
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMe3SI-promoted chemoselective deacetylation: a general and mild protocol†
Aakanksha Gurawa‡
,
Manoj Kumar‡ and
Sudhir Kashyap *
*
Carbohydrate Chemistry Research Laboratory (CCRL), Department of Chemistry, Malaviya National Institute of Technology Jaipur, (MNIT), Jaipur-302017, India. E-mail: skashyap.chy@mnit.ac.in; skr.kashyap@gmail.com
First published on 27th May 2021
Abstract
A Me3SI-mediated simple and efficient protocol for the chemoselective deprotection of acetyl groups has been developed via employing KMnO4 as an additive. This chemoselective deacetylation is amenable to a wide range of substrates, tolerating diverse and sensitive functional groups in carbohydrates, amino acids, natural products, heterocycles, and general scaffolds. The protocol is attractive because it uses an environmentally benign reagent system to perform quantitative and clean transformations under ambient conditions.
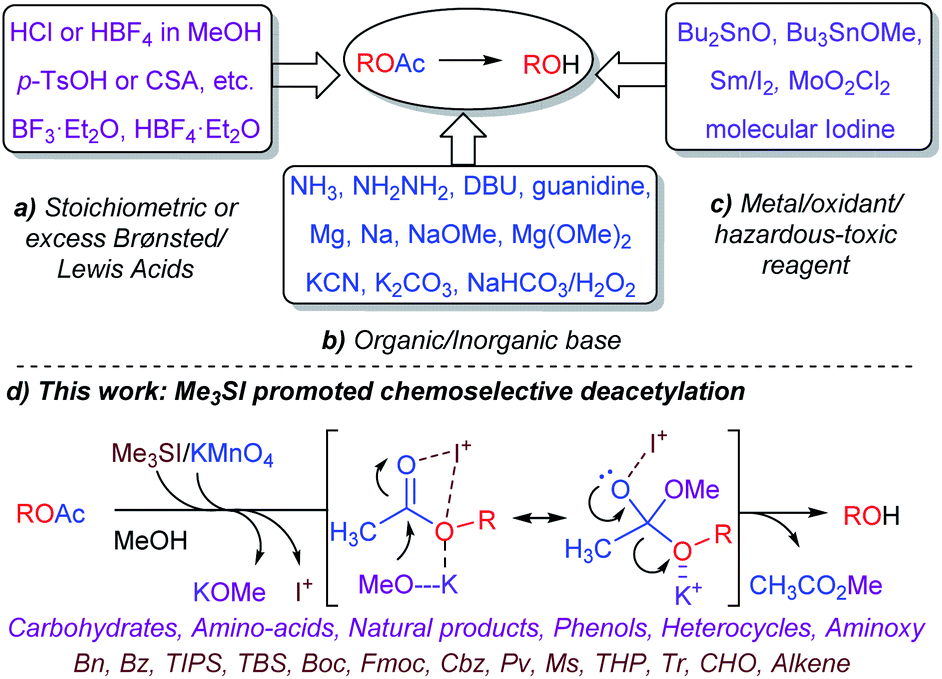
The divergent protection–deprotection approach and extensive functional group manipulation continues to serve as an important chemical tool to access biologically potent molecules and complex natural products.1 In particular, the hydroxyl moieties and their derivatives are ubiquitous in natural products and recognized as renowned scaffolds because of the overwhelming number of chemical and biological applications that they can be used in.2,3 The selective and orthogonal protection–deprotection of free hydroxyl group(s) are significant chemical transformations and frequently employed in target-oriented synthesis (TOS).4 Due to the widespread use and relative ease of protection–deprotection, introduction of an O-Ac group to mask the hydroxyl-moiety has remained as a highly reliable and convenient strategy, especially in synthetic carbohydrate chemistry.1–3 However, the chemoselective deprotection of O-Ac in the presence of analogous and sensitive O-protective groups such as benzoyl (Bz) or pivaloyl (Pv) is a notoriously challenging yet important task.1–3
Thus, considerable effort has been devoted to developing robust and selective methods for the deprotection of the acetate ester. As outlined in Scheme 1, the cleavage of O-Ac is conventionally performed under a homogeneous reagent system including: (a) Brønsted acids/Lewis acids such as HCl/MeOH,5 HBF4·Et2O,6 BF3·Et2O,7 p-TsOH or CSA,8 (b) inorganic/organic basic conditions employing Zemplén hydrolysis (NaOMe/MeOH),1 ammonia solution,9 hydrazine/AcOH/pyridine,10 DBU/PhH,11 guanidine/EtOH/DCM,12 Mg–metal or Mg(OMe)2,13 KCN/EtOH,14 K2CO3/MeOH/H2O,15 NaHCO3/H2O2,16 (c) metallic compounds or oxidants such as MoO2Cl2,17 molecular iodine/MeOH,18a Sm/I2,18b Bu2SnO/heating,19a Bu3SnOMe/DCE.19b Although, the use of heterogeneous catalysts such as CuFe2O4 nanoparticles20 and enzymes21 have also been demonstrated for deacetylation with limited substrate scope. Recently, a tetranuclear zinc cluster, Zn4(OCOCF3)6O has been investigated for use in trans-esterification and deacylation with discriminate selectivity.22
 | ||
| Scheme 1 Previous advances, and the Me3SI-catalyzed chemoselective deacetylation developed in this work. | ||
Despite the synthetic challenges and demands placed on modern chemistry, advancing green chemical syntheses employing milder and environmentally benign reagent systems remains a constant motivation and is actively pursued. In this context and our continued interest23 developing versatile and selective protocols inspired the development of a simple and chemoselective deacetylation method with remarkable and distinctive synthetic applicability. Recently the dual-reactivity of KMnO4 for selective deacetylation and one-pot deacetylation–oxidation of benzyl-O-acetates under controlled reaction conditions was successfully investigated.23a
