
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceGreen chemical and biological synthesis of cadaverine: recent development and challenges
Yuhong Huang *abcd,
Xiuling Jia,
Zhanling Mac,
Mateusz Łężyke,
Yaju Xuea and
Hai Zhaob
*abcd,
Xiuling Jia,
Zhanling Mac,
Mateusz Łężyke,
Yaju Xuea and
Hai Zhaob
aBeijing Key Laboratory of Ionic Liquids Clean Process, CAS Key Laboratory of Green Process and Engineering, State Key Laboratory of Multiphase Complex Systems, Institute of Process Engineering, Chinese Academy of Sciences, Beijing 100190, China. E-mail: yhhuang@ipe.ac.cn
bInnovation Academy for Green Manufacture, Chinese Academy of Sciences, Beijing 100190, China
cZhengzhou Institute of Emerging Industrial Technology, Zhengzhou City, Henan 450000, China
dZhongke Langfang Institute of Process Engineering, Langfang 065001, China
eWater Supply and Bioeconomy Division, Faculty of Environmental Engineering and Energy, Poznan University of Technology, Berdychowo 4, 60-965 Poznan, Poland
First published on 7th July 2021
Abstract
Cadaverine has great potential to be used as an important monomer for the development of a series of high value-added products with market prospects. The most promising strategies for cadaverine synthesis involve using green chemical and bioconversion technologies. Herein, the review focuses on the progress and strategies towards the green chemical synthesis and biosynthesis of cadaverine. Specifically, we address the specific biosynthetic pathways of cadaverine from different substrates as well as extensively discussing the origination, structure and catalytic mechanism of the key lysine decarboxylases. The advanced strategies for process intensification, the separation and purification of cadaverine have been summarized. Furthermore, the challenging issues of the environmental, economic, and applicable impact for cadaverine production are also highlighted. This review concludes with the promising outlooks of state-of-the-art applications of cadaverine along with some insights toward their challenges and potential improvements.
1. Introduction
Cadaverine (C5H14N2, molecular weight 102.18), a colorless viscous fuming liquid with a special odor, is also known as 1,5-diaminopentane, 1,5-pentanediamine and pentamethylene diamine. It is a natural polyamine with various chemical and biological activity in humans, animals, plants, bacteria, fungi and other microbes.1–3Recently, cadaverine has been found to be used as an important monomer to develop a series of high value-added products with commercial prospects. It can participate in plant physiological processes such as cell division and growth, promoting the development of pistils and stamens, regulating the senescence process, improving fruit development, and increasing yield.4 It also can be used to treat arrhythmia, relieve hypoglycemia, and effectively cure dysentery.5 Currently, much more attention is focused on the renewable polymers made by cadaverine, such as polyamide, polyurethane and polyurea.6 Among them, high-performance polyamide nylon 5X materials (for example, nylon 52, 5T, 54, 56, 510, 516 or 518, and so on) have excellent properties in terms of mechanical strength, air permeability, moisture absorption, and flame retardancy, and can be used in many important fields such as chemical fibers and engineering plastics, which have attracted much attention worldwide.7 Pentamethylene diisocyanate (PDI), a new aliphatic isocyanate, can be also synthesized from cadaverine and be applied in polyurethane coatings and adhesives. Both polyamide and polyurethanes are widely used in various fields globally as the multipurpose materials.8,9
The production of the above polyamide, polyurethane, polyurea and other polymers is restricted by the synthesis of cadaverine. Chemical synthesis of cadaverine usually faces serious equipment corrosion, low target product selectivity, poor catalyst stability, and incapability of continuous and stable production.10–24 Most importantly, the petroleum-based raw material has a significant impact on the environment. Synthesis of cadaverine through biocatalysis not only has good selectivity, but also can utilize renewable resources instead of fossil fuels, meeting the demand of the circular economy.25 However, bulk cadaverine is still not commercially available in the world due to the large expenditure of biosynthesis, which results from low enzyme stability and efficiency, complex cadaverine purification and wastewater treatment, addition of large amounts of acids and alkalis, and expensive inducers and co-factors.26–28
Recently, researchers and companies have set about solving the problems and challenges for efficient and economic cadaverine production. Significant progress has been achieved for the chemical and biological synthesis of cadaverine in the last 10 years, especially the fast development of synthetic biology in the construction of novel recombinant engineered strains with highly efficient and stable lysine decarboxylases and specific enzymes for in situ generation of co-factor pyridoxal phosphate (PLP), which can solve the challenges for efficient cadaverine production at industrial scale, even avoiding using large amounts of acids and alkalis for pH titration during the decarboxylation process. The combination of the advanced intensification process will further facilitate the downstream purification and wastewater treatment. Therefore, the significant progress for the biosynthesis of L-lysine to cadaverine has further promoted the efficient and economic production of cadaverine at industrial scale.
2. Chemical synthesis of cadaverine
Hydrogenation of glutaronitrile with the amorphous Ni alloy catalyst was first applied for cadaverine synthesis at early stage.10 It was widely accepted that glutaronitrile was first hydrogenated to 5-aminovaleronitrile and then further hydrogenated to cadaverine (Scheme 1A).11,12 However, the active intermediate 5-aminovaleronitrile can form undesired cyclic compound piperidine.12 Meanwhile, the synthesis of glutaronitrile required highly toxic cyanide as a precursor, bringing tremendous security challenges to mass production.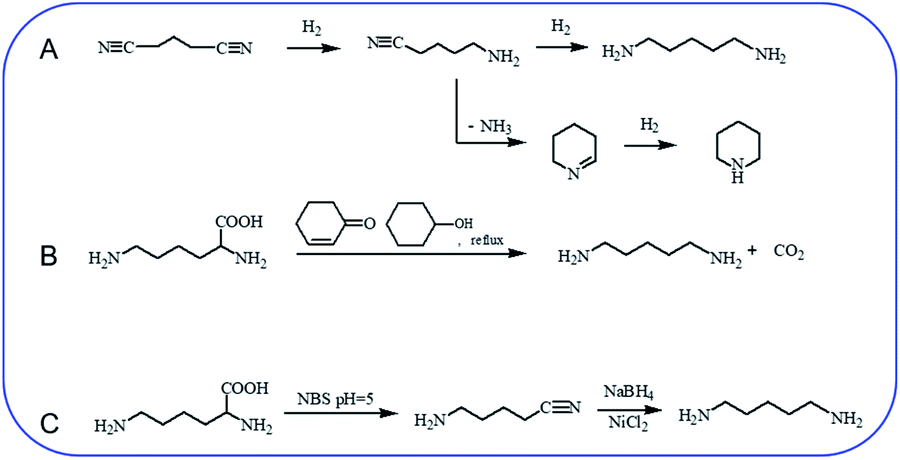 | ||
| Scheme 1 Chemical synthesis of cadaverine. (A) Hydrogenation of glutaronitrile for cadaverine synthesis,10,12 (B) decarboxylation of L-lysine to cadaverine by using 2-cyclohexen-1-one,14 (C) two-step decarboxylation of L-lysine to cadaverine.15 | ||
Chemo-catalytic decarboxylation of L-lysine is one alternative route to produce cadaverine with a high reaction efficiency. L-Lysine is one kind of high-valued renewable resources that can be easily obtained through a sustainable way of the microbial fermentation process. Its annual production has been reported to be 1210 tons and increases by about 4% per year.13 L-Lysine is now widely regarded as a key platform chemical due to its interesting functionality involving two amine groups and one carboxyl group to produce various value-added materials. In 1986, Hashimoto et al.14 were the first to report the decarboxylation of L-lysine to cadaverine with a yield of 87.8% by using 2-cyclohexen-1-one as the homogeneous catalyst (Scheme 1B). Nevertheless, 2-cyclohexen-1-one is toxic and has a similar boiling temperature to cadaverine, which seriously restricts its downstream purification and industrial application.
A two-step decarboxylation of L-lysine in a homogeneous phase was later proposed by Gilles,15 as shown in Scheme 1C. Firstly, L-lysine was converted to the corresponding nitrile in the presence of N-bromosuccinimide (NBS) in phosphate buffer at pH 5. Subsequently, the nitrile was reduced to cadaverine by nickel chloride hexahydrate/sodium borohydride. An overall yield of 77% cadaverine was finally obtained. However, NBS, as well as nickel chloride hexahydrate/sodium borohydride, was consumed and could not be reused, inhibiting the wide application of this two-step decarboxylation method.
Generally, homogeneous catalysis faces the challenge of product separation and purification. In contrast, heterogenous catalysis has the advantage of simple product recovery by centrifugation. Therefore, novel processes of heterogeneous chemo-catalytic decarboxylation of α-amino acid have been rapidly developed, among which heterogeneous catalysts supported by Pd or Ru are mostly studied. De Vos et al.16 studied Pd-catalyzed decarboxylation of pyroglutamic acid to 2-pyrrolidone. A wide range of supported Pd catalysts was evaluated and the highest yield (70%) was obtained with Pd/Al2O3 catalyst at 250 °C. Pd/Al2O3 catalyst also performed well in the decarboxylation of glutamic acid to 2-pyrrolidone. Jasper et al.17 modified supported Pd catalysts with Pb via two incipient wetness impregnation steps to catalyze the decarboxylation of proline to pyrrolidine. After modification with Pb, a much higher selectivity of pyrrolidine (95%) was obtained than using conventional Pd catalysts. The reason seemed to be that Pd became more electron-rich after modification with Pb, which facilitated desorption of N-containing chemical and subsequently avoided further side-reaction. However, supported Pd catalysts performed poorly in the production of primary amines from the amino acid.
Supported Ru was effective in the decarboxylation of amino acids to primary amines. It had been reported that hydrogenation-decarboxylation of L-lysine to cadaverine was successfully catalyzed by supported Ru with a yield of 32%.18 Although supported Ru was effective, the yield of cadaverine was still not ideal. The low yield of cadaverine could be attributed to the relatively high multi-functionality of L-lysine with two amino groups (–NH2) and one carboxyl group (–COOH). For example, Frost et al.19,20 investigated the conversion of L-lysine to caprolactam with a yield of 75% through the cyclization and deamination process. Hydrogenolysis of L-lysine to caprolactam was also studied in a one-pot catalytic reaction using a multifunctional heterogeneous catalyst Ir/HB-124.21 L-Lysine, as an alkaline amino acid, had four different dissociation states in an aqueous solution, which highly depended on pH (Fig. 1A).22,23 For pH from 1 to 8, L-lysine mainly existed as the cationic and dicationic forms. It shifted gradually to an anionic form as pH increased. A low pH was essential for decarboxylation of amino acids because hydrogen protons can protect amino groups and inhibit further deamination reaction.
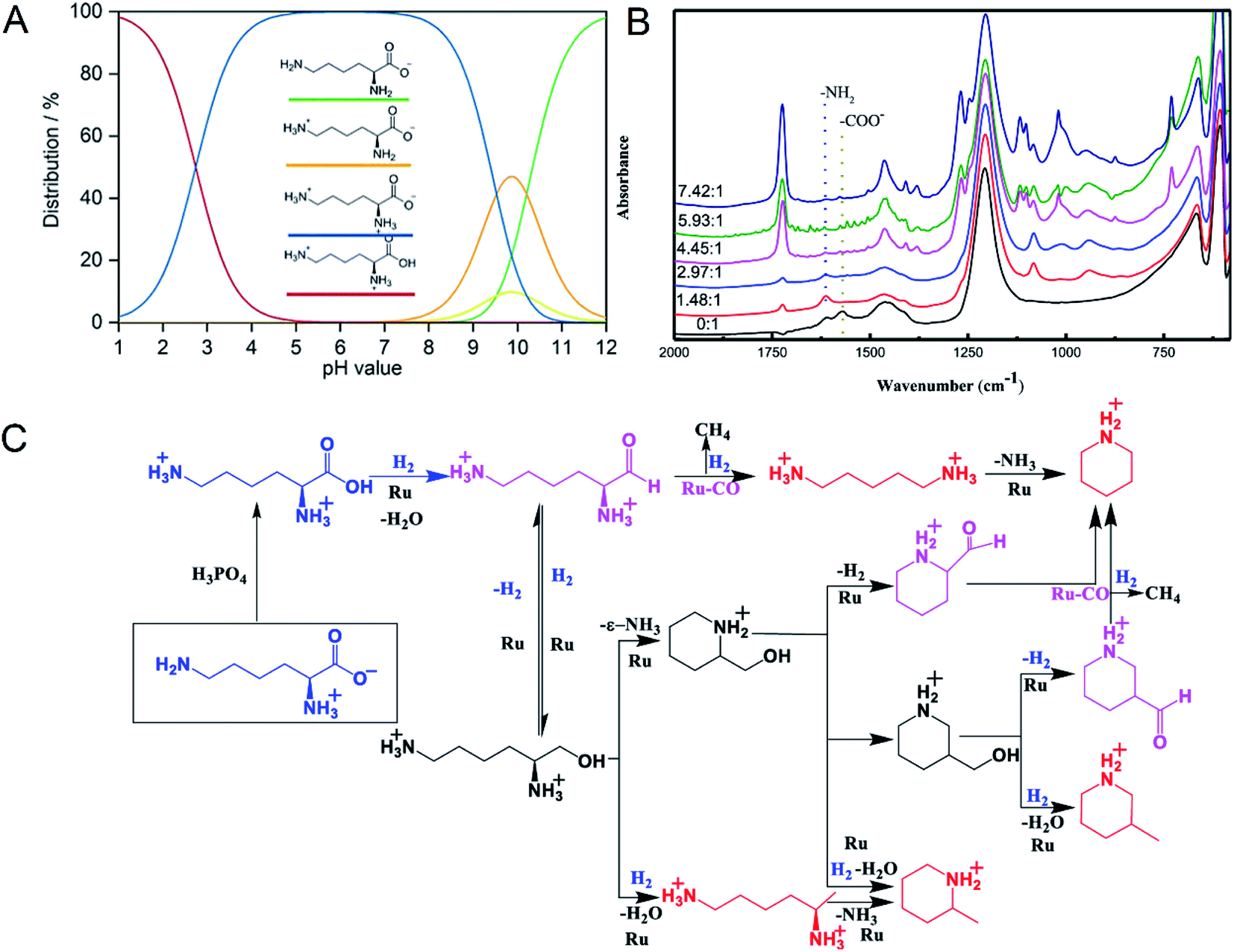 | ||
| Fig. 1 (A) L-lysine dissociation states at different pH values. Reprinted with permission,22 Copyright (2020) John Wiley and Sons. (B) In situ FTIR-ATR spectra of L-lysine in D2O at room temperature with different molar ratios of H3PO4 to L-lysine. Reprinted with permission,24 Copyright (2020) ACS publications. (C) Overall reaction process of the deoxygenation of L-lysine to diamines and other N-containing chemicals. Reprinted with permission,24 Copyright (2020) ACS publications. | ||
Recently, Xie et al.24 studied in detail the deoxygenation of L-lysine to prepare bio-based amines using Ru/C as a candidate catalyst. They found that without an acidic environment L-lysine mainly underwent deamination. However, it was also harmful for cadaverine production in a strong acid environment. Adequate acidity was necessary for highly selective deoxygenation or decarboxylation reaction to cadaverine. The yield of cadaverine was found to increase with the molar ratio of H3PO4 to L-lysine in a range of 0 to 6. The effect of different concentrations of H3PO4 on L-lysine dissociation states in D2O solution was analyzed by FTIR-ATR (Fig. 1B). When the molar ratio of H3PO4 to L-lysine was 0![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, no peak at 1724 cm−1 of –COOH group was observed, suggesting that the carboxyl group was deprotonated by neighboring amino groups. With the addition of a bit of H3PO4, peaks of carboxylate ions (–COO−) at 1570 cm−1 disappeared, indicating that H3PO4 was utilized to protonate –COO− to form –COOH. It was further verified by the growing peak intensity of the carboxyl group (–COOH) at 1724 cm−1. With a suitable amount of H3PO4, both amino groups and carboxyl groups in L-lysine were completely protonated, which was confirmed by the disappearance of –NH2 group peaks at 1614 cm−1. Complete protonation of all amino acids inhibited the deamination.
