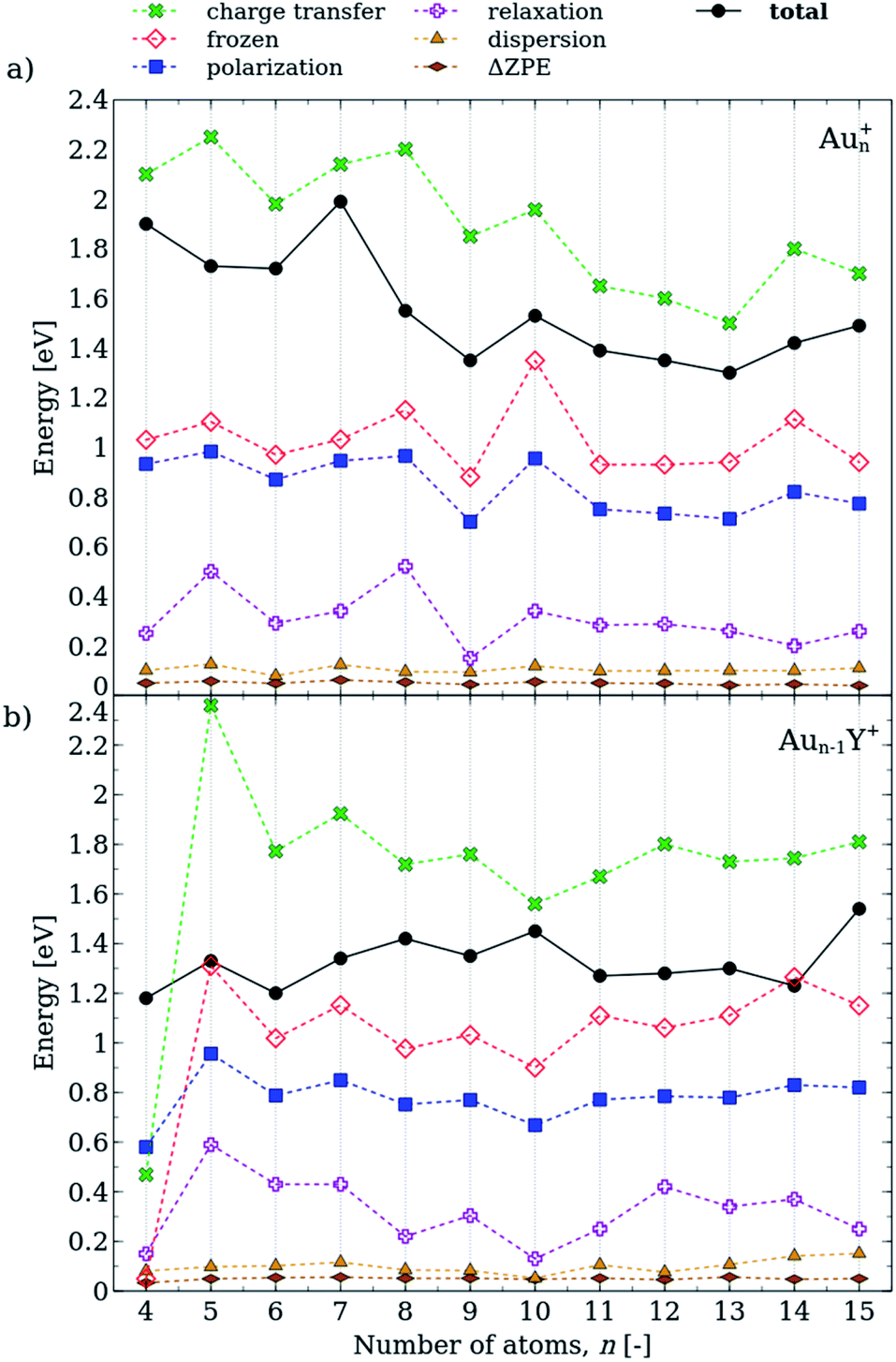 | ||
| Fig. 5 EDA of the propene adsorption energy (total, Eads) for (a) Aun+ and (b) Aun−1Y+ (n = 5–15) clusters. Full and hollow symbols are used for positive and negative contributions to Eads, respectively. | ||
4.5. Orbital interactions
The analysis of the Wiberg bond indices, natural charges, and the charge transfer energy suggests electron donation and back-donation during the propene adsorption. The total charge transfer energy can be decomposed in complementary occupied/virtual pairs (COVPs) contributions (δE), where each of the orbitals correspond either to the cluster or to the propene. Thus, COVPs can be used to interpret and visualize the electron donation and back donation taking place in the propene binding. The total electron transfer (δQ) corresponding to each COVP pair was also computed.Fig. 6 shows the main occupied-virtual pairs for selected sizes, representing the different propene binding modes. There are two main COVP pairs in the case of pure gold clusters. In the one denoted as “donation”, the π orbital of propene donates electrons to the s and p unoccupied orbitals of gold. In the “back-donation”, the d atomic orbitals of gold donate electrons to the π* orbital of propene. Both imply the weakening of the propene C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C double bond, in line with the analysis of the Wiberg bond indices (see the ESI† for the details). Similar COVPs are observed in the case of yttrium-doped adducts, where propene binds to a gold atom. The δE and the δQ values clearly show that the donation is more important than the back-donation. This is in line with the net electron transfer from propene to the cluster, found in the atomic charge analysis. The situation is different if propene binds to the Y atom. The two important COVPs of Au5Y+–C3H6 are shown in Fig. 6. As for the other clusters, the π orbital of propene donates electrons, however, in this case not to a gold atom, but to the d orbitals of the Y atom. The δQ and δE values of the back-donation are very small, and interestingly not the Y atom (where propene binds), but the gold atoms back-donate electrons. This can be explained by the unfavoured electron donation from the positively charged yttrium atom.