Emerging from these precedents, the Me3SI-catalyzed, simple and chemoselective removal of O-acetate, an elegant and promising alternative approach for the deprotection of the acetate ester (Scheme 1d) is presented herein. Inspired by recent research,23f an initial experiment employing Me3SI(OAc)2, generated in situ oxidative transfer of the acetyl groups from PhI(OAc)2 to Me3SI, which gave the regioselective 2-iodoglycosylation of enol ether functionality in 3,4,6-tri-O-acetyl-D-glucal (1a) with methanol together with traces of the deacetylated product 1 (Table 1, entry 1). The preliminary results led to the exploration of the possibility of using sulfonium iodate salts in the selective removal of the O-acetyl protection group. As summarized in Table 1, subsequent optimization of the reaction was performed by altering the reagents and solvent systems. Accordingly, the reactions of TOAc-glucal 1a using varying amounts of Me3SI and NaIO4 in the presence of MeOH facilitated the smooth deacetylation providing the desired product 1 with excellent efficiency under ambient conditions (Table 1, entries 2–5). It is worth noting, that the use of Me3SI in combination with NaIO4 in MeOH as the solvent, promoted the deprotection of acetates in a chemoselective manner, however the direct 1,2-addition product, 2-deoxy-glycoside was not detected.23f,g Meanwhile, the reaction using 0.2 equiv. of both Me3SI and NaIO4 was found to suffice for obtaining the complete deacetylation of 1a after 40 min (Table 1, entry 4).
|
|||||
|---|---|---|---|---|---|
| Entry | Catalyst (equiv.) | Additive (equiv.) | Solvent | Time | Yieldb |
| a Reaction conditions: 1a (1.0 equiv.), salt (0.1 to 1.0 equiv.), additive (0.1 to 1.0 equiv.), solvent (1 mL) in open-air at room temperature.b The isolated and unoptimized yields, based on the starting material 1a.c Increasing the amount of both salt and additive up to 1.0 equiv., 24 h. NR = no reaction. | |||||
| 1 | Me3SI (1.0) | PhI(OAc)2 (1.0) | MeOH | 24 h | Trace |
| 2 | Me3SI (1.0) | NaIO4 (1.0) | MeOH | 10 min | 100% |
| 3 | Me3SI (0.4) | NaIO4 (0.4) | MeOH | 30 min | 100% |
| 4 | Me3SI (0.2) | NaIO4 (0.2) | MeOH | 40 min | 100% |
| 5 | Me3SI (0.1) | NaIO4 (0.1) | MeOH | 12 h | 95% |
| 6 | n-Bu4NI (0.1) | NaIO4 (0.1) | MeOH | 12 h | 90% |
| 7 | KI (0.1) | NaIO4 (0.1) | MeOH | 12 h | 50% |
| 8 | NaI (0.1) | NaIO4 (0.1) | MeOH | 16 h | 90% |
| 9c | Me3SOI (0.1) | NaIO4 (0.1) | MeOH | 12 h | NR |
| 10c | Me3SBr (0.1) | NaIO4 (0.1) | MeOH | 12 h | NR |
| 11c | n-Bu4NBr (0.1) | NaIO4 (0.1) | MeOH | 12 h | NR |
| 12c | KBr (0.1) | NaIO4 (0.1) | MeOH | 12 h | NR |
| 13 | Me3SI (0.1) | KMnO4 (0.1) | MeOH | 5 min | 100% |
| 14 | Me3SI (0.1) | KMnO4 (0.1) | MeCN | 24 h | NR |
| 15 | Me3SI (0.1) | KMnO4 (0.1) | Toluene | 24 h | NR |
| 16 | Me3SI (0.1) | KMnO4 (0.1) | THF | 24 h | NR |
| 17 | Me3SI (0.1) | KMnO4 (0.1) | DCM | 24 h | NR |
| 18 | Me3SI (0.1) | K2S2O8 (0.1) | MeOH | 12 h | Trace |
| 19 | Me3SI (0.1) | Oxone (0.1) | MeOH | 12 h | Trace |
| 20 | Me3SI (0.1) | KBrO3 (0.1) | MeOH | 12 h | Trace |
| 21 | Me3SI (0.1) | NaBO3·H2O (0.1) | MeOH | 12 h | 40% |
| 22 | Me3SI (0.1) | Na3BO3 (0.1) | MeOH | 12 h | Trace |
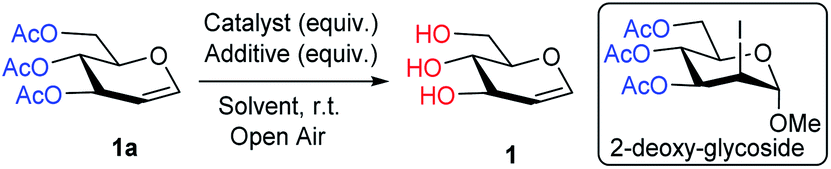
Consequently, TOAc-glucal 1a was subjected to various complementary halide salts employing NaIO4 (0.1 equiv.) in MeOH under ambient reaction conditions (Table 1, entries 6–12). Use of the catalytic n-Bu4NI and KI provided the desired product with good to modest transformation, 90% and 50% yields, respectively (Table 1, entries 6 and 7 vs. 5). It was found that NaI can efficiently promote the deacetylation in improved yields (90%) after 16 h under the present conditions (Table 1, entry 8). In contrast, Me3SOI and bromide salts such as Me3SBr, n-Bu4NBr, and KBr failed to deliver the deacetylated product even after increasing the amount of the salts and additive NaIO4 to 1.0 equiv. and a prolonged reaction time (Table 1, entries 9–12). Remarkably, switching to KMnO4 (0.1 equiv.) as the additive in a Me3SI-catalyzed reaction led to simple deacetylation of 1a with almost quantitative yields (100%), within 5 min (Table 1, entry 13). A rapid screening of common organic solvents including CH3CN, toluene, THF or CH2Cl2 were unsuccessful and did not give any desired transformations (Table 1, entries 14–17). Further optimization employing analogous additives such as K2S2O8, oxone, KBrO3, NaBO3·H2O, and Na3BO3 were found to be too sluggish or resulted in poor conversions (Table 1, entries 18–22).