:1, no peak at 1724 cm−1 of –COOH group was observed, suggesting that the carboxyl group was deprotonated by neighboring amino groups. With the addition of a bit of H3PO4, peaks of carboxylate ions (–COO−) at 1570 cm−1 disappeared, indicating that H3PO4 was utilized to protonate –COO− to form –COOH. It was further verified by the growing peak intensity of the carboxyl group (–COOH) at 1724 cm−1. With a suitable amount of H3PO4, both amino groups and carboxyl groups in L-lysine were completely protonated, which was confirmed by the disappearance of –NH2 group peaks at 1614 cm−1. Complete protonation of all amino acids inhibited the deamination.
The mechanism of L-lysine deoxygenation to cadaverine has been further proposed with the aid of GC and HPLC analysis, as described in Fig. 1C.24 L-Lysine in the zwitterionic form NH3+(CH2)4CH(NH2)COO− transformed into the divalent cationic form NH3+(CH2)4CH(NH3+)COOH in the presence of H3PO4. It was consequently reduced to the reactive aldehyde intermediate which could produce cadaverine through deoxygenation catalyzed by Ru. However, many side reactions of producing other N-containing chemicals competed fiercely with the target deoxygenation to cadaverine. Therefore, it is a significant challenge to directly synthesize cadaverine with 100% selectivity.
Although chemo-catalytic technology exhibited a high efficiency, the selectivity of cadaverine is not ideal because of the relatively high multi-functionality of L-lysine with two amino groups (–NH2) and one carboxyl group (–COOH). For chemo-catalytic synthesis of cadaverine, the key point is to urgently develop a catalyst that exhibits a significantly high selectivity. Presently, most studies focus on supported ruthenium catalyst, especially on Ru/C. Replacing amorphous activated carbon with other supports, such as hierarchical porous carbon materials, carbon nanotubes (single-walled or multi-walled), or molecular sieves with abundant channels, might be an alternative route to improve the selectivity of cadaverine because of the pore or channel confinement effect which inhibits the growth of large-sized byproduct. On the other hand, improving the dispersion degree of Ru atom on support which exposed more active sites was also worth trying. Therefore, improvement of the selectivity for cadaverine production remains the most significant for future industrial applications of this technology.
3. Biosynthesis of cadaverine
In 1942, Gale and Epps first found that Escherichia coli can induce different amino acid decarboxylases under acidic conditions,29 which brought new insight for the biosynthesis of cadaverine. Besides, renewable and sustainable resources are in urgent demand for producing cadaverine due to the petrol resource depletion and environmental pollution caused by the chemical process. The rapid development of biotechnology and synthetic biology in the past decade has provided a new chance to probe into the production of cadaverine (Table 1).| Host strain | Carbon source | Titer (g L−1) | Productivity (g L−1 h−1) | Yielda (g g−1) | Metabolic engineering strategies | Reference |
|---|---|---|---|---|---|---|
| a The isolated yield of cadaverine per substrate (carbon source). | ||||||
| C. glutamicum | Glucose | NA | NA | 0.13 | • Based on previously derived tailor-made diaminopentane producer | 30, 33 and 34 |
| • Deletion of act3 | ||||||
| C. glutamicum | Glucose | 2.75 | NA | 0.1 | • Overexpression of cadB from E. coli and lysine decarboxylase ldc from Hafnia alvei | 31 |
| C. glutamicum | Glucose | 88 | 1.76 | 0.29 | • Based on previously strain LYS-12 | 9 |
| • Overexpression of cg2893 and replacement of the native promoter by the sod promoter | ||||||
| • Overexpression of ldcC from E. coli and replacement of the native promoter by the tuf promoter | ||||||
| • Deletion of NCgl1469, lysE | ||||||
| C. glutamicum | Glucose | 103.78 | 1.6 | 1.04 | • Disruption of lysE | 35 |
| • Overexpression of ldcC from E. coli | ||||||
| C. glutamicum | Glucose | 0.74 | 0.045 | 0.076 | • Deletion of ldhA, pta-ack, cat, pqo, and cglNcgl1703-Ncgl1705 | 36 and 37 |
| • Modification and amplification of pyc (Ptuf-pycP458S) | ||||||
| • Attenuation of icd (icdA1G) | ||||||
| • Modification of lysC (lysCT311I) | ||||||
| • Introduction and overexpression of ldcC (Ptuf-ldcC) | ||||||
| • Replacement of the promoter of pgi by the ldhA promoter (PldhA-pgi) | ||||||
| E. coli | Galactose | 8.8 | 0.293 | 0.17 | • Introduction of a re-designed Leloir pathway (galE, galT, galK, galM, galP and pgm) and L-lysine formation pathway (asd, dapAfbr, dapB, ddh, lysA and lysCfbr) | 38 |
| • Overexpression of cadA | ||||||
| • Deletion of speE, speG, ygjG and puuPA | ||||||
| C. glutamicum and E. coli | Starch | 0.69 | 0.013 | 0.025 | • C. glutamicum: Deletion of pta-ackA, cat, aceAB, ldhA, nanR and snaA; overexpression of ldcC | 39 |
| • E. coli: Deletion of nagE, manXYZ and lysA; overexpression of amyA | ||||||
| C. glutamicum | Xylose | 103 | 1.37 | NA | • Based on strain DAP-Xyl139 | 40 and 41 |
| • icdGTG Peftufbp Psodtkt | ||||||
| • Deletion of act and lysE | ||||||
| E. coli | Glucose | 0.62 | 0.013 | 0.02 | • Based on strain JCM20137 | 42 |
| • Vector for CadA and BGL (Tfu0937 from T. fusca) coexpression using blc anchor protein | ||||||
| C. glutamicum | Cellobiose | 27 | 0.281 | 0.43 | • Deletion of cglMRR, pepck, NCgl1469, pyk2, iolR | 33 |
| • Substitution of pycP485S, homV59A, ppcN917G, zwf(A243T), lysCQ298G and pgi with start codon ATG→GTG | ||||||
| • Overexpression of ldcC and tfu0937 | ||||||
| B. methanolicus | Methanol | 11.3 | 0.376 | NA | • Based on wild-type B. methanolicus strain MGA3 | 43 |
| • Overexpression of cadA (pTH1mp-cadA) | ||||||
| B. methanolicus | Methanol | 17.5 | NA | NA | • Overexpression of cadA (pBV2mp-cadA) | 44 |
| C. glutamicum | Methanol | 1.5 | NA | NA | • Deletion of ald and fadH | 45 |
| • Overexpression of mdh, hxlAB, lysCfbr and ldcC (pEKEx3-mdh, hxlAB)(pVWEx1-lysCfbr-ldcC) | ||||||
| M. trichosporium | Methane | 0.28 | 0.002 | 0.91 | • Based on wild-type M. trichosporium strain OB3b | 46 |
| • Overexpression of lysA, pyc, ldcC and cadB (pAWP89-lysA-pyc-ldcC-cadB) | ||||||
| B. methanolicus | Mannitol | 6.3 | NA | 0.19 | • Overexpression of cadA (pBV2mp-cadA) | 47 |
| E. coli | L-Lysine | 221 | 13.813 | NA | • Overexpression of cadA and cadB (pETDuet-pelB-cadB-cadA) | 48 |
| E. coli | L-Lysine | 32.1 | 0.803 | NA | • Deletion of speE, puuA, speG and ygjG | 49 |
| • Overexpression of cadA and cadB(pSIT-Duet-cadA-pelB-cadB) | ||||||
| E. coli | L-Lysine | 102 | 17 | NA | • Overexpression of cadA, pdxY and ppk | 50 |
| • Addition of CTAB | ||||||
| E. coli | L-Lysine | 198 | 40 | NA | • Overexpression of CadA T88S | 51 |
| E. coli | L-Lysine | 157 | 16.526 | NA | • Overexpression of CadA F14C/K44C/L7M/N8G | 52 |
| E. coli | L-Lysine | 63.9 | 12.78 | NA | • Overexpression of gene ldc from H. alvei | 53 |
| E. coli | L-Lysine | 83.2 | 13.867 | NA | • Overexpression of cadA and pdxY (pSIT-cadA, pSUJ-pdxY) | 54 |
3.1. Glucose-based cadaverine
Corynebacterium glutamicum is an industrial microbe traditionally used for production of amino acids and the super L-lysine producer when using glucose as substrate (Fig. 2). These species have been comprehensively developed as an efficient microbial cell factory for cadaverine production by metabolic engineering strategy recently. It was found that the pathway N-acetyldiaminopentane of C. glutamicum can produce highly undesirable by-products when using glucose as substrate for producing cadaverine. The deletion of the responsible acetyltransferase gene in this competing pathway of C. glutamicum DAP-4 can produce 0.13 g cadaverine/g glucose, which increased cadaverine production by 11%.30 Further deletion of the lysine exporter lysE or overexpression of the cadaverine exporter/transporter might also improve the yield of cadaverine for the mutation.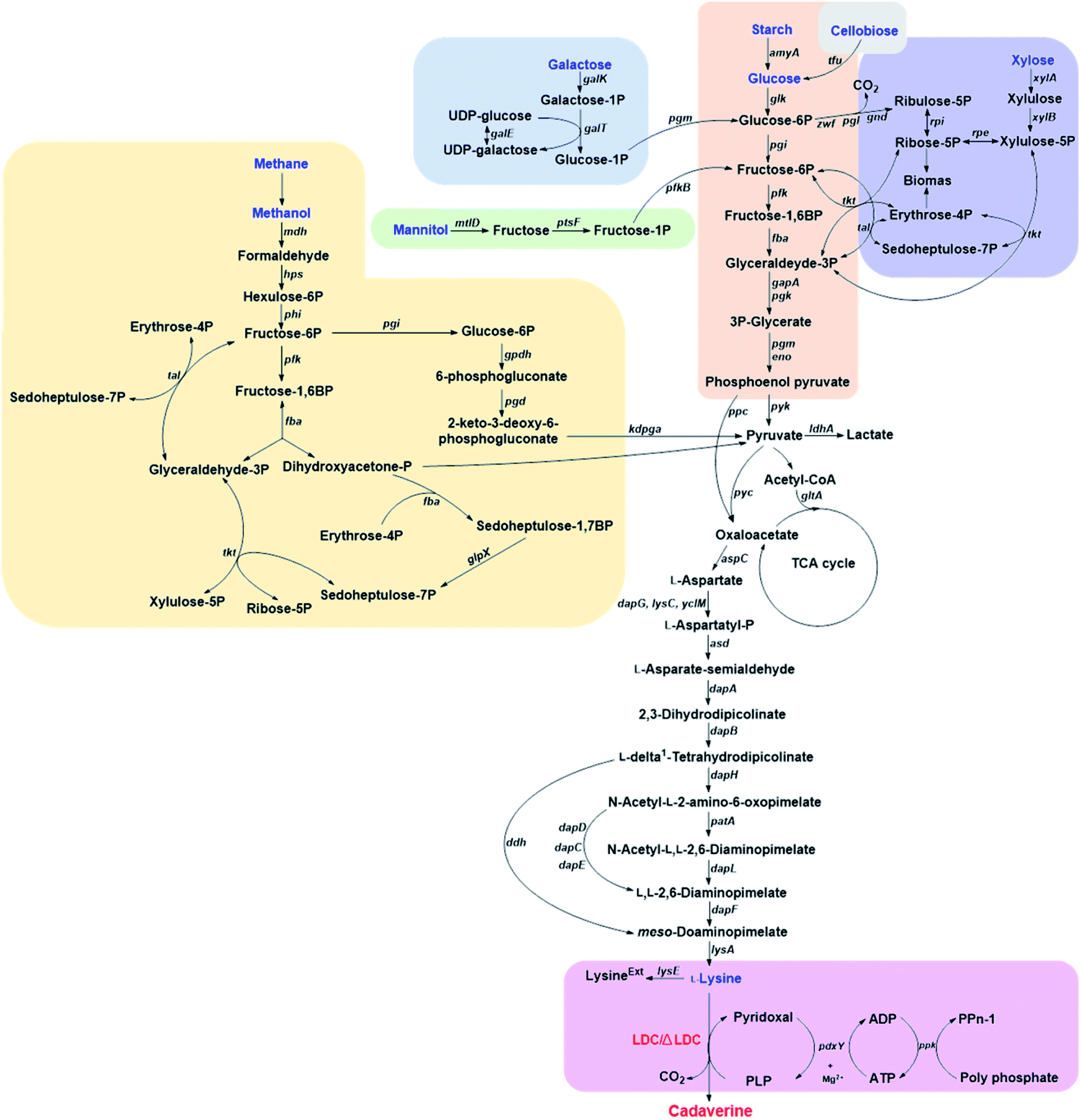 | ||
| Fig. 2 The pathways for producing cadaverine from glucose, galactose, starch, cellobiose, xylose, methane, mannitol and L-lysine. The following gene abbreviations have been used: gpdh, glucose-6-phosphate dehydrogenase; pgd, 6-phosphogluconate dehydratase, kdpga, 2-keto-3-deoxy-6-phosphogluconate aldolase; ppc, phosphoenolpyruvate carboxylase; pyk, pyruvate kinase; pyc, pyruvate carboxylase; ldhA, lactate dehydrogenase; gltA, citrate synthase; aspC, aspartate aminotransferase; dapG, aspartate kinase I; lysC, aspartate kinase II; yclM, aspartate kinase III; asd, aspartate semialdehyde dehydrogenase; dapA, dihydrodipicolinate synthase; dapB, dihydrodipicolinate reductase; dapH, tetrahydrodipicolinate N-acetyltransferase; patA, acetyl-diaminopimelate aminotransferase; dapL, N-acetyl-diaminopimelate deacetylase; dapF, diaminopimelate epimerase; lysA, meso-diaminopimelate decarboxylase; dapD, tetrahydrodipicolinate succinylase; dapC, succinyldiaminopimelate aminotransferase; dapE, succinyl-L-diaminopimelate desuccinylase; lysE, lysine exporter; ddh, meso-diaminopimelate dehydrogenase; pdxY, pyridoxal kinase; ppk, polyphosphate kinase; tfu β-glucosidase; glk, glucose kinase; pgi, glucose-6-phosphate isomerase; zwf, glucose-6-phosphate dehydrogenase; pgl, 6-phosphogluconolactonase; tkt, transketolase; tal, transaldolase; xylA, xylose isomerase; xylB, xylulokinase; rpe, ribulose-5-phosphate-3 epimerase; pfk, phosphofructokinase; fba, fructosediphosphate aldolase; mdh, methanol dehydrogenase; hps, 3-hexulose-6-phosphate synthase; phi, 6-phospho-3-hexuloisomerase; fba, fructose-1,6-bisphosphate aldolase; glpX, sedoheptulose-1,7-bisphosphatase; galK, galactokinase; galT, galactose 1-P uridylyltransferase; galE, UDP-galactose 4′-epimerase; pgm, phosphoglucomutase; mtlD, mannitol 2-dehydrogenase; ptsF, fructose-specific phosphotransferase system; pfkB, fructose 1phosphate kinase; gapA, D-glyceraldehyde-3-phosphate dehydrogenase; pgk, phosphoglycerate kinase; eno, enolase; amyA, α-amylase. | ||
Improving the secretion of cadaverine is another strategy to increase the conversion yield, which has been confirmed by expressing the fusion of cadaverine-lysine transporter cadB from E. coli and lysine decarboxylase ldc from Hafnia alvei in the host C. glutamicum ATCC 13032. The resulting engineered strain C. glutamicum CDV-2 can utilize glucose and increase the cadaverine transfer rate and thus improve the yield of cadaverine.31 Expired by the function of cadaverine-lysine transporter, Wittmann et al.32 have comprehensively identified the potential transporter for cadaverine by global transcription profiling, targeted gene deletion and physiological analysis. The results showed that the exporter permease Cg2893 can enhance the cadaverine production when using glucose as a substrate in C. glutamicum (yield: 0.14 g cadaverine/g glucose).32 The engineered strain C. glutamicum DAP-16 with the exporter permease Cg2893 (C. glutamicum LYS-12 (PtufldcCoptΔNCgl1469ΔlysE&Psodcg2893)) can reach the yield of 0.23 g cadaverine/g glucose.9 C. glutamicum DAP-16 was further evaluated in an industrial glucose medium and accumulated 88 g L−1 cadaverine within 50 h. The yield can be up to 0.29 g cadaverine/g glucose during the feeding phase.9
Suitable promoters are important for the efficient expression of lysine decarboxylase and improving the cadaverine yield from glucose. Strong promoter H30 has been used in the engineered C. glutamicum G-H30 to replace the tuf promoter for the regulation of lysine decarboxylase ldcC, which could produce 103.78 g L−1 cadaverine at 65 h cultivation with the yield of 1.04 g cadaverine/glucose by fed-batch fermentation.35 Moreover, fine-tuned strong inducible promoters could also be investigated, offering room for possible yield improvement and a convenient way to deal with production inhibition.55
Though the yield of cadaverine has been improved by metabolic engineering, secretion transporter, strong promoter, and efficient heterologous lysine decarboxylase, the carbon originating from glucose can pass through different pathways, such as glycolysis and pentose phosphate pathway depending on either aerobic or anaerobic conditions.36 Control of the split ratio of carbon flux could be another possibility to increase the cadaverine production. Recently, a new strategy has been developed for improving the production of cadaverine by automatic redirection of carbon flux between glycolysis and PPP under aerobic conditions in C. glutamicum YT14. The control process can be achieved by replacing the promoter of glucose-6-phosphate isomerase gene (pgi) with an anaerobic-specific promoter of the lactate dehydrogenase gene (ldhA). The new process can increase the yield of cadaverine by 4.4 times.36,37
3.2. Galactose-based cadaverine