C double bond, in line with the analysis of the Wiberg bond indices (see the ESI† for the details). Similar COVPs are observed in the case of yttrium-doped adducts, where propene binds to a gold atom. The δE and the δQ values clearly show that the donation is more important than the back-donation. This is in line with the net electron transfer from propene to the cluster, found in the atomic charge analysis. The situation is different if propene binds to the Y atom. The two important COVPs of Au5Y+–C3H6 are shown in Fig. 6. As for the other clusters, the π orbital of propene donates electrons, however, in this case not to a gold atom, but to the d orbitals of the Y atom. The δQ and δE values of the back-donation are very small, and interestingly not the Y atom (where propene binds), but the gold atoms back-donate electrons. This can be explained by the unfavoured electron donation from the positively charged yttrium atom.
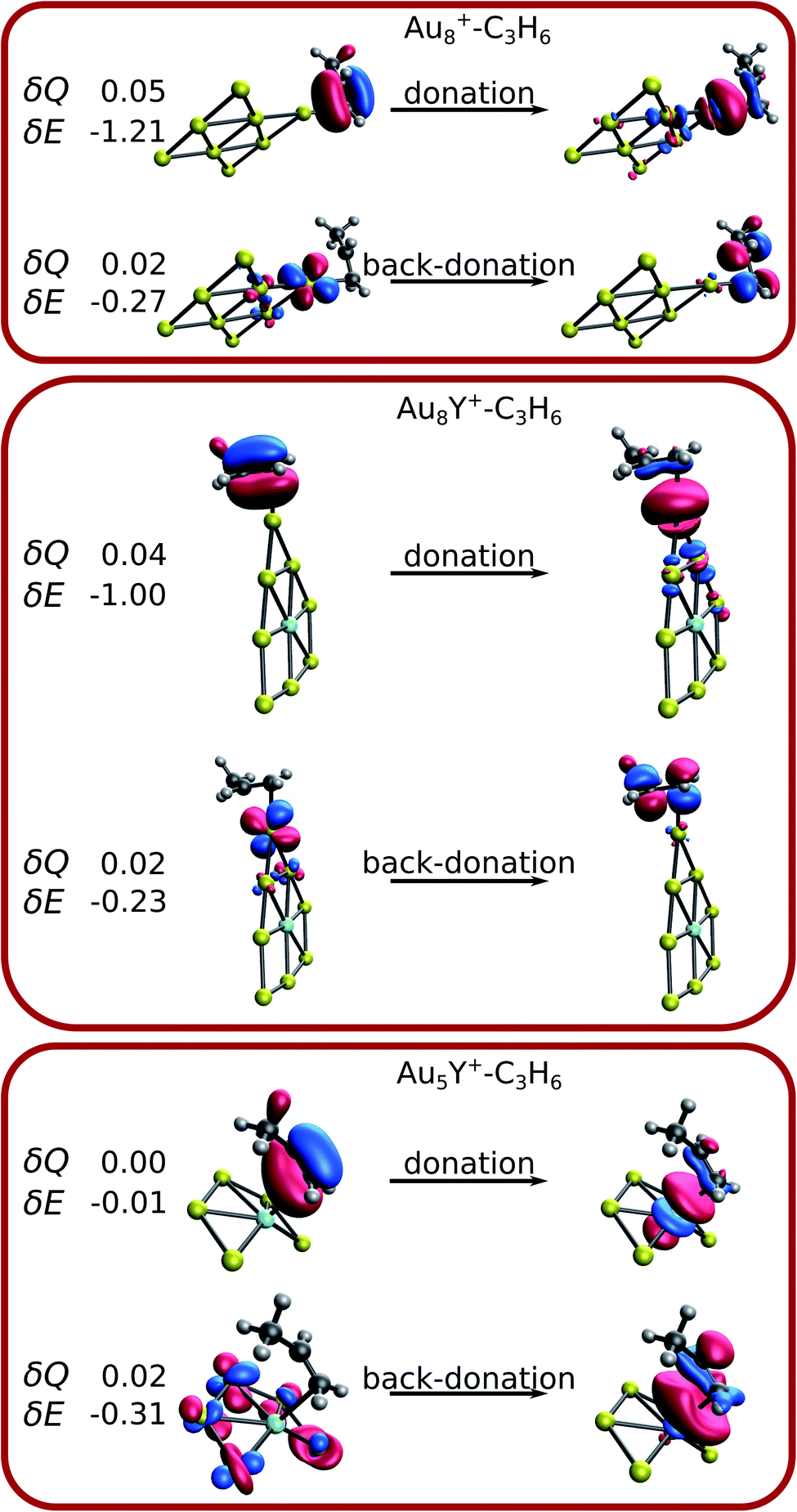 | ||
| Fig. 6 Complementary virtual-occupied orbital pairs of the selected Au8+–C3H6, Au8Y+–C3H6 and Au5Y+–C3H6 complexes. The energy differences (δE in eV) and the transferred charge (δQ in e−) corresponding to the COVPs are also shown. | ||
Thus, the electron donation from the propene to the cationic cluster is the main factor in the bonding, unlike the neutral adducts where the donation and the back-donation are of comparable importance for the binding of propene to a gold atom (Table S7†).
5 Conclusions
In summary, we studied the effect of the charge state on the propene adsorption of small bare and yttrium doped gold clusters by joint gas-phase reactivity measurements and DFT computations. The adsorption energies are clearly size, charge state, and dopant dependent. In contrast to the neutral Aun−1Y clusters, propene prefers to bind both to the gold and to the yttrium in the case of small clusters. The binding site preference is well explained by the natural atomic charges. It was found that for most sizes the yttrium doping lowers the adsorption energy of propene on the cationic