
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePreparation and characterization of polydopamine/melamine microencapsulated red phosphorus and its flame retardance in epoxy resin
Chen Chenga,
Yanling Lua,
Weining Maa,
Shaojie Lia,
Jun Yan*b and
Shiguo Du *a
*a
aArmy Engineering University of PLA-Shijiazhuang Campus, Shijiazhuang, Hebei 050003, P. R. China. E-mail: shiguoduaeu@163.com
bHebei Jiaotong Vocational and Technical College, Shijiazhuang, Hebei 050003, P. R. China. E-mail: yan-junjun@263.net
First published on 7th June 2021
Abstract
Polydopamine/melamine composite microencapsulated red phosphorus (RP@PDA/MA) was prepared and applied as the flame retardant for epoxy resin (EP) in this work. For comparison, polydopamine (PDA) coated red phosphorus (RP@PDA) was also prepared. The microstructure, chemical composition and thermal decomposition of the as prepared samples were systematically characterized. The results showed that PDA and PDA/MA shell structures were fabricated successfully via convenient water-based processes at room temperature. The flame retardance of red phosphorus (RP), RP@PDA, and RP@PDA/MA on EP was evaluated. The results showed that EP blending with 7 wt% RP@PDA/MA passed V-0 degree in the vertical burning test (UL-94), reached a limited oxygen index (LOI) of 30.9% and decreased the peak heat release rate of EP by 65.1% in the cone calorimeter test. The satisfactory flame retardance can be attributed to the intumescent flame retardant system consisting of RP@PDA/MA. The PDA and PDA/MA shell structures also improved the compatibility between RP and EP, thus RP@PDA and RP@PDA/MA had less significant impact on the tensile–strain properties of EP.
1. Introduction
Epoxy resin (EP) is widely used in aerospace industry, architecture, buildings and electric/electronic devices owing to its superior mechanical properties, strong cohesiveness and good solvent & chemical resistance.1–4 Nonetheless, the inherent inflammability and severe melt dripping problem of EP bring about potential fire hazards and limit its further application in specific fields.5,6 Hence, it is meaningful to improve the flame retardance of EP to meet the practical requirements.Nowadays, intumescent flame retardants (IFR) are extensively used for EP flame-retardant treatment.7,8 IFR generally consist of three components: an acid source, a carbon source and a blowing agent.9,10 Under high temperature, the acid source can catalyze the carbon source to form a char layer, the structure of which will be further enlarged by the inert gas emitting from the blowing agent. The char layer with an intumescent appearance can hinder the oxygen and heat from penetrating the organic substrates beneath and impede the evolution of flammable volatile more efficiently, leading to the satisfactory flame retardance performance.11 Compared to the traditional halogen-containing flame retardants, IFR hardly give rise to toxic gas during the burning period, hence the danger of choking and the damage to the environment can be significantly decreased.12
Red phosphorus (RP), a phosphorus-containing flame retardant (PFR), is capable of functioning as acid source for IFR system. Under high temperature, RP can react with the water vaper and generate phosphorus-containing acids, which will further catalyze the char-forming process of the carbon sources or organic substrates to form a carbon layer.13,14 As a concentrated source of phosphorus, RP possesses higher ratio of phosphorus element compared to the other PFR, meaning that it can function efficiently at a low weight loading.15 However, during application, the shortcomings of pristine RP, such as strong water absorbing proneness, high friction sensibility and poor thermal stability, are noteworthy. Besides, due to the inferior compatibility between RP and the organic matrixes, the addition of pure RP may significantly deteriorate the mechanical properties of the substrates. To overcome these shortcomings, RP is generally coated by an organic shell structure to fabricate microencapsulated RP (MRP) before it can be used as an applicable flame retardant.16–18 Additionally, when specific synergistic materials are applied as the shell structures, MRP can constitute an integrated IFR system, which is supposed to further improve the flame retardance of pristine RP.
Melamine (MA) is an efficient blowing agent which is commonly used together with PFR in the organic substrates at present.19–21 Because pristine MA has poor water resistance and tends to aggregate in the organic substrates, the researchers generally prepared MA-containing shell structures for PFR to get better application effect. For now, PFR with MA-containing shell structures (e.g., MA-formaldehyde resin,15 aluminum trihydrate/MA-formaldehyde resin,22 MA/pentaerythritol,23 MA/phenol,24 MA/4,4′-diphenylmethane25 and MA-containing polyphosphazene26) have been prepared successfully and applied in various polymers. The MA-containing shell structures, together with the cores of acid sources, constituted integrated IFR and improved the flame retardance of the organic substrates efficiently. However, it should be noted that the raw materials of MA-containing shell structures mentioned above, such as pentaerythritol, formaldehyde, phenol, 4,4′-diphenylmethane and 4,4′-diaminodipheyl ether, are all petroleum materials with certain toxicity, and specific preparation procedures need to be conducted in the organic solvent, which may do harm to the environment and personnel. Hence, taking the pollution of environment and consumption of energy sources into consideration, it is meaningful to substitute the petroleum materials by a “green” one to prepare MA-containing shell structures through a more environmentally friendly way.
For this purpose, in this work, polydopamine (PDA) was applied as the raw material to prepare MA-containing shell structure for MRP flame retardant. PDA is the first material-independent modifier that can adhere to virtually all material surfaces.27,28 In 1981, Waite and Tanzer noticed the superior adhesive property of mussels and isolated an acid-soluble protein from the phenol gland of a marine mussel. This basic material was rich in 3,4-dihydroxy-L-phenylalanine (DOPA) and lysine, which were the main origins of the superior adhesion.29,30 In 2007, Messersmith and coworkers found that generated by the self-assembly of the bio-based dopamine monomers, PDA was a synthetic analogue of melanin with a molecular structure similar to that of DOPA. This bionic structure enabled PDA to deposit on virtually all types of organic or inorganic surfaces, making PDA become a novel coating material henceforward.31,32 PDA is a totally bio-based material with excellent biocompatibility. Its chemical structure contains many functional groups, which can serve as the platform for various modification reactions, e.g., the Schiff base reaction and Michael addition with the amine and/or thiol-containing molecules. Additionally, PDA displays attractive properties in optics, electricity, and magnetics.33 With these advantages, PDA has been incorporated in the application across energy,34 catalytic field,35 environment,36 medical treatment37 and bio-chemistry.38 In recent years, the usage of PDA as bio-based flame retardant drawn the attention of the researchers. The abundant benzene rings in the PDA molecular structure indicated that it was a qualified carbon source in the IFR system,39 and the free radical scavenging ability of the PDA nanoparticles can also improve the flame retardance of the organic substrates.40,41
In this work, taking advantage of the non-toxicity, superior cohesiveness and chemical activity of PDA, PDA/MA composite coated RP (RP@PDA/MA) was prepared through Schiff base reaction for the first time to function as the flame retardant for EP. For comparison, PDA coated RP (RP@PDA) was also prepared with relative properties characterized. The whole processes for RP@PDA/MA and RP@PDA were totally water-based, and the application of bio-based PDA shell materials is consistent with the “green” tendency of the flame retardant. The flame retardance of the as prepared MRP samples, as well as the flame retardance mechanism and the impact on the mechanical properties, was investigated. This work aims to prepare an efficient MRP flame retardant with MA-containing shell structure through a more environmentally friendly way.
2. Experimental
2.1 Materials
Red phosphorus (AR), melamine (AR), and ethyl alcohol (Purity: 99.7%) were purchased from Tianjin Yongda Chemical Reagent Co., Ltd. Dopamine hydrochloride was obtained from Shanghai Aladdin Biochemical Technology Co., Ltd. 2-Amino-2-(hydroxymethyl)-propane-1,3-diol (Tris) was supplied by BioFroxx GmbH. Diglycidyl ether of biphenol A (DGEBA, E-44, epoxy equivalent = 0.44 mol/100 g) was purchased from Shandong Deyuan Epoxy Resin Co., Ltd.2.2 Preparation of RP@PDA
RP (0.75 g) was dispersed in 100 mL deionized water under room temperature. Then dopamine hydrochloride (0.35 g) was dissolved in the mixture with mechanical stirring, followed by the addition of Tris to adjust pH value to 8.5. The preparation system was stirred for 24 h at a constant speed under room temperature. During this time, the color of the suspension changed from red to brown, and finally exhibited a black appearance. The resulted RP@PDA was filtered, washed by water and ethyl alcohol, and dried in vacuum at 60 °C for 24 h.2.3 Preparation of RP@PDA/MA
MA (0.35 g) was dissolved in 100 mL deionized water under room temperature. Then RP (0.75 g) was dispersed in the MA solution with constant mechanical stirring. Dopamine hydrochloride (0.35 g) was added into the mixture and the pH value of the mixture was adjusted to 8.5 by Tris. The preparation system was stirred for 24 h at a constant speed under room temperature, which showed a similar color changing behavior as the RP@PDA preparation process did. The resulted RP@PDA/MA was filtered, washed by water and ethyl alcohol, and dried in vacuum at 60 °C for 24 h. The mechanisms of RP@PDA and RP@PDA/MA preparation processes were presented in Scheme 1.42,43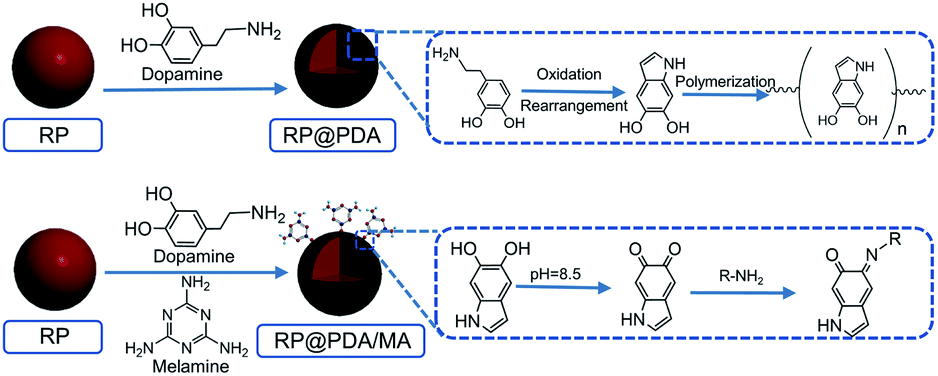 | ||
| Scheme 1 Preparation processes of RP@PDA and RP@PDA/MA. | ||
2.4 Preparation of flame-retardant EP composites
The bisphenol A based epoxy was blended with RP, RP@PDA and RP@PDA/MA by ultrasonic treatment for 30 min, respectively. Then the hardener was added dropwise and the mixture was stirred for 15 min (the weight ratio of the epoxy to hardener is 4![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1). Finally, the flame-retardant EP samples (FR-EP) were cured in a Teflon mold at 70 °C for 4 h. Based on the category of the flame retardants, the final products were denoted as EP, EP/RPX, EP/RP@PDAX, and EP/RP@PDA/MAX, respectively (X represents the weight ratio of the flame retardants).