Galactose is abundant in macroalgae and dairy waste, which is also found to be the alternative feedstock for cadaverine production.38 Kwak et al.38 has developed a novel recombinant strain with a redesigned synthetic expression cassettes for the Leloir pathway (galE, galT, galK, galM, galP and pgm) for galactose metabolism and the L-lysine formation pathway (asd, dapAfbr, dapB, ddh, lysA and lysCfbr) on the chromosome. The engineered strain was further constructed with overexpressed, endogenous lysine decarboxylase CadA and inactivated pathways related to the degradation/re-uptake of cadaverine. The final E. coli DHK4 can produce 8.80 g L−1 cadaverine with the yield of 0.170 g cadaverine/g galactose during fed-batch fermentation.38 This research has paved a new way for the cadaverine production from various feedstock by metabolic engineering and synthetic biology.3.3. Starch-based cadaverine
Mixed microbial consortia can be used for utilization of more complex substrates. Currently, the starch-based cadaverine has also been achieved by developing binary synthetic consortia consisting of C. glutamicum CgCAD4(pEKEx3-ldcC)(pECXT99A-crimson) and E. coli EcLYS1(pAmy)(pVWEx1-gfpuv).39 The engineered E. coli expressed the α-amylase, which hydrolyzed the starch to glucose for the engineered C. glutamicum to produce cadaverine. The co-cultured consortia can produce 0.69 g L−1 cadaverine with a yield of 0.025 g cadaverine/g glucose in 55 h.39 Strain modification could be a chance for further improving the low yield of starch-based cadaverine.3.4. Xylose-based cadaverine
Xylose-based cadaverine will be the milestone due to the use of non-food raw materials as the feedstock. Wittmann's group has created a recombinant strain C. glutamicum DAP-Xyl1 (pClik 5a MCS PgroXyl ldcC) which exhibited efficient production of the cadaverine from xylose.40,41 C. glutamicum with xylA and xylB from E. coli can utilize xylose or even industrially relevant hemicellulose fractions as sole carbon source.41 Based on the super strain C. glutamicum DAP-Xyl1, overexpression of the fructose bisphosphatase (fbp) gene and the tkt operon, attenuation of the isocitrate dehydrogenase (icd) gene, deletion of the diaminopentane N-acetyl transferase cg1722 and the lysine exporter lysE, resulted in highly tailored strain DAP-Xyl2 (icdGTG Peftufbp Psodtkt Δact ΔlysE), which can achieve 103 g L−1 of cadaverine with an outstanding yield of 32% from xylose in a fed-batch process.403.5. Cellobiose-based cadaverine
Except for xylose, cellobiose was also applied for cadaverine by the engineered E. coli. Cellobiose can be imported into the host cell by PEP-dependent phosphotransferase system and cel operon,56 or by hydrolyzing to glucose with β-glucosidase. Ikeda et al.42 has developed the one-step production of cadaverine from cellobiose by displaying efficient β-glucosidase Tfu0937 from Thermobifida fusca YX on the surface of E. coli (Jm-cadA-blc-Tfu) host combining with L-lysine decarboxylase (CadA) from E. coli with the anchor protein Blc. The recombinant strain E. coli (Jm-cadA-blc-Tfu) could produce 0.62 g L−1 cadaverine after 48 h from 28 g L−1 cellobiose with a yield of 0.02 g cadaverine/g glucose (1 g cellobiose corresponds to 1.1 g glucose).42 Matsuura et al.33 also used cellulosic materials for cadaverine production by constructing strain C. glutamicum MQN8-6L6B with well-known lysine decarboxylase (ldcC) and β-glucosidase Tfu0937 without pyruvate kinase pyk2 in the host. The metabolic engineering strain MQN8-6L6B can produce a relatively high amount of cadaverine (27 g L−1) from 57 g cellobiose through the fed-batch fermentation process with a yield of 0.43 g cadaverine/g glucose, which was superior to the above strain using glucose as substrate.33 All above-mentioned research developing novel strains utilizing non-food biomass such as lignocellulose, represents a significant achievement for green and sustainable production of cadaverine.3.6. Methanol-based cadaverine
Methanol as a highly attractive non-food source, is drawing a great deal of attention as feedstock for high value-added products or as the next generation of liquid fuels in the future. The natural methylotrophic Bacillus methanolicus has great potential for biotechnological applications as it can use methanol to produce L-lysine.57,58 Wendisch's group was the first to demonstrate that cadaverine can be produced from methanol by the methylotrophic and thermophilic bacterium B. methanolicus, engineered by heterologous expression of cadA and ldcC.43 The recombinant strain MGA3(pTH1mp-cadA) can produce 11.3 g L−1 cadaverine after 30 h fed-batch methanol cultivation.43 B. methanolicus was further improved by the establishment of the genome-based genetic tool-box through the addition of theta-replicating expression vectors and development of xylose- and mannitol-inducible promoter systems for cadA expression.44 The concentration of cadaverine was up to 17.5 g L−1 by using the new MAG3(pBV2mp-cadA), which was higher than the above MGA3(pTH1mp-cadA).44 Recently, it was reported that methanol can be used as substrate for cadaverine production by non-methylotrophic C. glutamicum. The engineered C. glutamicum harbored the heterogenous NAD+-dependent methanol dehydrogenase (Mdh) from B. methanolicus with the key enzymes hexulose-6-phosphate synthase and 6-phospho-3-hexuloisomerase of the ribulose monophosphate (RuMP) pathway, and deleted the endogenous aldehyde dehydrogenase genes ald and fadH.45 The resulted C. glutamicum strain ΔaldΔfadH(pEKEx3-mdh,hxlAB)(pVWEx1-lysCfbr-ldcC) can assimilate methanol by RuMP pathway when used sugar as co-substrate.45 Therefore, the synthetic biology approach has paved the first step towards cadaverine production from methanol by both methylotroph B. methanolicus and non-methylotrophic C. glutamicum. The yield, titer, and conversion efficiency can be further improved in the future for industrial methanol-based cadaverine strains.3.7. Methane-based cadaverine
Methane is currently used to primarily provide energy or heat via combustion. Almost 566 Tg methane per year is produced from anthropogenic sources, which constitutes 60% of annual global emissions. These, among others include the generation by the use of fossil fuels, landfilling and livestock farming. Increased emissions have negative consequences as methane is a greenhouse gas with approximately 20 times the impact of CO2. However, methane represents also an enormous resource and can be treated as a chemical platform for conversion into a range of higher value products. This is especially encouraging when methane from sustainable sources like landfills or anaerobic digesters are used or otherwise sources that are not economical to process for conventional heating. Microbial methane production and capture is an established and sustainable strategy for conversion of organic waste streams. These economic incentives and the changing attitude towards sustainable production of both fine and commodity chemicals result in the growing interest in the biological conversion of methane.Potentially, cadaverine can be produced by methanotrophic bacteria and methane as a feedstock. This concept has been recently demonstrated by group of Lee at Kyung Hee University.46 This approach reduces the use of expensive organic substrates in the process. Traditionally, methanotrophs are classified as type I (γ-proteobacteria) or type II (α-proteobacteria). This classification reflects mainly metabolic features (type I bacteria use the ribulose monophosphate pathway (RuMP) for formaldehyde assimilation while type II bacteria use serine-pathway), cell membrane composition and cell morphology.59 It is now known that methane can also be oxidized by certain thermoacidiphilic bacteria from phylum Verrucomicrobia60 and certain Archaea in consortia with sulfate-reducing bacteria.61 No methanotrophic archaea are capable of using oxygen. In general, type I methanotrophs are known to prefer lower CH4 and higher O2, while type II higher CH4 and lower O2 concentrations, respectively. Methanotrophs are able to assimilate CO2 (α-proteobacteria can assimilate up to 50%, while γ-proteobacteria up to 15% of their biomass from CO2) and are thus affected by CO2 concentrations.62
3.8. Mannitol-based cadaverine
Mannitol and laminarin are the major constituents in seaweed, especially in the brown macroalgae which are considered as source of the third generation of renewable energy. C. glutamicum cannot utilize mannitol naturally. However, the capability of producing L-lysine from mannitol was demonstrated by metabolically engineered C. glutamicum strain SEA-3.63 B. methanolicus MGA3 (pBV2mp-cadA) has also been constructed and could produce 6.3 g L−1 cadaverine under fed-batch mannitol fermentation. This process consumed energy and reduced power from TCA cycle but not from RuMP pathway.47 The tailor B. methanolicus with unique traits has a great chance of being engineered into cell factory for cadaverine production with different substrates.3.9. L-Lysine-based cadaverine
The direct microbial fermentation for cadaverine production with sugar (glucose, galactose, starch, lignocellulose), methanol, and mannitol as substrate is promising. However, it is uneconomical with unsuitable downstream purification process for industrial application due to the different microbial metabolites, low concentration of cadaverine (<100 g L−1),64 the cellular degradation of the product, product inhibition, and long period for fermentation.Recently, most research intends to construct the engineered strains with the CadA which can use the direct precursor L-lysine as substrate for producing the cadaverine by whole-cell conversion in one step (Fig. 3A). The reaction is the proton-dependent irreversible α-decarboxylation of L-lysine which is catalyzed by the lysine decarboxylase to generate CO2 and cadaverine (Fig. 3B).65,66 The product CO2 is disassociated from the cell and cadaverine is transported out of the cell by the inner membrane lysine-cadaverine antiporter CadB (Fig. 3C).67 This reaction process is convenient and does not require complex fermentation control. It can produce a high concentration of cadaverine without accumulation of by-products, resulting in the relatively pure product and economic downstream purification process. As described above, the global L-lysine market is expected to be about 3.28 million metric tons and the price of L-lysine has decreased to around $1.5 in 2020.68 Therefore, the whole-cell conversion of L-lysine to cadaverine is a promising and potentially economically-feasible process for industrial application.
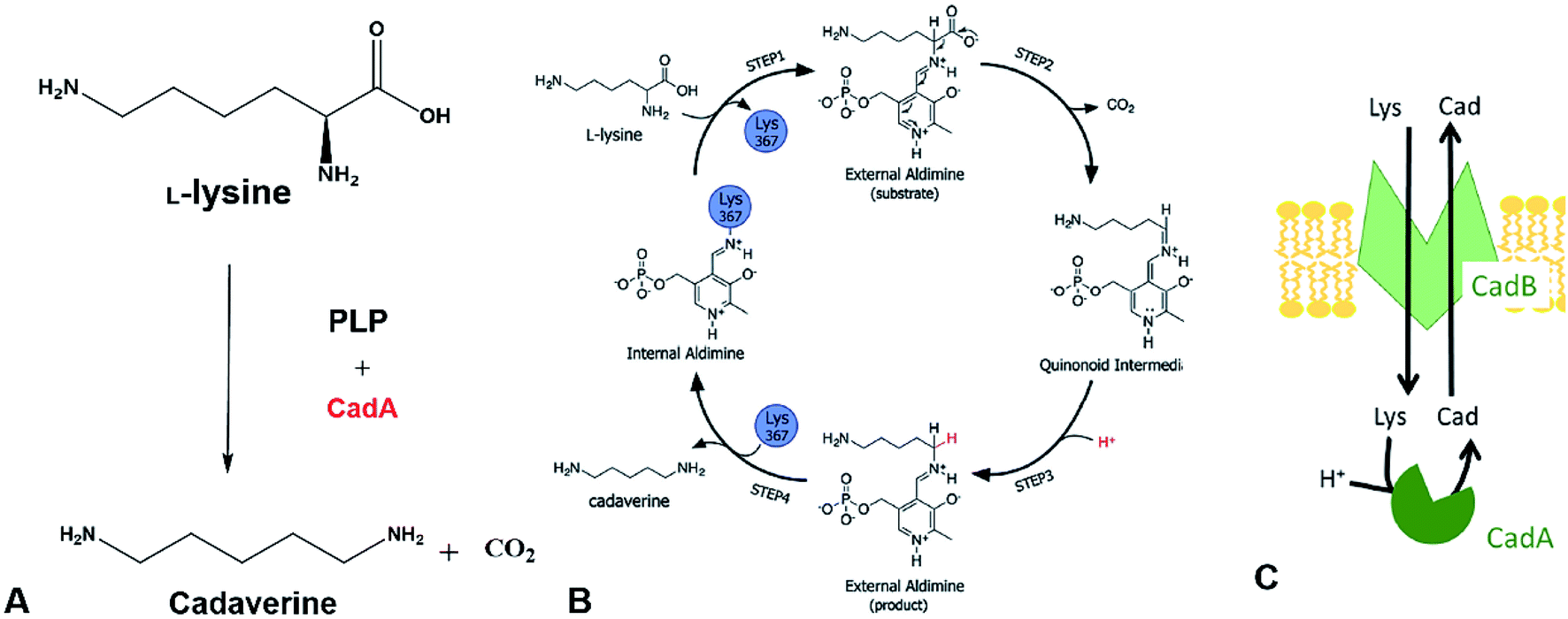 | ||
| Fig. 3 (A) Cadaverine production from L-lysine. (B) Possible reaction mechanism. Reprinted with permission,66 Copyright (2011) Royal Society of Chemistry. (C) The cadaverine transported out of the cell by the inner membrane lysine-cadaverine antiporter CadB. Reprinted with permission,67 Copyright (2019) Springer Nature. | ||
The one-step cadaverine production is usually carried out by the genetically engineered E. coli with the lysine decarboxylases which originate from E. coli, Aliivibrio salmonicida, subtropical soil, Vibrio vulnificus, Klebsiella pneumoniae, Selemonas ruminatium, Hafnia alvei, and Burkholderia sp.52,53,69–77 CadA from E. coli was confirmed with higher catalytic efficiency (kcat/Km: 71.43–120.1 s−1 mM−1) than lysine decarboxylases from other strains and higher expression level than the LdcC.41,56,78,79 Recently, Xue et al. have discovered two new lysine decarboxylases LdcEt and LdcAer from Edwardsiella tarda and Aeromonas sp. and the catalytic efficiency were increased by 201% and 242% compared to CadA from E. coli.80 CadA was the oligomer of five dimers that aggregated into a decamer as described in the following section on lysine decarboxylase structure. This large decamer structure (around 810 kDa) was sensitive and inactive for pH higher than 8.0.69 Directed evolution, protein engineering and computational enzyme design can lead to further improvement of the activity and stability for CadA. The screened mutants from the directed evolution of CadA had higher catalytic activity and thermal stability than CadA.81 The thermostability of the disulfide bond mutant (CadA F14C/K44C) was improved significantly and the half-life at 60 °C was increased by 216 folds over CadA. The further isolated mutant CadA F14C/K44C/L7M/N8G can produce 157 g L−1 cadaverine in 9.5 h with 76.7% conversion yield.52 Kou et al.51 have half-rationally designed the CadA by computational analysis and the target mutant CadA T88S has obviously improved the thermal and alkaline pH stability, increasing 2.9 folds in the half-life at 70 °C and 1.43 folds for the cadaverine productivity.