cluster.The experiments and the DFT computations both reveal that the positive charge state considerably increases the propene binding strength on bare as well as on yttrium-doped gold clusters, while the yttrium doping tends to lower the propene adsorption energy compared to the pure gold clusters, however, this effect has a size-dependency. EDA demonstrated that this is due to the enhanced charge transfer from propene to the cluster. It was concluded that in contrast to the neutrals, in the propene adducts of the cationic clusters not only the frozen and the charge transfer, but also the relaxation and the polarization term determine the size-dependent adsorption energy. LOL analysis, characterization of the bond critical points of the electron density, and the Wiberg bond indices showed that there is non-covalent interaction between the propene and the cluster.
These findings open the way towards the fine-tuning of the propene reactivity in its catalytic conversions that make use of gold nanoparticles.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work is supported by the KU Leuven–Budapest University of Technology and Economics joint research funding (CELSA/18/032). P. F. and J. V. acknowledge the FWO (Research Foundation Flanders) for a postdoctoral grant and a PhD fellowship, respectively.References
- Z. Luo, A. W. Castleman and S. N. Khanna, Reactivity of Metal Clusters, Chem. Rev., 2016, 116(23), 14456–14492 CrossRef CAS PubMed.
- M. Haruta, Size- and support-dependency in the catalysis of gold, Catal. Today, 1997, 36(1), 153–166 CrossRef CAS.
- A. Halder, L. A. Curtiss, A. Fortunelli and S. Vajda, Perspective: Size selected clusters for catalysis and electrochemistry, J. Chem. Phys., 2018, 148(11), 110901 CrossRef PubMed.
- L. Liu and A. Corma, Metal Catalysts for Heterogeneous Catalysis: From Single Atoms to Nanoclusters and Nanoparticles, Chem. Rev., 2018, 118(10), 4981–5079 CrossRef CAS PubMed.
- S. Vajda and M. G. White, Catalysis Applications of Size-Selected Cluster Deposition, ACS Catal., 2015, 5(12), 7152–7176 CrossRef CAS.