:1). Finally, the flame-retardant EP samples (FR-EP) were cured in a Teflon mold at 70 °C for 4 h. Based on the category of the flame retardants, the final products were denoted as EP, EP/RPX, EP/RP@PDAX, and EP/RP@PDA/MAX, respectively (X represents the weight ratio of the flame retardants).
2.5 Characterization
Fourier transform infrared (FTIR) spectra were recorded on the TENSOR II Fourier transform spectrophotometer (Bruker, Germany). The wavenumber range was set from 400 to 4000 cm−1 and the resolution of the spectra was 4 cm−1.X-ray photoelectron spectroscopy (XPS) was carried out with a ThermoFischer ESCALAB 250Xi spectrometer. The residual pressure in the analysis chamber was about 10−9 Pa. The range of kinetic energy for full spectra was between 0 and 1100 eV. Binding energies were calibrated with respect to the C1s core level peak at 284.6 eV.
The scanning electron microscope (SEM) micrographs of were observed with JSM-7800F scanning electron microscopy. Samples were metal sprayed in advance. The energy dispersive spectrometer (EDS) was tested by X-max 80.
Thermogravimetric analysis (TGA) tests were conducted by SDT Q600 thermal analyzer (TA Instrument, USA) under nitrogen or synthesis air atmosphere at the gas flow of 100 mL min−1, with a heating rate at 15 K min−1.
The limiting oxygen index (LOI) test was carried out on HC-2 oxygen index instrument (Nanjing Jiangning Testing Equipment Company, Ltd, China) according to ASTM D2863. The dimensions of the tested samples were 130 × 6.5 × 3.2 mm3. The vertical burning (UL-94) test was conducted on CFZ-2 horizontal and vertical flame tester (Nanjing Jiangning Testing Equipment Company, Ltd, China) according to the UL-94 ASTM D3801 test standard. The dimensions of the tested samples were 130 × 13 × 3.2 mm3.
The cone calorimeter test, according to ISO 5660 standard procedure, was conducted on cone calorimeter (Stanton Redcroft Type, England) to identify the combustion performance of the EP samples. The heat flux was set as 35 kW m−2 and the specimens used were 100 × 100 × 3 mm3.
Thermogravimetric analysis/infrared spectrometry (TG-IR) measurements of EP samples were performed in STA 449F3 thermal analysis-infrared combined system (Netzsch, Germany) in nitrogen, with a heating rate at 15 K min−1. The spectra were obtained with a scan interval of 2.23 s, and the resolution of the spectra was 4 cm−1.
Raman spectroscopy measurements were performed under room temperature with LabRAM HR Evolution laser Raman spectrometer (HORIBA Jobin Yvon, France) with excitation provided in backscattering geometry by a 532 nm argon laser line.
The tensile-strain tests of EP samples were conducted on an electronic universal mechanical testing mechanical testing machine (Instron 5892) according to the ISO 527.2 standard. The tests were conducted at 25 °C. The constant crosshead speed is 1 mm min−1.
3. Results and discussion
3.1 FTIR spectra of RP samples
The FTIR spectra of RP, RP@PDA and RP@PDA/MA were presented in Fig. 1. The spectrum of pristine RP exhibited typical water absorption peaks at 3430 cm−1 and 1637 cm−1 due to the high moisture absorption rate of RP, and the broad absorption peak at 1184 cm−1 can be attributed to the P![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretching vibration of phosphoric acid and metaphosphoric acid.22 In the spectrum of RP@PDA, the emerging characteristic peaks at 1511 cm−1 and 1280 cm−1 can be attributed to the deformation vibration of benzene rings and the bending vibration of C–N bonds, respectively. Furthermore, the absorption peak at 1060 cm−1 was generated by the C–O bonds of PDA.44 For the FTIR spectrum of RP@PDA/MA, besides the characteristic peaks of PDA, the new peaks that presented at 1662 cm−1, 1558 cm−1 and 814 cm−1 were all associated with the triazine ring structure,26 which indicated the existence of MA in the shell structure of RP@PDA/MA.
O stretching vibration of phosphoric acid and metaphosphoric acid.22 In the spectrum of RP@PDA, the emerging characteristic peaks at 1511 cm−1 and 1280 cm−1 can be attributed to the deformation vibration of benzene rings and the bending vibration of C–N bonds, respectively. Furthermore, the absorption peak at 1060 cm−1 was generated by the C–O bonds of PDA.44 For the FTIR spectrum of RP@PDA/MA, besides the characteristic peaks of PDA, the new peaks that presented at 1662 cm−1, 1558 cm−1 and 814 cm−1 were all associated with the triazine ring structure,26 which indicated the existence of MA in the shell structure of RP@PDA/MA.
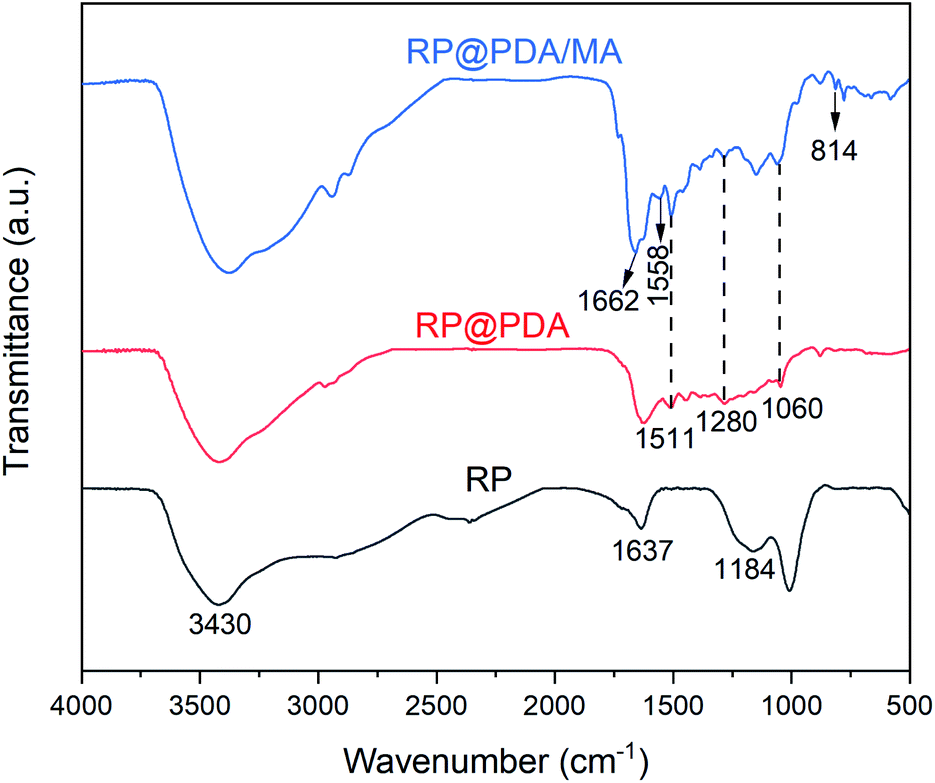 | ||
| Fig. 1 FTIR spectra of RP, RP@PDA and RP@PDA/MA. | ||
3.2 XPS spectra
XPS was utilized to characterize the composition of the surface elements of RP samples. The survey spectra were presented in Fig. 2, and the relative data of atomic concentration were listed in Table 1. As can be observed, pristine RP showed the strongest P2p and P2s peaks and the highest phosphorus atomic concentration. However, owing to the wrapping effect of the shell structures, the P2p and P2s peak intensities of RP@PDA and RP@PDA/MA, as well as the phosphorus atomic concentration, were decreased significantly, while the C and N atomic concentrations were increased on the contrary.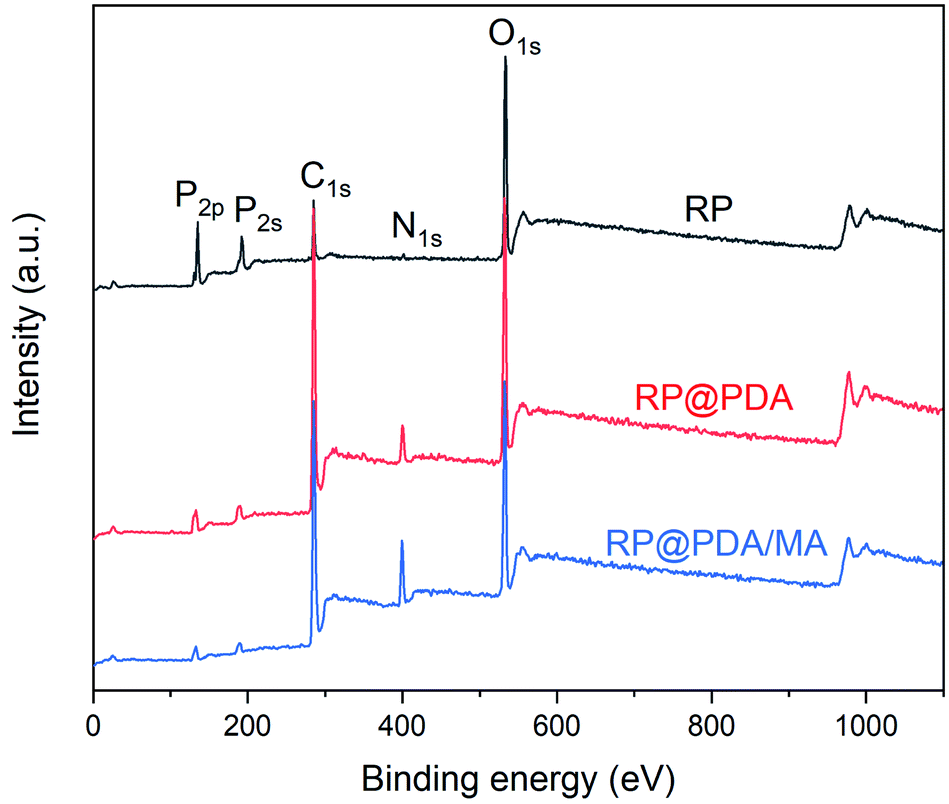 | ||
| Fig. 2 XPS spectra of RP, RP@PDA and RP@PDA/MA. | ||
| Sample | C (at.%) | O (at.%) | N (at.%) | P (at.%) |
|---|---|---|---|---|
| RP | 24.4 | 55.3 | 1.21 | 19.01 |
| RP@PDA | 68.1 | 21.7 | 5.66 | 4.53 |
| RP@PDA/MA | 67.3 | 20.1 | 9.45 | 3.05 |
The high resolution XPS was employed to analyze the element chemical states. As shown in Fig. 3(a) and (b), the C1s peaks of RP@PDA and RP@PDA/MA can be divided into three peaks with the binding energy of 284.3 eV, 286.1 eV and 288.5 eV, corresponding to CC, C–O & CN and CO & C–N, respectively, indicating the existence of heteroatoms (N and O) in the shell structures.45 The N1s spectra of RP@PDA and RP@PDA/MA can be divided into three peaks at 398.6 eV, 400.0 eV and 401.2 eV, which were assigned to pydinc N (CN–C), pyrrolic N (N–C3) and graphitic N (C–N–H), respectively.46 It can be seen from the N1s spectra that the peak area percentage of pydinc N in RP@PDA/MA was higher than that in RP@PDA (45.8% vs. 9.0%). The reason may be that the pydinc N group is a typical chemical bond in the triazine rings of MA, hence the MA-containing shell structure of RP@PDA/MA increased the concentration of it.