Different from the C. glutamicum, the wild-type E. coli uses CadB for L-lysine importing and cadaverine secretion under the acid stress. Therefore, the introduction of the CadB in the engineered host could increase the transfer rate of the substrate and the product. The engineered co-expressed CadA and CadB strain E. coli BL-BADE (pETDuet-pelB-cadB-cadA) can increase the yield of cadaverine by 12%. The optimized feeding can produce 221 g L−1 cadaverine with a molar yield of 92% in 16 h.48 The other engineered strain E. coli BL21ΔspeEΔpuuAΔspeGΔygjG (pSITDuet-cadA-pelB-cadB) can also increase the yield of cadaverine.49 Even though the CadB can increase the transfer rate of the L-lysine and cadaverine, other intensification should be developed to further improve the catalytic productivity.
The in vivo reaction for converting L-lysine to cadaverine is highly dependent on the pyridoxal phosphate (PLP) cofactor as the CadA is the PLP-fold type I lysine decarboxylase. Therefore, the concentration of PLP in the catalytic active site microenvironment is important for improving the catalytic efficiency. Recently, introducing an intracellular ATP regeneration system to provide the PLP in situ was developed by constructing the polyphosphate kinase (ppk) into systems containing CadA and PdxY or by integrating the intracellular PLP synthesis gene yaaDE to the E. coli BL-BADE (pETDuet-pelB-cadB-cadA). This novel strategy showed relatively high productivity of CadA (62.5 g L−1 h−1).43,50 Xue et al.54 also constructed the recombinant strain APK BL21(DE3) (pSIT-CadA, pSUJ-pdxY), which can reach 7008 nmol g−1-DCW of PLP. Through a combination of cold-shock treatment, the cadaverine productivity could reach 121 g L−1 h−1.
However, the enzyme needs further improvement for industrial application. Discovery of novel efficient lysine decarboxylases and rational design of interesting enzymes are especially needed for high-level cadaverine production.
4. Lysine decarboxylases
4.1. Phylogenetic diversity of lysine decarboxylases
Lysine decarboxylase constitutes an example of non-homologous isofunctional enzymes (NISEs), i.e. non-homologous enzymes that evolved to catalyze the same reaction.82 There are three known NISEs for lysine decarboxylation.83 Two of lysine decarboxylases (LDCs) evolved from the same enzyme aspartate aminotransferase superfamily, although one is homologous to arginine (ADC) and ornithine (ODC) decarboxylases and the second one to 2,4-diaminobutyric acid decarboxylase (DABA DC). The first one is found in both biosynthetic84 and acid-responsive forms.69 The DABA DC homologue was first discovered in S. coelicolor and closely related proteins were reported to be confined solely to bacterial siderophore biosynthetic pathways.85 This LDC form therefore appears to be tightly linked to the evolution of hydroxamate-type NIS biosynthetic pathways. The third LDC arose from the alanine racemase (AR) superfamily, which is a bifunctional enzyme specific to both lysine and ornithine. It is found in both bacteria74 and quinolizidine alkaloid-producing plants such as Lupinus angustifolius, Sophora flavescens, Echinosophora koreensis, Thermopsis chinensis, and Baptisia australis.86 The most studied CadA belongs to the aspartate amino-transferase fold (AAT-fold) of PLP-dependent Lysine–Arginine–Ornithine decarboxylases (LAOdcs).87 This enzyme possesses the long wing domain as CheY-like response regulator receiver for the formation of higher-order oligomers.Distinct evolutionary origin of most known non-homologous isofunctional enzymes for lysine decarboxylation can be observed in Fig. 4A.88–90 Sequences annotated as belonging to the same taxons are not grouped within the same clades. Clearly, depicted phylogenetic distances between sequences are influenced by both taxonomic origins of the host but also different structural origin of the ancestor analogues. Carriel et al.87 have comprehensively analyzed the phylogeny of LAOdcs and reported their ancestral short forms in Cyanobacteria and Firmicutes. Distinct subfamilies of long LAOdcs emerged in Proteobacteria. Moroever, the phylogenic tree indicated that the CadA was ancestral in some gammaproteobacterial lineages, and the constitute LdcC was derived from CadA (Fig. 4B).
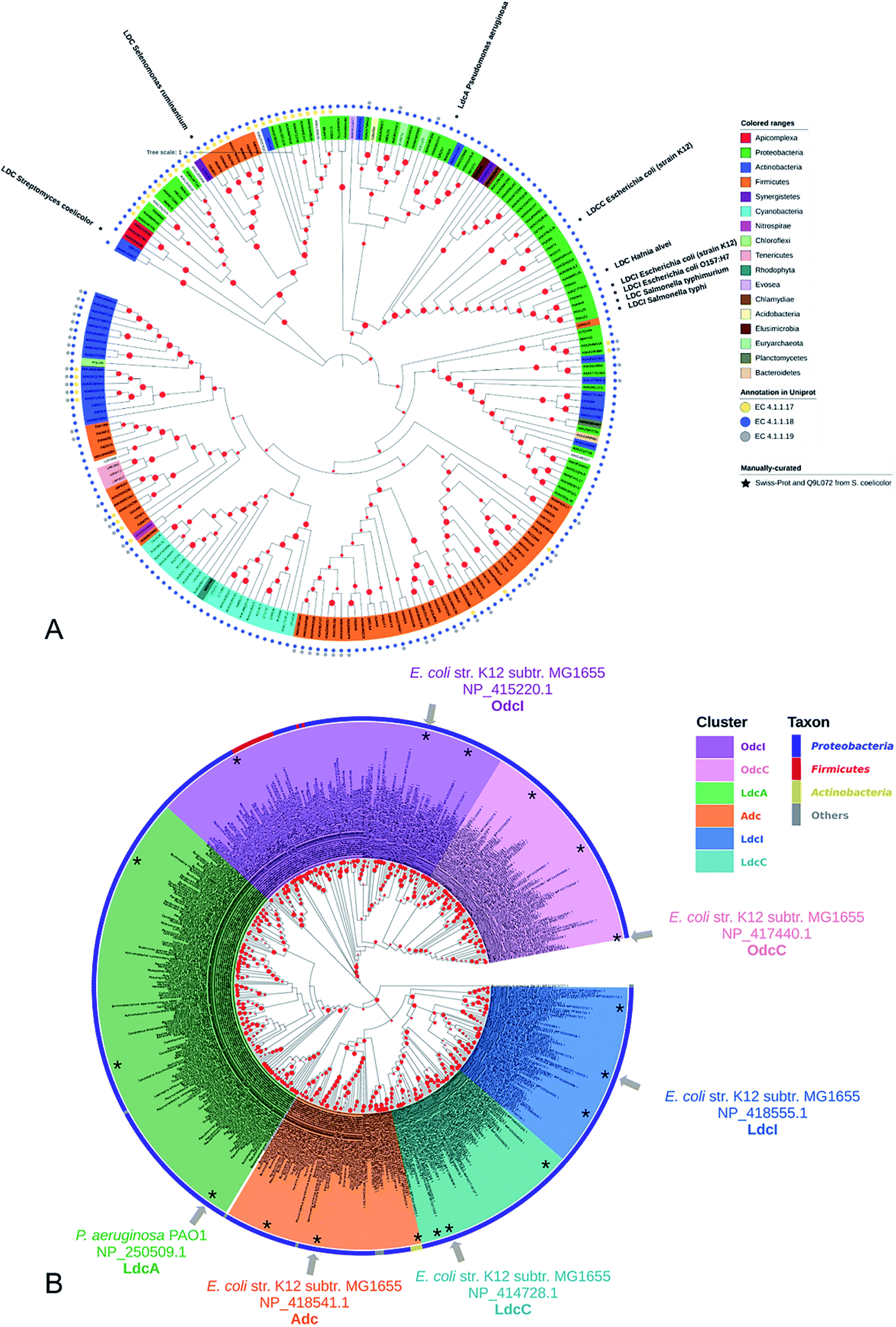 | ||
| Fig. 4 (A) Phylogenic tree of enzymes annotated with lysine decarboxylase activity in Swiss-Prot and TrEMBL databases. For TrEMBL database only AA sequences between 300 and 1000 AAs and representatives of clusters at 70% AA identity are shown. A phylogenetic tree was constructed using the neighbor-joining method with MEGA X.88,89 Branches with bootstrap values below 0.9 were removed. The evolutionary distances were computed using the Poisson correction method90 and were in the units of the number of amino acid substitutions per site. Swiss-Prot sequences and LDC from S. coelicolor were marked with colored squares according to their evolutionary origin: ADC/ODC from AAT-fold (red), DABA DC from AAT-fold (blue), and AR-fold (green). (B) Phylogenic tree of the Lysine–Arginine–Ornithine decarboxylases (LAOdcs) Cluster II. Reprinted with permission,87 Copyright (2018) Oxford University Press. | ||
4.2. Structure of lysine decarboxylase
Gale and Epps29 first found that E. coli could induce different amino acid decarboxylases under acid conditions. The cell had an arsenal of defense mechanisms for acid stress as the lysine decarboxylase was induced obviously in anaerobic growth conditions. In E. coli cadA gene is present as part of the cadBA operon with lysine–cadaverine antiporter cadB. This operon is regulated by the transcriptional factor CadC enabling response to acid and oxidative stresses, integrating signals, such as low pH, high lysine and low Cad levels. This genomic context of inducible lysine decarboxylase is strongly conserved within Enterobacteria. CadC, anchored in the inner membrane will replace H-NS proteins and bind the Cad1 and Cad2 sites to activate cadBA operon transcription and expression when the strains are subjected to acidic stress or high L-lysine level (Fig. 5A and B).87,91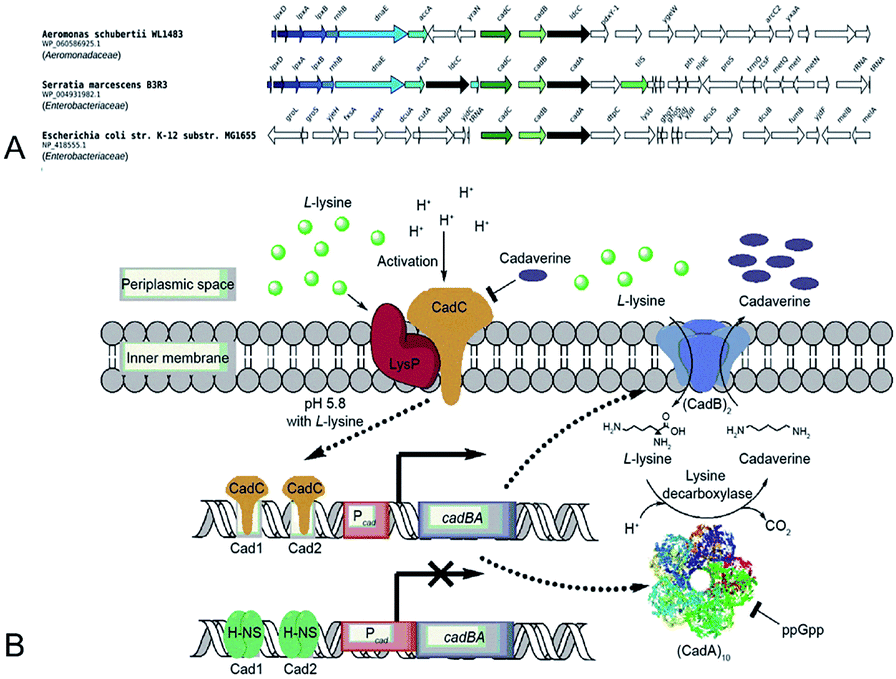 | ||
| Fig. 5 (A) Conserved genomic context of genes encoding CadA. Reprinted with permission,87 Copyright (2018) Oxford University Press. (B) The regulation of cadaverine production in Proteobacteria. Reprinted with permission,91 Copyright (2017) Elsevier. | ||
Lysine decarboxylases form decamer with wing domain and C-terminal domain.92 The negative staining electron microscope (EM) first showed that the decamer of CadA had the shape of two pentameric rings, tightly stacked together back-to-back and turned around 35° with each other (Fig. 6A and B). The five regulatory ATPase variant A (RavA) oligomers were demonstrated to be highly specific to two CadA decamers. Consequently, RavA–CadA strongly interacted together, forming an extremely large cage-like complex (∼3.3 MDa).93 The EM further showed that CadA was dimeric at pH 8.0 and low ionic strength, and that the dimers could form as decamers when pH dropped to 7.0 and ionic strength increased.93
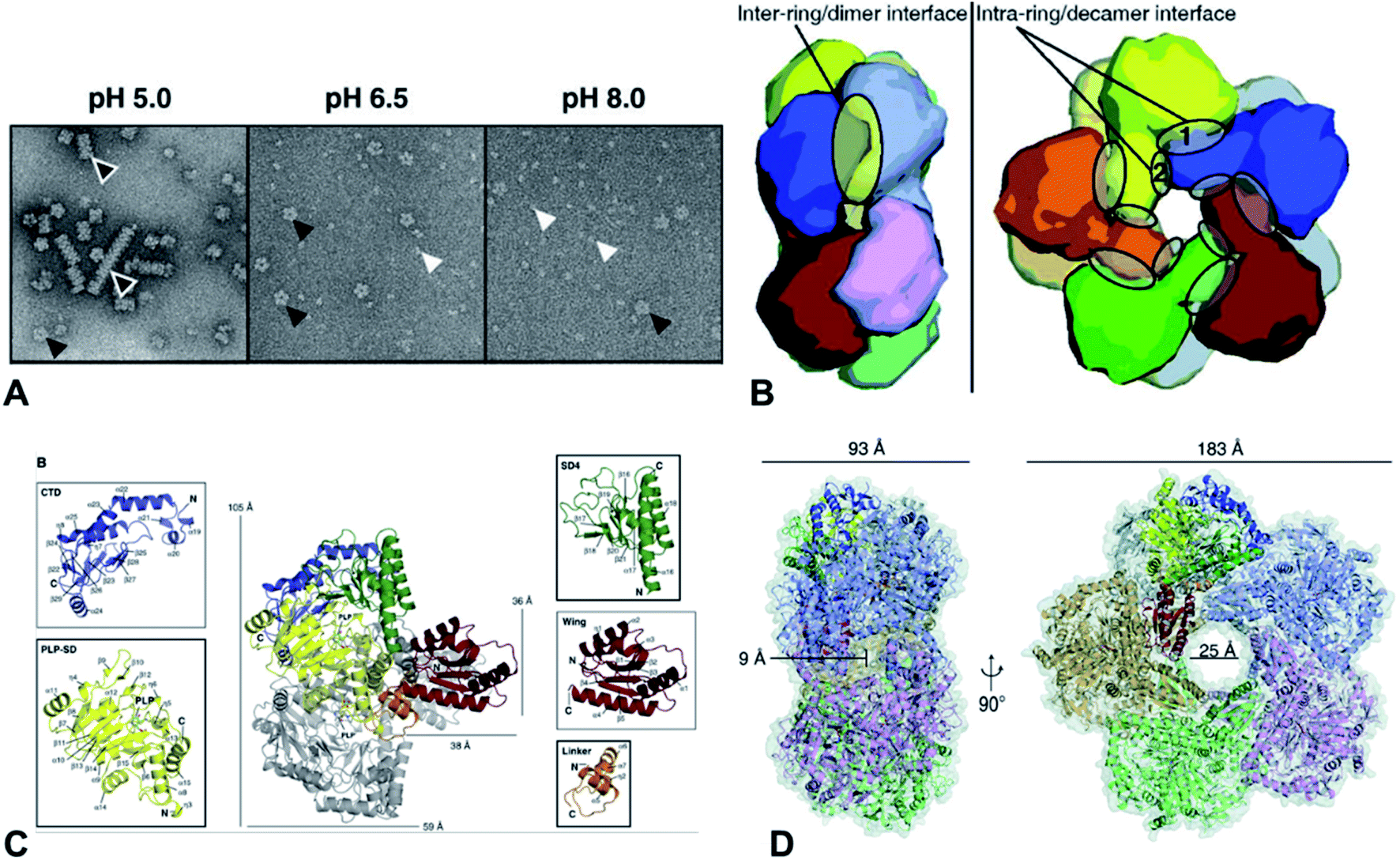 | ||
| Fig. 6 (A) Molecular structure of CadA. Negative-staining EM of CadA (LdcI) at pH 5.0, 6.5, and 8.0, (B) the dimensions of one of the side pores and the central pore for CadA decamer, (C) CadA dimer structure with the C-terminal domain (CTD), PLP binding subdomain (PLP-SD), subdomain 4 (SD4), wing and linker domains, and (D) the inter-ring/dimer interface and the intra-ring/decamer interface. Reprinted with permission,69 Copyright (2011) John Wiley and Sons. | ||
The large cage-like complex had been comprehensively investigated by X-ray structure analysis of RavA94 and CadA,69 respectively. The CadA monomer (81.2 kDa) had been divided into N-terminal wing domain (residues 1–129) responsible for decamerization of CadA, a core domain (residues 130–563) including a linker region (residues 130–183), PLP binding subdomain (PLP-SD) (residues 184–417) and subdomain 4 (SD4) (residues 418–563), and a C-terminal domain (CTD) (residues 564–715), which corresponded to the PLP-fold type I enzymes (Fig. 6C).69 CadA dimer was the primary unit and the active form of CadA was the decamer which was 183 Å wide and 93 Å high with a 25 Å central pore (Fig. 6D). The core domain of each dimer interacted together tightly to form a narrow cleft (9 Å diameter channel) that led into the PLP-binding site (T220, S221, H245, D330, A332, W333, S364, H366, K367, L368 in one monomer and T398, T399, S400 from the other monomer of the dimer). PLP was covalently bonded to the Ɛ-amino acid of the K367 by Schiff base.69 However, the substrate L-lysine binding site has not been clearly identified. The oligomeric states of CadA were found to be highly dependent on the pH and enzyme protein concentration.