- M. Schlangen and H. Schwarz, Effects of Ligands, Cluster Size, and Charge State in Gas-Phase Catalysis: A Happy Marriage of Experimental and Computational Studies, Catal. Lett., 2012, 142(11), 1265–1278 CrossRef CAS.
- S. Cao, F. F. Tao, Y. Tang, Y. Li and J. Yu, Size- and shape-dependent catalytic performances of oxidation and reduction reactions on nanocatalysts, Chem. Soc. Rev., 2016, 45(17), 4747–4765 RSC.
- H. Schwarz, “Doping Effects in Cluster-Mediated Bond Activation, Angew. Chem., 2015, 54(35), 10090–10100 CrossRef CAS PubMed.
- P. Ferrari, J. Vanbuel, E. Janssens and P. Lievens, Tuning the Reactivity of Small Metal Clusters by Heteroatom Doping, Acc. Chem. Res., 2018, 51(12), 3174–3182 CrossRef CAS PubMed.
- S. M. Lang and T. M. Bernhardt, Gas phase metal cluster model systems for heterogeneous catalysis, Phys. Chem. Chem. Phys., 2012, 14(26), 9255–9269 RSC.
- J. Vanbuel, P. Ferrari and E. Janssens, Few-atom cluster model systems for a hydrogen economy, Adv. Phys.: X, 2020, 5(1), 1754132 CAS.
- Y. Chen, G. Li, H. Qian and R. Jin, Catalysis by Atomically Precise Gold Nanoclusters, Catalysis by materials with well-defined structures, Elsevier, 2015, pp. 239–262 Search PubMed.
- S. Tosoni and G. Pacchioni, Oxide-Supported Gold Clusters and Nanoparticles in Catalysis: A Computational Chemistry Perspective, Chemcatchem, 2019, 11(1), 73–89 CrossRef CAS.
- S. Lee, L. M. Molina, M. J. López, J. A. Alonso, B. Hammer, B. Lee, S. Seifert, R. E. Winans, J. W. Elam, M. J. Pellin and S. Vajda, Selective propene epoxidation on immobilized au(6-10) clusters: the effect of hydrogen and water on activity and selectivity, Angew. Chem., 2009, 48(8), 1467–1471 CrossRef CAS PubMed.
- J. Huang, T. Akita, J. Faye, T. Fujitani, T. Takei and M. Haruta, Propene epoxidation with dioxygen catalyzed by gold clusters, Angew. Chem., 2009, 48(42), 7862–7866 CrossRef CAS PubMed.
- J. Terzan, M. Hus, B. Likozar and P. Djinovic, Propylene epoxidation using molecular oxygen over copper- and silver-based catalysts: A review, ACS Catal., 2020, 10, 13415–13436 CrossRef CAS.
- T. Nijhuis, T. Visser and B. Weckhuysen, Mechanistic study into the direct epoxidation of propene over gold/titania catalysts, J. Phys. Chem. B, 2005, 109(41), 19309–19319 CrossRef CAS PubMed.
- M. Boronat, A. Pulido, P. Concepción and A. Corma, Propene epoxidation with O2 or H2–O2 mixtures over silver catalysts: theoretical insights into the role of the particle size, Phys. Chem. Chem. Phys., 2014, 16(48), 26600–26612 RSC.
- L. Cheng, C. Yin, F. Mehmood, B. Liu, J. Greeley, S. Lee, B. Lee, S. Seifert, R. E. Winans, D. Teschner, R. Schlögl, S. Vajda and L. A. Curtiss, Reaction Mechanism for Direct Propylene Epoxidation by Alumina-Supported Silver Aggregates. The Role of the Particle/Support Interface, ACS Catal., 2014, 4(1), 32–39 CrossRef CAS.
- T. Hayashi, K. Tanaka and M. Haruta, Selective Vapor-Phase Epoxidation of Propylene over Au/TiO2Catalysts in the Presence of Oxygen and Hydrogen, J. Catal., 1998, 178, 566–575 CrossRef CAS.
- J. Lu, X. Zhang, J. J. Bravo-Suárez, K. K. Bando, T. Fujitani and S. T. Oyama, Direct propylene epoxidation over barium-promoted Au/Ti-TUD catalysts with H2 and O2: Effect of Au particle size, J. Catal., 2007, 250(2), 350–359 CrossRef CAS.
- N. Lopez, T. Janssens, B. Clausen, Y. Xu, M. Mavrikakis, T. Bligaard and J. Nørskov, On the origin of the catalytic activity of gold nanoparticles for low-temperature CO oxidation, J. Catal., 2004, 223(1), 232–235 CrossRef CAS.