Having improved and optimized the reagent system, the generality and limitation of Me3SI-catalyzed selective deacetylation protocol was investigated next, and the results are summarized in Scheme 2. Of the particular note, diverse and commonly used O-protecting groups in D-glucal comprising benzoyl (1b), benzyl (1c), methyl (1d), and tert-butyldimethylsilyl ether (1e) were fully tolerated, thus unambiguously establishing the potential synthetic applicability of the representative chemoselective procedure. Subsequently, the selective deacetylation of several per-O-acetylated glycals: D-galactal (2a), D-rhamnal (3a), L-rhamnal (4a), D-xylal (5a), D-arabinal (6a) and L-arabinal (7a), also disaccharide derived glycals bearing 1,4-glycosidic linkages, for example D-lactal (8a) and D-maltal (9a), were performed successfully to obtain the corresponding products 2–9 in excellent yields.
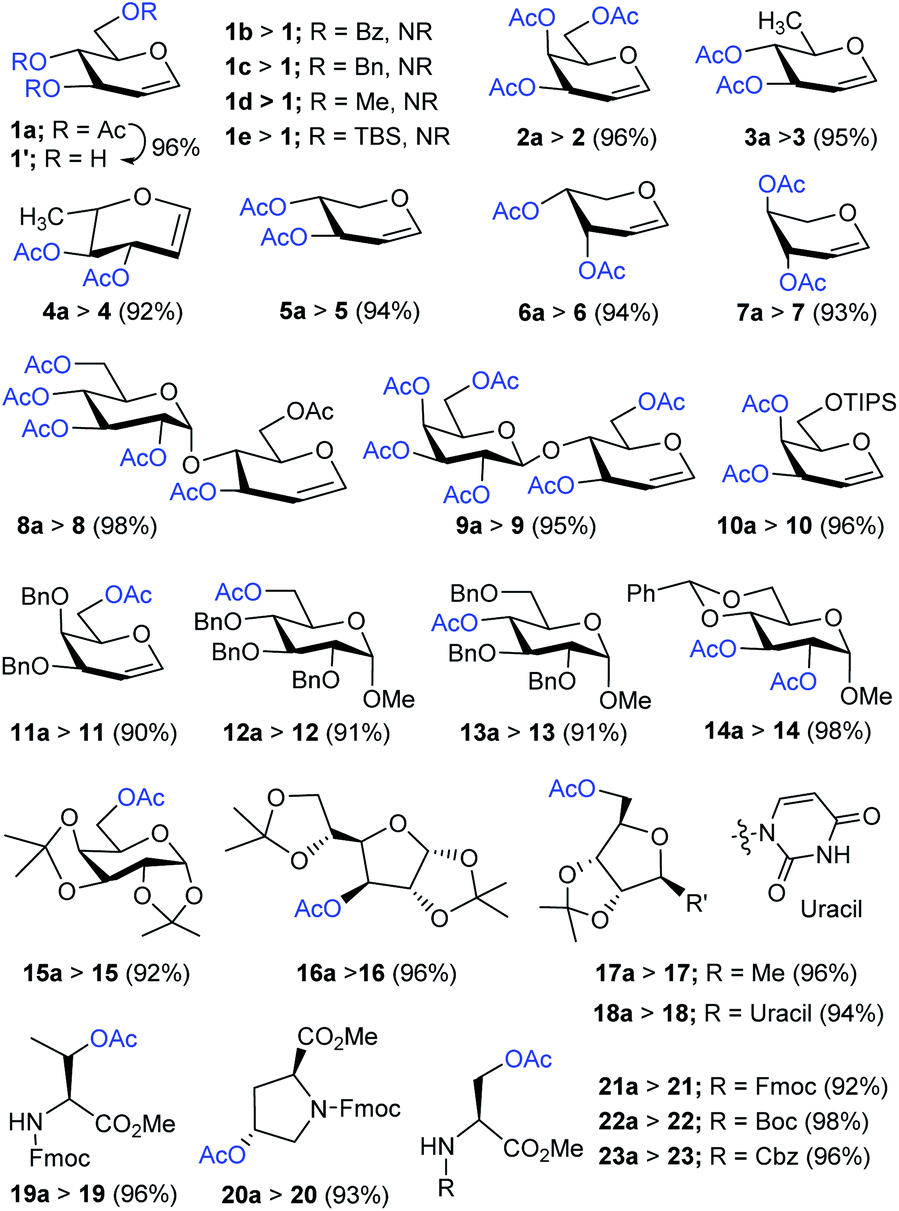 | ||
| Scheme 2 The study of the substrate scope and a functional group compatibility investigation for the chemoselective deacetylation. Reaction conditions: 1a–23a (1.0 equiv.), Me3SI (0.1 equiv.), additive (0.1 equiv.), MeOH (1 mL), 25 °C under atmospheric pressure, 1–12 h. The isolated and unoptimized yields are shown. | ||
Furthermore, the substrates bearing O-TIPS (10a), O-Bn (11a–13a), and 4,6-O-benzylidene (14a) in carbohydrate substrates were uniformly sustained under selective deacetylation conditions giving the hydroxyl products 10–14, respectively, (up to 98% yields). Indeed, the glycosides comprising sensitive O-isopropylidene linkages in D-galactopyranose (15a), D-glucofuranose (16a), D-ribose (17a), and unprotected uridine (18a) were selectively deacetylated to generate the corresponding free-hydroxyl compounds 15–18 in good yields. Importantly, the amino acid derivatives 19a–23a including relatively subtle or labile substituents such as tert-butylcarbamate (Boc), fluorenylmethoxycarbonyl (Fmoc) carbobenzoxy (Cbz), and methyl ester (–CO2Me), underwent efficient chemoselective deacetylation giving the desired products 19–23 in good to excellent yields.
