DOI:
10.1039/D1RA03197J
(Paper)
RSC Adv., 2021,
11, 30078-30087
Catalytic hydrogenolysis of larix bark proanthocyanidins in ionic liquids produces UV blockers with potential for use in cosmetics
Received
30th April 2021
, Accepted 20th August 2021
First published on 9th September 2021
Abstract
The bark of larix, a major tree species in the coniferous forests of China's Greater Khingan Mountains, is typically treated as waste. The bark is, however, rich in flavonoids, known as proanthocyanidins, although their high degree of polymerization and high molecular weight reduce their biological activity and potential applications. Ionic liquids, a new type of “green solvent”, characterized by low vapor pressure and good stability, have been developed and used as new solvents for naturally occurring macromolecules. Here, we used 1-butyl-3-methylimidazole chloride ([BMIM]Cl) as the ionic solvent to reduce the degree of polymerization of larix bark proanthocyanidins by Pd/C-catalyzed hydrogenolysis. The optimal reaction conditions, determined using an orthogonal experimental design, were: reaction temperature, 90 °C; reaction time, 1.5 h; catalyst loading, 4 g L−1 (Pd/C: [BMIM]Cl); and hydrogen pressure, 2.5 MPa. Characterization of the reaction products by UV-Vis and IR spectroscopy and gel permeation chromatographys showed that they retained the proanthocyanidin structure. We showed that whilst both the native and depolymerized proanthocyanidins were able to block UV light when added to commercially available skin creams and sunscreens, the depolymerized proanthocyanidins were more effective at a given concentration. This study expands the applications of a new “green” ionic liquid solvent, provides a technical foundation for the low-cost depolymerization of larix bark proanthocyanidins, and also explores a potential high-value use for waste larix bark as the source of a UV-blocking additive for cosmetics.
1 Introduction
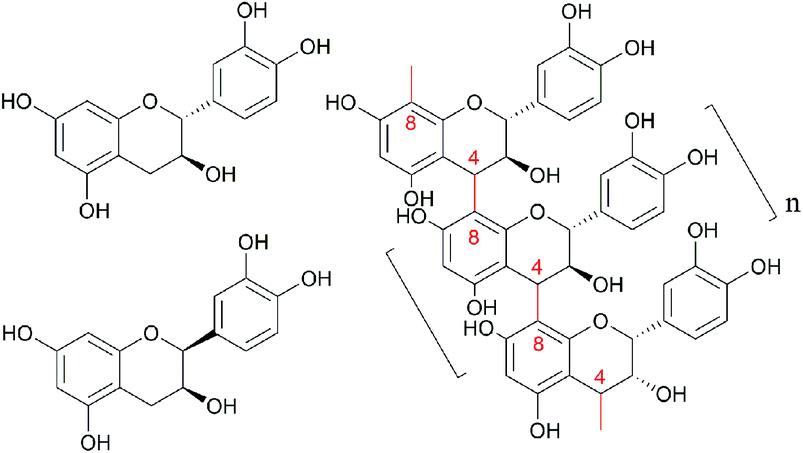
Larix, a long-lived, fast-growing tree, is the main species in the coniferous forests of China's Greater Khingan Mountains and is also an important species for forest regeneration and afforestation. The harvesting and processing of larix produces a large amount of residues, such as bark and wood powder, which are typically viewed as a low value resource and used as fuel. Preparation of highly active polyphenols from the by-products of the pulping industry is a green strategy to realize the high value utilization of agricultural and forestry wastes.1 These phenolic structures, such as lignin, have broad application prospects in the field of materials, and also inspire our interest in the research of bioactive polyphenols.1–3 Larix bark is, however, rich in proanthocyanidins, which account for 10–16% of the bark mass. Proanthocyanidins are formed by polymerization of catechin and epicatechin subunits via bonds between C4 in the C ring and C8 or C6 in the A ring (C4–C8 or C4–C6 bonds, Fig. 1). The average degree of polymerization (DP) of proanthocyanidins in larix bark is 9–10, and the molecular weight is ∼2800.4,5 Proanthocyanidins, a type of flavonoid comprising “flavan-3-alcohol” monomers, are the main component of larix bark tannin extracts, which have been widely used in the leather tanning industry.6,7 Although the average DP of proanthocyanidins in larix bark is 9–10, lower molecular weight polymers are also present and larix proanthocyanidins can be further classified as larix polymeric proanthocyanidins (LPPCs, DP ≥5) or larix oligomeric proanthocyanidins (LOPCs, DP <5). Because of their large molecular weight, LPPCs cannot easily penetrate biofilms, resulting in low biological activity and, consequently, low value. LOPCs, on the other hand, have strong biological activity. As well as showing strong anti-oxidant properties,8,9 LOPCs have been reported to protect retinal cells.10 suppress tumor growth,11 combat the damage done by inflammation,12 and protect against cardiovascular and cerebrovascular diseases.13 Because of their aromatic structure, proanthocyanidins strongly absorb UV light and have a good prospects for use in sunscreens.14 One study15 showed that the sun protection factor (SPF) of high-concentration proanthocyanidins from red raspberry seeds was much higher than that of commercially available sunscreens, confirming the potential of proanthocyanidins as natural ingredients for highly UV-blocking sunscreens.
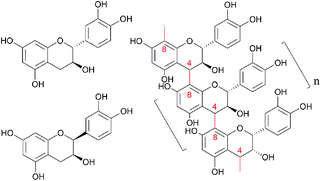 |
| | Fig. 1 The molecular structure scheme of catechin, epicatechin and proanthocyanidins LPPCs, n ≥5; LOPC n <5. | |
At present, the applications of cellulose and lignin in various fields have been reviewed,2,3,16–18 but due to the influence of polymerization degree on the activity, the application of proanthocyanidins is relatively less. So far, depolymerization of low-value LPPCs to produce high-value LOPCs and investigation of their UV-blocking properties thus has important practical significance for improving the range of application of proanthocyanidins. Depolymerization of LPPCs is typically achieved using chemical degradation,19 biodegradation,20 oxidation21,22 or hydrogenolysis.23 but all of these methods have drawbacks. Chemical degradation requires the use of acids, alkalis and other chemical reagents, which pose a potential risk to the environment. In biodegradation, which uses microbial or biological enzymes, the reaction conditions are strict, the reaction takes a long time, and the degree of depolymerization is uncertain. Oxidation typically uses light, electricity and heat to depolymerize proanthocyanidins into smaller molecules and generates CO2 and H2O. There are also many by-products, which makes it difficult to obtain pure LOPCs. Hydrogenolysis is commonly used to break C–C bonds in biological macromolecules and is one of the most important strategies for directional cleavage of C4–C8 (or C4–C6) bonds in proanthocyanidins. Under the right conditions, hydrogenolysis does not destroy the structure of polyphenols and selectively produces LOPCs with higher purity and better activity than other methods. Du et al.23 studied the catalytic hydrogenolysis of LPPCs in ethanol, using Pd/C as the catalyst, and Jiang et al.24 found that hydrogenolysis of LPPCs can be catalyzed by HP-2MGL resin.