The X-ray further clearly showed that the RavA hexamer displayed a six-leg shape and each two of them interacted with a CadA dimer at the top or the bottom of the complex, respectively and the apparent binding constant of RavA–CadA was 0.56 μM.94 The binding sites on RavA–CadA had been also identified. The results showed that the complex cage of two CadA decamers and five RavA hexamers interactions and the RavA–RavA interactions were mediated by LARA domain of RavA, which had been confirmed by the X-ray structure, negative staining EM, bioinformatics, and the experimental results. The binding site between LARA domain and CadA and the function of ATPase might also reduce the inhibition of the inhibitor ppGpp.69,94 The structural principles of RavA–CadA interaction were then clearly elucidated by cryo-electron microscope (CryoEM) to 11 Å resolution.95 The CryoEM results confirmed that the loop at the N-terminus of the LARA domain was the main determinant of the CadA–RavA cage formation, positioning the foot with respect to the rest of the leg, binding to CadA, and binding to the triple helical bundle of the adjacent RavA.95 The 3D CryoEM reconstruction of LdcC and CadA was also applied to explain why the RavA was highly specific to bind the induced CadA but not the constitution lysine decarboxylase LdcC under the acid stress, which was because the RavA notably bound to the C-terminal β-sheet of CadA.96
Another novel lysine decarboxylase LdcA from Pseudomonas aeruginosa has currently been solved to 3.7 Å resolution by CryoEM.87,97 The PLP-binding domain of LdcA was similar to CadA but there was no visible covalent bond with the active site K393 and PLP for LdcA. Meanwhile, the ppGpp binding pocket in LdcA indicated that LdcA was different from CadA and not inhibited by ppGpp. According to the phylogenic and structural analysis of LdcA, this enzyme could be another potential candidate for cadaverine production by an engineered host.
5. Process intensification for cadaverine production
Development of a novel fermentation or whole-cell conversion process for cadaverine production is an advanced strategy to improve productivity, streamline the operation, reduce energy consumption and the cost, which is extremely important for large-scale application.Whole-cell conversion for cadaverine production is a promising process for cadaverine production as described above. However, the low reusability and productivity of the cells remain big challenges for cadaverine production on a large scale. Therefore, it is urgent to improve lysine decarboxylases' activity and stability. Enzyme or cell immobilization technology is one possibility to improve the enzymatic efficiency, stability, and recyclability. Bhatia et al.98 were the first to immobilize the recombinant E. coli YH91 cells by barium alginate beads. The immobilized cells have a notably half-life of 131 h and 3-time higher thermostability compared with free cells. The immobilized cells still retained 56% activity after 18 cycles.98 However, the productivity of cadaverine still has great room for improvement. It was reported that E. coli AST3 can be entrapped and immobilized successfully in a mixture of 3.62% sodium alginate and 4.71% polyvinyl alcohol by dripping into 4.21% CaCl2.99 The results showed that the activity of the cells was reduced by the high concentration of cadaverine. Then the cell protectant polyvinylpyrrolidone (PVP) was found to improve the stability of the immobilized cells in 2 h (1.8-fold higher than the free cells). In the reactor, the PVA-SA-PVP-immobilized E. coli AST3 could produce 146 g L−1 cadaverine with an over 98% molar conversion yield of L-lysine.99 The immobilization of free lysine decarboxylase could be another strategy to improve the catalytic efficiency. It is interesting to see that the CadA can be in situ immobilized by the molecular biology technology.100 The poly(3-hydroxybutyrate) (P(3HB)) and PhaP1 (P(3HB)) granule-associated protein have been used to fuse CadA, resulting in the phasin-fused CadA bound to the intracellular P(3HB) granules. The immobilized CadA exhibited high thermal stability and achieved a 75–80% conversion yield over five reaction cycles.100 The fusion technology has also been used to develop the fusion protein by chitin-binding domain ChBD–CadA, which can be immobilized on the renewable biomass chitin (Fig. 7A).101 The fusion protein was characterized and showed high alkali stability, 97% molar conversion yield with 135.6 g L−1 cadaverine and reusability. This immobilization process was very efficient as more than 90% CadA can be specifically immobilized on the chitin in only 10 min under the mild condition.101 Park et al.102 have developed a cross-linked enzyme aggregate (CLEA) for CadA in vitro. The CadACLEA has significantly high thermostability than the free CadA. The 100 mM L-lysine can be 100% converted to cadaverine in 2 h. After 10 cycles, it still had 53% activity even there was no cell protective barrier. This was a super property as the free enzyme cannot be recovered for further recycle use.52 Another innovative and GMO-free approach was using catalytically active inclusion bodies (CatIBs) to immobilize the constitutive LdcC.103 The respective EcLDCc-CatIBs exhibited high stability (half-life was around 54 h) and could produce 88.4 g L−1 cadaverine.103 However, the process for preparing the CatIBs was time and energy consuming, which was difficult for scaling up.
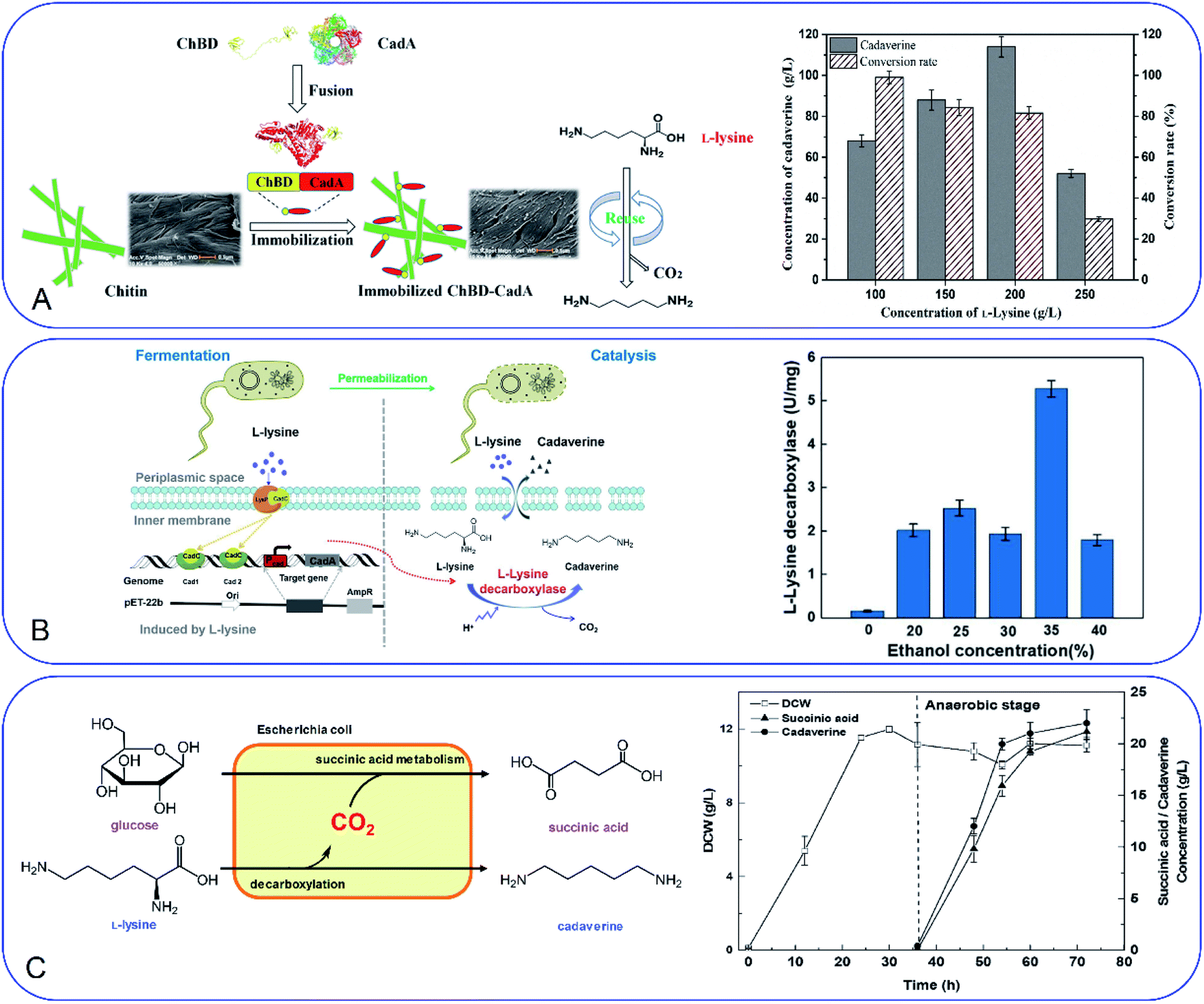 | ||
| Fig. 7 (A) Fusion technology for converting L-lysine to cadaverine via chitin binding domain ChBD. Reprinted with permission,101 Copyright (2020) Frontiers. (B) High production of cadaverine by the permeabilized cells by 35% ethanol. Reprinted with permission,105 Copyright (2020) Elsevier. (C) Coproduction of succinic acid and cadaverine using L-lysine as a neutralizer and CO2 from decarboxylation reactions. Reprinted with permission,108 Copyright (2018) Royal Society of Chemistry. | ||
Improvement of the cell permeability at the appropriate level would increase the transfer rate of L-lysine and cadaverine significantly during the whole-cell conversion process. Different surfactants have been tested for the effect on cadaverine production by the engineered strain E. coli ymAYK in Yang's group.50 The results indicated that 0.01% hexadecyltrimethylammonium bromide (CTAB) can increase 57.8% conversion compared with the negative control.50 The non-ionic detergent Brij 56 was also confirmed to increase the yield of cadaverine by Hafnia alvei.104 Rui et al.105 have studied the permeabilized cells of engineered strain E. coli BL21 (Pcad–CadA) by 35% (v/v) ethanol, which can produce 205 g L−1 cadaverine after 20 h by using the industrial materials in 2 L fermenter (Fig. 7B). Therefore, little amount of chemical surfactants or additives would make the whole-cell conversion of cadaverine much easier and more economic. The process has great potential for industrial application for cadaverine production.
Optimization of the reaction conditions and reduction of the additive use during the enzyme induction and reaction processing are also important for the efficient and economic production of cadaverine. After investigating the initial pH, buffer concentration, biocatalyst concentration, L-lysine concentrations, and PLP concentration for E. coli BL21(DE3) (pET-28a(+)-cadA), the whole-cell conversion showed an outstanding efficiency and can produce 144.74 g L−1 cadaverine with the productivity of 72.37 g L−1h−1.76 Kim et al.104 also statistically optimized the cadaverine production system and found that the H. alvei strain had the highest conversion rate under the optimum conditions (125.1 mM L-lysine, 71.5 g L−1 acetone at 35.2 °C for 8.4 h). Kim et al.106 have developed an advanced expression system for E. coli XL1-Blue (pKE112-HaLdcC) which can produce 123.2 g L−1 cadaverine after 12 h of reaction without the addition of the inducer IPTG and co-factor PLP. The IPTG- and PLP free whole-cell bioconversion system will absolutely improve the cost efficiency for the industrial cadaverine production.
Novel fermentation process with the mixed culture could be an advanced strategy for efficient cadaverine production. Recently, the synergetic microbial consortia with the two engineered E. coli NT1004 and CAD03 was applied to improve the cadaverine yield.107 The E. coli NT1004 can use glucose for L-lysine production and the E. coli CAD03 can use glycerol for growth and convert L-lysine to cadaverine without intracellular degradation. After optimization of the culture conditions and multi-stage constant-speed feeding strategy, the microbial consortia cooperated well and exhibited high productivity of cadaverine (28.5 g L−1) during the fermentation process.107
CO2 is also the product after the decarboxylation. The higher yield of cadaverine will result in releasing a higher amount of CO2, which is usually discharged like waste-gas in the bench-scale experiment. It will be of great significance if the CO2 can be used reasonably. In Ouyang's group, they have investigated different environmentally friendly strategies for CO2 utilization in the cadaverine production process (Fig. 7C).108,109 Succinic acid fermentation with cadaverine production was combined as a novel system for the first time. The disassociated CO2 during the cadaverine production process can be fixed in the succinic acid synthetic pathway by the engineered E. coli SC01 under the anaerobic stage, which can achieve 21.2 g L−1 of succinic acid and 22 g L−1 of cadaverine.108 The disassociated CO2 can be also used for self-controlling the pH during the cadaverine producing process. The novel in situ pH control system by sealing the reactor can maintain the reaction pH below 7.5 during the whole process.109 This system can reduce the extra acid for controlling the pH of the reactor, maintain the cell shape and activity and produce a high yield of cadaverine (208.2 g L−1) in 3 h.109 The use of the disassociated CO2 is a green and efficient method for industrial cadaverine.