- R. Pal, L.-M. Wang, Y. Pei, L.-S. Wang and X. C. Zeng, Unraveling the Mechanisms of O2 Activation by Size-Selected Gold Clusters: Transition from Superoxo to Peroxo Chemisorption, J. Am. Chem. Soc., 2012, 134(22), 9438–9445 CrossRef CAS PubMed.
- A. Lyalin and T. Taketsugu, Reactant-Promoted Oxygen Dissociation on Gold Clusters, J. Phys. Chem. Lett., 2010, 1(12), 1752–1757 CrossRef CAS.
- A. P. Woodham, G. Meijer and A. Fielicke, Charge separation promoted activation of molecular oxygen by neutral gold clusters, J. Am. Chem. Soc., 2013, 135(5), 1727–1730 CrossRef CAS PubMed.
- D. H. Wells, W. N. Delgass and K. T. Thomson, Density functional theory investigation of gold cluster geometry and gas-phase reactivity with O2, J. Chem. Phys., 2002, 117(23), 10597–10603 CrossRef CAS.
- S. Chrétien, M. S. Gordon and H. Metiu, Binding of propene on small gold clusters and on Au(111): simple rules for binding sites and relative binding energies, J. Chem. Phys., 2004, 121(8), 3756–3766 CrossRef PubMed.
- A. Lyalin and T. Taketsugu, Adsorption of Ethylene on Neutral, Anionic, and Cationic Gold Clusters, J. Phys. Chem. C, 2010, 114(6), 2484–2493 CrossRef CAS.
- P. Weis, O. Welz, E. Vollmer and M. M. Kappes, Structures of mixed gold-silver cluster cations (AgmAun+,m+n<6): Ion mobility measurements and density-functional calculations, J. Chem. Phys., 2004, 120(2), 677–684 CrossRef CAS PubMed.
- J. D. Haeck, N. Veldeman, P. Claes, E. Janssens, M. Andersson and P. Lievens, Carbon monoxide adsorption on silver doped gold clusters, J. Phys. Chem. A, 2011, 115(11), 2103–2109 CrossRef PubMed.
- M. Neumaier, F. Weigend, O. Hampe and M. M. Kappes, Binding energy and preferred adsorption sites of CO on gold and silver-gold cluster cations: adsorption kinetics and quantum chemical calculations, Faraday Discuss., 2008, 138, 393–406 RSC.
- S. Chrétien, M. S. Gordon and H. Metiu, Density functional study of the adsorption of propene on mixed gold-silver clusters, AunAgm: Propensity rules for binding, J. Chem. Phys., 2004, 121(20), 9931–9937 CrossRef.
- L. Lin, P. Lievens and M. T. Nguyen, “Theoretical Study of CO Adsorption on Yttrium-doped Gold Clusters AunY (n = 1 – 9), Chem. Phys. Lett., 2010, 498(4), 296–301 CrossRef CAS.
- L. Lin, P. Claes, P. Gruene, G. Meijer, A. Fielicke, M. T. Nguyen and P. Lievens, “Far-Infrared Spectra of Yttrium-Doped Gold Clusters AunY (n=1–9), ChemPhysChem, 2010, 11(9), 1932–1943 CAS.
- L. Lin, P. Claes, T. Höltzl, E. Janssens, T. Wende, R. Bergmann, G. Santambrogio, G. Meijer, K. R. Asmis, M. T. Nguyen and P. Lievens, The structure of Au6Y+ in the gas phase, Phys. Chem. Chem. Phys., 2010, 12(42), 13907–13913 RSC.
- L. Lin, T. Höltzl, P. Gruene, P. Claes, G. Meijer, A. Fielicke, P. Lievens and M. T. Nguyen, Fluxionality and σ-Aromaticity in Small Yttrium-Doped Gold Clusters, ChemPhysChem, 2008, 9(17), 2471–2474 CrossRef CAS PubMed.
- N. Veldeman, E. Janssens, K. Hansen, J. D. Haeck, R. E. Silverans and P. Lievens, Stability and dissociation pathways of doped AunX+ clusters (X = Y, Er, Nb), Faraday Discuss., 2008, 138, 147–162 RSC.
- J. Barabás, J. Vanbuel, P. Ferrari, E. Janssens and T. Höltzl, Non-covalent Interactions and Charge Transfer between Propene and Neutral Yttrium-Doped and Pure Gold Clusters, Chem. - Eur. J., 2019, 25(69), 15795–15804 CrossRef PubMed.