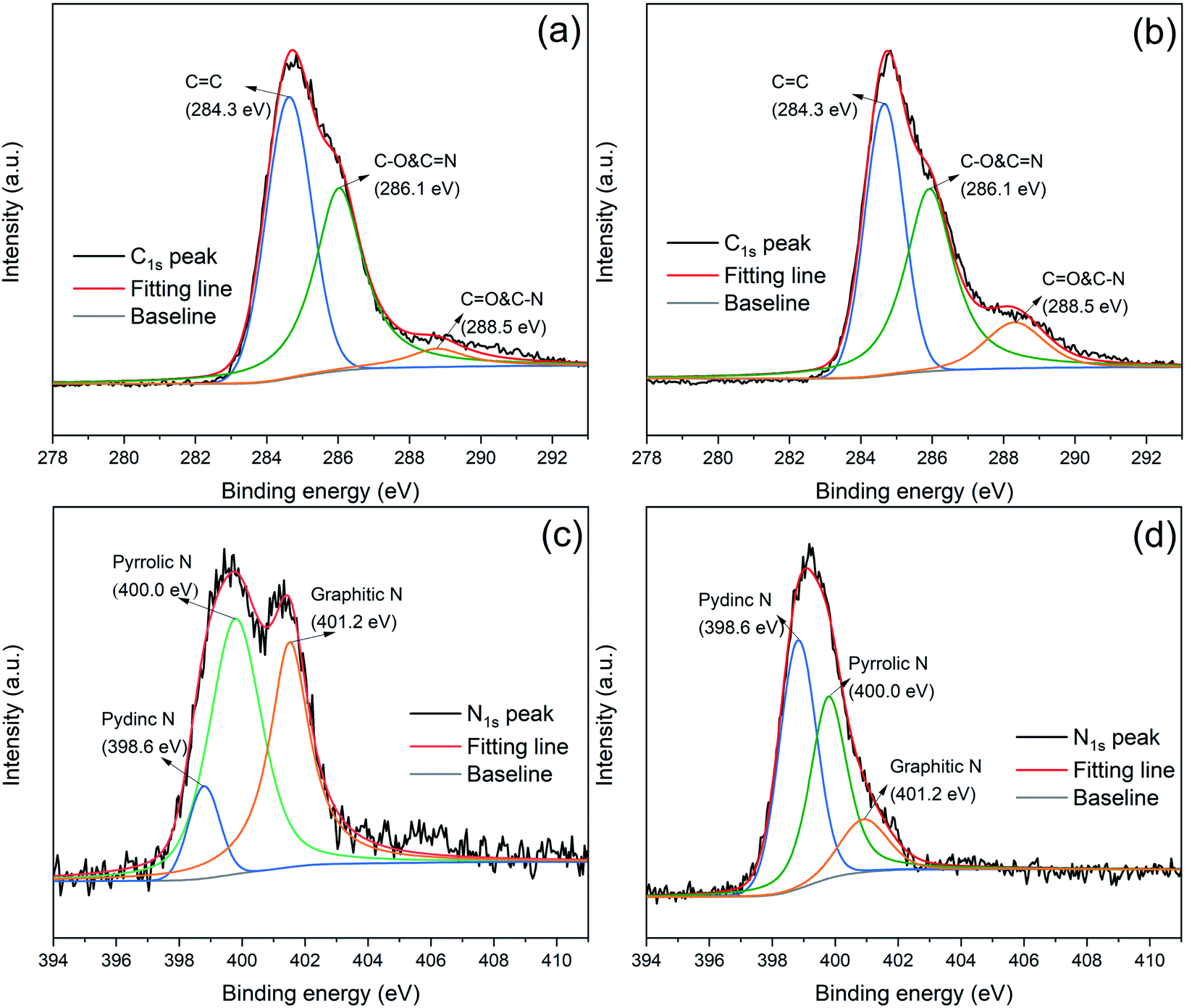 | ||
| Fig. 3 C1s peaks of (a) RP@PDA and (b) RP@PDA/MA; N1s peaks of (c) RP@PDA and (d) RP@PDA/MA. | ||
3.3 SEM micrographs
The SEM micrographs of RP, RP@PDA and RP@PDA/MA were presented in Fig. 4. As shown in Fig. 4(a1) and (a2), pristine RP exhibited a quite smooth surface with an angular overall appearance. The surface morphologies of RP@PDA and RP@PDA/MA were changed visibly. As shown in Fig. 4(b1) and (b2), RP@PDA exhibited a rougher appearance, which can be attributed to the PDA nanoparticle accumulation on the surface.47 The nanoparticles indicated the possible formation mechanism of PDA films: as illustrated in Scheme 1, in the basic aqueous medium, dopamine can be oxidated by the oxygen in air, and generate a mass of melanin-like PDA particles and oligomers. With the assistance of mechanical stirring, the nanoparticles and oligomers with superior adhesive property aggregated on the surface of RP and formed PDA films. However, because the PDA film formed in a single reaction was thin, the overall appearance of RP@PDA was not changed significantly. For RP@PDA/MA, a distinct shell structure can be observed in Fig. 4(c1) and (c2), and the surface morphology of RP was changed significantly. Similarly, the PDA/MA shell structure was fabricated by the accumulation of the adhesive PDA/MA nanoparticles. The difference is that a Schiff base reaction can be conducted between PDA particles and MA as shown in Scheme 1. In theory, the three amino groups of MA can all react with PDA nanoparticles, which means that MA can function as a chemical binder and render more PDA deposit on the surface of RP, resulting in the significant core–shell structure of RP@PDA/MA. The morphologies presented by SEM micrographs, together with the results of FTIR and XPS mentioned above, indicated the successful fabrication of PDA and PDA/MA shell structures. | ||
| Fig. 4 SEM micrographs of (a1–a2) RP, (b1–b2) RP@PDA, and (c1–c2) RP@PDA/MA. | ||
3.4 Thermogravimetric analysis of RP samples
The TG and DTG curves of RP, RP@PDA and RP@PDA/MA were presented in Fig. 5 with the detail data listed in Table 2, wherein Tonset was referred to the temperature of 5% weight loss, and Tmax represented the temperature at maximum weight loss rate. As shown in Fig. 5(a), RP showed a dramatical weight decay at 400–520 °C with almost no residual left at 600 °C, indicating its thorough sublimation under high temperature. Due to the decomposition of the shell structures, RP@PDA and RP@PDA/MA showed a weight loss prior to the sublimation of RP cores, which resulted in the significantly lowered Tonset. However, because of the char-forming ability of PDA and MA, a certain amount of char residuals can be fabricated at this period, which can provide shield effect for the RP cores beneath. As shown in Fig. 5(b), RP@PDA and RP@PDA/MA showed lower Tmax and mass loss rate at Tmax compared to those of pristine RP. The reason may be that RP can react with the shell structures to fabricated thermally stable phosphonate/phosphonamidate derivatives with the shell structures at high temperature.41 This reaction, together with the char residuals formed earlier, provided shield effect and suppressed the sublimation of RP efficiently, causing the lower Tmax and mass loss rate. During TGA tests, RP@PDA/MA exhibited the highest char weight ratio and the lowest mass loss rate at Tmax, indicating its superior char-forming ability and thermal stability.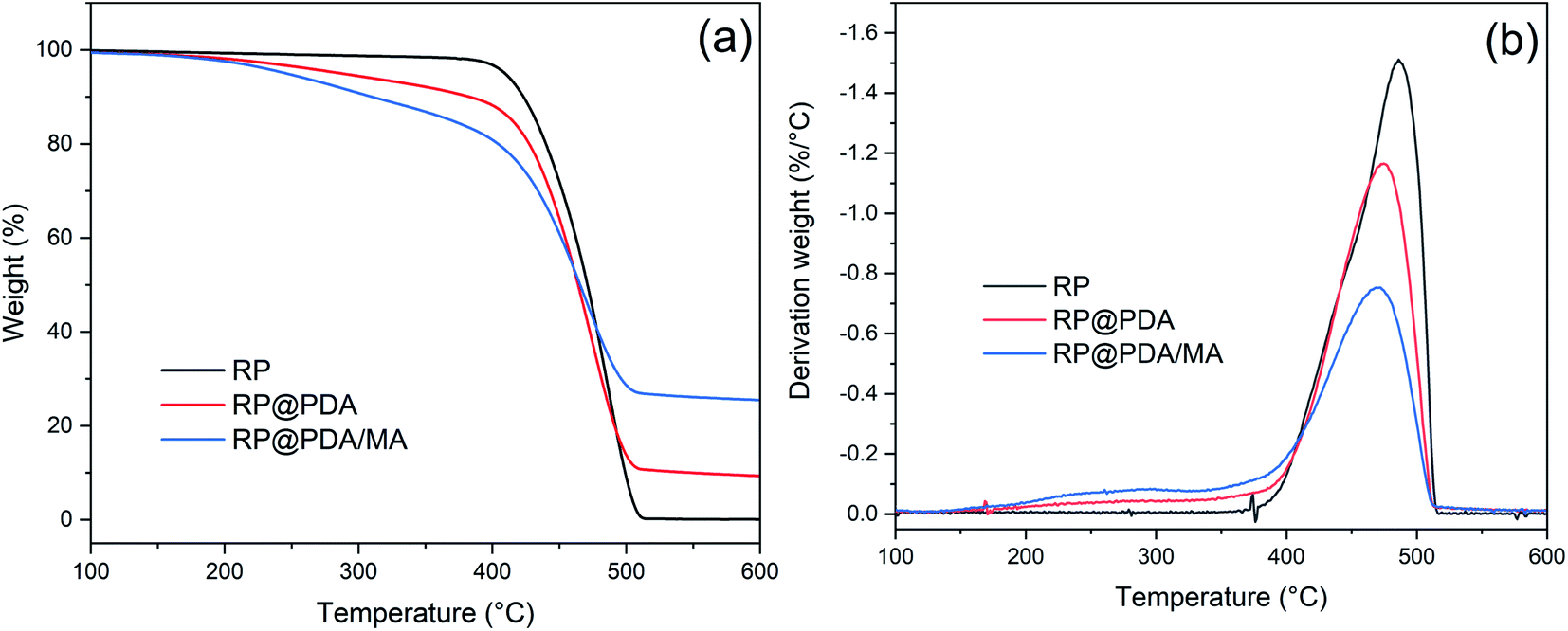 | ||
| Fig. 5 (a) TG and (b) DTG curves of RP, RP@PDA, and RP@PDA/MA. | ||
| Samples | Tonset (°C) | Tmax (°C) | Mass loss rate at Tmax (%/°C) | Residual mass (%) |
|---|---|---|---|---|
| RP | 410 | 486 | 1.51 | 0.08 |
| RP@PDA | 287 | 474 | 1.16 | 9.32 |
| RP@PDA/MA | 242 | 470 | 0.75 | 25.46 |
3.5 LOI and UL-94 tests of EP samples