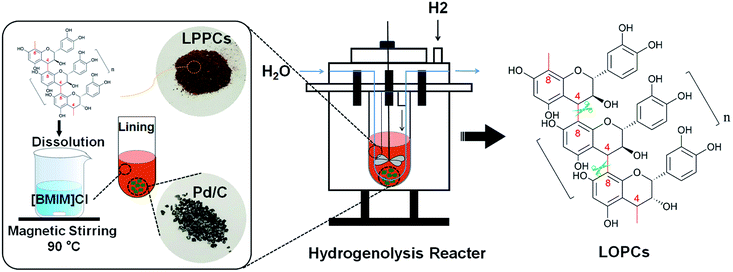
So far, most catalytic hydrogenolysis reactions have been carried out in organic solvents. This limits the reaction temperature because organic solvents have a high vapor pressure and evaporate above a certain temperature. The environmental pollution caused by volatilization of organic matter is also a concern. In terms of resource recycling, environmental protection and realization of “green chemistry”,25 it is, therefore, important to identify non-volatile, stable, green solvents in which to carry out hydrogenolysis of LPPCs. Ionic liquids are salts that are liquids at temperatures below 100 °C and are usually composed of a combination of organic cations and organic/inorganic anions.26 Compared with organic solvents, ionic liquids, which are frequently referred to as “green” solvents,27 have excellent thermodynamic stability and low vapor pressures, with almost no volatilization. Ionic liquids are excellent solvents and reaction mediums for natural macromolecules. If the economic problem of solvent recovery is solved, the recovery of ionic liquid can have a positive impact on the environment. At present, ionic liquids are widely used as solvents for natural lignocellulose.28 Yang et al.29 prepared high-strength cellulose from corn straw using 1-butyl-3-methylimidazole chloride ([BMIM]Cl) as the solvent. [BMIM]Cl is a relatively common ionic liquid, which has been shown to be good solvent for the extraction of proanthocyanidins.30 Using [BMIM]Cl as the solvent for catalytic hydrogenolysis of LPPCs will provide a more thermodynamically stable environment for the reaction and will effectively achieve “zero emissions”, thus reducing environmental pollution. It will also expand the range of applications of ionic liquids as solvents for reactions involving natural macromolecules. In this study, LPPCs (DP = 13.19) and Larix bark natural oligomeric proanthocyanidins (NOPCs, DP = 4.65) were extracted from larix bark by solvent extraction. Then, the hydrogenolysis reaction was carried out under the catalyst we synthesized (Fig. 2). The method for synthesizing the catalyst is simple and green, and formaldehyde can gently reduce palladium to a reduced state, which is beneficial to catalyze the hydrogenolysis reaction. An orthogonal experimental design was used to determine the effects of reaction temperature, hydrogen pressure, reaction time and catalyst loading on the yield of hydrogenolysis products and to optimize the process conditions. The reaction products were characterized by UV-Vis and FTIR spectroscopy and gels permeation chromatography (GPC). The effects of adding LPPCs and LOPCs to skin creams or sunscreens was then evaluated, in terms of UV transmission, stability to UV light and sensory evaluation. This work not only broadens the scope of applications of “green” ionic liquids, but also provides a technical basis for the low-cost depolymerization, development and utilization of proanthocyanidins from larix barks.
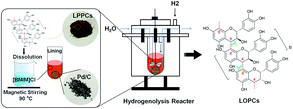 |
| | Fig. 2 Schematic diagram of hydrogenolysis of LPPCs in ionic liquid. | |
2 Results and discussion
2.1 Determination of standard curves
Larix bark proanthocyanidins are macromolecular polymers comprising catechin (or epicatechin) monomers. The standard curves of Abs vs. catechin mass concentration and Abs vs. catechin molar concentration were, therefore, drawn using the Vanillin-HCl assay,31 with catechin as the standard. The regression equation of Abs and M was Abs = 4.83558 × 10−4M + 0.00676, with a correlation coefficient R2 = 0.999 by linear fitting of the obtained data. In a similar way, the linear regression equation of Abs and C was Abs = 6.45827 × C + 0.01176, with R2 = 0.999. Using this method, we determined the DP of LPPCs and NOPCs to be 13.19 and 4.65 respectively. In a previous report,5 the average DP of proanthocyanidins in larix bark was usually between 9 and 10. The difference is mainly due to removal of NOPCs in the process of extracting the LPPCs.
2.2 Properties of Pd/C catalyst
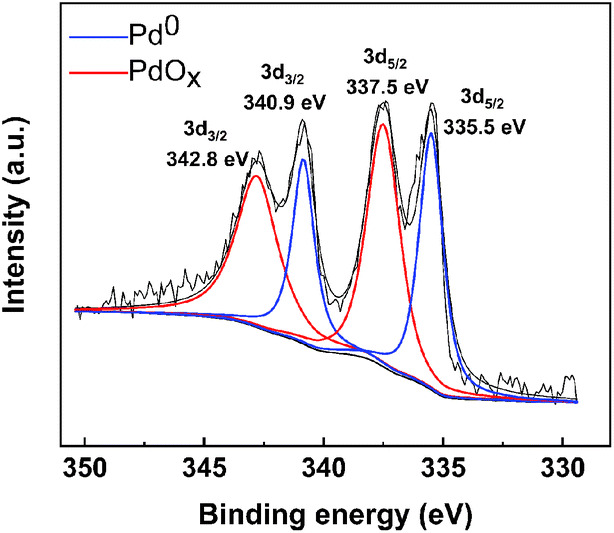
2.2.1 Palladium valence. XPS is an advanced analytical technique used in the microscopic analysis of electronic materials and components. The principle is to use X-rays to radiate the sample and excite the valence electrons of the surface elements of the sample. By plotting the binding energy of photoelectrons as the x-coordinate and the relative intensity (pulse per s) as the y-coordinate, drawing images can be used to characterize the elemental composition and valence state of the sample surface.XPS analysis and characterization of the Pd/C catalyst can provide information about the elements on the surface of the catalyst and about the valence of the Pd. In Fig. 3, the binding energies of 335.5 eV and 340.9 eV correspond to Pd 3d5/2 and Pd 3d3/2, respectively, of the reduced state, Pd0. The binding energies of Pd0 is consistent with the reports in the literature.32 The difference is, the binding energies of 337.5 eV and 342.8 eV correspond to Pd 3d5/2 and Pd 3d3/2, respectively, of the oxidized state, PdOx. Since the catalyst prepared by this method has not been calcined, PdCx will not be produced. On the one hand, PdOx is due to the fact that Pd is not completely reduced in the preparation process; on the other hand, it is due to the fact that Pd is exposed to the air for too long after reduction. The XPS diagram reveals the presence of two valence states of elemental palladium, Pd(0) and Pd(2), on the surface of the activated carbon. Because of the excellent ability of metallic palladium to adsorb hydrogen,33 we speculate that Pd(0) plays a major role in the catalytic hydrogenolysis of proanthocyanidins. Therefore, the synthesis method of this catalyst is feasible and gentle.
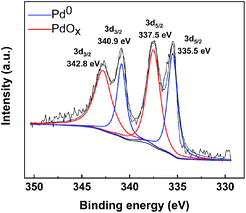 |
| | Fig. 3 XPS diagram of Pd/C catalyst. | |
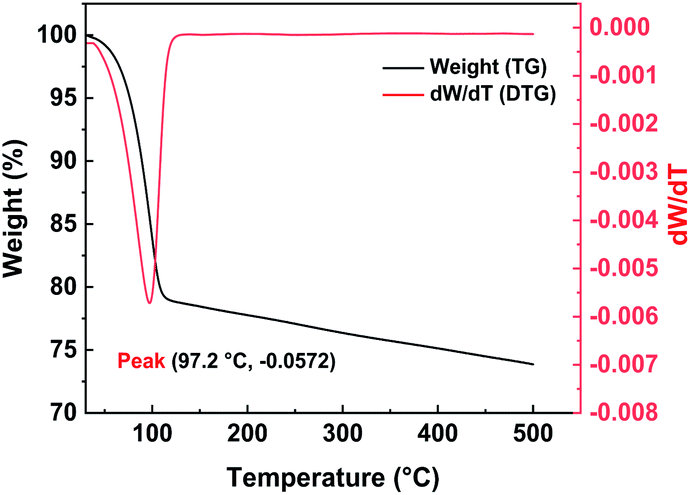
2.2.2 Thermal stability of Pd/C catalyst. TGA is an analytical method that analyzes and characterizes the thermal stability of samples. The TG curve, which has temperature on the x-axis and sample mass on the y-axis, reflects the relationship between the mass of the sample and temperature. The DTG curve is the first derivative of the TG curve and, over a particular range, the peak value of the DTG curve indicates that the rate of weight loss of the test sample has reached its maximum value. Since catalytic hydrogenolysis needs to take place at a particular temperature, the thermal stability of the Pd/C catalyst is very important. The Pd/C catalyst was characterized over the temperature range 30–500 °C. The TG and DTG curves of the Pd/C catalyst (Fig. 4) show that the rate of weight loss gradually increases with increasing temperature in one weightlessness peak, until a temperature of 97.2 °C is reached. The TG and DTG curve showed that the catalyst has significant weight loss (∼20%) from 50 °C to 110 °C, which is mainly caused by water evaporation. At 110 °C, the rate of weight loss of the Pd/C significantly decreased, and the weight increased linearly as the temperature decreased. The main cause of weightlessness may be the result of sintering the activated carbon at too high a temperature, which causes the internal pore structure to collapse. Higher temperatures will thus cause a decline in catalytically active sites and adversely affect the catalytic reaction. Throughout the testing process, the total weight loss of the Pd/C catalyst was 26.13%. TGA thus showed that our Pd/C catalyst was relatively stable over the temperature range that is suitable for catalytic hydrolysis. Excessively high temperatures would, however, adversely affect the activity of the catalyst and reduce the yield of the hydrogenolysis reaction.