6. Separation and purification of cadaverine
As mentioned above, cadaverine is a colorless viscous fuming liquid with a boiling point of 178–180 °C. According to its physicochemical properties, the existing separation and purification methods of cadaverine are developed based on experience with other diamine compounds. Considering the raw production mixture of cadaverine may contain nonvolatile inorganic/organic impurities, which may significantly interfere with cadaverine evaporation and distillation, causing high energy consumption, solvent extraction has replaced direct distillation and become the most popular and economic separation method. Suitable solvent extraction can achieve energy-efficient separation of cadaverine from fermentation broth. Krzyżaniak et al.110 compared the partition coefficients of cadaverine in several extractants including 4-nonylphenol, 3,4-bis((2-ethylhexyl)oxy)phenol, di-2-ethylhexyl phosphoric acid (D2EHPA), versatic acid 1019, di-nonyl-naphthalene-sulfonic acid (DNNSA) and 4-octylbenzaldehyde. The results showed that 4-nonylphenol was a suitable extractant with an extraction yield of over 90% for cadaverine from fermentation broth. Meanwhile, Kind et al.9 investigated the partition coefficients of cadaverine in n-butanol, 2-butanol, 2-octanol, and cyclohexanol to determine the best extractant. The result showed that n-butanol was more effective for recovering the cadaverine. Shortly afterwards, Hong et al.111 screened 10 different organic solvents and determined methyl ethyl ketone (MEK) to be a suitable extractant with a cadaverine extraction efficiency of over 70.1% (Fig. 8A).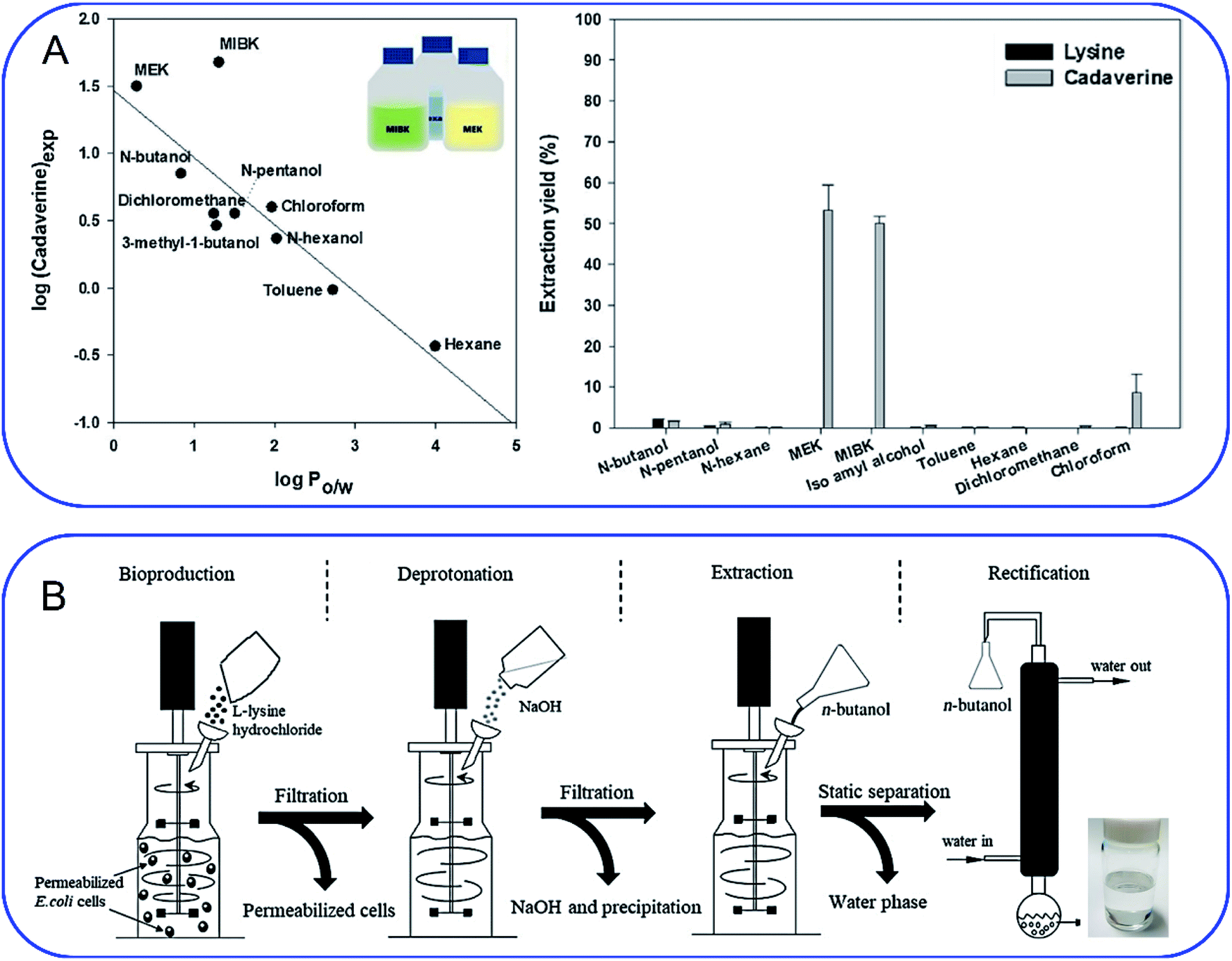 | ||
| Fig. 8 (A) Screening of extractants by comparing (left) the correlation of logP with cadaverine recovery yield and (right) cadaverine extraction yield. Reprinted with permission,111 Copyright (2018) Elsevier. (B) Schematic overview of an integrated process for the production of high-purity cadaverine from lysine decarboxylase. Reprinted with permission,112 Copyright (2020) John Wiley and Sons. | ||
When solvent extraction is completed, subsequent cadaverine purification from the cadaverine-containing phase can be performed by distillation, rectification, precipitation, or chromatography (Fig. 8B).112 Shanghai Cathay Biotechnology R&D Center Ltd recovered nearly 100% of cadaverine through distillation using high boiling solvents including C14 alkanes, C11/C14 alkane mixtures, diphenyl ethers, and dodecanol at a low heating temperature of 130 °C, which is 50 °C lower than the boiling point of cadaverine.113 They further employed a 732 strong acid cation exchange resin (styrene-based macroporous type) to adsorb cadaverine from the fermentation broth, followed by elution with 3% sodium hydroxide solution at 60 °C. This strategy can avoid the interference from impurities (e.g. tetrahydropyridine, bacteria and proteins) during distillation, and thus improve the recovery yield of cadaverine. Then the eluted cadaverine solution was further distilled at 70–150 °C. As a result, a cadaverine recovery yield of 89.3% was obtained after adsorption and distillation. Meanwhile, the cadaverine purity of 99.2% was obtained from the fraction at 92 °C through rectification.114 Ying et al.115 used the ultra-high crosslinking resin (polystyrene-divinylbenzene type) as the skeleton for the adsorption and separation of cadaverine, and the recovery yield of cadaverine adipic acid was over 96% with the purity of 97.5%. Ajinomoto Corporation77 employed an adipic acid to neutralize cadaverine from fermentation broth to prepare cadaverine dicarboxylate, which was decolorized and concentrated with 20% activated carbon and reduced-pressure distillation. The concentrated cadaverine was further precipitated and centrifuged to obtain the cadaverine dicarboxylate crystals with a purity of over 99%.
In recent years, many advances have been made in the cadaverine separation and purification device and technical process. Nanjing Tech University has contributed a lot to this aspect. Ouyang et al.116 designed a device for the continuous production and extraction of cadaverine using immobilized cells. Cadaverine recovery yield can reach 90.43% when calcium phytate column adsorbed cadaverine at the flow rate of 100 mL min−1. They also designed a rectification apparatus including a rectifying column, a tower kettle and a foam separator, to purify high concentrations of cadaverine. As a result, the cadaverine purity could reach 98.5% and its primary yield could reach 95%.39 Shanghai Cathay Biotechnology R&D Center Ltd provided a purification system designed for the biosynthesis of cadaverine, which comprised a reaction tank, a solid–liquid separator, an evaporator, a condenser and a collection tank. Such a purification device not only can directly and effectively purify the crude cadaverine and obtain a high cadaverine recovery yield with the purity of over 67%, but also is corrosion resistant, low energy consumption and low cost, especially suitable for industrial mass production.117 Besides, they also designed a separation device, which directly targeted the alkaline extraction of cadaverine in the fermentation broth and can significantly reduce the solid residue in the subsequent treatment.118 Ningxia Eppen Biotech Co., Ltd developed an environmentally friendly cadaverine purification technical process applied for lysine sulfate fermentation broth, which obtained both high purity cadaverine and high value-added by-products without solid waste generation. This improved technology is suitable for both partial use to retrofit parts of existing lines and for full-scale use to set up completely new lines.119
Although the current methods of solvent extraction, distillation, rectification, precipitation, and adsorption can improve the purity of cadaverine on the laboratory/small-scale, organic solvents as product extractants will destroy the integrity of cell membranes and are toxic to cells. The extensive use of organic solvents will be a burden for the environment. In addition, an autocatalytic reaction might produce a pentacyclic ring during the separation of cadaverine, which will affect the subsequent polymerization and the quality of nylon 5X significantly. Most importantly, some organic solvents with low selectivity need multiple steps to separate cadaverine, making the separation process complicated and increasing the production cost. Therefore, there is an urgent need to develop a green and efficient technology for cadaverine purification and separation.
7. The application of cadaverine
The global output of plastics and chemical fibers is about 500 million tons per year. However, these plastics and chemical fiber raw materials mainly rely on petroleum, which will lead to significant resource depletion and environmental problems. At present, the use of petroleum-based materials is gradually transitioning to the use of bio-based materials as their green, environmentally friendly, and renewable raw material characteristics, which has important social and economic benefits. It is expected that 80% of the world's petroleum-based materials will be replaced by bio-based materials by 2035. Cadaverine production from bio-based materials is becoming increasingly important for the biochemical engineering industry.Bio-based nylon 5X is a polymer material polymerized by bio-based cadaverine and a variety of dibasic acids (Fig. 9). Different types of nylon have various properties such as high temperature resistance, medium high temperature, medium low temperature and low temperature characteristics, and can be widely used in fibers and engineering plastics. It has been confirmed that bio-nylon 510 has excellent melting temperature and mechanical strength. Meanwhile, bio-nylon 510 is softer and its surface quality is better than the nylon 6 and nylon 66. The elongation at break of bio-nylon 510 was comparable for nylon 6 and nylon 66.9 Bio-nylon 56 also has advantages in mechanical strength, air permeability, moisture absorption, and flame retardancy. Other varieties of bio-nylon 5X are on the way for development.
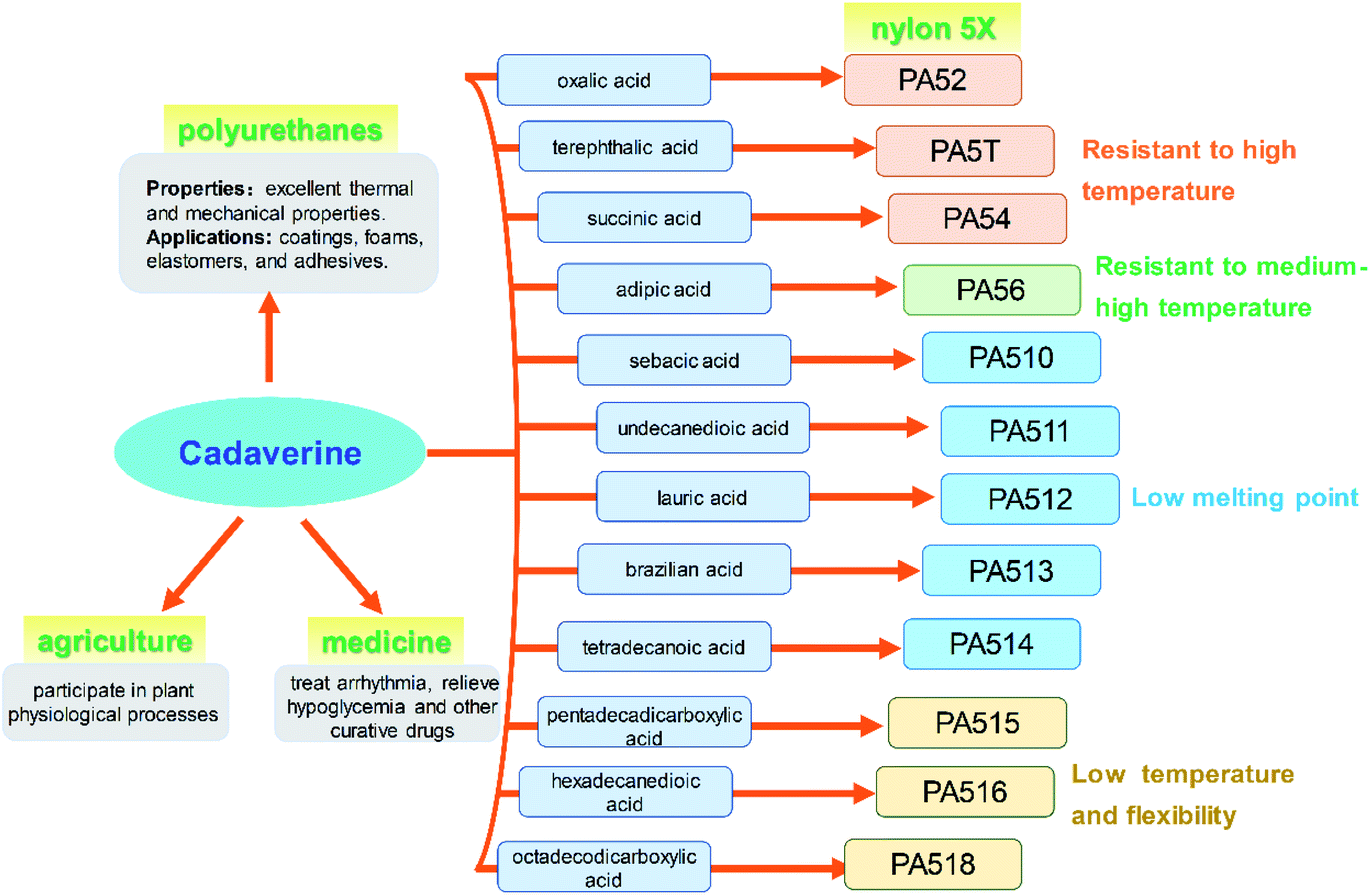 | ||
| Fig. 9 Applications of cadaverine. | ||
Polyurethanes are also the most widely used materials in various fields such as coatings, foams, elastomers, and adhesives, for their excellent thermal and mechanical properties. The polyurethanes are produced by polyisocyanates and polyols. Most of the polyisocyanates are synthesized by diisocyanates, including the aromatic isocyanates and aliphatic isocyanates. Bio-based PDI, a new aliphatic isocyanate, can be synthesized from cadaverine and be used for polyurethane coatings and adhesive (Fig. 9). This new product with high reaction activity and low consumption not only can replace hexamethylene diisocyanate (HDI), but also has advantages in weather resistance.
Cadaverine can participate in plant physiological processes such as cell division and growth, promoting the development of pistil and stamen, regulating the senescence process, improving plant fruit development, and increasing fruit yield (Fig. 9). It has great potential to be used in the agriculture area.
It has been reported that cadaverine can be used to treat arrhythmia, relieve hypoglycemia and for other curative effects, and effectively treat dysentery. This outstanding effect will make cadaverine to achieve even higher value for medicine (Fig. 9).
8. Suggestions for future development of the cadaverine production
8.1. The comparison of chemical and biological synthesis of cadaverine from L-lysine
Both chemical and biological strategies can convert the L-lysine to cadaverine through the decarboxylation process (Table 2). The conversion rate and selectivity of biological process, especially the whole cell conversion, can reach up to 100% with the high productivity of 204 g L−1 h−1. Bio-synthesis method, through fermentation or whole-cell conversion manufacturing technique, exhibits an absolutely dominant advantage in the directional synthesis of cadaverine. The mild reaction conditions of bio-synthesis method at room temperature cause a low energy consumption, resulting in a low production cost. Whereas, it still needs great efforts to have the stable industrial enzymes and recombinant microbes.| Strategy | Conversion rate (%) | Selectivity (%) | Productivity (g L−1 h−1) | Advance technology | Advantage | Shortage | Challenge |
|---|---|---|---|---|---|---|---|
| Bio-synthesis | >99 | 100 | 10–204 | Synthetic biology & fermentation/whole-cell conversion | Low energy consumption; environmentally friendly | Low enzyme activity strongly caused by the poisonous cadaverine | Instability and the deficiency of enzyme source;Food-competition |
| Chemo-catalysis | >99 | 30–60 | 2–5 | Batch/continuous production | Fast separation through simple centrifugation; good reuse of heterogeneous catalysts | Unavoidable corrosion because of the strong acid | Low selectivity of cadaverine |
The selectivity and productivity of chemical synthesis of cadaverine are lower than the biological process. However, the catalyst has a good stability which is not affected by cadaverine, especially in the heterogeneous catalysis process. Product can be readily separated from catalysts just by simple centrifugation, which greatly facilitates the production process. In general, the separated catalysts remain a good reaction activity after simple pre-treatment, which can be reused in a next reaction cycle. Different from the traditional batch production of bio-synthesis method, chemo-catalysis has an obvious advantage of the continuous manufacturing technique, for example, the continuous fixed-bed, fluidized-bed and moving-bed reactor, which fits well with the industrial production in a large scale. Unfortunately, the decarboxylation of L-lysine to cadaverine through chemo-catalysis has to use strong acid to protect L-lysine from severe deamination process which is accompany with an unavoidable corrosion problem. Meanwhile, the low selectivity of cadaverine in the chemo-catalysis is the great challenge for large scale application.