The LOI and UL-94 tests were applied to assess the flammability of EP composites. As presented in Table 3, neat EP showed a low LOI value of 19.4% with no rating in UL-94 test, indicating its high inflammability and sever dripping problems. The addition of RP-containing flame retardants increased the LOI value of EP to different extent. However, blending with RP or RP@PDA individually still cannot make EP pass V-0 degree in UL-94 test. As for EP/RP@PDA/MA7.0, it got the highest LOI value (30.9%) and passed V-0 degree in UL-94 tests, meaning that the IFR system formed by RP@PDA/MA can efficiently improve the flame retardance and anti-dripping property of EP.| Samples | LOI (%) | Dripping | UL-94 |
|---|---|---|---|
| EP | 19.4 | Yes | No rating |
| EP/RP3.0 | 22.5 | Yes | No rating |
| EP/RP5.0 | 25.5 | Yes | No rating |
| EP/RP7.0 | 27.7 | Yes | V-1 |
| EP/RP@PDA3.0 | 24.4 | Yes | No rating |
| EP/RP@PDA5.0 | 27.6 | Yes | No rating |
| EP/RP@PDA7.0 | 28.5 | Yes | V-1 |
| EP/RP@PDA/MA3.0 | 27.3 | Yes | No rating |
| EP/RP@PDA/MA5.0 | 28.9 | Yes | V-1 |
| EP/RP@PDA/MA7.0 | 30.9 | No | V-0 |
3.6 Cone calorimeter test
Cone calorimeter test (CC) is a primary method to simulate the burning behavior of the samples in large-scale fire. The relative key parameters, such as peak heat release rate (PHRR), total heat release (THR), time to ignition (TTI), and residual mass were listed in Table 4, and the curves of heat release rate (HRR) and THR versus time were presented in Fig. 6. As shown in Fig. 6(a) and Table 4, PHRR of neat EP reached to 1183.7 kW m−2, indicating its violent burning behavior under the real fireground circumstance. The addition of RP-containing flame retardants significantly decreased the PHRR of neat EP, and the lowest PHRR was got by EP/RP@PDA/MA7.0. For THR curves, the addition of RP-containing flame retardants lowered the THR value of EP, meaning that partial EP has participated in the fabrication of char residuals instead of burning up.48| Sample | EP | EP/RP7.0 | EP/RP@PDA7.0 | EP/RP@PDA/MA7.0 |
|---|---|---|---|---|
| PHRR (kW m−2) | 1183.7 ± 29.8 | 839.8 ± 37.5 | 583.3 ± 30.4 | 412.6 ± 47.6 |
| THR (MJ m−2) | 101.0 ± 3.5 | 67.7 ± 0.3 | 64.8 ± 0.3 | 53.1 ± 0.5 |
| TTI (s) | 52 ± 3 | 36 ± 2 | 40 ± 5 | 64 ± 4 |
| Residual mass (wt%) | 1.7 ± 0.3 | 10.4 ± 1.7 | 12.1 ± 2.6 | 23.1 ± 2.2 |
| FGI (s m2 kW−1) | 0.044 | 0.042 | 0.068 | 0.155 |
| PSPR (m2 s−1) | 0.31 ± 0.01 | 0.58 ± 0.03 | 0.36 ± 0.01 | 0.27 ± 0.01 |
| TSP (m2) | 29.5 ± 0.2 | 44.1 ± 2.0 | 42.7 ± 0.9 | 35.7 ± 0.4 |
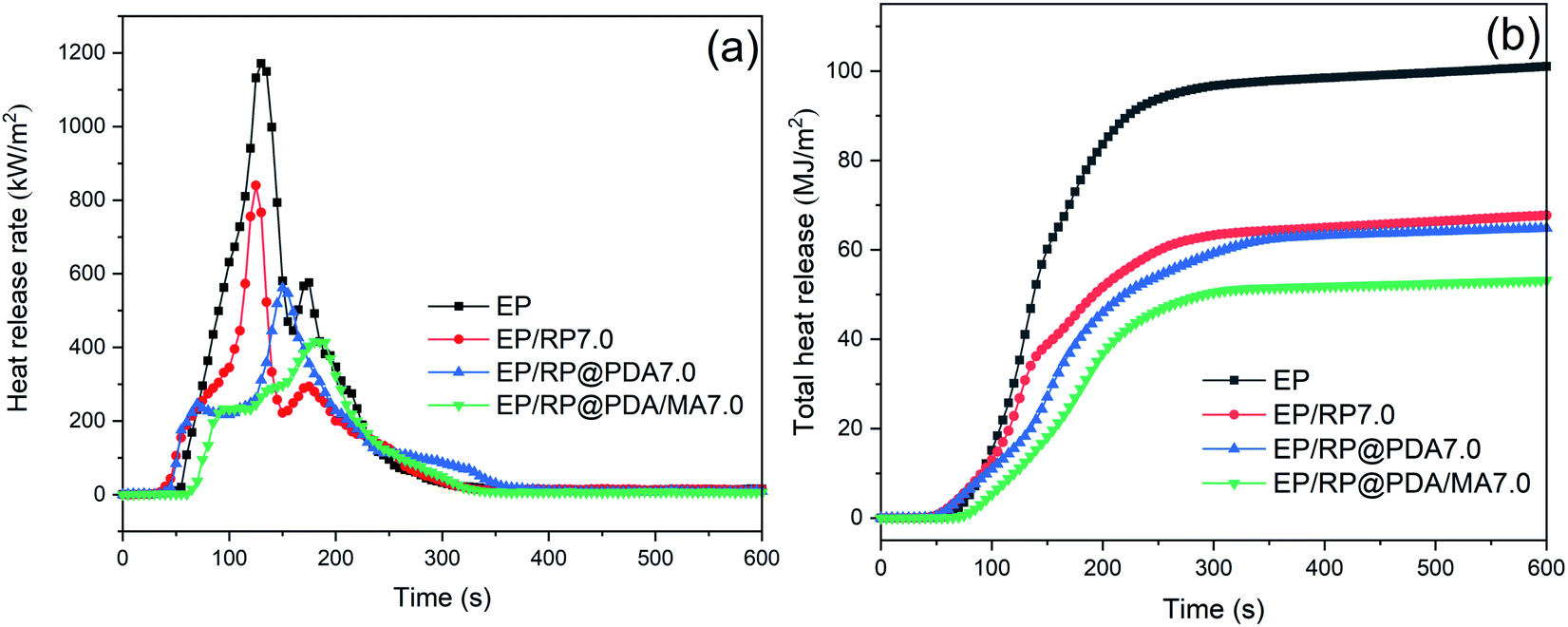 | ||
| Fig. 6 (a) HRR and (b) THR curves of EP samples. | ||
Fire propagation index (FPI), defined as the ratio of TTI and PHRR, was applied to evaluate the difficult degree of flashover. Larger FPI generally means for lower fire hazard. It can be seen in Table 4 that EP/RP@PDA/MA7.0 got the largest FGI during CC. Together with the HRR and THR curves, it can be concluded that RP@PDA/MA possessed better flame retardance than that of RP or RP@PDA.
The curves of smoke production rate (SPR) and total smoke production (TSP) were presented in Fig. 7 to evaluate the smoke suppression performance of the flame retardants. The data of peak smoke production rate (PSPR) and total smoke production (TSP) were listed in Table 4. It can be observed in Fig. 7(a) that EP/RP7.0 got the highest PSPR, and the TSP of EP/RP7.0 was significantly increased compared to that of pristine EP as shown in Fig. 7(b). This phenomenon may be caused by the gas phase flame retardance of RP.49 In the gas phase, RP can transfer into volatile phosphorous compounds, such as P˙, PO˙, PO2˙, and HPO˙, which will further trap the active radicals (H˙ and HO˙) and break down the chain reaction in the flame. In this process, a mass of incomplete-oxidation products can be generated thus caused the increased smoke emission. However, as illustrated by the results of TGA mentioned above, PDA/MA shell structure significantly suppressed the sublimation rate of RP, meaning that when RP@PDA/MA acted as the flame retardant, more phosphorus elements tended to function in condensed phase to fabricate thermostable char residuals instead of emitting to the gas phase, which was beneficial to the smoke suppression. The results of cone calorimeter tests demonstrated that RP@PDA/MA possessed better smoke suppression and flame retardance performance simultaneously, making it more suitable for EP flame-retardant treatment.