- A. Becke, Density-functional exchange-energy approximation with correct asymptotic behavior, Phys. Rev. A, 1988, 38(6), 3098–3100 CrossRef CAS PubMed.
- J. P. Perdew, Density-functional approximation for the correlation energy of the inhomogeneous electron gas, Phys. Rev. B: Condens. Matter Mater. Phys., 1986, 33(12), 8822–8824 CrossRef PubMed.
- H. F. Schaefer, Methods of Electronic Structure Theory, 1977 Search PubMed.
- P. J. Hay and W. R. Wadt, Ab initio effective core potentials for molecular calculations. Potentials for K to Au including the outermost core orbitals, J. Chem. Phys., 1985, 82(1), 299–310 CrossRef CAS.
- W. R. Wadt and P. J. Hay, Ab initio effective core potentials for molecular calculations. Potentials for main group elements Na to Bi, J. Chem. Phys., 1985, 82(1), 284–298 CrossRef CAS.
- L. Ferrighi, B. Hammer and G. K. H. Madsen, 2D-3D transition for cationic and anionic gold clusters: a kinetic energy density functional study, J. Am. Chem. Soc., 2009, 131(30), 10605–10609 CrossRef CAS PubMed.
- E. M. Fernández, J. M. Soler, I. L. Garzón and L. C. Balbás, Trends in the structure and bonding of noble metal clusters, Phys. Rev. B: Condens. Matter Mater. Phys., 2004, 70(16), 165403 CrossRef.
- S. Gilb, P. Weis, F. Furche, R. Ahlrichs and M. M. Kappes, Structures of small gold cluster cations (Aun+, n<14): Ion mobility measurements versus density functional calculations, J. Chem. Phys., 2002, 116(10), 4094–4101 CrossRef CAS.
- P. Ferrari, H. A. Hussein, C. J. Heard, J. Vanbuel, R. L. Johnston, P. Lievens and E. Janssens, Effect of palladium doping on the stability and fragmentation patterns of cationic gold clusters, Phys. Rev. A, 2018, 97(5), 52508 CrossRef CAS.
- P. Ferrari, G.-L. Hou, O. V. Lushchikova, F. Calvo, J. M. Bakker and E. Janssens, The structures of cationic gold clusters probed by far-infrared spectroscopy, Phys. Chem. Chem. Phys., 2020, 22(20), 11572–11577 RSC.
- Y. Wang, J. Lv, L. Zhu and Y. Ma, Crystal structure prediction via particle-swarm optimization, Phys. Rev. B: Condens. Matter Mater. Phys., 2010, 82(9), 94116 CrossRef.
- Y. Wang, J. Lv, L. Zhu and Y. Ma, CALYPSO: A method for crystal structure prediction, Comput. Phys. Commun., 2012, 183(10), 2063–2070 CrossRef CAS.
- B. Gao, P. Gao, S. Lu, J. Lv, Y. Wang and Y. Ma, Interface structure prediction via CALYPSO method, Chin. Sci. Bull., 2019, 64(5), 301–309 CAS.
- T. Höltzl, N. Veldeman, J. D. Haeck, T. Veszprémi, P. Lievens and M. T. Nguyen, Growth Mechanism and Chemical Bonding in Scandium-Doped Copper Clusters: Experimental and Theoretical Study in Concert, Chem. - Eur. J., 2009, 15(16), 3970–3982 CrossRef PubMed.
- M. A. Rohrdanz and J. M. Herbert, Simultaneous benchmarking of ground- and excited-state properties with long-range-corrected density functional theory, J. Chem. Phys., 2008, 129(3), 34107 CrossRef PubMed.
- F. Weigend and R. Ahlrichs, Balanced basis sets of split valence, triple zeta valence and quadruple zeta valence quality for H to Rn: Design and assessment of accuracy, Phys. Chem. Chem. Phys., 2005, 7(18), 3297–3305 RSC.
- A. D. Becke and E. R. Johnson, A density-functional model of the dispersion interaction, J. Chem. Phys., 2005, 123(15), 154101 CrossRef PubMed.
- A. D. Becke and E. R. Johnson, Exchange-hole dipole moment and the dispersion interaction, J. Chem. Phys., 2005, 122(15), 154104 CrossRef PubMed.
- K. Raghavachari, G. W. Trucks, J. A. Pople and M. Head-Gordon, A fifth-order perturbation comparison of electron correlation theories, Chem. Phys. Lett., 1989, 157(6), 479–483 CrossRef CAS.