Encouraged by these results, the scope of the selective protocol was extended by performing the systematic deacetylation of substrates involving benzylic, alkylic, allylic, phenolic, naphthyl, hetereocyclic, alicyclic, spirocyclic, natural-products, long chain aliphatic, and aminoxy moieties. As illustrated in Scheme 3, a series of substrates enclosing comparatively susceptible and electronically diverse O-protecting groups (O-Ac; 24a vs. O-Piv; 24b, O-Boc; 24c, O-Ms; 24d, O-THP; 24e, O-Tr; 24f) were well protected under present conditions, further confirming the effectiveness of the chemoselective method. The Me3SI-catalyzed simple removal of acetyl groups, in menthyl (25a), geranyl (26a), cholesteryl (27a), and diversely substituted aryl substrates 28a–35a, was conducted to efficiently obtain the desired products 25–35 in good-to-excellent yields (90–99%). It is worth noting, that the selective deacetylation for a substrate consisting of two electronically different acetates was accomplished successfully (cleavage of O-acetate vs. N-acetate in 29a).
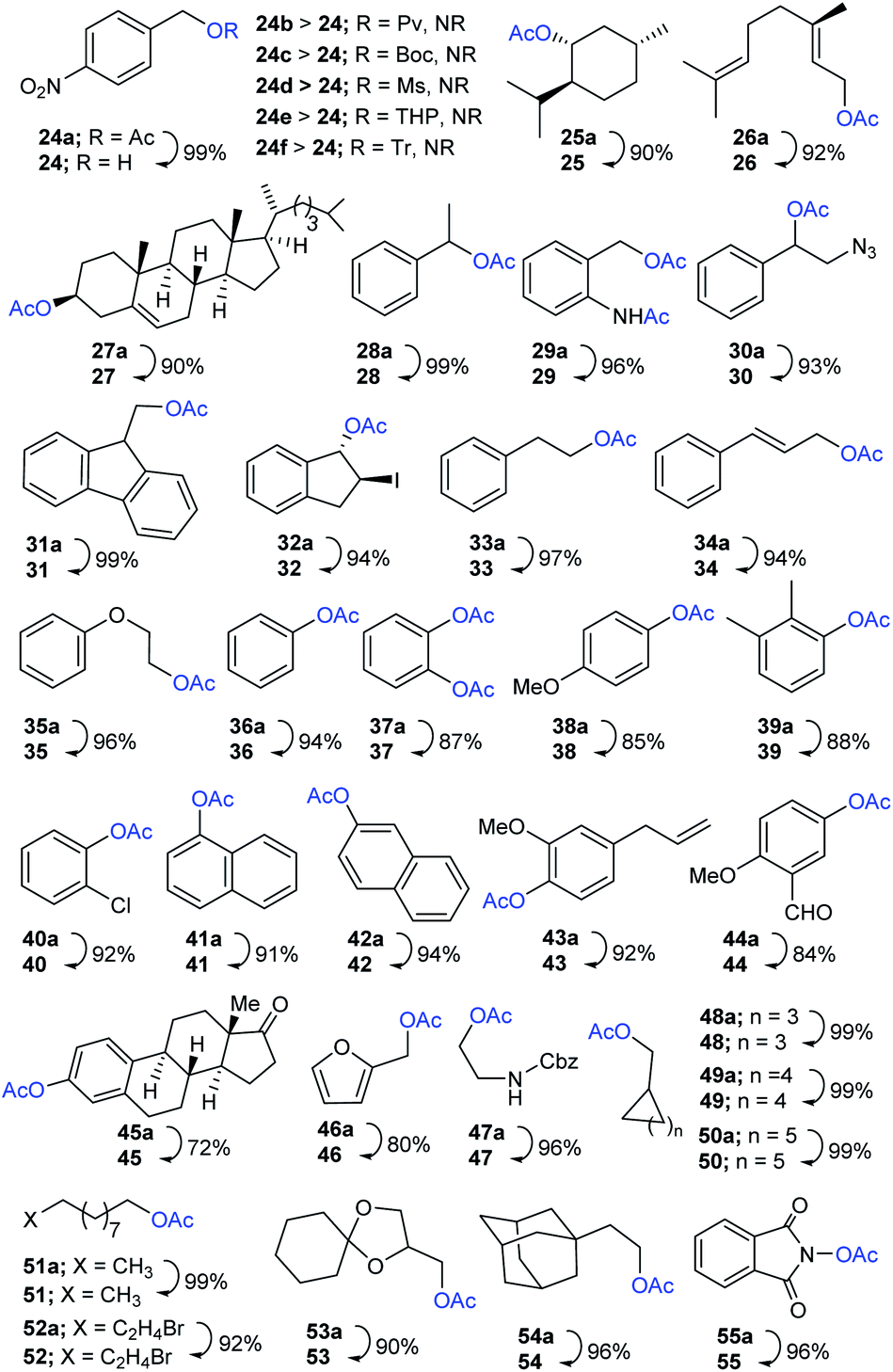 | ||
| Scheme 3 Extended scope of the Me3SI-catalyzed selective deacetylation. Reaction conditions: 24a–55a (1.0 equiv.), Me3SI (0.1 equiv.), additive (0.1 equiv.), MeOH (1 mL), 25 °C under atmospheric pressure, 1–12 h. The isolated and unoptimized yields are shown. | ||
Next, the diversely substituted phenolic or naphthylic O-acetates 36a–42a, eugenyl 43a, vanillyl 44a, and estrone acetate 45a were subjected, under ambient conditions, to access the corresponding free-phenols 36–45 in acceptable yields (72–96%). Likewise, the reaction employing heterocyclic furfuryl acetate (46a), 2-amino-(Cbz)-ethyl acetate (47a), and alicyclic substrates with different ring sizes (five, six, or seven) 48a–50a proceeded smoothly providing the corresponding products 46–50 (80–99% yields). Additionally, long chain aliphatic acetates 51a and 52a, a spirocycle bearing sensitive dioxa moiety 53a, sterically hindered adamantyl 54a, and aminoxy-O-acetate 55a were competent, when used in the applied protocol, of producing their corresponding hydroxyls 51–55 in satisfactory yields (90–99%).
To gain further insight into the Me3SI-promoted deacetylation, the reaction of TOAc-glucal 1a was performed with equimolar amounts of trimethylsulfonium iodide and an additive (0.1 equiv. each) employing deuterated methanol (CD3OD) as the solvent (Fig. 1). The 1H-NMR analyses of the crude reaction mixture revealed the presence of a distinctive resonance which was due to an anomeric proton (δ 6.42 dd, J1-2 = 6.10, 1.4 Hz, H-1) and other characteristic chemical shifts which conformed with that of the desired product 1′ (tri-O-2H-D-glucal).
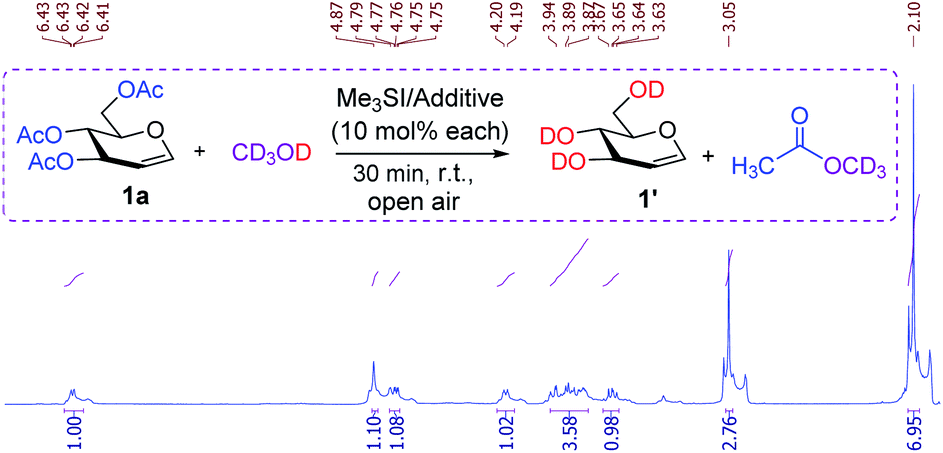 | ||
| Fig. 1 Experiments with deuterated methanol and 1H-NMR analyses, used to provide mechanistic rationalization for Me3SI-promoted deacetylation. | ||