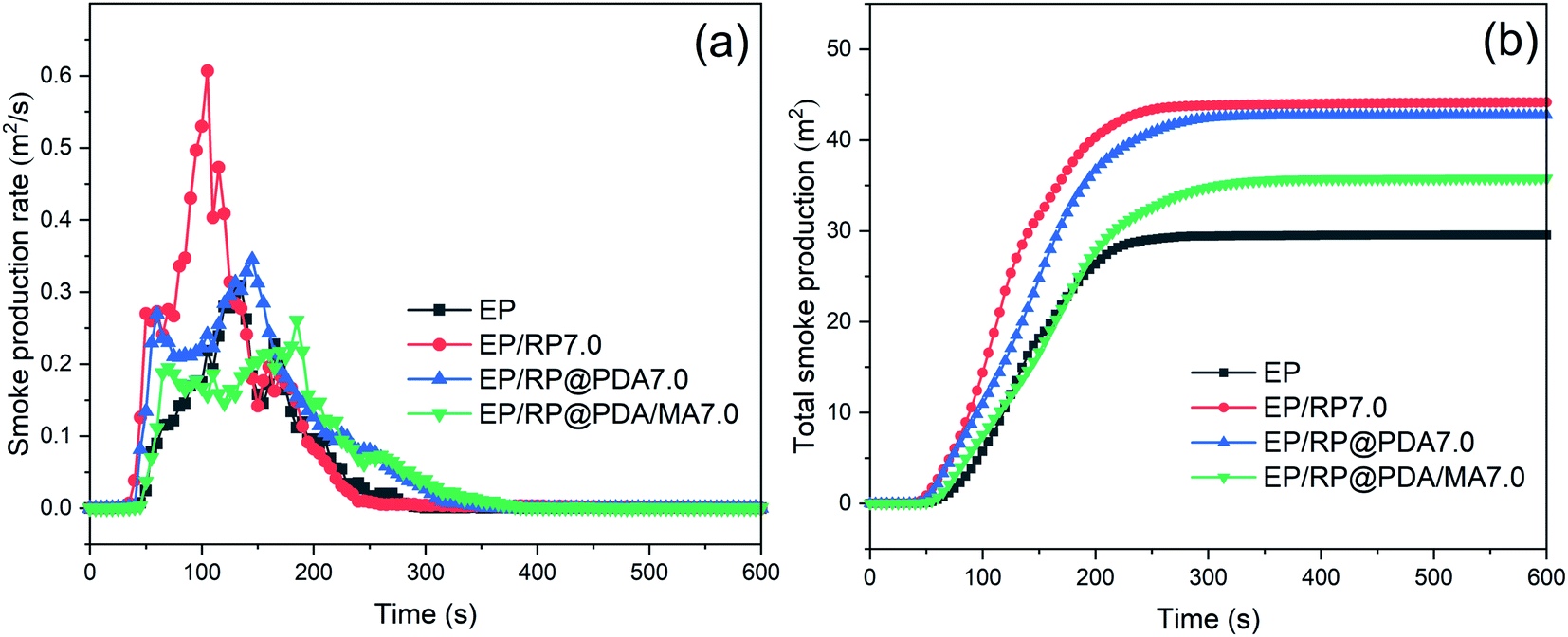 | ||
| Fig. 7 (a) SPR and (b) TSP curves of EP samples. | ||
3.7 Characterization of the char residuals
To investigate the flame retardance mechanism, the char residuals formed by FR-EP after CC tests were characterized. As shown in Fig. 8(a1)–(c1), owing to the IFR system formed by RP@PDA/MA, EP/RP@PDA/MA7.0 fabricated intumescent char residuals after CC tests. The SEM micrographs of the char residuals were shown in Fig. 8(a2)–(c2). After CC tests, three FR-EP samples formed porous char residuals. The reason for this morphology is that under high temperature, EP decomposed and generated a mass of gaseous products. Meanwhile, the RP-containing flame retardants catalyzed the EP substrates or shell structures to form char residuals, which were supposed to retard the emission of the gaseous products. However, if the char residuals were not compact enough, a mass of gaseous products can first form bubble-like structures on the char residuals, then disrupted them and formed porous structures.50 The bubble-like structures observed in the SEM micrographs verified this formation mechanism. Based on this, the distinct porous structures shown in Fig. 8(a2) and (b2) indicated that the char residuals formed by EP/RP7.0 and EP/RP@PDA7.0 cannot retard the escape of flammable gaseous products to the flame zone efficiently. Additionally, the holes on the char residuals can act as the channel for the exchange of heat and oxygen, which may also do harm to the flame retardance performance of EP composites.51 In Fig. 8(c2), though EP/RP@PDA/MA still fabricated the char residuals with a porous structure, the diameter of the holes was decreased at the same magnification, meaning that the EP beneath has been protected properly. This was because RP@PDA/MA constituted integrated IFR system, which can fabricate more compact char residuals and retard the emission of gaseous products efficiently, as well as isolate the oxygen and heat from the EP beneath. The intumescent morphology and compact structures of the char residuals can be regarded as the reason for the superior flame retardance of EP/RP@PDA/MA7.0.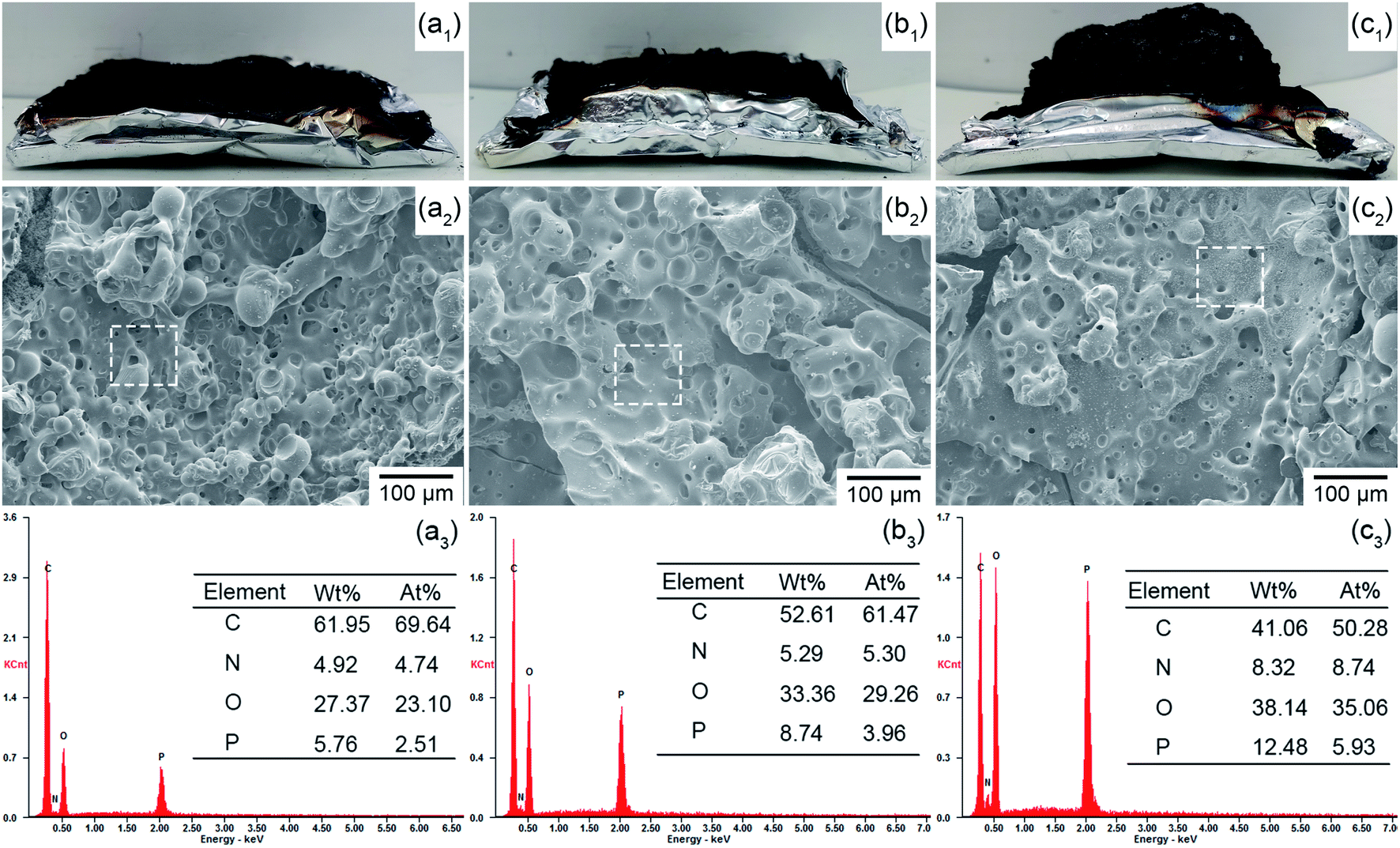 | ||
| Fig. 8 Char residuals analysis. (a1–c1) Digital photos, (a2–c2) SEM micrographs, (a3–c3) EDS results; (a1–a3) EP/RP7.0, (b1–b3) EP/RP@PDA7.0, (c1–c3) EP/RP@PDA/MA7.0. | ||
Fig. 8(a3)–(c3) present the EDS results of the char residuals with the selected areas shown in Fig. 8(a2)–(c2). The EDS results demonstrated that the three FR-EP samples fabricated phosphorus-containing char residuals after CC, wherein the char residuals formed by EP/RP@PDA/MA7.0 got the highest phosphorus weight ratio (wt%) and atomic ratio (at%), meaning that more phosphorus elements of RP@PDA/MA functioned in the condensed phase instead of sublimating. This result was consistent with the better smoke suppression performance of EP/RP@PDA/MA7.0.
To investigate the graphitic structures and the graphitization degree of the char residuals, the Raman spectra were characterized and the results were shown in Fig. 9. Two typical peaks corresponding to the carbon materials, i.e., the D-band (1360 cm−1) and the G-band (1585 cm−1), were presented in the Raman spectra of the char residuals. Generally, the graphitization degree of the char residuals can be estimated by the ratio of the integrated area of D-band and G-band (AD/AG). Lower AD/AG means for better carbon structure.52 Hence, according to the results of the Raman spectra, it can be concluded that EP/RP@PDA/MA7.0 formed a char residual with higher graphitization degree that can protect the underlying substrates more efficiently.