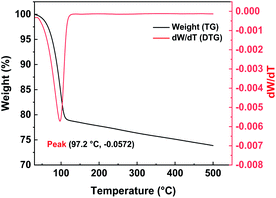 |
| | Fig. 4 TG/DTG curves of Pd/C catalyst. | |
2.3 Optimization by orthogonal experimental design
An orthogonal experimental design was adopted, with hydrogenolysis yield (φ) as the investigation index, and reaction time, reaction temperature, catalyst loading and hydrogen pressure as the four investigation factors. Five levels were designed for each factor, and an L25 (56) orthogonal experimental table (Table 1) was established. The experimental results were shown in Table 2.
Table 1 Table of orthogonal experimental horizontal factors
| Level |
Factor |
| Reaction time, h |
Reaction temperature, °C |
Catalyst loading, g L−1 |
Hydrogen pressure, MPa |
| 1 |
1 |
70 |
2 |
1 |
| 2 |
1.5 |
80 |
3 |
2 |
| 3 |
2 |
90 |
4 |
2.5 |
| 4 |
3 |
110 |
5 |
3 |
| 5 |
4 |
130 |
6 |
4 |
Table 2 Orthogonal experimental table for hydrogenolysis of LPPCs in ionic liquid
| Project |
Reaction time, h |
Reaction temperature, °C |
Catalyst loading, g L−1 |
Hydrogen pressure, MPa |
DP |
φ, % |
| 1 |
1 |
130 |
5 |
1 |
9.02 |
31.61 |
| 2 |
1 |
110 |
6 |
3 |
7.23 |
45.19 |
| 3 |
1 |
90 |
2 |
2 |
8.84 |
32.98 |
| 4 |
1 |
80 |
3 |
4 |
10 |
24.18 |
| 5 |
1 |
70 |
4 |
2.5 |
8.48 |
35.71 |
| 6 |
1.5 |
130 |
4 |
3 |
8.45 |
35.94 |
| 7 |
1.5 |
110 |
5 |
2 |
9.16 |
30.55 |
| 8 |
1.5 |
90 |
6 |
4 |
7.3 |
44.66 |
| 9 |
1.5 |
80 |
2 |
2.5 |
7.43 |
43.67 |
| 10 |
1.5 |
70 |
3 |
1 |
8.37 |
36.54 |
| 11 |
2 |
130 |
3 |
2 |
10.66 |
19.18 |
| 12 |
2 |
110 |
4 |
4 |
8.35 |
36.69 |
| 13 |
2 |
90 |
5 |
2.5 |
3.98 |
69.83 |
| 14 |
2 |
80 |
6 |
1 |
9.46 |
28.28 |
| 15 |
2 |
70 |
2 |
3 |
10.31 |
21.84 |
| 16 |
3 |
130 |
2 |
4 |
8.45 |
35.94 |
| 17 |
3 |
110 |
3 |
2.5 |
9.6 |
27.22 |
| 18 |
3 |
90 |
4 |
1 |
9.4 |
28.73 |
| 19 |
3 |
80 |
5 |
3 |
9.72 |
26.31 |
| 20 |
3 |
70 |
6 |
2 |
8.2 |
37.83 |
| 21 |
4 |
130 |
6 |
2.5 |
10 |
24.18 |
| 22 |
4 |
110 |
2 |
1 |
10.44 |
20.85 |
| 23 |
4 |
90 |
3 |
3 |
6.87 |
47.92 |
| 24 |
4 |
80 |
4 |
2 |
8.37 |
36.54 |
| 25 |
4 |
70 |
5 |
4 |
10.9 |
17.36 |
2.4 Influence of each factor on yield of hydrogenolysis
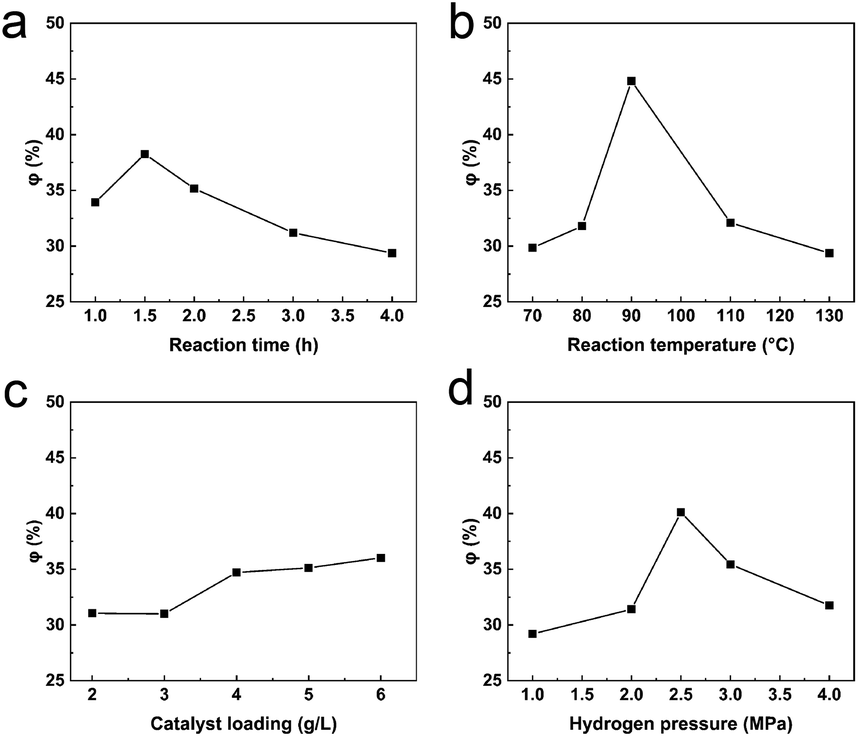
2.4.1 Reaction time. φ increased slowly from 1 h to 1.5 h and reached a maximum (38.27%) at 1.5 h, before falling to 29.37% after 4 h (Fig. 5a). The reaction reached equilibrium at 1.5 h and, with increasing reaction time, the newly formed LOPCs may undergo unwanted side reactions which reduce the amount of the desired products. The optimal reaction time for hydrogenolysis of LPPCs was thus 1.5 h.
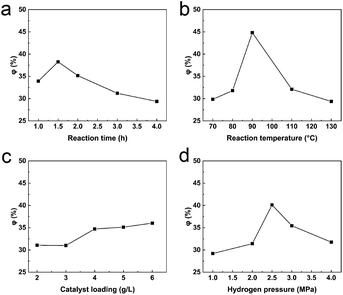 |
| | Fig. 5 (a) Plot of φ plotted vs. hydrogenolysis time; (b) plot of φ vs. reaction temperature; (c) plot of φ vs. catalyst loading; (d) plot of φ vs. hydrogen pressure. | |
2.4.2 Reaction temperature. Reaction temperature has one of the biggest impacts on chemical reactions. φ increased rapidly over the temperature range 80–90 °C (Fig. 5b), reaching 44.82% at 90 °C. The DP of the LPPCs decreased significantly at this temperature for two reasons. Firstly, increased temperature significantly reduces the viscosity of ionic liquids, thus improving the mass transfer rate of hydrogen and, secondly, the rate of C–C bond fracture is accelerated at higher temperatures. Between 90 °C and 130 °C, φ fell rapidly to 29.37%, likely because of side reactions, such as pyrolysis of the LPPCs, at high temperatures. Since these by-products are not LOPCs, the yield of the hydrogenolysis reaction decreases. As a result, 90 °C was determined to be the optimal temperature for hydrogenolysis.