8.2. Green chemical process for cadaverine production
In chemical process, the relatively high reaction activity of L-lysine causes big difficulties in improving the selectivity of cadaverine. L-Lysine has been reported to take part in the deamination process to give piperidine and its derivatives, or deeply hydrogenation to prepare L-lysinol, or ring-closing reaction through dehydration to form 3-amio-2-azepanone. To inhibit the growth of large-sized byproduct which mostly contains nitrogen heterocycle in decarboxylation process of L-lysine, it is greatly meaningful to utilize the confinement effect of the supports, for example hierarchical porous carbon materials, or molecular sieves with abundant channels.On the other hand, cadaverine is not stable, especially with the presence of acidic sites and transition metals, which can easily undergo deamination to form piperidine. The highest selectivity of cadaverine was reported to be just 51% in heterogeneous phase catalysis. Studies of cadaverine synthesis were predominantly carried out in a batch reactor, following with the problem that cadaverine cannot be removed out in time from the reaction system. The continuous reactor, such as fixed-bed, fluided-bed or moving-bed reactor, might be the candidate manufacturing technique in future for cadaverine production. More work is urgently needed to give a high selectivity of cadaverine.
8.3. Bio-process for cadaverine production
The low catalytic efficiency and instability of lysine decarboxylase are key problems for the industrial production of cadaverine. The activity of lysine decarboxylase is highly inhibited by the substrate L-lysine and the product cadaverine. Phan and Krithika120,121 found that lysine decarboxylase was inhibited when the concentration of L-lysine was higher than 5 mM. When the concentration of L-lysine was higher than 1 M, the catalytic efficiency of lysine decarboxylase was reduced significantly.50 The structural stability of lysine decarboxylase was also affected by acid conditions. Lysine decarboxylase mainly existed as decamers and aggregated at pH 5.0. However, when the pH increased to 8.0, 95% of decamers gradually depolymerized into dimers.69 In the decarboxylation process of L-lysine to cadaverine, the pH of the reaction system rose continuously, causing the enzyme to become unstable and unavailable for reuse.69,122 Therefore, the activity and instability of lysine decarboxylase have severely restricted the industrial application of free enzyme or whole-cell conversion of cadaverine.Recently, significant progress has been achieved for the biological synthesis of cadaverine by using metabolically engineered microbial strains, especially with the host of Escherichia coli and Corynebacterium glutamicum. These genetic strains mainly use the food-based glucose, galactose, starch, mannitol as the carbon source, or use hemicellulose-based substrate xylose and cellobiose, which requiring an efficient and complex pretreatment process for the biomass. However, the low yield (<100 g L−1) and productivity (24–72 h) of the cadaverine is obtained by using the food-sugar based substrate and hemicellulose-based substrate. Therefore, the L-lysine is considered as the most potential substrate for efficient cadaverine production at large scale. The concentration of cadaverine can reach up to 200 g L−1. However, the price for L-lysine is still the challenge for the economic production of the cadaverine. Most of the L-lysine originates from corn or glucose through the fermentation process, and competes with the food production, which is ultimately undesirable. Meanwhile, the released large amount of CO2 in this process also poses negative environmental effects. Therefore, there is a strong need for consideration of more sustainable substrates such as methane and methanol as described above. Potentially, cadaverine can be produced by methanotrophic bacteria using methane as feedstock. This approach reduces the use of expensive organic substrates in the process. However, achieving relevant yields for large scale production is a problem. A more fundamental understanding of methane biosynthetic pathways and their regulation is necessary.
The process for cadaverine production is complicated and time-consuming (Fig. 10A and B). The medium composition for the recombinant engineered strain growth and enzyme induction is very diverse, which includes organic and inorganic chemicals. The unreacted medium components including the substrate, by-products of strain, and the released protein, result in the difficulty in the separation and purification of cadaverine. Whole-cell conversion can obtain a higher concentration of cadaverine without the influence of extracellular metabolic byproducts and medium components, which is relatively conducive to the subsequent separation of cadaverine. However, a centrifugation process is required before the whole-cell catalysis, which will complicate and waste a large amount of fermentation water. Although the free enzyme catalytic system avoids the interference of fermentation medium components and cell barrier for transferring substrate and product, the reutilization rate of enzymes is poor. Free enzyme also requires centrifugation and breaking cells, which will cause complex metabolic by-products in the cell, L-lysine, PLP, and buffer, increasing the difficulty of scaling up and purification. Therefore, it is an immediate requirement to develop a green and efficient one-pot whole-cell conversion process (Fig. 10C) and efficient separation process for cadaverine production.
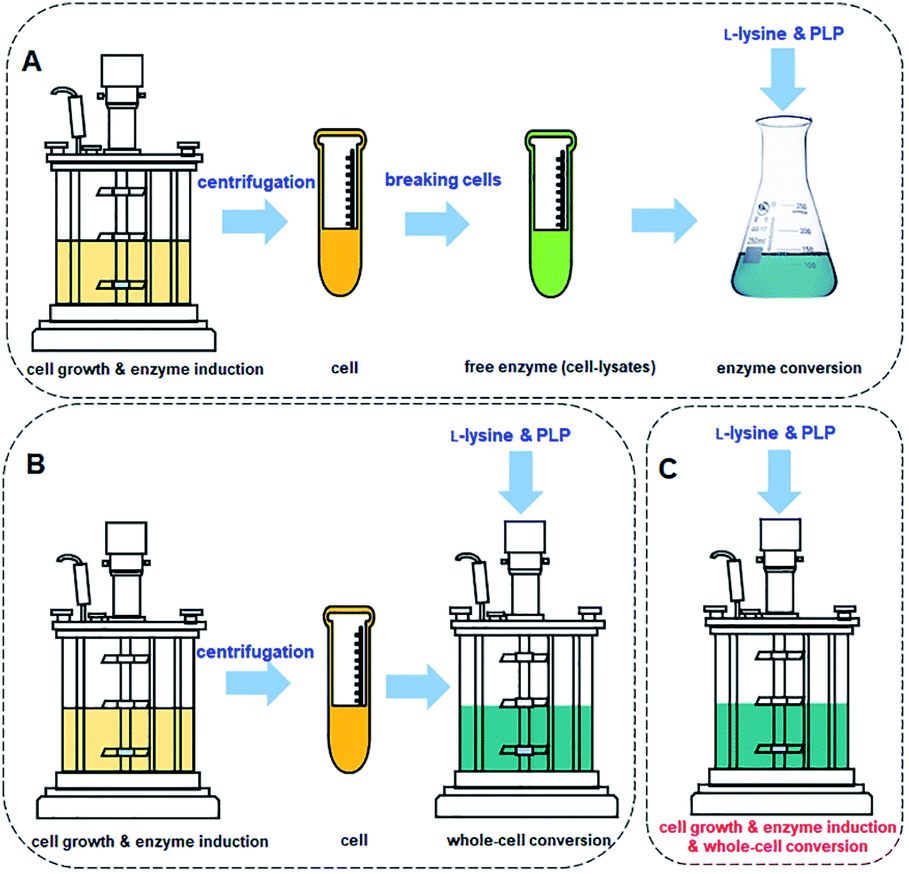 | ||
| Fig. 10 (A) Cadaverine synthesis process from L-lysine: free enzyme conversion, (B) traditional whole-cell conversion, and (C) one-pot whole-cell conversion process. | ||
Cadaverine is an important monomer to develop a series of high value-added products with market prospects. Regarding the green chemical and biological synthesis of cadaverine described in this review, especially the biological process, it has been developed rapidly these years. However, the production of cadaverine and cadaverine-based materials is still on bench and pilot scale. There is an incentive need for further improving the enzyme alkaline and thermo stability of the key lysine decarboxylases and for establishing an efficient and green purification process to recover the high quality cadaverine in the near future. Meanwhile, the market for the new bio-based materials, such as nylon 5X and polyurethanes, which using cadaverine as the core monomer, is significant to guide the development tendency of the cadaverine. Therefore, cadaverine has enormous potential for future development and application by the advocation of circular economy.
Author contributions
The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the Beijing Nova Program of Science and Technology (Z201100006820141), National Natural Science Foundation of China (grant number 22078346), Innovation Academy for Green Manufacture, CAS (IAGM2020C19), Henan Key Research and Development Project (202102210046), Hebei Natural Science Foundation (B2020103010) and the CAS Pioneer Hundred Program. Sincerely appreciate Prof. Suojiang Zhang (IPE, CAS) for his careful academic guidance and great support.References
- S. Negrel and J. M. Brunel, Curr. Med. Chem., 2021, 28, 3406–3448 CrossRef CAS PubMed.
- M. Mindt, T. Walter, P. Kugler and V. F. Wendisch, Biotechnol. J., 2020, 15, 15 Search PubMed.
- T. Eisenberg, M. Abdellatif, S. Schroeder, U. Primessnig, S. Stekovic, T. Pendl, A. Harger, J. Schipke, A. Zimmermann, A. Schmidt, M. M. Tong, C. Ruckenstuhl, C. Dammbrueck, A. S. Gross, V. Herbst, C. Magnes, G. Trausinger, S. Narath, A. Meinitzer, Z. H. Hu, A. Kirsch, K. Eller, D. Carmona-Gutierrez, S. Buttner, F. Pietrocola, O. Knittelfelder, E. Schrepfer, P. Rockenfeller, C. Simonini, A. Rahn, M. Horsch, K. Moreth, J. Beckers, H. Fuchs, V. Gailus-Durner, F. Neff, D. Janik, B. Rathkolb, J. Rozman, M. H. de Angelis, T. Moustafa, G. Haemmerle, M. Mayr, P. Willeit, M. von Frieling-Salewsky, B. Pieske, L. Scorrano, T. Pieber, R. Pechlaner, J. Willeit, S. J. Sigrist, W. A. Linke, C. Muhlfeld, J. Sadoshima, J. Dengjel, S. Kiechl, G. Kroemer, S. Sedej and F. Madeo, Nat. Med., 2016, 22, 1428–1438 CrossRef CAS.
- A. Kielkowska and M. Dziurka, Physiol. Plantarum, 2021, 171, 48–65 CrossRef CAS PubMed.
- B. A. McCormick, M. I. Fernandez, A. M. Siber and A. T. Maurelli, Cell. Microbiol., 1999, 1, 143–155 CrossRef CAS.
- V. Sessini, B. Haseeb, A. Boldizar and G. Lo Re, RSC Adv., 2021, 11, 637–656 RSC.
- K. Jo, H. J. Kim and H. H. Lee, Fibers Polym., 2019, 20, 63–68 CrossRef CAS.
- T. M. Carole, J. Pellegrino and M. D. Paster, Twenty-Fifth Symposium on Biotechnology for Fuels and Chemicals, Totowa, NJ, 2003 Search PubMed.
- S. Kind, S. Neubauer, J. Becker, M. Yamamoto, M. Volkert, G. Abendroth, O. Zelder and C. Wittmann, Metab. Eng., 2014, 25, 113–123 CrossRef CAS PubMed.
- B. W. Hoffer and J. A. Moulijn, Appl. Catal. A: Gen., 2009, 352, 193–201 CrossRef CAS.
- C. Li, L. Chen and T. Lei, Petrochem. Technol., 2010, 39, 524–527 CAS.
- d. B. Claude and F. Pierre, Catal. Rev., 1994, 36, 459–506 CrossRef.
- V. Froidevaux, C. Negrell, S. Caillol, J. P. Pascault and B. Boutevin, Chem. Rev., 2016, 116, 14181–14224 CrossRef CAS PubMed.
- H. Mitsunori, E. Yutaka, O. Yasutomo, I. Toshiaki and A. Seiichi, Chem. Lett., 1986, 15, 893–896 CrossRef.
- G. Laval and B. T. Golding, Synlett, 2003, 34, 542–546 Search PubMed.
- F. De Schouwer, L. Claes, N. Claes, S. Bals, J. Degrève and D. E. De Vos, Green Chem., 2015, 17, 2263–2270 RSC.
- J. Verduyckt, M. Van Hoof, F. De Schouwer, M. Wolberg, M. Kurttepeli, P. Eloy, E. M. Gaigneaux, S. Bals, C. E. A. Kirschhock and D. E. De Vos, ACS Catal., 2016, 6, 7303–7310 CrossRef CAS.
- J. Verduyckt, R. Coeck and D. E. De Vos, ACS Sustain. Chem. Eng., 2017, 5, 3290–3295 CrossRef CAS.
- Board of Trustees of Michigan State University, Invention, WO2005/123669A1, 2005.
- Board of Trustees of Michigan State University, US2010/0145003A1, 2010.
- J. Sebastian, M. Zheng, Y. Jiang, Y. Zhao, H. Wang, Z. Song, X. Li, J. Pang and T. Zhang, Green Chem., 2019, 21, 2462–2468 RSC.
- J. Deischter, N. Wolter and R. Palkovits, ChemSusChem, 2020, 13, 3614–3621 CrossRef CAS PubMed.
- G. L. Gambino, G. M. Lombardo, A. Grassi and G. Marletta, J. Phys. Chem. B, 2004, 108, 2600–2607 CrossRef CAS.
- S. Xie, C. Jia, Z. Wang, S. S. G. Ong, M.-j. Zhu and H. Lin, ACS Sustain. Chem. Eng., 2020, 8, 11805–11817 CrossRef CAS.
- V. F. Wendisch, M. Mindt and F. Perez-Garcia, Appl. Microbiol. Biot., 2018, 102, 3583–3594 CrossRef CAS.
- J. W. Lee, T. Y. Kim, Y. S. Jang, S. Choi and S. Y. Lee, Trends Biotechnol., 2011, 29, 370–378 CrossRef CAS.
- S. Y. Lee, H. U. Kim, T. U. Chae, J. S. Cho, J. W. Kim, J. H. Shin, D. I. Kim, Y. S. Ko, W. D. Jang and Y. S. Jang, Nat. Catal., 2019, 2, 18–33 CrossRef CAS.
- K. R. Choi, W. D. Jang, D. Yang, J. S. Cho, D. Park and S. Y. Lee, Trends Biotechnol., 2019, 37, 817–837 CrossRef CAS.
- E. F. Gale and H. M. R. Epps, Biochem. J., 1942, 36, 600–618 CrossRef CAS PubMed.
- S. Kind, W. K. Jeong, H. Schröder, O. Zelder and C. Wittmann, Appl. Environ. Microbiol., 2010, 76, 5175–5180 CrossRef CAS PubMed.
- M. Li, D. Li, Y. Huang, M. Liu, H. Wang, Q. Tang and F. Lu, J. Ind. Microbiol. Biotechnol., 2014, 41, 701–709 CrossRef CAS PubMed.
- S. Kind, S. Kreye and C. Wittmann, Metab. Eng., 2011, 13, 617–627 CrossRef CAS PubMed.
- R. Matsuura, M. Kishida, R. Konishi, Y. Hirata, N. Adachi, S. Segawa, K. Imao, T. Tanaka and A. Kondo, Biotechnol. Bioeng., 2019, 116, 2640–2651 CrossRef CAS.
- S. Kind, W. K. Jeong, H. Schroder and C. Wittmann, Metab. Eng., 2010, 12, 341–351 CrossRef CAS PubMed.
- H. T. Kim, K.-A. Baritugo, Y. H. Oh, S. M. Hyun, T. U. Khang, K. H. Kang, S. H. Jung, B. K. Song, K. Park, I.-K. Kim, M. O. Lee, Y. Kam, Y. T. Hwang, S. J. Park and J. C. Joo, ACS Sustain. Chem. Eng., 2018, 6, 5296–5305 CrossRef CAS.
- S. Kobayashi, H. Kawaguchi, T. Shirai, K. Ninomiya and Y. Tsuge, ACS Synth. Biol., 2020, 9, 814–826 CrossRef CAS PubMed.
- K. Murai, D. Sasaki, S. Kobayashi, A. Yamaguchi, H. Uchikura, T. Shirai, K. Sasaki, A. Kondo and Y. Tsuge, ACS Synth. Biol., 2020, 9, 1615–1622 CrossRef CAS PubMed.
- D. H. Kwak, H. G. Lim, J. Yang, S. W. Seo and G. Y. Jung, Biotechnol. Biofuels, 2017, 10, 20 CrossRef PubMed.
- E. Sgobba, A. K. Stumpf, M. Vortmann, N. Jagmann, M. Krehenbrink, M. E. Dirks-Hofmeister, B. Moerschbacher, B. Philipp and V. F. Wendisch, Bioresource Technol, 2018, 260, 302–310 CrossRef CAS PubMed.