With no variance, the acetyl protons of methyl acetate (CH3CO-OCD3) were found at δ 2.10 (s, 9H) which potentially resulted from the methanolysis of the acetate groups with CD3OD. In addition, substantial amounts of trimethylsulfonium hydroxide species were observed consistently at δ 3.05 as a singlet (vs. Me3SI at δ 1.56),24 and this established the role of trimethylsulfonium iodide in selective and simple cleavage of acetates. A set of control experiments were performed to identify the possible intermediate iodine species in this transformation. The studies revealed the absence of molecular iodine in the present reaction conditions as confirmed by a negative starch solution test (the color did not change to an intense blue color).24 Indeed, the standard Me3SI-mediated deacetylation reaction employing a common radical scavenger such as TEMPO (a stable aminooxy radical) or 2,6-di-tert-butyl-4-methylphenol (BHT) preceded smoothly, and further ruled out a radical pathway in the process. Although, attempting a reaction of TOAc-glucal 1a using a catalytic amount of iodine in methanol solvent was inconsistent (50–60% conversion) resulting in the mono-deacetylation of the primary acetate (6-OAc) even after a prolonged reaction time (48 h).18a It is worth noting that the characteristic Lewis acidity of molecular iodine (I2) has been exploited in the selective O-Ac as well N-Boc protection of diverse substrates.25
Based on the previously mentioned experimental observations and with reference to precedents in the literature, a plausible mechanistic rationalization for a Me3SI-catalyzed reaction is postulated in Scheme 4. Initially, KMnO4 showed rapid disproportionation, resulting in permanganic acid (HMnO4 ↔ H+ + MnO4−) and potassium methoxide (KOMe ↔ K+ + MeO−) ions in the presence of methanol, which was attributed to the reducing behavior of methanol in the present system.26 The main oxidizing reagent Mn(VII)O4− was capable of inverting the polarity of the reactivity of the halide salt Me3SI in slightly acidic conditions which resulted in the iodate (I+) species [A], and was disproportionate to Mn(IV)O2 and H2O. The reduced Mn(IV) species would then readily oxidize back to the higher Mn(VI)O4− in a slightly basic medium (KOMe/H2O) under the normal atmosphere26c and this was likely to facilitate the release of trimethylsulfonium hydroxide. Meanwhile, the incipient electrophilic intermediate [A] initiated from the oxidation of Me3SI coordinated with the oxygen atoms of the carbonyl functionality (CH3CO2–) in the acetate compound [1a] to generate a transient species [B].25b,25c,27 This significantly induced the polarization of the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O double bond and increased the electrophilicity of the carbonyl carbon as well contributing towards the bond lengthening of the carbonyl CO moiety in [B].