2.4.3 Catalyst loading. The catalyst provides the main active site for catalytic hydrogenolysis and, within limits, increased catalyst loading accelerates the reaction. As shown in Fig. 5c, φ increased from 31.06% to 34.72% when the catalyst loading was increased from 3 g L−1 (Pd/C: [BMIM]Cl) to 4 g L−1, and then stabilized. When the catalyst loading was increased further to 6 g L−1, φ was 36.03%, indicating that the overall yield did not change significantly within this range of catalyst loadings. From an economic perspective, we believe that a catalyst loading of 4 g L−1 is the best option for large-scale processing.
2.4.4 Hydrogen pressure. Under our reaction conditions, the hydrogen pressure is proportional to the hydrogen molecular concentration. This means that, within limits, higher pressure is more favorable for gas–liquid–solid phase contact and thus the hydrogenolysis reaction. The curve of φ vs. hydrogen pressure is shown in Fig. 5d. Within the range 1–2.5 MPa, φ increased rapidly and reached a maximum value of 40.12% at 2.5 MPa. When the hydrogen pressure was further increased to 4 MPa, φ gradually decreased to 31.77%. There are two possible reasons for the decrease in φ. Firstly, increased hydrogen pressure may lead to hydrogenation/hydrogenolysis of other groups in the LPPCs, thus destroying the original polyphenolic structure. Secondly, further hydrogenation/hydrogenolysis may lead to degradation of the newly formed LOPCs. A hydrogen pressure of 2.5 MPa is thus most suitable for hydrogenolysis of LPPCs.
2.5 Significance and optimization of each factors
Using Pd/C as the catalyst, the optimal conditions for hydrogenolysis of LPPCs in the ionic liquid [BMIM]Cl were: reaction time, 1.5 h; reaction temperature, 90 °C; catalyst loading, 4 g L−1; and hydrogen pressure, 2.5 MPa. It is worth noting that the orthogonal experiment contains some errors, which will reduce the significance of each factor. As shown in the analysis results of Tables 2 and 3, it can be seen that the mutual influence of factors is very obvious. Firstly, the interval level selected for each factor needs to be expanded and, secondly, interactions between factors interfere with the experimental results and the interactions between factors needs to be further investigated.
Table 3 Variance analysis table for hydrogenolysis of LPPCs in ionic liquid
| Factor |
DOF |
Adj SS |
Adj MS |
F |
P |
| Reaction time |
4 |
240 |
60.01 |
0.32 |
0.858 |
| Reaction temperature |
4 |
817 |
204.24 |
1.08 |
0.426 |
| Catalyst loading |
4 |
113.5 |
28.36 |
0.15 |
0.746 |
| Hydrogen pressure |
4 |
367 |
91.75 |
0.49 |
0.958 |
| Error |
8 |
1507.9 |
188.49 |
|
|
| SUM |
24 |
3045.4 |
|
|
|
2.6 Structural characterization of proanthocyanidins
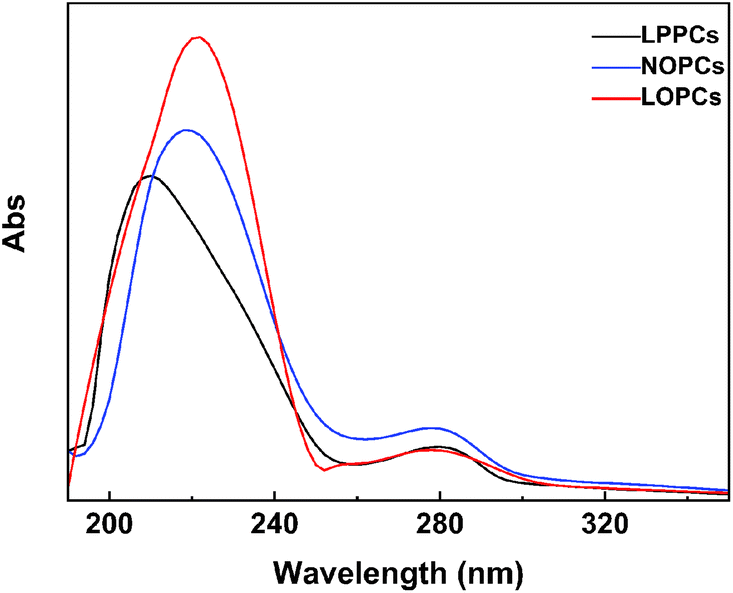
2.6.1 UV-Vis spectroscopy. UV-Vis spectroscopy measures the absorption by a sample of light at wavelengths between 190–800 nm and can be used for the qualitative and quantitative detection of proanthocyanidins. The spectra of LPPCs, NOPCs and LOPCs were recorded using a UV-Vis spectrophotometer over the range 190–800 nm. There were no absorption peaks between 350 nm and 800 nm and the absorption spectra at 190–350 nm are shown in Fig. 6. The maximum absorption wavelength of LPPCs, NOPCs and LOPCs is ∼278 nm, which is consistent with literature values for the characteristic absorption wavelength of proanthocyanidins.34 LOPCs and NOPCs showed maximum absorption peaks at ∼220 nm, whereas the maximum absorption peak of LPPCs was at 208 nm, indicating that, like NOPCs, LOPCs obtained by hydrogenolysis have the characteristics of oligomeric proanthocyanidins. This indicates that Pd/C catalyzed hydrogenolysis of LPPCs in an ionic liquid produces LOPCs, which have a similar absorption spectrum to NOPCs.
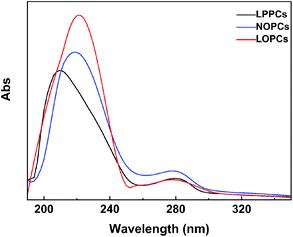 |
| | Fig. 6 UV-Vis spectra of LPPCs, NOPCs and LOPCs. | |
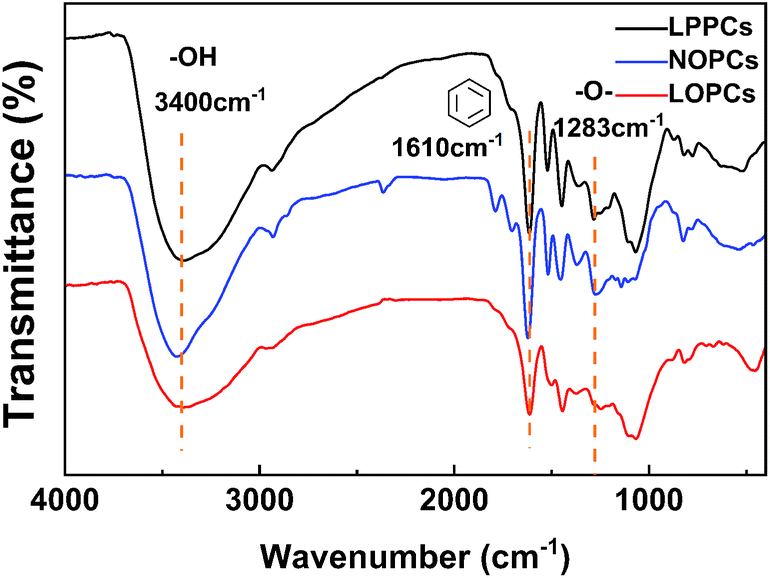
2.6.2 IR spectroscopy. IR spectroscopy provides information about the functional groups present in a sample compound and is most often used for qualitative detection. The FTIR spectra of the LPPCs, NOPCs and LOPCs are shown in Fig. 7 The stretching vibrations of hydroxyl groups can be seen as wide, strong peaks at 3400 cm−1. The strong peaks at 1610 cm−1 are characteristic of benzene ring and the peaks at 1616 cm−1, 1517 cm−1 and 1453 cm−1 are characteristic of the “breathing” vibrations of benzene rings. The peak at 1282 cm−1 is characteristic of ether bonds. This is consistent with the literature.35 Compared with the spectra of the LPPCs and NOPCs from larix barks, the spectrum of the LOPCs retains the infrared absorption peaks corresponding to each functional group of proanthocyanidins, confirming that the LOPCs still have the structural characteristics of proanthocyanidins. The catalytic hydrogenolysis of LPPCs in [BMIM]Cl also shows good selectivity. In other words, the reaction breaks C–C bond between proanthocyanidin monomers, thus reducing the DP and molecular weight of the LPPCs, but does not destroy the structure of the polyphenols, providing the required LOPCs, which have higher biological activity.