- N. Buschke, J. Becker, R. Schäfer, P. Kiefer, R. Biedendieck and C. Wittmann, Biotechnol. J., 2013, 8, 557–570 CrossRef CAS PubMed.
- N. Buschke, H. Schröder and C. Wittmann, Biotechnol. J., 2011, 6, 306–317 CrossRef CAS.
- N. Ikeda, M. Miyamoto, N. Adachi, M. Nakano, T. Tanaka and A. Kondo, AMB Express, 2013, 3, 67 CrossRef PubMed.
- I. Naerdal, J. Pfeifenschneider, T. Brautaset and V. F. Wendisch, Microb. Biotechnol., 2015, 8, 342–350 CrossRef CAS PubMed.
- M. Irla, T. M. B. Heggeset, I. Nærdal, L. Paul, T. Haugen, S. B. Le, T. Brautaset and V. F. Wendisch, Front. Microbiol., 2016, 7, 1481 CrossRef PubMed.
- L. Leßmeier, J. Pfeifenschneider, M. Carnicer, S. Heux, J.-C. Portais and V. F. Wendisch, Appl. Microbiol. Biotechnol., 2015, 99, 10163–10176 CrossRef PubMed.
- T. T. Nguyen, O. K. Lee, S. Naizabekov and E. Y. Lee, Green Chem., 2020, 22, 7803–7811 RSC.
- S. Hakvåg, I. Nærdal, T. Heggeset, K. Kristiansen, I. M. Aasen and T. Brautaset, Front. Microbiol., 2020, 11, 680 CrossRef PubMed.
- W. C. Ma, W. J. Cao, H. Zhang, K. Q. Chen, Y. Li and P. K. Ouyang, Biotechnol. Lett., 2015, 37, 799–806 CrossRef CAS PubMed.
- C. Y. Huang, W. W. Ting, Y. C. Chen, P. Y. Wu, C. D. Dong, S. F. Huang, H. Y. Lin, S. F. Li, I. S. Ng and J. S. Chang, Biochem. Eng. J., 2020, 156, 107514 CrossRef CAS.
- Y. M. Moon, S. Y. Yang, T. R. Choi, H. R. Jung, H. S. Song, Y. h. Han, H. Y. Park, S. K. Bhatia, R. Gurav, K. Park, J. S. Kim and Y. H. Yang, Enzyme Microb. Technol., 2019, 127, 58–64 CrossRef CAS PubMed.
- F. Kou, J. Zhao, J. Liu, C. Sun, Y. Guo, Z. Tan, F. Cheng, Z. Li, P. Zheng and J. Sun, Biotechnol. Lett., 2018, 40, 719–727 CrossRef CAS PubMed.
- E. Y. Hong, S. G. Lee, B. J. Park, J. M. Lee, H. Yun and B. G. Kim, Biotechnol. J., 2017, 12, 1700278 CrossRef PubMed.
- C. Wang, K. Zhang, C. Zhongjun, H. Cai, W. Honggui and P. Ouyang, Biotechnol. Bioproc. Eng., 2015, 20, 439–446 CrossRef CAS.
- C. Xue, K. M. Hsu, W. W. Ting, S. F. Huang, H. Y. Lin, S. F. Li, J. S. Chang and I. S. Ng, Biochem. Eng. J., 2020, 161, 107659 CrossRef CAS.
- J. Rytter, S. Helmark, J. Chen, M. Lezyk, C. Solem and P. Jensen, Appl. Microbiol. Biotechnol., 2019, 98, 2617–2623 CrossRef.
- B. G. Hall and P. W. Betts, Genetics, 1987, 115, 431–439 CrossRef CAS.
- I. Nærdal, R. Netzer, M. Irla, A. Krog, T. M. B. Heggeset, V. F. Wendisch and T. Brautaset, J. Biotechnol., 2017, 244, 25–33 CrossRef PubMed.
- T. Brautaset, Ø. M. Jakobsen, K. D. Josefsen, M. C. Flickinger and T. E. Ellingsen, Appl. Microbiol. Biotechnol., 2007, 74, 22–34 CrossRef CAS PubMed.
- P. J. Strong, S. Xie and W. P. Clarke, Environ. Sci. Technol., 2015, 49, 4001–4018 CrossRef CAS.
- A. Pol, K. Heijmans, H. R. Harhangi, D. Tedesco, M. S. Jetten and H. J. Op den Camp, Nature, 2007, 450, 874–878 CrossRef CAS PubMed.
- A. Boetius, K. Ravenschlag, C. J. Schubert, D. Rickert, F. Widdel, A. Gieseke, R. Amann, B. B. Jorgensen, U. Witte and O. Pfannkuche, Nature, 2000, 407, 623–626 CrossRef CAS.
- Y. A. Trotsenko and J. C. Murrell, Adv. Appl. Microbiol., 2008, 63, 183–229 CAS.
- S. L. Hoffmann, L. Jungmann, S. Schiefelbein, L. Peyriga, E. Cahoreau, J.-C. Portais, J. Becker and C. Wittmann, Metab. Eng., 2018, 47, 475–487 CrossRef CAS PubMed.
- T. U. Chae, J. H. Ahn, Y. S. Ko, J. W. Kim, J. A. Lee, E. H. Lee and S. Y. Lee, Metab. Eng., 2019, 58, 2–16 CrossRef.
- D. L. Sabo, E. A. Boeker, B. Byers, H. Waron and E. H. Fischer, Biochemistry, 1974, 13, 662–670 CrossRef CAS PubMed.
- J. F. Rocha, A. F. Pina, S. F. Sousa and N. Cerqueira, Catal. Sci. Technol., 2019, 9, 4864–4876 RSC.
- E. M. Krammer and M. Prévost, J. Membr. Biol., 2019, 252, 465–481 CrossRef CAS PubMed.
- J. Cheng, P. Chen, A. Song, D. Wang and Q. Wang, J. Ind. Microbiol. Biotechnol., 2018, 45, 719–734 CrossRef CAS PubMed.
- U. Kanjee, I. Gutsche, E. Alexopoulos, B. Zhao, M. El Bakkouri, G. Thibault, K. Liu, S. Ramachandran, J. Snider, E. F. Pai and W. A. Houry, EMBO J., 2011, 30, 931–944 CrossRef CAS PubMed.
- F. Kou, J. Zhao, J. Liu, J. Shen, Q. Ye, P. Zheng, Z. Li, J. Sun and Y. Ma, J. Mol. Catal. B-Enzym., 2016, 133, S88–S94 CrossRef.
- J. Deng, H. Gao, Z. Gao, H. Zhao, Y. Yang, Q. Wu, B. Wu and C. Jiang, PloS One, 2017, 12, e0185060 CrossRef.
- L. Han, J. Yuan, X. Ao, S. Lin, X. Han and H. Ye, Front. Microbiol., 2018, 9, 3082 CrossRef PubMed.
- J. H. Kim, H. J. Kim, Y. H. Kim, J. M. Jeon, H. S. Song, J. Kim, S. Y. No, J. H. Shin, K. Y. Choi, K. M. Park and Y. H. Yang, J. Microbiol. Biotechnol., 2016, 26, 1586–1592 CrossRef CAS.
- Y. Takatsuka, Y. Yamaguchi, M. Ono and Y. Kamio, J. Bacteriol., 2000, 182, 6732–6741 CrossRef CAS.
- A. Sugawara, D. Matsui, N. Takahashi, M. Yamada, Y. Asano and K. Isobe, J. Biosci. Bioeng., 2014, 118, 496–501 CrossRef CAS PubMed.
- Y. K. Leong, C.-H. Chen, S.-F. Huang, H.-Y. Lin, S.-F. Li, I. S. Ng and J.-S. Chang, Biochem. Eng. J., 2020, 157, 107547 CrossRef CAS.
- Ajinomoto Co., Inc., Inc., Invention, US2005/0003497A1, 2004.
- Z. G. Qian, X. X. Xia and S. Y. Lee, Biotechnol. Bioeng., 2011, 108, 93–103 CrossRef CAS.
- S. Kind and C. Wittmann, Appl. Microbiol. Biotechnol., 2011, 91, 1287–1296 CrossRef CAS.
- Y. Xue, Y. Zhao, X. Ji, J. Yao, P. K. Busk, L. Lange, Y. Huang and S. Zhang, Green Chem., 2020, 22, 8656–8668 RSC.
- Ajinomoto Co., Inc. Tokyo 104-8315 (JP), Invention, EP3118312B1, 2015.
- M. V. Omelchenko, M. Y. Galperin, Y. I. Wolf and E. V. Koonin, Biology Direct, 2010, 5, 31 CrossRef.
- A. J. Michael, Biochem. J., 2017, 474, 2277–2299 CrossRef CAS PubMed.
- Y. Yamamoto, Y. Miwa, K. Miyoshi, J. Furuyama and H. Ohmori, Genes Genet. Syst., 1997, 72, 167–172 CrossRef CAS PubMed.
- M. Burrell, C. Hanfrey, L. Kinch, K. Elliott and A. Michael, Mol. Microbiol., 2012, 86, 485–499 CrossRef CAS.
- S. Bunsupa, K. Katayama, E. Ikeura, A. Oikawa, K. Toyooka, K. Saito and M. Yamazaki, Plant Cell, 2012, 24, 1202–1216 CrossRef CAS PubMed.
- D. Carriel, P. Simon Garcia, F. Castelli, P. Lamourette, F. Fenaille, C. Brochier-Armanet, S. Elsen and I. Gutsche, Genome Biol. Evol., 2018, 10, 3058–3075 CAS.
- M. Nei, Mol. Biol. Evol., 1987, 24, 189–204 Search PubMed.
- S. Kumar, G. Stecher, M. Li, C. Knyaz and K. Tamura, Mol. Biol. Evol., 2018, 35, 1547–1549 CrossRef CAS.
- E. Zuckerkandl and L. Pauling, in Evolving Genes and Proteins, ed. V. Bryson and H. J. Vogel, Academic Press, 1965, pp. 97–166 Search PubMed.
- W. Ma, K. Chen, Y. Li, N. Hao, X. Wang and P. Ouyang, Engineering, 2017, 3, 308–317 CrossRef.
- J. Andréll, M. G. Hicks, T. Palmer, E. P. Carpenter, S. Iwata and M. J. Maher, Biochemistry, 2009, 48, 3915–3927 CrossRef.
- J. Snider, I. Gutsche, M. Lin, S. Baby, B. Cox, G. Butland, J. Greenblatt, A. Emili and W. A. Houry, J. Biol. Chem., 2006, 281, 1532–1546 CrossRef CAS.
- M. El Bakkouri, I. Gutsche, U. Kanjee, B. Zhao, M. Yu, G. Goret, G. Schoehn, W. P. Burmeister and W. A. Houry, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 22499–22504 CrossRef CAS PubMed.
- H. Malet, K. Liu, M. El Bakkouri, S. W. Chan, G. Effantin, M. Bacia, W. A. Houry and I. Gutsche, eLife, 2014, 3, e03653 CrossRef PubMed.
- E. Kandiah, D. Carriel, J. Perard, H. Malet, M. Bacia, K. Liu, S. W. S. Chan, W. A. Houry, S. Ollagnier de Choudens, S. Elsen and I. Gutsche, Sci. Rep., 2016, 6, 24601 CrossRef CAS PubMed.
- E. Kandiah, D. Carriel, P. S. Garcia, J. Felix, M. Banzhaf, G. Kritikos, M. Bacia-Verloop, C. Brochier-Armanet, S. Elsen and I. Gutsche, Structure, 2019, 27, 1842–1854.e1844 CrossRef CAS PubMed.
- S. K. Bhatia, Y. H. Kim, H. J. Kim, H. M. Seo, J. H. Kim, H. S. Song, G. Sathiyanarayanan, S. H. Park, K. Park and Y. H. Yang, Bioprocess Biosyst. Eng., 2015, 38, 2315–2322 CrossRef CAS.
- G. Wei, W. Ma, A. Zhang, X. Cao, J. Shen, Y. Li, K. Chen and P. Ouyang, Appl. Microbiol. Biotechnol., 2018, 102, 7837–7847 CrossRef CAS PubMed.
- H.-M. Seo, J.-H. Kim, J.-M. Jeon, H.-S. Song, S. K. Bhatia, G. Sathiyanarayanan, K. Park, K. J. Kim, S. H. Lee, H. J. Kim and Y.-H. Yang, Process Biochem., 2016, 51, 1413–1419 CrossRef CAS.
- N. Zhou, A. Zhang, G. Wei, S. Yang and P. Ouyang, Front. Bioeng. Biotech., 2020, 8, 103 CrossRef.
- S. H. Park, F. Soetyono and H. K. Kim, J. Microbiol. Biotechnol., 2017, 27, 289–296 CrossRef CAS PubMed.
- R. Kloss, M. H. Limberg, U. Mackfeld, D. Hahn, A. Grunberger, V. D. Jager, U. Krauss, M. Oldiges and M. Pohl, Sci. Rep., 2018, 8, 5856 CrossRef.
- H. Kim, H. Y. Yoo, N. Park, H. Kim, J. Lee, Y. Baek, T. Lee, J. M. Oh, J. Cho and C. Park, Polymers, 2019, 11, 1372 CrossRef CAS.
- J. Rui, S. You, Y. Zheng, C. Wang, Y. Gao, W. Zhang, W. Qi, R. Su and Z. He, Bioresour. Technol., 2020, 302, 122844 CrossRef CAS PubMed.
- H. T. Kim, K. A. Baritugo, Y. H. Oh, K. H. Kang, Y. J. Jung, S. Jang, B. K. Song, I. K. Kim, M. O. Lee, Y. T. Hwang, K. Park, S. J. Park and J. C. Joo, Polymers, 2019, 11, 1184 CrossRef CAS.
- J. Wang, X. Lu, H. Ying, W. Ma, S. Xu, X. Wang, K. Chen and P. Ouyang, Front. Microbiol., 2018, 9, 1312 CrossRef.
- J. Wang, J. Mao, W. Tian, G. Wei, S. Xu, W. Ma, K. Chen, M. Jiang and P. Ouyang, Green Chem., 2018, 20, 2880–2887 RSC.
- G. Wei, A. Zhang, X. Lu, F. He, H. Li, S. Xu, G. Li, K. Chen and P. Ouyang, J. CO2 Util., 2020, 37, 278–284 CrossRef CAS.
- A. Krzyżaniak, B. Schuur and A. B. de Haan, J. Chem. Technol. Biot., 2013, 88, 1937–1945 CrossRef.
- Y. G. Hong, H. J. Kim, J. M. Jeon, Y. M. Moon, J. W. Hong, J. C. Joo, B. K. Song, K. M. Park, S. H. Lee and Y. H. Yang, J. Ind. Eng. Chem., 2018, 64, 167–172 CrossRef CAS.
- Y. Liu, Y. Zheng, H. Wu, W. Zhang, T. Ren, S. You, W. Qi, R. Su and Z. He, J. Chem. Technol. Biot., 2020, 95, 1542–1549 CrossRef CAS.
- Shanghai Cathay Biotechnology R&D Center Ltd., Invention, CN105612257A, 2013.
- Shanghai Cathay Biotechnology R&D Center Ltd, Invention, CN 108276292 A, 2017.
- Nanjing Tech University, Invention, CN106861236B, 2017.
- Nanjing Tech University, Invention, CN106367326B, 2017.
- Shanghai Cathay Biotechnology R&D Center Ltd, Utility model, CN 204400883 U 2014.
- Shanghai Cathay Biotechnology Co., Utility model, CN 212610365 U, 2020.
- Ningxia Eppen Biotech Co., Ltd, Invention, CN 109402189 A, 2018.
- A. P. H. Phan, T. T. Ngo and H. M. Lenhoff, Anal. Biochem., 1982, 120, 193–197 CrossRef CAS PubMed.
- G. Krithika, J. Arunachalam, H. Priyanka and K. Indulekha, 11 International Conference in Thermal Energy Storage, 2010 Search PubMed.
- U. Kanjee, I. Gutsche, S. Ramachandran and W. A. Houry, Biochemistry, 2011, 50, 9388–9398 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2021 |