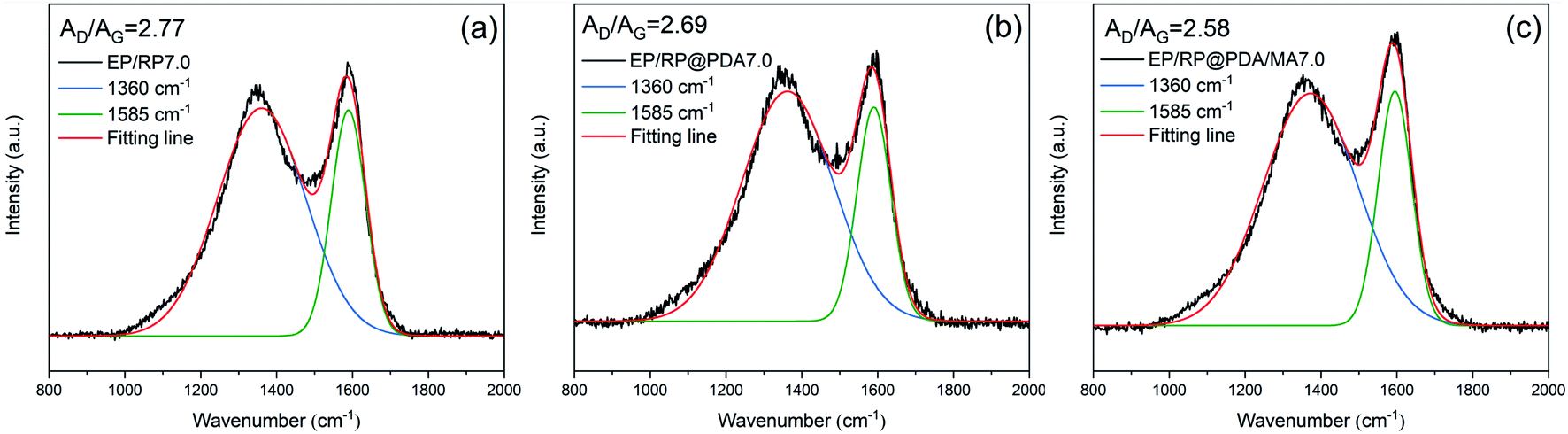 | ||
| Fig. 9 Raman spectra of the char residuals of (a) EP/RP7.0, (b) EP/RP@PDA7.0, and (c) EP/RP@PDA/MA7.0. | ||
3.8 Thermogravimetric analysis of EP samples
TGA were applied to investigate degradation behavior of EP samples. The thermal properties of EP samples in nitrogen were characterized with the TG and DTG curves presented in Fig. 10(a) and (b), respectively. As shown in Fig. 10(a), EP and FR-EP showed dramatical one-step weight loss at the range of 350–450 °C. The char residual weight ratio of FR-EP at 800 °C was increased, meaning that partial EP participated in the char-forming process. In Fig. 10(b), it can be observed that EP/RP@PDA/MA7.0 got the lowest weight loss rate at Tmax, which can be attributed to the high thermal stability and efficient char-forming ability of RP@PDA/MA. Additionally, at the range of 410–475 °C, the weight loss rate of FR-EP was significantly lower than that of neat EP. The reason may be that RP reacted with water vaper at this temperature range and generated H3PO4, which further catalyzed the char-forming process of the decomposing EP thus suppressed its degradation rate.16 This phenomenon was consistent with the increased char residuals of FR-EP shown in Fig. 10(a), and the highest char residual weight ratio and lowest weight loss rate at Tmax of EP/RP@PDA/MA7.0 indicated that RP@PDA/MA can improve the thermal stability of EP efficiently.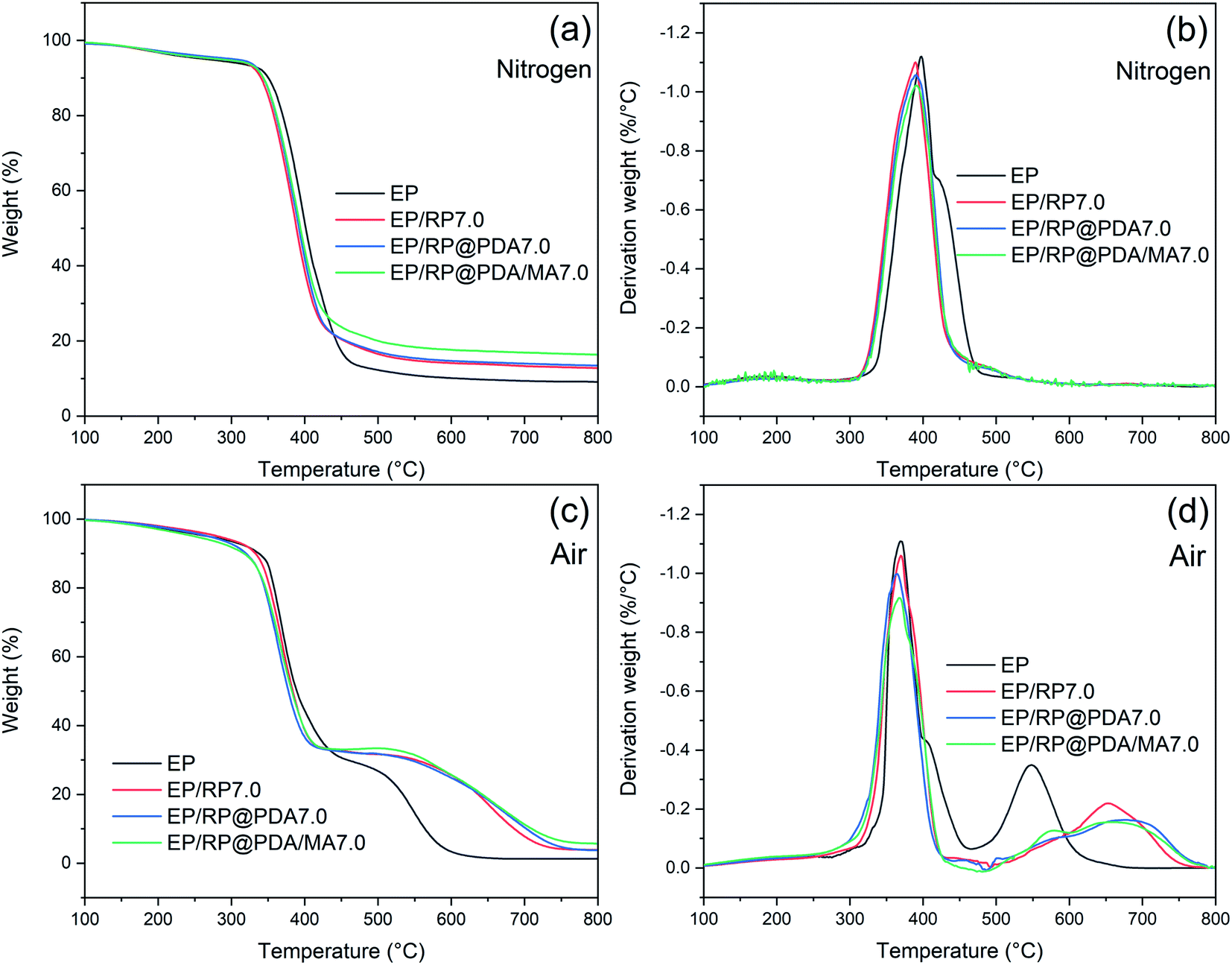 | ||
| Fig. 10 (a) TG and (b) DTG curves of EP samples in nitrogen; (c) TG and (d) DTG curves of EP samples in air. | ||
The decomposition behavior of EP samples in air was also investigated with the TG and DTG curves presented in Fig. 10(c) and (d), respectively. As shown in Fig. 10(c), EP exhibited a typical two-step weight loss behavior. The first weight loss step at 300–400 °C can be attributed to the decomposition of the main chain of EP, and the second one at 500–600 °C was due to the oxidation of the carbon layer.53 FR-EP showed similar weight loss behavior as neat EP did. However, it should be noted that at the range of 400–500 °C, the decomposition rate of FR-EP samples was dramatically slowed down. This phenomenon was caused by the rapid oxidation of RP, a weight increasing reaction that turned it into phosphoric oxide and phosphoric acid in air.54 The TG and DTG curves at 400–800 °C reflected the thermal stability of the char residuals. As shown in Fig. 10(c), at approximately 600 °C, the char residuals formed by EP were almost totally collapsed down, and a distinct minor weight loss peak emerged at 550 °C in Fig. 10(d). However, the char residuals of FR-EP can still exist at approximately 750 °C, and the temperature for the minor weight loss peaks was higher, which indicated the improved thermal stability of the phosphorus-containing char residuals. Furthermore, compared to EP/RP7.0, the minor peaks of EP/RP@PDA7.0 and EP/RP@PDA/MA7.0 were less significant. The reason may be that the abundant aromatic structures of PDA further improved the thermal stability of the char residuals and suppressed the degradation at high temperature.55
3.9 Analysis of the pyrolysis in the gas phase
To further investigate the decomposition behavior of the EP samples, TG-IR was conducted to track the production of volatiles under nitrogen atmosphere. Fig. 11 exhibited the FTIR spectra of the pyrolysis products at different temperature. The main characteristic peaks of EP and EP/RP@PDA/MA7.0, i.e., H2O or phenol (3650 cm−1), hydrocarbons (2930 cm−1), CO2 (2360 cm−1), carbonyl compounds (1740 cm−1), and aromatic compounds (1510 cm−1) were similar, indicating that these pyrolysis products were mainly caused by the decomposition of EP substrates.48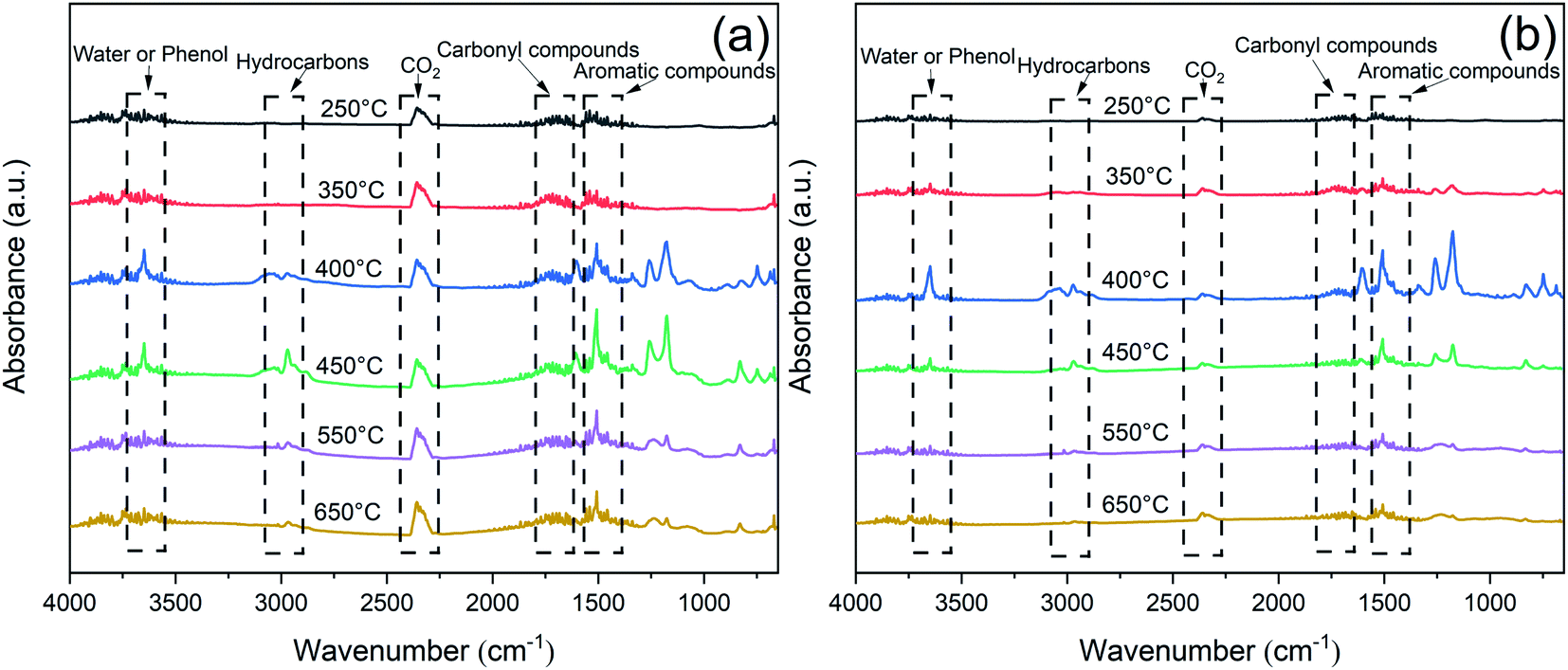 | ||
| Fig. 11 FTIR spectra of the pyrolysis products of (a) EP and (b) EP/RP@PDA/MA7.0. | ||
The curves of absorbance intensity vs. time/temperature were presented in Fig. 12. The intensities of the absorbance peaks of EP/RP@PDA/MA7.0, including the total gas, hydrocarbons, aromatic compounds, carbonyl compounds, and CO2, were decreased significantly compared to those of EP. The results indicated that RP@PDA/MA was capable of suppressing the release of the gas components, which can be attributed to retardation effect of the carbon layer formed by the IFR. Actually, most of the organic pyrolysis products can act as the fuel to support burning process, hence the suppressed organic gas emission can be the reason for the improved flame retardance of EP/RP@PDA/MA7.0.