- M. Kállay, P. R. Nagy, D. Mester, Z. Rolik, G. Samu, J. Csontos, J. Csóka, P. B. Szabó, L. Gyevi-Nagy, B. Hégely, I. Ladjánszki, L. Szegedy, B. Ladóczki, K. Petrov, M. Farkas, P. D. Mezei and Á. Ganyecz, The MRCC program system: Accurate quantum chemistry from water to proteins, J. Chem. Phys., 2020, 152(7), 74107 CrossRef PubMed.
- Mrcc, a quantum chemical program suite written by M. Kállay, P. R. Nagy, D. Mester, Z. Rolik, G. Samu, J. Csontos, J. Csóka, P. B. Szabó, L. Gyevi-Nagy, B. Hégely, I. Ladjánszki, L. Szegedy, B. Ladóczki, K. Petrov, M. Farkas, P. D. Mezei, and Á. Ganyecz, 2020. see, www.mrcc.hu Search PubMed.
- T. Yanai, D. P. Tew and N. C. Handy, “A new hybrid exchange–correlation functional using the Coulomb-attenuating method (CAM-B3LYP), Chem. Phys. Lett., 2004, 393(1), 51–57 CrossRef CAS.
- V. N. Staroverov, G. E. Scuseria, J. Tao and J. P. Perdew, Comparative assessment of a new nonempirical density functional: Molecules and hydrogen-bonded complexes, J. Chem. Phys., 2003, 119(23), 12129–12137 CrossRef CAS.
- S. Simon, M. Duran and J. J. Dannenberg, How does basis set superposition error change the potential surfaces for hydrogen-bonded dimers?, J. Chem. Phys., 1996, 105(24), 11024–11031 CrossRef CAS.
- S. Boys and F. Bernardi, The calculation of small molecular interactions by the differences of separate total energies. Some procedures with reduced errors, Mol. Phys., 1970, 19(4), 553–566 CrossRef CAS.
- Y. Shao, Z. Gan, E. Epifanovsky, A. T. B. Gilbert, M. Wormit, J. Kussmann, A. W. Lange, A. Behn, J. Deng, X. Feng, D. Ghosh, M. Goldey, P. R. Horn, L. D. Jacobson, I. Kaliman, R. Z. Khaliullin, T. Kuś, A. Landau, J. Liu, E. I. Proynov, Y. M. Rhee, R. M. Richard, M. A. Rohrdanz, R. P. Steele, E. J. Sundstrom, H. L. Woodcock, P. M. Zimmerman, D. Zuev, B. Albrecht, E. Alguire, B. J. Austin, G. J. O. Beran, Y. A. Bernard, E. Berquist, K. Brandhorst, K. B. Bravaya, S. T. Brown, D. Casanova, C.-M. Chang, Y. Chen, S. H. Chien, K. D. Closser, D. L. Crittenden, M. Diedenhofen, R. A. DiStasio, H. Do, A. D. Dutoi, R. G. Edgar, S. Fatehi, L. Fusti-Molnar, A. Ghysels, A. Golubeva-Zadorozhnaya, J. Gomes, M. W. D. Hanson-Heine, P. H. P. Harbach, A. W. Hauser, E. G. Hohenstein, Z. C. Holden, T.-C. Jagau, H. Ji, B. Kaduk, K. Khistyaev, J. Kim, J. Kim, R. A. King, P. Klunzinger, D. Kosenkov, T. Kowalczyk, C. M. Krauter, K. U. Lao, A. D. Laurent, K. V. Lawler, S. V. Levchenko, C. Y. Lin, F. Liu, E. Livshits, R. C. Lochan, A. Luenser, P. Manohar, S. F. Manzer, S.-P. Mao, N. Mardirossian, A. V. Marenich, S. A. Maurer, N. J. Mayhall, E. Neuscamman, C. M. Oana, R. Olivares-Amaya, D. P. O'Neill, J. A. Parkhill, T. M. Perrine, R. Peverati, A. Prociuk, D. R. Rehn, E. Rosta, N. J. Russ, S. M. Sharada, S. Sharma, D. W. Small, A. Sodt, T. Stein, D. Stück, Y.-C. Su, A. J. W. Thom, T. Tsuchimochi, V. Vanovschi, L. Vogt, O. Vydrov, T. Wang, M. A. Watson, J. Wenzel, A. White, C. F. Williams, J. Yang, S. Yeganeh, S. R. Yost, Z.-Q. You, I. Y. Zhang, X. Zhang, Y. Zhao, B. R. Brooks, G. K. L. Chan, D. M. Chipman, C. J. Cramer, W. A. Goddard, M. S. Gordon, W. J. Hehre, A. Klamt, H. F. Schaefer, M. W. Schmidt, C. D. Sherrill, D. G. Truhlar, A. Warshel, X. Xu, A. Aspuru-Guzik, R. Baer, A. T. Bell, N. A. Besley, J.-D. Chai, A. Dreuw, B. D. Dunietz, T. R. Furlani, S. R. Gwaltney, C.-P. Hsu, Y. Jung, J. Kong, D. S. Lambrecht, W. Liang, C. Ochsenfeld, V. A. Rassolov, L. V. Slipchenko, J. E. Subotnik, T. V. Voorhis, J. M. Herbert, A. I. Krylov, P. M. Gill and M. Head-Gordon, Advances in molecular quantum chemistry contained in the Q-Chem 4 program package, Mol. Phys., 2015, 113(2), 184–215 CrossRef CAS.