O double bond and increased the electrophilicity of the carbonyl carbon as well contributing towards the bond lengthening of the carbonyl CO moiety in [B].
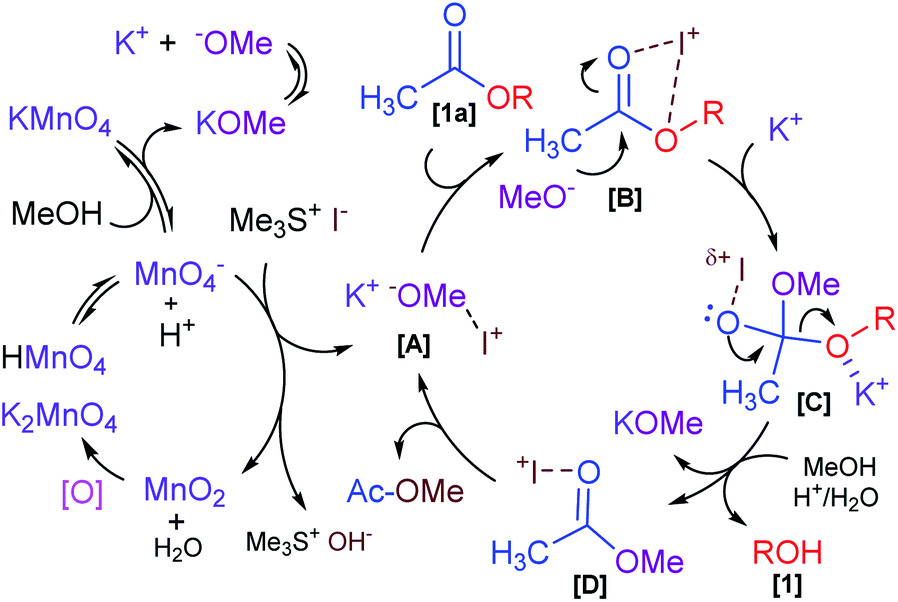 | ||
| Scheme 4 A plausible mechanism for Me3SI-promoted selective deacetylation. | ||
Subsequent coordination of the K+ ion with alkyl-oxygen (RO−) and attack of methoxide (MeO−) as the nucleophile on the electrophilic carbon in [B], would then eventually produce the tetrahedral intermediate [C]. Following the elimination of the penultimate transient species [D] and simultaneously trapping of the respective alkoxide ion (RO−) in a hydrolytic environment would unambiguously lead to the formation of the respective alkanol (ROH) [1] with the liberation of potassium methoxide. The proposed intermediate [D] finally displaced the corresponding methyl acetate (CH3CO2Me) by releasing the required iodate species [A] which presumably participated in another catalytic cycle en route to the selective deacetylation process.
In summary, a catalytic and practical procedure for chemoselective deacetylation with a general substrate scope employing environmentally benign reagents under ambient reaction conditions is reported. It is worth noting that the catalytic protocol is broadly applicable to numerous substrates, including carbohydrates, amino acids, and natural products, tolerating orthogonal and sensitive groups (esters, ethers, silyl ethers, carbamates, carboxybenzyl, mesyl, 2-tetrahydropyranyl, trityl, aldehydes, and also alkenes). Furthermore, the method is advantageous as it involves a safe and convenient reaction, ensuring smooth and quantitative conversion as well as preventing trans-esterification.
Experimental
Representative procedure for chemoselective deacetylation
To a previously prepared solution of acetate substrate (50 mg, 1.0 equiv.) in MeOH (1 mL) was added Me3SI (0.1 equiv.) and KMnO4 (0.1 equiv.). The mixture was stirred at room temperature in an open-air environment and the reaction progress was monitored by TLC. After the complete consumption of the starting material had occurred, typically 5 min to 12 h, the reaction mixture was filtered and washed with EtOAc (10 mL). The filtrate was treated with saturated aqueous NaHCO3 (5 mL), and the aqueous layer was extracted with EtOAc (3 × 30 mL). The combined organic layers were then washed with water and brine, dried over anhydrous Na2SO4, and concentrated in vacuo to obtain the analytically pure products.Conflicts of interest
There are no conflicts to declare.Acknowledgements
SK gratefully acknowledges the Department of Science and Technology, India for the Early Career Research Award and Research Grant for Young Scientists (SERB-ECR/2017/001477). SK thanks TEQIP-III for their generous support and MRC MNIT (Advanced Analytical and Characterization Centre) for providing the NMR spectroscopic and analytical data. The authors are also grateful to the Director of MNIT for providing the necessary infrastructure for this research. AG acknowledges a UGC Fellowship.Notes and references
- (a) T. W. Greene and P. G. M. Wuts, Protective Groups in Organic Synthesis, Wiley, New York, 4th edn, 2006 Search PubMed; (b) P. J. Kocienski, Protecting Groups, Thieme, New York, 1994 Search PubMed.