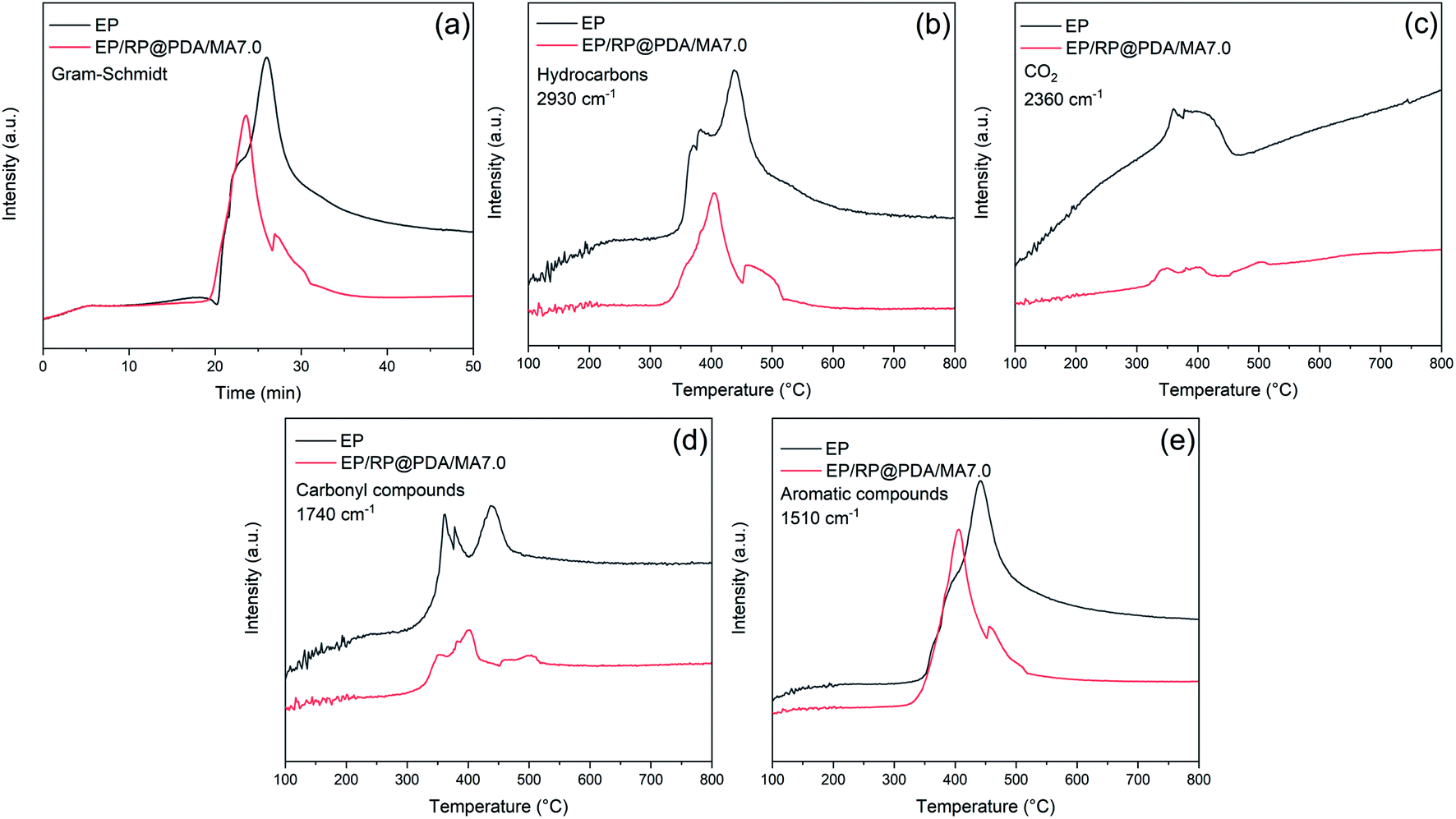 | ||
| Fig. 12 Absorbance intensity of pyrolysis products for EP and EP/RP@PDA/MA7.0 vs. time/temperature: (a) total gas; (b) hydrocarbons; (c) CO2; (d) carbonyl compounds; (e) aromatic compounds. | ||
3.10 Flame retardance mechanism
Based on the results of the tests mentioned above, the flame retardance mechanism of RP@PDA/MA is proposed in Scheme 2. The core–shell structures of RP@PDA/MA fabricated an integrated IFR system, in which RP functioned as the acid source, and the PDA/MA shell structure acted as the carbon source and blowing agent simultaneously. Due to the wrapping effect of the shell structures, more phosphorus elements of RP@PDA/MA tended to function in the condensed phase and fabricate thermostable phosphorus-containing char residuals. Meanwhile, the abundant aromatic structures of PDA further improved the thermal stability of the char residuals, and MA endowed them with an intumescent appearance. The intumescent char residuals can efficiently impede the oxygen and heat from penetrating the underlying EP substrates, as well as retard the emission of the flammable pyrolysis products, leading to the satisfactory flame retardance of EP/RP@PDA/MA7.0.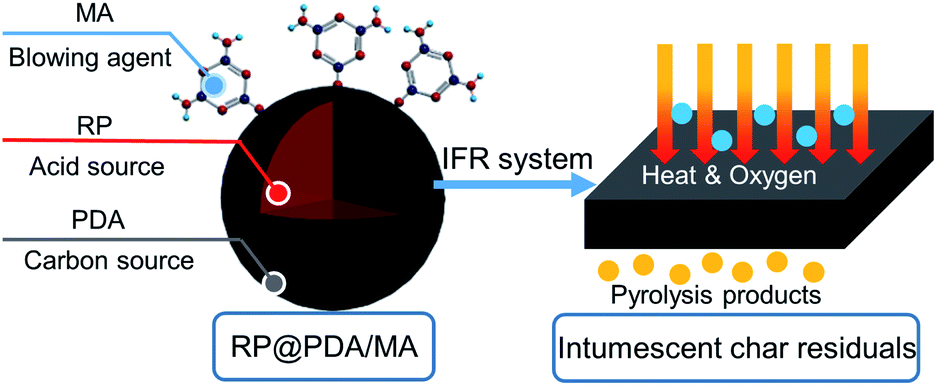 | ||
| Scheme 2 Flame retardance mechanism of RP@PDA/MA. | ||
3.11 Tensile properties of EP composites
To investigate the impact of the flame retardants on the mechanical properties of EP, the tensile tests were conducted with detail results presented in Fig. 13 and Table 5. It can be observed that due to the poor compatibility between RP and EP substrates, EP/RP7.0 exhibited the lowest tensile strength, elongation at break and Young's modulus in this test. However, EP/RP@PDA7.0 and EP/RP@PDA/MA7.0 showed better mechanical properties, which can be attributed to that the inorganic–organic interfaces between RP and EP were transferred into the organic/organic ones by the PDA and PDA/MA shell structures. The results of the tensile tests indicated that the PDA and PDA/MA shell structures improved the compatibility between RP and the organic substrates.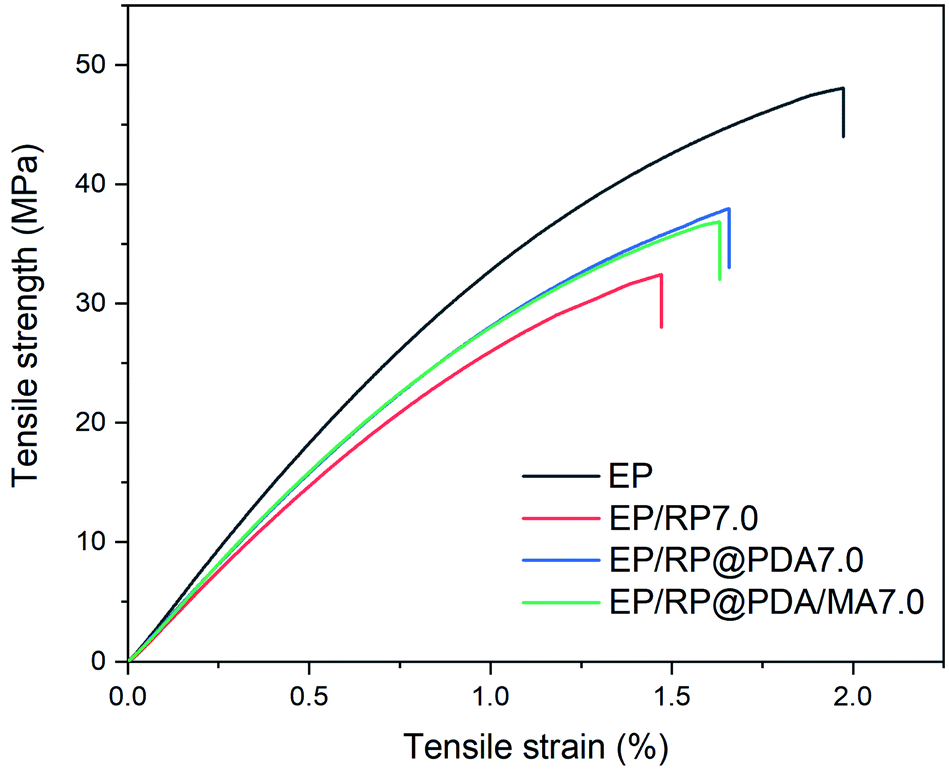 | ||
| Fig. 13 Typical stress–strain curves of EP samples. | ||
| Sample | Elongation at break (%) | Tensile strength (MPa) | Young's modulus (GPa) |
|---|---|---|---|
| EP | 1.83 ± 0.21 | 46.9 ± 4.3 | 3.77 ± 0.15 |
| EP/RP7.0 | 1.33 ± 0.20 | 31.0 ± 2.6 | 2.98 ± 0.11 |
| EP/RP@PDA7.0 | 1.66 ± 0.21 | 37.7 ± 3.5 | 3.26 ± 0.09 |
| EP/RP@PDA/MA7.0 | 1.63 ± 0.09 | 36.9 ± 1.5 | 3.16 ± 0.20 |
4. Conclusions
Substituting part of the petroleum raw materials by bio-based PDA, this work has developed a “green” approach to fabricate MRP flame retardant with MA-containing shell structures. RP@PDA/MA constituted an integrated IFR system, and EP blending with 7wt% RP@PDA/MA got satisfactory flame retardance and smoke suppression performance, simultaneously. As far as it goes, the PDA/MA shell structures improved the flame retardance performance and compatibility of pristine RP. Furthermore, the superior adherence of PDA and the easy-to-implement method used in this work mean that the PDA/MA shell structure can be easily fabricated on the surface of other phosphorus-containing acid source to broaden their application field. Additionally, due to the feasible reaction between PDA and amino groups, other nitrogen type blowing agents may fabricate composite shell structure with PDA and have potential value. To comprehensively investigate the practicability of RP@PDA/MA, contrast experiments with traditional MA-containing shell structures microencapsulated RP are required in the future work. Moreover, this work preliminarily verified the feasibility of the preparation and application of RP@PDA as flame retardant for EP. To get better practical performance of RP@PDA/MA, the influence of the composition ratio on the morphology and properties needs to be investigated in the future work to determine the appropriate condition.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work wants to thank Shiyanjia Lab for the support of XPS analysis.Notes and references
- A. Shah, A. Ding, Y. Wang, L. Zhang, D. Wang, J. Muhammad, H. Huang, Y. Duan, X. Dong and Z. Zhang, Carbon, 2016, 96, 987–997 CrossRef CAS.