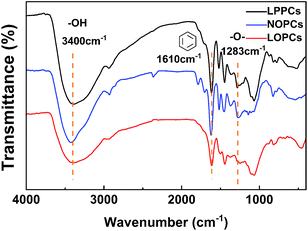 |
| | Fig. 7 IR spectra of LPPCs, NOPCs and LOPCs. | |
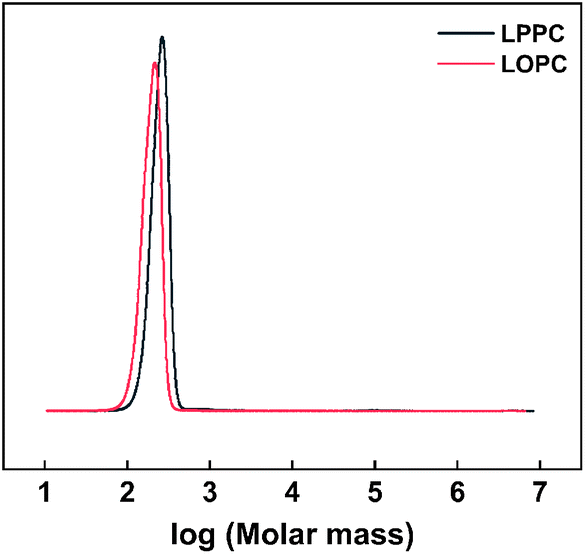
2.6.3 Gel permeation chromatography. The molecular weights of LPPCs before hydrogenolysis and LOPCs after hydrogenolysis were determined by GPC. The molecular weight distribution curves (Fig. 8) show that the molecular weight of the LOPCs is significantly lower than that of the LPPCs, confirming the efficiency of Pd/C for catalytic depolymerization of LPPCs.
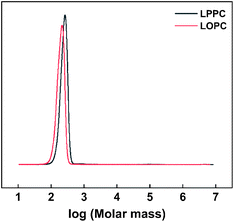 |
| | Fig. 8 Molecular weight distribution curves of LOPCs and LPPCs. | |
2.7 Ultraviolet blocking by proanthocyanidins
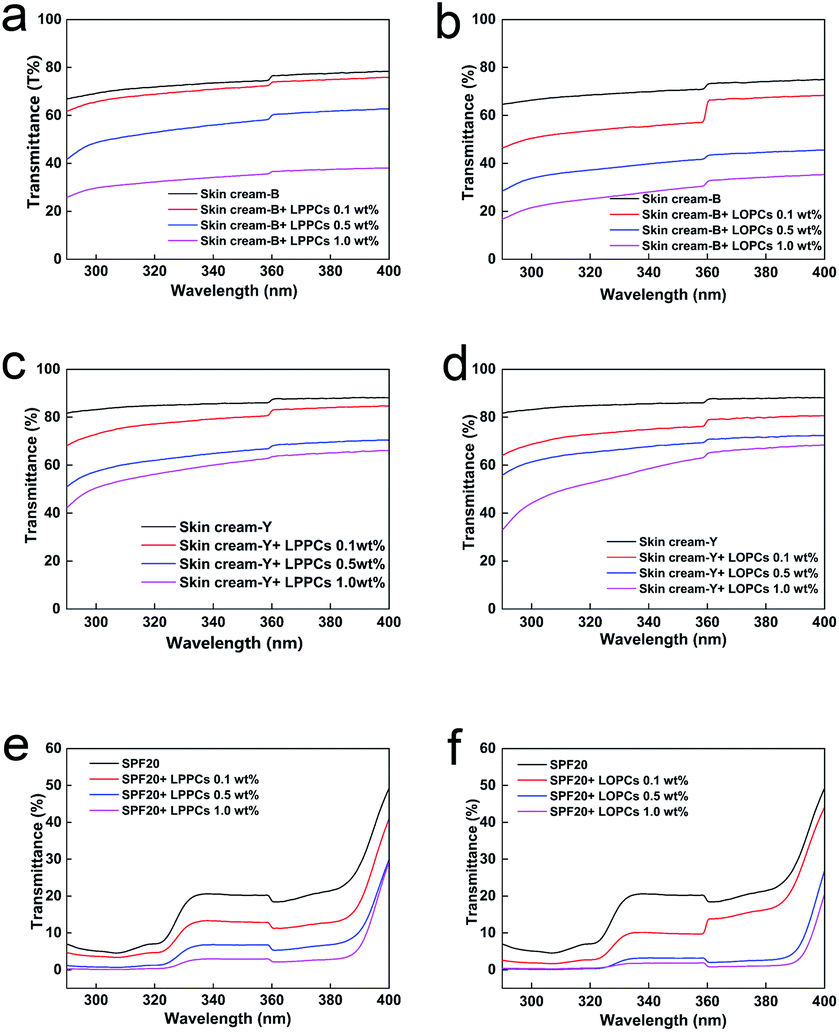
2.7.1 Ultraviolet blocking by skin creams and sunscreen containing different amounts of proanthocyanidins. Proanthocyanidins are aromatic compounds that contain a large number of phenolic hydroxyl groups and thus strongly absorb light in the UV region of the spectrum (Fig. 9). LOPCs and LPPCs were mixed with two different skin creams and a sunscreen to explore their effect on the UV blocking performance of the skin creams and sunscreen. The UV absorbance of LOPCs was superior to that of LPPCs over the range 290–400 nm. As well as enhancing the UV absorbance, the hydrogenolysis reaction also serves as an additional purification process so the purity of the LOPCs was improved compared with that of the LPPCs. Over the wavelength range 320–400 nm, skin cream-B transmits close to 80% of UV light and, over the range 290–320 nm, transmittance is close to 70% (Fig. 9a and b). Transmission by skin cream-Y exceeded 80% in both UVA and UVB regions (Fig. 9c and d). These experiments indicate that commercially available skin creams absorb UV light only weakly. The sunscreen performed better than the skin creams, although transmittance increased above 390 nm (Fig. 9e and f). In all cases, UV transmittance decreased significantly after addition of proanthocyanidins and the decrease was concentration-dependent over the range 0.1–1.0 wt% mass fraction of proanthocyanidins. At the same mass concentration, the UV transmittance of both skin creams and sunscreen decreased more markedly after addition of LOPCs than after addition of LPPCs.
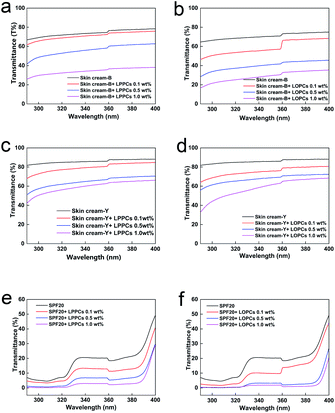 |
| | Fig. 9 UVB and UVA transmittance of skin creams and sunscreen mixed with different mass concentrations of LOPCs and LPPCs. (a and b) skin cream-B with different mass concentrations of LPPCs and LOPCs; (c and d) skin cream-Y with different mass concentrations of LPPCs and LOPCs; (e and f) SPF20 sunscreen with different mass concentrations of LPPCs and LOPCs. | |
The synergistic effect of proanthocyanidins with sunscreen (Fig. 9e and f), may be due to π–π* superposition of aromatic rings in the proanthocyanidins with aromatic rings in the sunscreen. Addition of either LPPCs or LOPCs to sunscreen thus reduces transmission of UV light and enhances protection. At the same concentration, LOPCs enhanced the performance of the sunscreen more than LPPCs.
2.7.2 Sensory evaluation. The skin creams and sunscreen purchased in the market are all white creams. After adding proanthocyanidins, the color of the samples became light brown and deepened with increasing mass concentrations of proanthocyanidins (Fig. 10). Apart from the color change, other properties, such as smell, texture and feel on the skin, were unchanged after addition of proanthocyanidins. The brown color may, however, be rejected by consumers and reduce the commercial potential of proanthocyanidin-containing sunscreen products. Methods to reduce the color of proanthocyanidins whilst retaining their UV-blocking properties, should be further studied.