- H. Jacobsen, Localized-orbital locator (LOL) profiles of chemical bonding, Can. J. Chem., 2008, 86(7), 695–702 CrossRef CAS.
- H. Schmider and A. Becke, Chemical content of the kinetic energy density, J. Mol. Struct.: THEOCHEM, 2000, 527(1), 51–61 CrossRef CAS.
- R. Z. Khaliullin, E. A. Cobar, R. C. Lochan, A. T. Bell and M. Head-Gordon, Unravelling the origin of intermolecular interactions using absolutely localized molecular orbitals, J. Phys. Chem. A, 2007, 111(36), 8753–8765 CrossRef CAS PubMed.
- R. F. W. Bader, Atoms in molecules : a quantum theory, 1990 Search PubMed.
- T. Lu and F. Chen, Multiwfn: a multifunctional wavefunction analyzer, J. Comput. Chem., 2012, 33(5), 580–592 CrossRef CAS PubMed.
- A. E. Reed and F. Weinhold, “Natural bond orbital analysis of near-Hartree–Fock water dimer, J. Chem. Phys., 1983, 78(6), 4066–4073 CrossRef CAS.
- NBO 6.0 E.D. Glendening, J. K. Badenhoop, A. E. Reed, J. E. Carpenter, J. A. Bohmann, C. M. Morales, C. R. Landis and F. Weinhold, Theoretical Chemistry Institute, University of Wisconsin, Madison, 2013 Search PubMed.
- The PyMOL Molecular Graphics System, Version 1.7 Schrödinger, LLC Search PubMed.
- http://iqmol.org/.
- P. Ferrari, J. Vanbuel, Y. Li, T. Liao, E. Janssens and P. Lievens, The Double-Laser Ablation Source Approach, 2017, pp. 59–78 Search PubMed.
- P. Ferrari, J. Vanbuel, N. M. Tam, M. T. Nguyen, S. Gewinner, W. Schöllkopf, A. Fielicke and E. Janssens, Effects of Charge Transfer on the Adsorption of CO on Small Molybdenum-Doped Platinum Clusters, Chem. - Eur. J., 2017, 23(17), 4120–4127 CrossRef CAS PubMed.
- P. Ferrari, L. M. Molina, V. E. Kaydashev, J. A. Alonso, P. Lievens and E. Janssens, Controlling the Adsorption of Carbon Monoxide on Platinum Clusters by Dopant-Induced Electronic Structure Modification, Angew. Chem., 2016, 55(37), 11059–11063 CrossRef CAS PubMed.
- B. R. Goldsmith, J. Florian, J.-X. Liu, P. Gruene, J. T. Lyon, D. M. Rayner, A. Fielicke, M. Scheffler and L. M. Ghiringhelli, Two-to-three dimensional transition in neutral gold clusters: The crucial role of van der Waals interactions and temperature, Phys. Rev. Mater., 2019, 3(1), 16002 CrossRef CAS.
- S. M. Lang, T. M. Bernhardt, V. Chernyy, J. M. Bakker, R. N. Barnett and U. Landman, Selective C-H Bond Cleavage in Methane by Small Gold Clusters, Angew. Chem., 2017, 56(43), 13406–13410 CrossRef CAS PubMed.
- H. Schwarz and K. R. Asmis, Identification of Active Sites and Structural Characterization of Reactive Ionic Intermediates by Cryogenic Ion Trap Vibrational Spectroscopy, Chem. –Eur. J., 2019, 25(9), 2112–2126 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1ra03262c |
| This journal is © The Royal Society of Chemistry 2021 |