- (a) K.-T. Huang, B.-C. Wu, C.-C. Lin, S.-C. Luo, C. Chen, C.-H. Wong and C.-C. Lin, Carbohydr. Res., 2006, 341, 2151–2155 CrossRef CAS PubMed; (b) J. B. Schwarz, S. D. Kuduk, X.-T. Chen, D. Sames, P. W. Glunz and S. J. Danishefsky, J. Am. Chem. Soc., 1999, 121, 2662–2673 CrossRef CAS; (c) X. Mao, P. Li, T. Li, M. Zhao, C. Chen, J. Liu, Z. Wang and L. Yu, Chin. Chem. Lett., 2020, 31, 3276–3278 CrossRef CAS; (d) X. Xiao, Z. Shao and L. Yu, Chin. Chem. Lett., 2021 DOI:10.1016/j.cclet.2021.03.047.
- (a) J. Žemlička, J. Beránek and J. Smrt, Collect. Czech. Chem. Commun., 1962, 27, 2784–2795 CrossRef; (b) C. B. Reese and J. C. M. Stewart, Tetrahedron Lett., 1968, 9, 4273–4276 CrossRef; (c) V. Schwarz, Collect. Czech. Chem. Commun., 1962, 27, 2567–2574 CrossRef CAS; (d) T. B. Winholdz and D. B. R. Johnston, Tetrahedron Lett., 1967, 8, 2555–2557 CrossRef; (e) B. Srinivas, T. R. Reddy and S. Kashyap, Carbohydr. Res., 2015, 406, 86–92 CrossRef CAS PubMed; (f) T. R. Reddy, S. K. Battina and S. Kashyap, J. Carbohydr. Chem., 2015, 34, 133–144 CrossRef CAS.
- (a) J. K. Baskin, Chem. Rev., 2000, 100, 4265–4266 CrossRef PubMed; (b) R. W. Dugger, J. A. Ragan and D. H. B. Ripin, Org. Process Res. Dev., 2005, 9, 253–258 CrossRef CAS; (c) J. S. Carey, D. Laffan, C. Thomson and M. T. Williams, Org. Biomol. Chem., 2006, 4, 2337–2347 RSC.
- (a) V. Pozsgay, J. Am. Chem. Soc., 1995, 117, 6673–6681 CrossRef CAS; (b) N. Yamamoto, T. Nishikawa and M. Isobe, Synlett, 1995, 505–506 CrossRef CAS; (c) E. J. Corey, D. A. Clark, G. Goto, A. Marfat, C. Mioskowski, B. Samuelsson and C. Hammarström, J. Am. Chem. Soc., 1980, 102, 1436–1439 CrossRef CAS; (d) N. E. Byramova, M. V. Ovchinnikov, L. Backinowsky and N. K. Kochetkov, Carbohydr. Res., 1983, 124, C8–C11 CrossRef CAS.
- V. Pozsgay and B. Coxon, Carbohydr. Res., 1993, 257, 189–215 CrossRef.
- D. Askin, C. Angst and S. Danishefsky, J. Org. Chem., 1987, 52, 622–635 CrossRef CAS.
- A. G. González, I. Brouard, F. León, J. I. Padrón and B. Jaime, Tetrahedron Lett., 2001, 42, 3187–3188 CrossRef.
- T. Neilson and E. S. Werstiuk, Can. J. Chem., 1971, 49, 493–499 CrossRef CAS.
- Y. Ishido, N. Nakazaki and N. Sakairi, J. Chem. Soc., Perkin Trans. 1, 1979, 2088–2098 RSC.
- L. H. B. Baptistella, J. F. dos Santos, K. C. Ballabio and A. J. Marsaioli, Synthesis, 1989, 436–439 CrossRef CAS.
- (a) N. Kunesch, C. Meit and J. Poisson, Tetrahedron Lett., 1987, 28, 3569–3572 CrossRef CAS; (b) U. Ellervik and G. Magnusson, Tetrahedron Lett., 1997, 38, 1627–1628 CrossRef CAS.
- (a) Y.-C. Xu, A. Bizuneh and C. Walker, J. Org. Chem., 1996, 61, 9086–9089 CrossRef CAS; (b) Y.-C. Xu, E. Lebeau and C. Walker, Tetrahedron Lett., 1994, 35, 6207–6210 CrossRef; (c) Y.-C. Xu, A. Bizuneh and C. Walker, Tetrahedron Lett., 1996, 37, 455–458 CrossRef CAS.
- J. Herzig, A. Nudelman, H. E. Gottlieb and B. Fischer, J. Org. Chem., 1986, 51, 727–730 CrossRef CAS.
- (a) N. C. Barua and R. R. Schmidt, Synthesis, 1986, 11, 891–893 CrossRef; (b) J. J. Plattner, R. D. Gless and H. Rapoport, J. Am. Chem. Soc., 1972, 94, 8613–8615 CrossRef CAS PubMed.
- Q. Y. Zheng, L. G. Darbie, X. Cheng and C. K. Murray, Tetrahedron Lett., 1995, 36, 2001 CrossRef CAS.