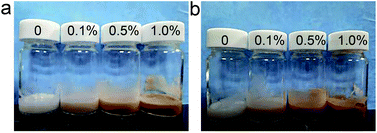 |
| | Fig. 10 Photographs of skin cream with different mass concentrations of proanthocyanidins: (a) LOPCs, (b) LPPCs. | |
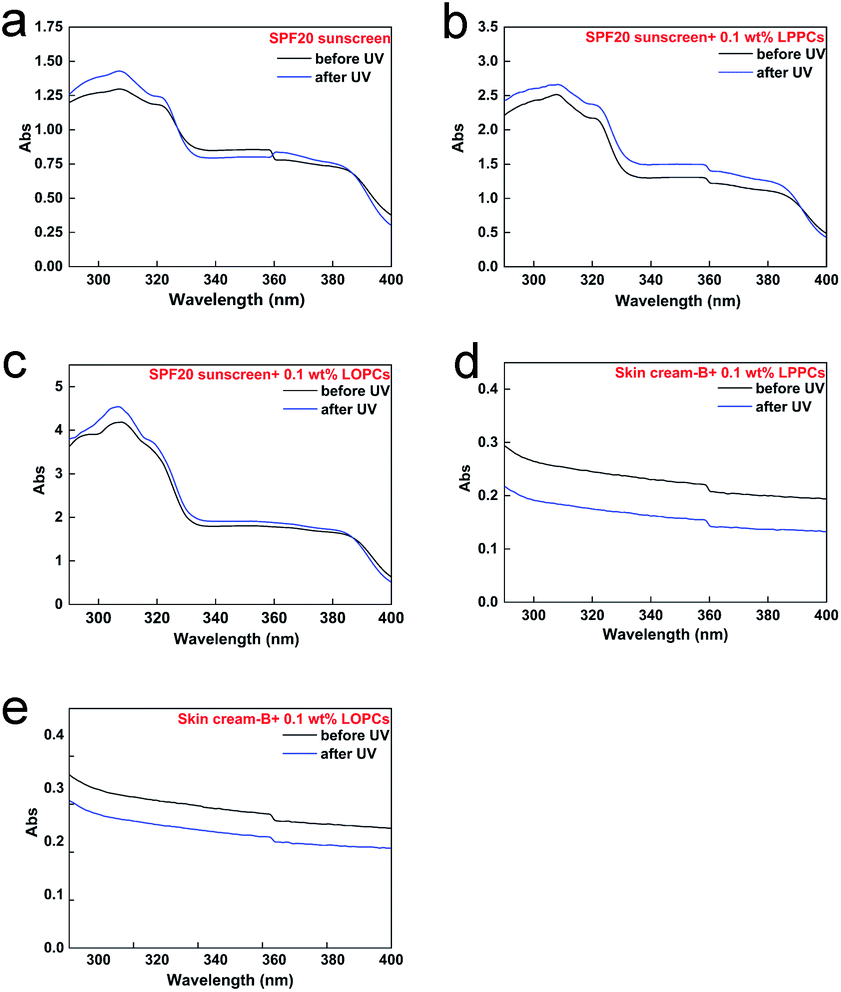
2.7.3 Stability to UV radiation. Commercial sunscreens showed little change in capacity to absorb UV light in the region 290–400 nm after exposure to UV light for 2 h (Fig. 11a). When sunscreens containing proanthocyanidins were exposed to UV radiation for 2 h, however, their UV absorbance in both UVA and UVB regions increased (Fig. 11b and c), indicating an improvement in protection. After irradiation with UV light for 2 h, the UV absorbance of skin cream-B containing 0.1 wt% LPPCs or 0.1 wt% LOPCs decreased in both UVA and UVB regions (Fig. 11d and e), indicating poor stability to UV light. In order to explore how to improve the sun protection afforded by sunscreen containing proanthocyanidins under UV radiation, we used the light stability of skin cream-B as a reference. Unlike sunscreen, skin cream-B does not contain active sunscreen ingredients. When added to sunscreen, proanthocyanidins may interact with some of the active ingredients during 2 h of UV exposure, forming better UV-blocking agents. The active ingredients in sunscreens, such as butylmethoxydibenzoylmethane, have poor light stability and readily undergo photodegradation. Proanthocyanidins are effective natural antioxidants and their ability to scavenge free radicals may protect the active ingredients in sunscreens from degradation. There is thus a theoretical basis for using proanthocyanidins in the cosmetics industry to enhance the reliability and safety of sunscreens.
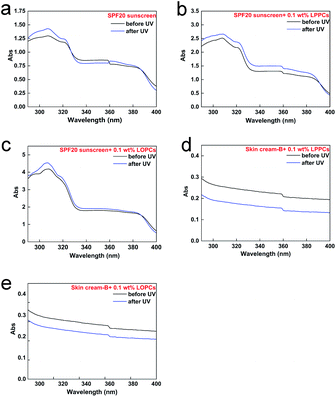 |
| | Fig. 11 Diagrams of UV absorption before and after UV irradiation for 2 h. (a) SPF20 sunscreen; (b) SPF20 sunscreen with 0.1 wt% LPPCs; (c) SPF20 sunscreen with 0.1 wt% LOPCs; (d) skin cream-B with 0.1 wt% LPPCs; (e) skin cream-B with 0.1 wt% LOPCs. | |
3 Conclusion
In this work, we used Pd/C as the catalyst to hydrogenolyze LPPCs in a non-organic solvent, the ionic liquid [BMIM]Cl. An orthogonal experimental design was used to optimize the conditions for the hydrogenolysis reactions. The best conditions were found to be: temperature, 90 °C; reaction time, 1.5 h; catalyst loading 4 g L−1 (Pd/C: [BMIM]Cl); and hydrogen pressure, 2.5 MPa. ANOVA showed that the significance of the interactions between different experimental factors needs further investigation. UV-Vis and IR spectroscopy, together with GPC analysis, showed that the LOPCs produced during the hydrogenolysis reaction retained the structural characteristics of proanthocyanidins. Compared with reaction in traditional organic solvents, reaction in the ionic liquid achieves direct hydrogenolysis of LPPCs. Because the ionic liquid is also stable and non-volatile, it causes less environmental pollution than organic solutions and provides a sustainable alternative for hydrogenolysis of LPPCs.
To demonstrate potential applications of the proanthocyanidins, we added LPPCs and LOPCs to both skin cream and sunscreen. The UV transmittance of skin cream with added proanthocyanidins was lower than that of pure skin cream, confirming the ability of proanthocyanidins to block UV light. LOPCs provided a better UV barrier than LPPCs at the same concentration. Addition of proanthocyanidins to a shop-bought sunscreen also improved sun protection, with UV absorbance increasing with increasing UV radiation time. The specific mechanisms underlying the improved performance, however, need further study and clarification. One potential downside of adding proanthocyanidins to cosmetic products is that the products take on a brown color, which may be unattractive to consumers. To achieve better acceptance, it may be necessary to develop methods for decolorizing proanthocyanidins.
This study provides a new solvent for the study of directed depolymerization of proanthocyanidins, and also provides a commercially relevant example of the benefits of using ionic liquids, which are safe, sustainable and easy to recycle, for reactions of biomolecules. We have also explored value-added applications of proanthocyanidins and showed that they are good UV-blockers, with great potential for use in sunscreens and other products in which protection from the harmful effects of UV light is needed.