- J. Wan, B. Gan, C. Li, J. M. Aldareguia, X. Wang and D. Wang, Chem. Eng. J., 2016, 284, 1080–1093 CrossRef CAS.
- J. Wang, L. Qian, Z. Huang, Y. Fang and Q. Yong, Polym. Degrad. Stab., 2016, 130, 173–181 CrossRef CAS.
- Y. Shuang, J. Wang, S. Huo, L. Cheng and W. Mei, Polym. Degrad. Stab., 2015, 119, 251–259 CrossRef.
- Z. Zhu, L. Wang and L. Dong, Polym. Degrad. Stab., 2019, 162, 129–137 CrossRef CAS.
- Z. Shao, M. Zhang, Y. Li, Y. Han, L. Ren and D. Cong, Chem. Eng. J., 2018, 345, 471–482 CrossRef CAS.
- X. Li, F. Zhang, R. Jian, Y. Ai, J. Ma, G. Hui and D. Wang, Composites, Part B, 2019, 176, 107200 CrossRef CAS.
- R. Jian, Y. Ai, L. Xia, L. Zhao and H. Zhao, J. Hazard. Mater., 2019, 371, 529–539 CrossRef CAS PubMed.
- D. Enescu, A. Frache, M. Lavaselli, O. Monticelli and F. Marino, Polym. Degrad. Stab., 2013, 98, 297–305 CrossRef CAS.
- Y. Wang, X. Zhang, A. Li and M. Li, Chem. Commun., 2015, 51, 14801–14804 RSC.
- Y. Liu, B. Xu, L. Qian, Y. Chen and Y. Qiu, Polym. Adv. Technol., 2020, 31, 3316–3327 CrossRef CAS.
- W. Zhang, X. He, T. Song, Q. Jiao and R. Yang, Polym. Degrad. Stab., 2014, 109, 209–217 CrossRef CAS.
- J. Liu, H. Li, H. Chang, Y. He, A. Xu and B. Pan, Fire Mater., 2019, 43, 971–980 CrossRef CAS.
- B. Szolnoki, A. Toldy and G. Marosi, Phosphorus, Sulfur Silicon Relat. Elem., 2019, 194, 309–312 CrossRef CAS.
- Z. C ao, D. Xue, T. Fu, S. Deng, W. Liao and Y. Wang, Polym. Degrad. Stab., 2017, 136, 103–111 CrossRef CAS.
- J. Liu, Y. Guo, H. Chang, H. Li, A. Xu and B. Pan, Fire Mater., 2018, 42, 958–966 CrossRef CAS.
- L. A. Savas, T. K. Deniz, U. Tayfun and M. Dogan, Polym. Degrad. Stab., 2017, 135, 121–129 CrossRef CAS.
- D. Wang, H. He and P. Yu, J. Appl. Polym. Sci., 2016, 133, 43225 Search PubMed.
- A. Bifulco, D. Parida, K. A. Salmeia, S. Lehner and S. Gaan, Composites Part C: Open Access, 2020, 2, 100022 Search PubMed.
- Z. Zhu, P. Lin, H. Wang, L. Wang and F. Fang, J. Mater. Sci., 2020, 55, 12836–12847 CrossRef CAS.
- T. Ma and C. Guo, J. Anal. Appl. Pyrolysis, 2017, 124, 239–246 CrossRef CAS.
- S. Chang, Z. Chao, W. Yuan and J. Ren, J. Appl. Polym. Sci., 2012, 125, 3014–3022 CrossRef CAS.
- J. Gu, G. Zhang, S. Dong, Q. Zhang and K. Jie, Surf. Coat. Technol., 2007, 201, 7835–7841 CrossRef CAS.
- G. F. Levchik, S. A. Vorobyova, V. V. Gorbarenko, S. V. Lechik and E. D. Weil, J. Fire Sci., 2000, 18, 172–182 CrossRef CAS.
- J. Liu, Y. He, H. Chang, Y. Guo, H. Li and B. Pan, Polym. Degrad. Stab., 2020, 171, 109051 CrossRef CAS.
- S. Qiu, C. Ma, X. Wang, X. Zhou, X. Feng, R. K. K. Yuen and H. Yuan, J. Hazard. Mater., 2018, 344, 839–848 CrossRef CAS PubMed.
- H. Lee, E. Park and H. Lee, Adv. Mater., 2020, 32, e1907505 CrossRef PubMed.
- Y. Liu, K. Ai and L. Lu, Chem. Rev., 2014, 114, 5057–5115 CrossRef CAS PubMed.
- J. Waite and M. Tanzer, Science, 1981, 212, 1038–1040 CrossRef CAS PubMed.
- J. Waite, Int. J. Adhes. Adhes., 1987, 7, 9–14 CrossRef CAS.
- J. Korbel, A. Urban, J. Affourtit, B. Godwin, F. Grubert and J. Simons, Science, 2007, 318, 420–426 CrossRef CAS PubMed.
- J. Ryu, P. Messersmith and H. Lee, ACS Appl. Mater. Interfaces, 2018, 10, 7523–7540 CrossRef CAS PubMed.
- J. Simon and D. Peles, Acc. Chem. Res., 2010, 43, 1452–1460 CrossRef CAS PubMed.
- Q. Huang, J. Chen, M. Liu, H. Huang, X. Zhang and Y. Wei, Chem. Eng. J., 2020, 387, 124019 CrossRef.
- Á. Molnár, ChemCatChem, 2020, 12, 2649–2689 CrossRef.
- Z. Yan, Y. Zhang, H. Yang, G. Fan, A. Ding, H. Liang, G. Li, N. Ren and B. Bruggen, Chem. Eng. Res. Des., 2020, 157, 195–214 CrossRef CAS.
- X. Yu, X. Tang, J. He, X. Yi, G. Xu, L. Tian, R. Zhou, C. Zhang and K. Yang, Advanced Science News, 2017, 34, 1600296 Search PubMed.
- H. Yang, J. Luo, Y. Lv, P. Shen and Z. Xu, J. Membr. Sci., 2015, 483, 42–59 CrossRef CAS.
- S. Zhu, W. Wang, Z. Islam, Y. Fu and Y. Dong, J. Appl. Polym. Sci., 2021, 138, 49696 CrossRef CAS.
- W. Yang, S. Wu, W. Yang, A. Yuen, Y. Zhou, G. Yeoh, C. Boyer and C. Wang, Composites, Part B, 2020, 186, 107828 CrossRef CAS.
- L. Zhang, Q. Wang, R. Jian and D. Wang, J. Mater. Chem. A, 2020, 8, 2529–2538 RSC.
- Y. Wang, Y. Zhang, C. Hou and M. Liu, J. Taiwan Inst. Chem. Eng., 2016, 61, 292–298 CrossRef CAS.
- A. Postma, Y. Yan, Y. Wang, A. N. Zelikin, E. Tjipto and F. Caruso, Chem. Mater., 2009, 21, 3042–3044 CrossRef CAS.
- W. Cai, W. Guo, Y. Pan, J. Wang and X. Mu, Composites, Part A, 2018, 111, 94–105 CrossRef CAS.
- B. Feng, X. Wu, Y. Niu, W. Li, Y. Yao, W. Hu and C. Li, Sustainable Energy Fuels, 2019, 3, 3455–3461 RSC.
- M. Zhu, Y. Muhammad, P. Hu, B. Wang, Y. Wu, X. Sun, Z. Tong and Z. Zhao, Appl. Catal., B, 2018, 232, 182–193 CrossRef CAS.
- J. Jiang, L. Zhu, L. Zhu, B. Zhu and Y. Xu, Langmuir, 2011, 27, 14180–14187 CrossRef CAS PubMed.
- Z. Xiong, Y. Zhang, X. Du, P. Song and Z. Fang, ACS Sustainable Chem. Eng., 2019, 7, 8954–8963 CrossRef CAS.
- U. Braun and B. Schartel, Macromol. Chem. Phys., 2004, 205, 2185–2196 CrossRef CAS.
- Y. Zhang, B. Yu, B. Wang, K. Liew, L. Song, C. Wang and Y. Hu, Ind. Eng. Chem. Res., 2017, 56, 1245–1255 CrossRef CAS.
- Y. Sui, L. Qu, X. Dai, P. Li, J. Zhang, S. Luo and C. Zhang, RSC Adv., 2020, 10, 12492 RSC.
- Y. Pan, J. Zhan, H. Pan, W. Wang, G. Tang, L. Song and Y. Hu, ACS Sustainable Chem. Eng., 2016, 4, 1431–1438 CrossRef CAS.
- Y. Xu, J. Wang, Y. Tan, M. Qi, L. Chen and Y. Wang, Chem. Eng. J., 2018, 337, 30–39 CrossRef CAS.
- J. Liu, S. Peng, Y. Zhang, H. Chang, Z. Yu, B. Pan, L. Chang, J. Ma and Q. Niu, Polym. Degrad. Stab., 2015, 121, 208–221 CrossRef CAS.
- W. Lu, Q. Li, Y. Zhang, H. Yu, S. Hirose, H. Hatakeyama, Y. Matsumoto and Z. Jin, J. Wood Sci., 2018, 64, 287–293 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2021 |