- C.-Y. Liu, H.-L. Chen, C.-M. Ko and C.-T. Chen, Tetrahedron, 2011, 67, 872–876 CrossRef CAS.
- (a) B. Ren, L. Cai, L.-R. Zhang, Z.-J. Yang and L.-H. Zhang, Tetrahedron Lett., 2005, 46, 8083–8086 CrossRef CAS; (b) R. Yanada, N. Negoro, K. Bessho and K. Yanada, Synlett, 1995, 1261–1263 CrossRef CAS.
- (a) M. G. Perez and M. S. Maier, Tetrahedron Lett., 1995, 36, 3311–3314 CrossRef CAS; (b) A. Nudelman, J. Herzig, H. E. Gottlieb, E. Keinan and J. Sterling, Carbohydr. Res., 1987, 162, 145–152 CrossRef CAS.
- J. E. Tasca, A. Ponzinibbio, G. Diaz, R. D. Bravo, A. Lavat and M. G. Gonzalez, Top. Catal., 2010, 53, 1087–1090 CrossRef CAS.
- (a) K. Naemura, N. Takahashi and H. Chikamatsu, Chem. Lett., 1988, 30, 1717–1720 CrossRef; (b) C. R. Johnson and C. H. Senanayake, J. Org. Chem., 1989, 54, 735–736 CrossRef CAS; (c) O. Houille, T. Schmittberger and D. Uguen, Tetrahedron Lett., 1996, 37, 625–628 CrossRef CAS; (d) R. López, E. Montero, F. Sánchez, J. Cañada and A. Fernández-Mayoralas, J. Org. Chem., 1994, 59, 7027–7032 CrossRef; (e) E. W. Holla, V. Sinnwell and W. Klaffke, Synlett, 1992, 413–414 CrossRef CAS; (f) T. Itoh, A. Uzu, N. Kanda and Y. Takagi, Tetrahedron Lett., 1996, 37, 91–92 CrossRef CAS; (g) K. Takabe, N. Mase, T. Hisano and H. Yoda, Tetrahedron Lett., 2003, 44, 3267–3269 CrossRef CAS; (h) T. Hisano, K. Onodera, Y. Toyabe, N. Mase, H. Yoda and K. Takabe, Tetrahedron Lett., 2005, 46, 6293–6295 CrossRef CAS.
- T. W. Lin, A. K. Adak, H. J. Lin, A. Das, W. C. Hsiao, T. C. Kuan and C. C. Lin, RSC Adv., 2016, 6, 58749–58754 RSC.
- (a) A. Gurawa, M. Kumar, D. S. Rao and S. Kashyap, New J. Chem., 2020, 44, 16702–16707 RSC; (b) D. S. Rao, T. R. Reddy, A. Gurawa, M. Kumar and S. Kashyap, Org. Lett., 2019, 21, 9990–9994 CrossRef CAS PubMed; (c) T. R. Reddy, D. S. Rao and S. Kashyap, Chem. Commun., 2019, 55, 2833–2836 RSC; (d) D. S. Rao, T. R. Reddy and S. Kashyap, Org. Biomol. Chem., 2018, 16, 1508–1518 RSC; (e) D. S. Rao, T. R. Reddy, K. Babachary and S. Kashyap, Org. Biomol. Chem., 2016, 14, 7529–7543 RSC; (f) T. R. Reddy, D. S. Rao, K. Babachary and S. Kashyap, Eur. J. Org. Chem., 2016, 291–311 CrossRef CAS; (g) G. Kundoor, D. S. Rao and S. Kashyap, Asian J. Org. Chem., 2016, 5, 264–270 CrossRef CAS; (h) S. Chittela, T. R. Reddy, P. R. Krishna and S. Kashyap, J. Org. Chem., 2015, 80, 7108–7116 CrossRef CAS PubMed; (i) T. R. Reddy, D. S. Rao and S. Kashyap, RSC Adv., 2015, 5, 28338–28343 RSC; (j) S. Chittela, T. R. Reddy, P. R. Krishna and S. Kashyap, RSC Adv., 2014, 4, 46327–46331 RSC.
- See the ESI.†.
- (a) G. Basumatary and G. Bez, Tetrahedron Lett., 2017, 58, 4312–4315 CrossRef CAS; (b) R. Varala, S. Nuvula and S. R. Adapa, J. Org. Chem., 2006, 71, 8283–8286 CrossRef CAS PubMed; (c) K. P. R. Kartha and R. A. Field, Tetrahedron, 1997, 53, 11753–11766 CrossRef CAS.
- (a) P. K. Sen, P. R. Samaddar and K. Das, Transition Met. Chem., 2005, 30, 261–267 CrossRef CAS; (b) S. Dash, S. Patel and B. K. Mishra, Tetrahedron, 2009, 65, 707–739 CrossRef CAS , and references cited therein; ; (c) N. Singh and D. G. Lee, Org. Process Res. Dev., 2000, 5, 599–603 CrossRef.
- The electrophilic iodine (I+) species generated in the proposed pathway likely to coordinate with oxygen atoms of carbonyl group under basic conditions. See: (a) W. A. Szarek, A. Zamojski, K. N. Tiwari and E. R. Ison, Tetrahedron Lett., 1986, 27, 3827–3830 CrossRef CAS; (b) J. L. Wahlstrom and R. C. Ronald, J. Org. Chem., 1998, 63, 6021–6022 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: General procedures, characterization data, and spectral charts. See DOI: 10.1039/d1ra03209g |
| ‡ Aakanksha Gurawa and Manoj Kumar contributed equally. |
| This journal is © The Royal Society of Chemistry 2021 |