4 Experimental
4.1 Materials and reagents
Larix bark was crushed to a powder with particle diameter 0.5–1.0 mm as raw material. Catechin was purchased from Beijing Kool Chemical Co, Ltd. 1-Butyl-3-methylimidazole chloride ([BMIM]Cl) was purchased from Shanghai Chengjie Chemical Co, Ltd. PdCl2, vanillin, ferric chloride, ethanol, silver nitrate, PdCl2 and catechin were purchased from Tianjin Mesco Chemical Co, Ltd. Methanol, ethanol, petroleum ether, ethyl acetate, silver nitrate, catechin, vanillin, formaldehyde (37–40%) and acetic acid were purchased from Tianjin Tianli chemical reagent Co, Ltd. Hydrochloric acid (36–38wt%), nitric acid, sodium hydroxide and hydrogen peroxide were purchased from Tianjin Kemiou Chemical Regent Co, Ltd. acetic acid, methanol, ferric chloride, 1-butyl-3-methylimidazole chloride ([BMIM]Cl) and all of the above reagents are analytically pure. Methanol (chromatographically pure) was purchased from Tianjin Tianli chemical reagent Co, Ltd. Potassium bromide (spectral pure) was purchased from Tianjin Kemiou Chemical Reagent Co, Ltd. Marketed cosmetics, skin cream-B (produced by a company in Suzhou), skin cream-Y (produced by a company in Tianjin), and SPF20 sunscreen (SPF20), were purchased from supermarket in Harbin.
4.2 Preparation of LPPCs
Larix bark proanthocyanidins were extracted using the ethanol extraction method, as previously described.23
4.3 Characterization of proanthocyanidins
4.3.1 Determination of degree of polymerization. The average DP of the proanthocyanidins was determined by Vanillin-HCl assay,31 using catechin as the standard. Accurately weighed samples of the LPPCs and NOPCs respectively, obtained in Section 2.2 (0.010 g) were dissolved in [BMIM]Cl (1 mL) and then accurately diluted to 10 mL with methanol in volumetric flasks to provide mother solutions. Aliquots (0.5 mL) of the mother solutions were diluted to 10 mL with methanol in volumetric flasks and aliquots of these diluted solutions (1 mL) were used to determine Abs, and calculate the mass concentration (M). Aliquots (0.5 mL) of the diluted solutions described above were also transferred to 10 mL light-impermeable tubes with constant volumes of acetic acid. Abs was determined, and the molar concentration (C) was calculated. The DP of the proanthocyanidins was calculated using formula (1).Here, Mr is the relative molecular weight of catechin (290.27).
4.3.2 UV-Vis spectroscopy. The proanthocyanidin samples to be tested were dissolved in 70% (v/v) aqueous ethanol (40 μg mL−1). 70% (v/v) aqueous ethanol was used as the blank control. The samples were scanned over the range 200–800 nm using a Tu-1950a dual-beam UV-Vis spectrophotometer (Beijing General Analysis Instrument Co, Ltd, China).
4.3.3 IR spectroscopy. The KBr embossing method was used. The proanthocyanidin samples to be tested (2 mg) were ground together with KBr (200 mg) in an agate mortar under an infrared lamp and the resulting powder was pressed into thin discs. The spectra were recorded using an FTIR-650 Fourier infrared spectrometer (Tianjin Port East Science and Technology Development Co, Ltd.).
4.3.4 Gel permeation chromatography. The proanthocyanidins (25 mg) were dissolved in 50% (v/v) aqueous methanol (10 mL) and the solutions were filtered through a 0.45 μm pore diameter filtration membrane. The molecular weight distribution of the proanthocyanidins was determined by gel chromatography using an Agilent 1100 HPLC system (Agilent Technologies, Inc., USA). The samples (50 μL) were injected for tandem gel chromatography on a double column (79911GF-084 + 79911GF-083) at a temperature of 30 °C. A diode array detector was used, with a detection wavelength of 270 nm. The mobile phase was 50% aqueous methanol and the flow rate was 1.0 mL min−1.
4.4 Preparation of Pd/C catalyst
The catalyst was prepared as previously described.33 Activated carbon (AC, 20–40 mesh, 6.0 g) was placed in a 250 mL round bottom flask, soaked in 12% hydrochloric acid solution, boiled at 50 °C for 10 h, washed until free from Cl− ions and then dried. The pre-treated AC was mixed with 12% nitric acid and refluxed at 80 °C for 3 h. After washing, the newly formed AC-HNO3 was dried. AC-HNO3 was immersed in H2O2 (5 M, completely covered), refluxed for 5 h, washed and dried at 110 °C to provide AC-HNO3-H2O2. PdCl2 (0.4743 g), water (15 mL) and concentrated hydrochloric acid (3 mL) were heated until the PdCl2 was fully dissolved. The solution was then added dropwise with stirring to the AC-HNO3-H2O2 at 80 °C in a water bath. The pH was adjusted to 10 and the reaction mixture was kept at 90 °C for 2 h. Formaldehyde solution (37–40%, 10 mL) was added and the reaction mixture was aged for 30 h. The resulting Pd/C catalyst was removed by suction filtration, washed and dried.
4.5 Characterization of Pd/C catalyst
X-ray photoelectron spectroscopy. X-ray photoelectron spectroscopy (XPS) of the Pd/C catalyst was performed using a K-Alpha X-ray photoelectron spectrometer (Thermo Fisher Scientific, Inc., USA), under the following conditions: Ag 3D, 5/2 resolution, half peak width 0.85 eV, binding energy range 0–1350 eV, Al target, X-ray maximum values of 12 kV and 30 mA.
4.5.1 Thermogravimetric analysis (TGA). TGA of the Pd/C catalyst was carried out using a Pyris1 thermogravimetric analyzer (PerkinElmer, Inc., USA), with a temperature range of 30–500 °C and a heating rate of 10 °C min−1.
4.5.2 Catalytic hydrogenolysis of LPPCs. LPPCs (0.50 g) were magnetically stirred with [BMIM]Cl (53.6 g, ∼50 mL) at 90 °C to prepare a solution (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :100, w/v). The solution, together with an aliquot of the Pd/C catalyst, was placed in a hydrogenolysis reactor. Hydrogen was then fed into the reactor and the reaction temperature was set. Once the set temperature was reached, the reactor agitator was started and the reaction was stirred at a rotational speed of 500 rad min−1. At the end of the reaction, the reaction mixture was taken out of the reactor and the solid catalyst Pd/C was removed by filtration to provide an ionic liquid solution of the product. Transformed this solution (1 mL) in 10 mL volumetric flask, then diluted with methanol to volume and determined DP according to (1) the efficiency of the hydrogenolysis of the proanthocyanidins was evaluated using the yield (φ) of desired products. The DP of the product was determined and φ was calculated using (2).
:100, w/v). The solution, together with an aliquot of the Pd/C catalyst, was placed in a hydrogenolysis reactor. Hydrogen was then fed into the reactor and the reaction temperature was set. Once the set temperature was reached, the reactor agitator was started and the reaction was stirred at a rotational speed of 500 rad min−1. At the end of the reaction, the reaction mixture was taken out of the reactor and the solid catalyst Pd/C was removed by filtration to provide an ionic liquid solution of the product. Transformed this solution (1 mL) in 10 mL volumetric flask, then diluted with methanol to volume and determined DP according to (1) the efficiency of the hydrogenolysis of the proanthocyanidins was evaluated using the yield (φ) of desired products. The DP of the product was determined and φ was calculated using (2).| | |
Φ = (DP0 − DP1)/DP0 × 100%
| (2) |
Where, DP0 and DP1 is the average degree of polymerization of LPPCs and hydrogenolysis products respectively. The residual product solution was dialysed for 72 h. The solid products were obtained after washing, centrifugation and drying. The total solids were dissolved in 70% (v/v) aqueous ethanol. After removing ethanol by rotary evaporation, LOPCs were extracted with ethyl acetate. The LOPCs obtained at 110 °C, with a Pd/C loading of 5 g L−1, a hydrogen pressure of 2.5 MPa and a 2 h reaction time were used for subsequent characterization and UV blocking studies.
4.6 Sun protection testing of proanthocyanidins
4.6.1 Preparation of proanthocyanidin-containing skin cream and determination of UV transmittance. The proanthocyanidins were mixed with either skin cream or sunscreen (mass fractions of proanthocyanidins, 0, 0.1, 0.5 and 1.0%) and stirred at 600 rpm for 24 h at room temperature in the dark. To test whether the proanthocyanidins were completely dissolved, or were suspended, in the skin cream or sunscreen, the samples were centrifuged at 10000 rpm for 1 h. If there was no precipitate after centrifugation, the proanthocyanidins were fully soluble in the skin cream and sunscreen. Skin creams and sunscreens containing different concentrations of proanthocyanidins were spread evenly on clean quartz glass (40 × 40 × 1 mm3) and then dried in a dark room for 20 min. The UV transmittance (T%) was measured using a TU-1950 UV spectrophotometer, equipped with a solid sample holder. Each scan was from UVB (290–320 nm) to UVA (320–400 nm).
4.6.2 Sensory evaluation. The color sensation of the skin creams and sunscreens containing different concentrations of proanthocyanidins were evaluated by sight, smell, and feel on skin.
4.6.3 UV radiation assay. Sunscreens, with and without proanthocyanidins, were exposed to UV light to investigate the effect of UV radiation. The samples were prepared as described in Section 2.7.1 and the coated quartz glass sheets were irradiated with UV light for 2 h. The UV light source were two 15 W UV lamps together with an illumination intensity of 1330.4 W cm−2. The UV absorbance (Abs) of the samples from UVB (290–320 nm) to UVA (320–400 nm) was measured before and after UV exposure to determine the light stability of these sunscreens.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
This work was supported by the National Undergraduate Training Programs for Innovations (201910225069) and the Natural Science Foundation of Hei-longjiang Province of China (Grant No. LH2019C009).
Notes and references
- L. An, C. Si, G. Wang, W. Sui and Z. Tao, Ind. Crops Prod., 2019, 128, 177–185 CrossRef CAS.
- X. Li, R. Xu, J. Yang, S. Nie, D. Liu, Y. Liu and C. Si, Ind. Crops Prod., 2019, 130, 184–197 CrossRef CAS.
- H. Liu, T. Xu, K. Liu, M. Zhang, W. Liu, H. Li, H. Du and C. Si, Ind. Crops Prod., 2021, 165, 113425 CrossRef CAS.
- R. T. Neto, S. A. O. Santos, J. Oliveira and A. J. D. Silvestre, Ind. Crops Prod., 2020, 151, 112450 CrossRef CAS.
- L. Ni, F. Zhao, B. Li, T. Wei, H. Guan and S. Ren, Molecules, 2018, 23, 2445 CrossRef PubMed.
- E. E. Bayramoğlu, Ind. Crops Prod., 2013, 41, 53–56 CrossRef.
- B. Teng, J. Wu, Y. Wang and W. Chen, Pol. J. Environ. Stud., 2017, 26, 2249–2257 CrossRef CAS.
- I. Ben El Hadj Ali, R. Bahri, M. Chaouachi, M. Boussaïd and F. Harzallah-Skhiri, Ind. Crops Prod., 2014, 62, 188–195 CrossRef CAS.
- N. Diwani, J. Fakhfakh, K. Athmouni, D. Belhaj, A. El Feki, N. Allouche, H. Ayadi and H. Bouaziz-Ketata, Ind. Crops Prod., 2020, 144, 112040 CrossRef CAS.
- L. Linlin, G. Xing, T. Lili, W. Dabo and W. Qin, Arch. Pharmacal Res., 2020, 43, 1056–1066 CrossRef PubMed.
- T. Shusuke, R. Preethi, G. Jinghua, C. Jacob, Y. Madelaine and G. Ajay, Sci. Rep., 2018, 8, 1–13 Search PubMed.
- C. Chen, P. Somavat, V. Singh and E. Gonzalez de Mejia, Ind. Crops Prod., 2017, 109, 464–475 CrossRef CAS.
- M. J. Kruger, N. Davies, K. H. Myburgh and S. Lecour, Food Res. Int., 2014, 59, 41–52 CrossRef CAS.
- J. Song, S. Chen, X. Zhao, J. Cheng, Y. Ma, S. Ren and S. Li, RSC Adv., 2021, 11, 6374–6382 RSC.
- T. Jiao, Y. Lu, L. Ye, J. Yao, Y. Zhuang, Y. Huang, M. Zhao, C. Lu and J. Li, J. Chin. Inst. Food Sci. Technol., 2019, 19, 98–105 Search PubMed.
- H. Du, W. Liu, M. Zhang, C. Si, X. Zhang and B. Li, Carbohydr. Polym., 2019, 209, 130–144 CrossRef CAS PubMed.
- K. Liu, H. Du, T. Zheng, H. Liu, M. Zhang, R. Zhang, H. Li, H. Xie, X. Zhang, M. Ma and C. Si, Carbohydr. Polym., 2021, 259, 117740 CrossRef CAS PubMed.
- W. Liu, H. Du, M. Zhang, K. Liu, H. Liu, H. Xie, X. Zhang and C. Si, ACS Sustainable Chem. Eng., 2020, 8, 7536–7562 CrossRef CAS.
- H. Suo, R. Tian, J. Li, S. Zhang, Y. Cui, L. Li and B. Sun, Food Res. Int., 2019, 123, 440–449 CrossRef CAS PubMed.
- S. Deprez, C. Brezillon, S. Rabot, C. Philippe, I. Mila, C. Lapierre and A. Scalbert, J. Nutr., 2000, 130, 2733–2738 CrossRef CAS PubMed.
- J. Lee, Food Chem., 2010, 123, 51–56 CrossRef CAS.
- E. M. Jorgensen, A. B. Marin and J. A. Kennedy, J. Agric. Food Chem., 2004, 52, 2292–2296 CrossRef CAS PubMed.
- D. U. Xiao, L. U. Zhongbing, T. A. O. Yi, L. Xuepin and S. H. I. Bi, J. Sichuan Union Univ., Eng. Sci. Ed., 2005, 37, 65–70 Search PubMed.
- G. Q. Jiang, G. Z. Fang, L. L. Li, Z. X. Shi and Z. R. Zhang, Bioresources, 2014, 9, 662–672 CAS.
- H. C. Erythropel, J. B. Zimmerman, T. M. de Winter, L. Petitjean, F. Melnikov, C. H. Lam, A. W. Lounsbury, K. E. Mellor, N. Z. Janković, Q. Tu, L. N. Pincus, M. M. Falinski, W. Shi, P. Coish, D. L. Plata and P. T. Anastas, Green Chem., 2018, 20, 1929–1961 RSC.
- X. Meng, R. Zhang, H. Liu, X. Zhang, Z. Liu, C. Xu and A. A. Klusener Peter, Sci. Sin.: Chim., 2018, 48, 387–396 Search PubMed.
- C. J. Clarke, W. C. Tu, O. Levers, A. Brohl and J. P. Hallett, Chem. Rev., 2018, 118, 747–800 CrossRef CAS PubMed.
- C. G. Yoo, Y. Pu and A. J. Ragauskas, Current Opinion in Green and Sustainable Chemistry, 2017, 5, 5–11 CrossRef.
- J. Yang, X. Lu, Y. Zhang, J. Xu, Y. Yang and Q. Zhou, Green Energy Environ., 2020, 5, 223–231 CrossRef.
- L. Ran, C. Yang, M. Xu, Z. Yi, D. Ren and L. Yi, Sep. Purif. Technol., 2019, 226, 154–161 CrossRef CAS.
- C. Li, S. Xu and Z. Wang, Food Sci., 2004, 25, 157–161 CAS.
- M. Brun, A. Berthet and J. C. Bertolini, J. Electron Spectrosc. Relat. Phenom., 1999, 104, 55–60 CrossRef CAS.
- L. Liu, S. Zhang and G. Fang, Chem. Ind. For. Prod., 2014, 34, 13–20 Search PubMed.
- L.-Z. Lin and J. M. Harnly, J. Agric. Food Chem., 2012, 60, 5832–5840 CrossRef CAS PubMed.
- S. Luo, X. Zhang, X. Zhang and L. Zhang, Nat. Prod. Res., 2014, 28, 1116–1120 CrossRef CAS PubMed.
|
| This journal is © The Royal Society of Chemistry 2021 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence b,
Xiaoxia Chenb,
Chong Gaob,
Liwen Nib,
Xuechun Wangb,
Wudi Zhangb and
Shixue Ren
b,
Xiaoxia Chenb,
Chong Gaob,
Liwen Nib,
Xuechun Wangb,
Wudi Zhangb and
Shixue Ren